Potential and Limitations of Cross-Protective Vaccine against Malaria by Blood-Stage Naturally Attenuated Parasite



Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Parasites and Infection
2.3. Anti-Malaria Drug Treatment
2.4. Histology
2.5. Flow Cytometry
2.6. Cell Depletion
2.7. Prime–Boost Live Vaccination and Cell Transfer Experiments
2.8. Statistical Analysis
3. Results
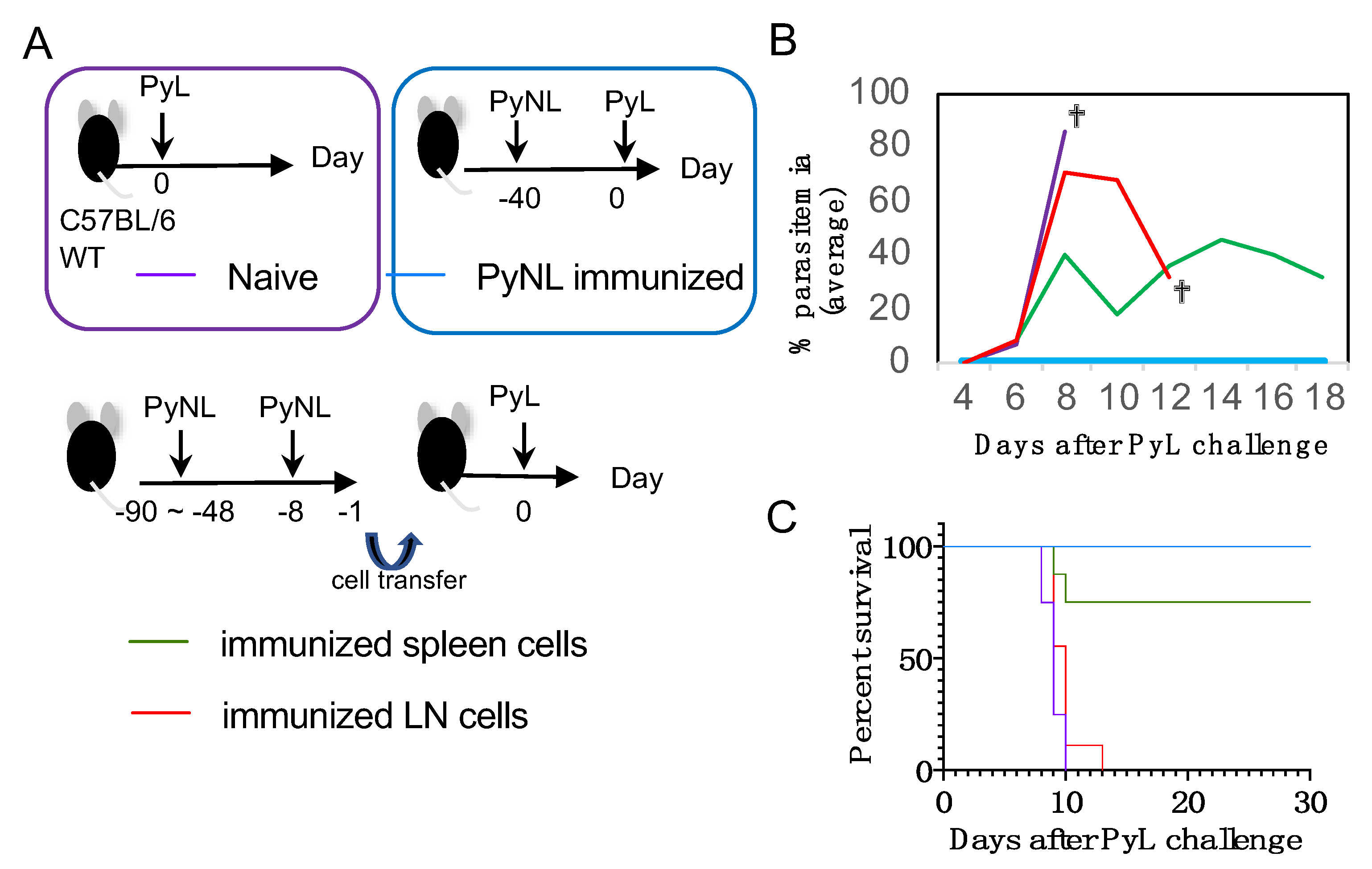
3.1. Blood-Stage PyNL Infection as a Whole Organism Vaccine
3.2. Cross-Strain Protection by Blood-Stage PyNL Infection in Immunodeficient Mice
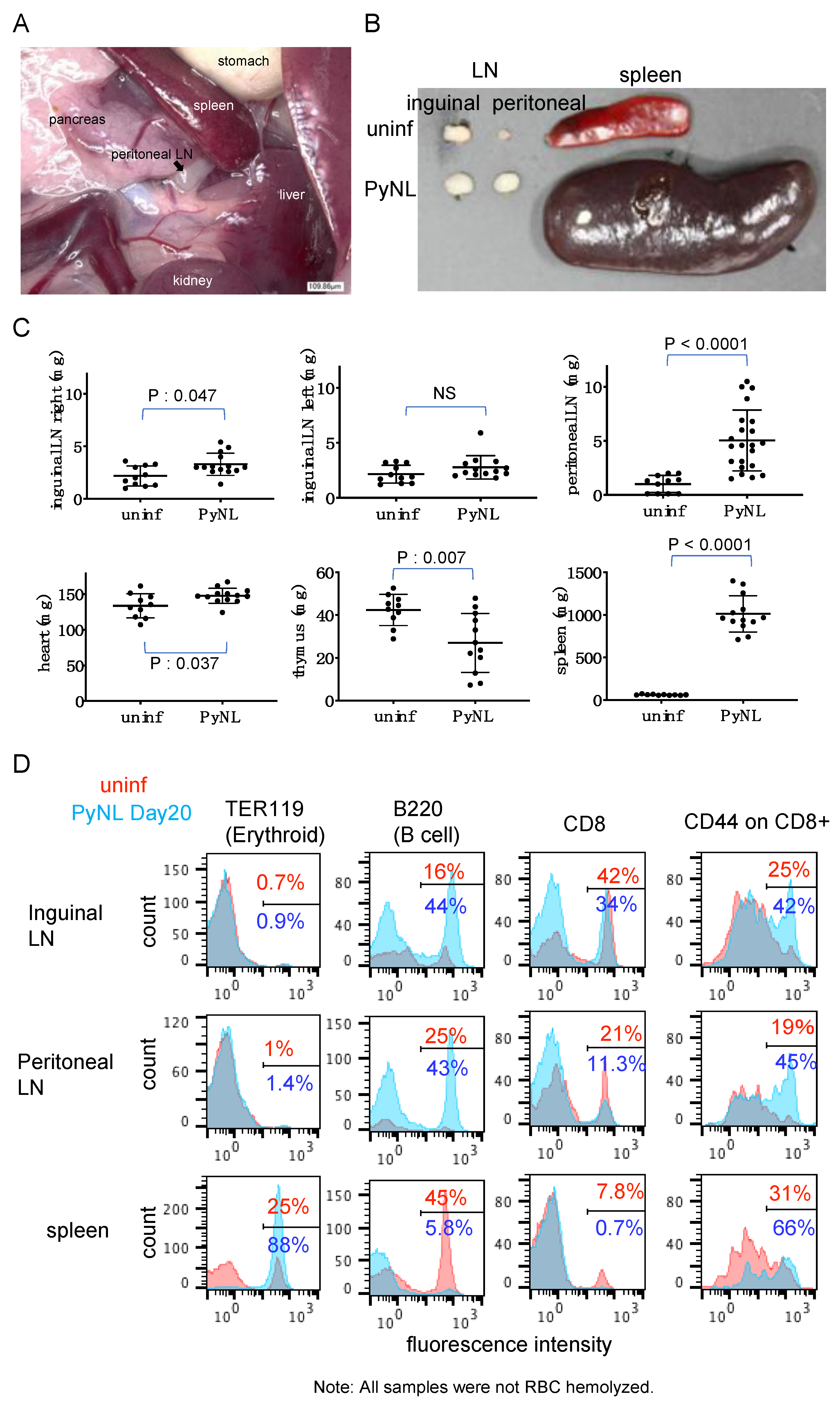
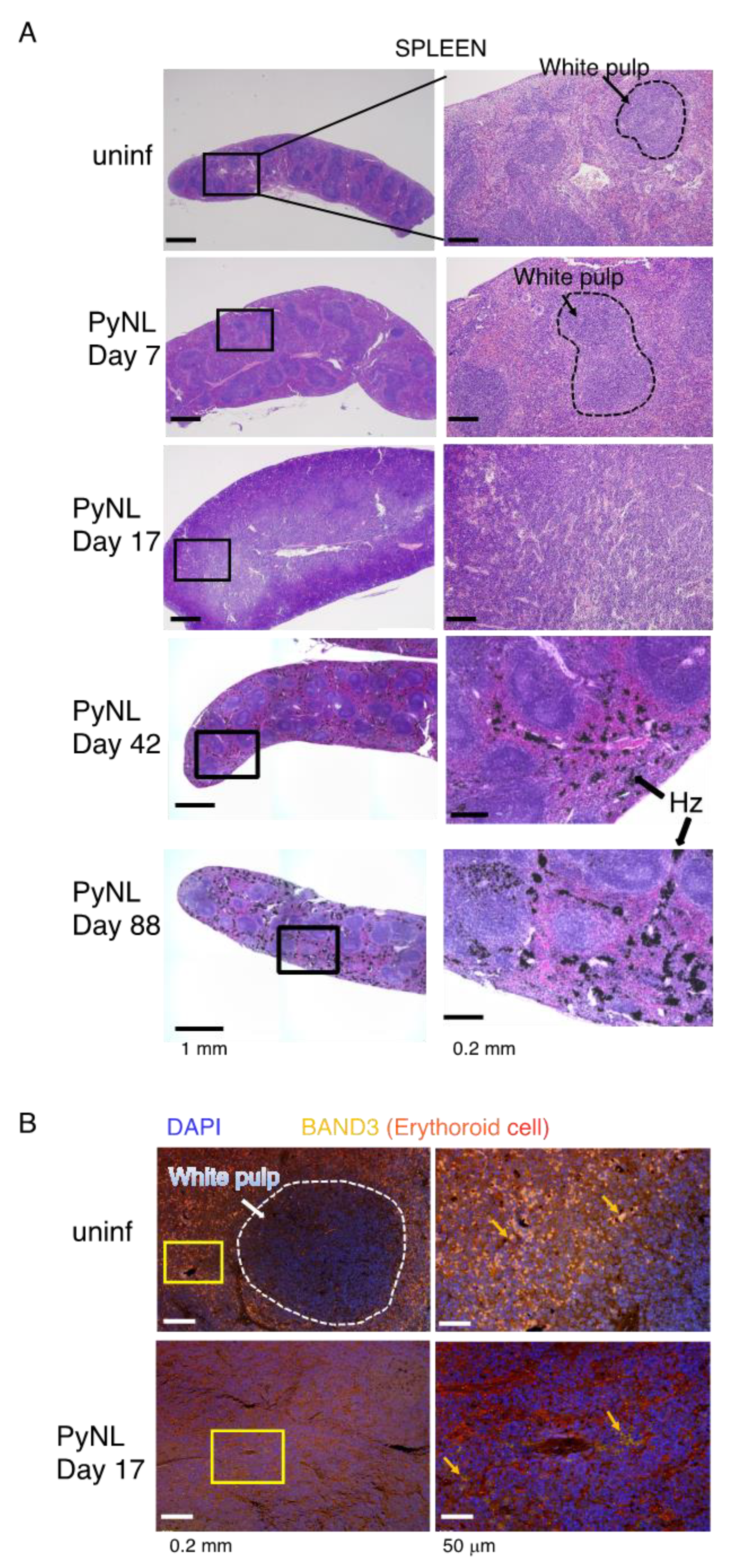
3.3. PyNL Infection Induced Splenomegaly, non-Reversible Changes in the Spleen and Expansion of the Peritoneal LN
3.4. PyNL Induced Activation of Immune Cells in Peritoneal LN but Did not Confer Protection
3.5. Limited Cross-Species Protection by PyNL Infection
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Garcia, L.S. Malaria. Clin. Lab. Med. 2010, 30, 93–129. [Google Scholar] [CrossRef]
- Grau-Bove, X.; Tomlinson, S.; O’Reilly, A.O.; Harding, N.J.; Miles, A.; Kwiatkowski, D.; Donnelly, M.J.; Weetman, D.; The Anopheles gambiae 1000 Genomes Consortium. Evolution of the insecticide target rdl in african anopheles is driven by interspecific and interkaryotypic introgression. Mol. Biol. Evol. 2020. [Google Scholar] [CrossRef]
- Ashley, E.A.; Pyae Phyo, A.; Woodrow, C.J. Malaria. Lancet 2018, 391, 1608–1621. [Google Scholar] [CrossRef]
- Imwong, M.; Hien, T.T.; Thuy-Nhien, N.T.; Dondorp, A.M.; White, N.J. Spread of a single multidrug resistant malaria parasite lineage (pfpailin) to vietnam. Lancet Infect. Dis. 2017, 17, 1022–1023. [Google Scholar] [CrossRef]
- Verma, R.; Khanna, P.; Chawla, S. Malaria vaccine can prevent millions of deaths in the world. Hum. Vaccines Immunother. 2013, 9, 1268–1271. [Google Scholar] [CrossRef]
- Hollingdale, M.R.; Sedegah, M. Development of whole sporozoite malaria vaccines. Expert Rev. Vaccines 2017, 16, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Itsara, L.S.; Zhou, Y.; Do, J.; Grieser, A.M.; Vaughan, A.M.; Ghosh, A.K. The development of whole sporozoite vaccines for plasmodium falciparum malaria. Front. Immunol. 2018, 9, 2748. [Google Scholar] [CrossRef]
- Frimpong, A.; Kusi, K.A.; Ofori, M.F.; Ndifon, W. Novel strategies for malaria vaccine design. Front. Immunol. 2018, 9, 2769. [Google Scholar] [CrossRef] [PubMed]
- Draper, S.J.; Sack, B.K.; King, C.R.; Nielsen, C.M.; Rayner, J.C.; Higgins, M.K.; Long, C.A.; Seder, R.A. Malaria vaccines: Recent advances and new horizons. Cell Host Microbe 2018, 24, 43–56. [Google Scholar] [CrossRef]
- Cockburn, I.A.; Seder, R.A. Malaria prevention: From immunological concepts to effective vaccines and protective antibodies. Nat. Immunol. 2018, 19, 1199–1211. [Google Scholar] [CrossRef]
- Wilby, K.J.; Lau, T.T.; Gilchrist, S.E.; Ensom, M.H. Mosquirix (rts,s): A novel vaccine for the prevention of plasmodium falciparum malaria. Ann. Pharmacother. 2012, 46, 384–393. [Google Scholar] [CrossRef] [PubMed]
- Laurens, M.B. Rts,s/as01 vaccine (mosquirix): An overview. Hum. Vaccines Immunother. 2020, 16, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Morrison, C. Landmark green light for mosquirix malaria vaccine. Nat. Biotechnol. 2015, 33, 1015–1016. [Google Scholar] [CrossRef] [PubMed]
- Rts, S.C.T.P. Efficacy and safety of rts,s/as01 malaria vaccine with or without a booster dose in infants and children in africa: Final results of a phase 3, individually randomised, controlled trial. Lancet 2015, 386, 31–45. [Google Scholar]
- White, M.T.; Verity, R.; Griffin, J.T.; Asante, K.P.; Owusu-Agyei, S.; Greenwood, B.; Drakeley, C.; Gesase, S.; Lusingu, J.; Ansong, D.; et al. Immunogenicity of the rts,s/as01 malaria vaccine and implications for duration of vaccine efficacy: Secondary analysis of data from a phase 3 randomised controlled trial. Lancet Infect Dis. 2015, 15, 1450–1458. [Google Scholar] [CrossRef]
- WHO. Malaria: Q&a on the Malaria Vaccine Implementation Programme (Mvip). Available online: https://www.who.int/malaria/media/malaria-vaccine-implementation-qa/en/ (accessed on 6 June 2020).
- Palacpac, N.M.; Ntege, E.; Yeka, A.; Balikagala, B.; Suzuki, N.; Shirai, H.; Yagi, M.; Ito, K.; Fukushima, W.; Hirota, Y.; et al. Phase 1b randomized trial and follow-up study in uganda of the blood-stage malaria vaccine candidate bk-se36. PLoS ONE 2013, 8, e64073. [Google Scholar] [CrossRef] [PubMed]
- Clyde, D.F. Immunization of man against falciparum and vivax malaria by use of attenuated sporozoites. Am. J. Trop. Med. Hyg. 1975, 24, 397–401. [Google Scholar] [CrossRef]
- Luke, T.C.; Hoffman, S.L. Rationale and plans for developing a non-replicating, metabolically active, radiation-attenuated plasmodium falciparum sporozoite vaccine. J. Exp. Biol. 2003, 206, 3803–3808. [Google Scholar] [CrossRef]
- Lyke, K.E.; Ishizuka, A.S.; Berry, A.A.; Chakravarty, S.; DeZure, A.; Enama, M.E.; James, E.R.; Billingsley, P.F.; Gunasekera, A.; Manoj, A.; et al. Attenuated pfspz vaccine induces strain-transcending t cells and durable protection against heterologous controlled human malaria infection. Proc. Natl. Acad. Sci. USA 2017, 114, 2711–2716. [Google Scholar] [CrossRef]
- Epstein, J.E.; Paolino, K.M.; Richie, T.L.; Sedegah, M.; Singer, A.; Ruben, A.J.; Chakravarty, S.; Stafford, A.; Ruck, R.C.; Eappen, A.G.; et al. Protection against plasmodium falciparum malaria by pfspz vaccine. JCI Insight 2017, 2, e89154. [Google Scholar] [CrossRef]
- van Schaijk, B.C.; Ploemen, I.H.; Annoura, T.; Vos, M.W.; Foquet, L.; van Gemert, G.J.; Chevalley-Maurel, S.; van de Vegte-Bolmer, M.; Sajid, M.; Franetich, J.F.; et al. A genetically attenuated malaria vaccine candidate based on p. Falciparum b9/slarp gene-deficient sporozoites. eLife 2014, 3, e03582. [Google Scholar] [CrossRef] [PubMed]
- Roestenberg, M.; Walk, J.; van der Boor, S.C.; Langenberg, M.C.C.; Hoogerwerf, M.A.; Janse, J.J.; Manurung, M.; Yap, X.Z.; Garcia, A.F.; Koopman, J.P.R.; et al. A double-blind, placebo-controlled phase 1/2a trial of the genetically attenuated malaria vaccine pfspz-ga1. Sci. Transl. Med. 2020, 12, eaaz5629. [Google Scholar] [PubMed]
- Butler, N.S.; Schmidt, N.W.; Vaughan, A.M.; Aly, A.S.; Kappe, S.H.; Harty, J.T. Superior antimalarial immunity after vaccination with late liver stage-arresting genetically attenuated parasites. Cell Host Microbe 2011, 9, 451–462. [Google Scholar] [CrossRef] [PubMed]
- Imai, T.; Ishida, H.; Suzue, K.; Hirai, M.; Taniguchi, T.; Okada, H.; Suzuki, T.; Shimokawa, C.; Hisaeda, H. Cd8 t cell activation by murine erythroblasts infected with malaria parasites. Sci. Rep. 2013, 3, 1572. [Google Scholar] [CrossRef] [PubMed]
- Imai, T.; Ishida, H.; Suzue, K.; Taniguchi, T.; Okada, H.; Shimokawa, C.; Hisaeda, H. Cytotoxic activities of cd8 t cells collaborate with macrophages to protect against blood-stage murine malaria. eLife 2015, 4, e04232. [Google Scholar] [CrossRef]
- Imai, T.; Iwawaki, T.; Akai, R.; Suzue, K.; Hirai, M.; Taniguchi, T.; Okada, H.; Hisaeda, H. Evaluating experimental cerebral malaria using oxidative stress indicator okd48 mice. Int. J. Parasitol. 2014, 44, 681–685. [Google Scholar] [CrossRef]
- Imai, T.; Shen, J.; Chou, B.; Duan, X.; Tu, L.; Tetsutani, K.; Moriya, C.; Ishida, H.; Hamano, S.; Shimokawa, C.; et al. Involvement of cd8+ t cells in protective immunity against murine blood-stage infection with plasmodium yoelii 17xl strain. Eur. J. Immunol. 2010, 40, 1053–1061. [Google Scholar]
- Imai, T.; Suzue, K.; Ngo-Thanh, H.; Ono, S.; Orita, W.; Suzuki, H.; Shimokawa, C.; Olia, A.; Obi, S.; Taniguchi, T.; et al. Fluctuations of spleen cytokine and blood lactate, importance of cellular immunity in host defense against blood stage malaria plasmodium yoelii. Front. Immunol. 2019, 10, 2207. [Google Scholar] [CrossRef]
- Chotivanich, K.; Udomsangpetch, R.; McGready, R.; Proux, S.; Newton, P.; Pukrittayakamee, S.; Looareesuwan, S.; White, N.J. Central role of the spleen in malaria parasite clearance. J. Infect. Dis. 2002, 185, 1538–1541. [Google Scholar] [CrossRef]
- Del Portillo, H.A.; Ferrer, M.; Brugat, T.; Martin-Jaular, L.; Langhorne, J.; Lacerda, M.V. The role of the spleen in malaria. Cell Microbiol. 2012, 14, 343–355. [Google Scholar] [CrossRef]
- Fehling, H.J.; Swat, W.; Laplace, C.; Kuhn, R.; Rajewsky, K.; Muller, U.; von Boehmer, H. Mhc class i expression in mice lacking the proteasome subunit lmp-7. Science 1994, 265, 1234–1237. [Google Scholar] [CrossRef] [PubMed]
- Hanayama, R.; Tanaka, M.; Miyasaka, K.; Aozasa, K.; Koike, M.; Uchiyama, Y.; Nagata, S. Autoimmune disease and impaired uptake of apoptotic cells in mfg-e8-deficient mice. Science 2004, 304, 1147–1150. [Google Scholar] [CrossRef] [PubMed]
- Nakae, S.; Komiyama, Y.; Nambu, A.; Sudo, K.; Iwase, M.; Homma, I.; Sekikawa, K.; Asano, M.; Iwakura, Y. Antigen-specific t cell sensitization is impaired in il-17-deficient mice, causing suppression of allergic cellular and humoral responses. Immunity 2002, 17, 375–387. [Google Scholar] [CrossRef]
- Clemmer, L.; Martins, Y.C.; Zanini, G.M.; Frangos, J.A.; Carvalho, L.J. Artemether and artesunate show the highest efficacies in rescuing mice with late-stage cerebral malaria and rapidly decrease leukocyte accumulation in the brain. Antimicrob. Agents Chemother. 2011, 55, 1383–1390. [Google Scholar] [CrossRef]
- Wautier, J.L.; Wautier, M.P. Molecular basis of erythrocyte adhesion to endothelial cells in diseases. Clin. Hemorheol. Microcirc. 2013, 53, 11–21. [Google Scholar] [CrossRef]
- Sprent, J. Turnover of memory-phenotype cd8+ t cells. Microbes Infect. 2003, 5, 227–231. [Google Scholar] [CrossRef]
- Carlton, J.M.; Adams, J.H.; Silva, J.C.; Bidwell, S.L.; Lorenzi, H.; Caler, E.; Crabtree, J.; Angiuoli, S.V.; Merino, E.F.; Amedeo, P.; et al. Comparative genomics of the neglected human malaria parasite plasmodium vivax. Nature 2008, 455, 757–763. [Google Scholar] [CrossRef]
- Neafsey, D.E.; Galinsky, K.; Jiang, R.H.; Young, L.; Sykes, S.M.; Saif, S.; Gujja, S.; Goldberg, J.M.; Young, S.; Zeng, Q.; et al. The malaria parasite plasmodium vivax exhibits greater genetic diversity than plasmodium falciparum. Nat. Genet. 2012, 44, 1046–1050. [Google Scholar] [CrossRef]
- Guimaraes, L.O.; Bajay, M.M.; Wunderlich, G.; Bueno, M.G.; Rohe, F.; Catao-Dias, J.L.; Neves, A.; Malafronte, R.S.; Curado, I.; Kirchgatter, K. The genetic diversity of plasmodium malariae and plasmodium brasilianum from human, simian and mosquito hosts in brazil. Acta Trop. 2012, 124, 27–32. [Google Scholar] [CrossRef]
- Tewari, R.; Rathore, D.; Crisanti, A. Motility and infectivity of plasmodium berghei sporozoites expressing avian plasmodium gallinaceum circumsporozoite protein. Cell Microbiol. 2005, 7, 699–707. [Google Scholar] [CrossRef]
- Tewari, R.; Spaccapelo, R.; Bistoni, F.; Holder, A.A.; Crisanti, A. Function of region i and ii adhesive motifs of plasmodium falciparum circumsporozoite protein in sporozoite motility and infectivity. J. Biol. Chem. 2002, 277, 47613–47618. [Google Scholar] [CrossRef] [PubMed]
- Triller, G.; Scally, S.W.; Costa, G.; Pissarev, M.; Kreschel, C.; Bosch, A.; Marois, E.; Sack, B.K.; Murugan, R.; Salman, A.M.; et al. Natural parasite exposure induces protective human anti-malarial antibodies. Immunity 2017, 47, 1197–1209 e1110. [Google Scholar] [CrossRef] [PubMed]
- Marin-Mogollon, C.; van Pul, F.J.A.; Miyazaki, S.; Imai, T.; Ramesar, J.; Salman, A.M.; Winkel, B.M.F.; Othman, A.S.; Kroeze, H.; Chevalley-Maurel, S.; et al. Chimeric plasmodium falciparum parasites expressing plasmodium vivax circumsporozoite protein fail to produce salivary gland sporozoites. Malar. J. 2018, 17, 288. [Google Scholar] [CrossRef] [PubMed]
- Mogollon, C.M.; van Pul, F.J.; Imai, T.; Ramesar, J.; Chevalley-Maurel, S.; de Roo, G.M.; Veld, S.A.; Kroeze, H.; Franke-Fayard, B.M.; Janse, C.J.; et al. Rapid generation of marker-free p. Falciparum fluorescent reporter lines using modified crispr/cas9 constructs and selection protocol. PLoS ONE 2016, 11, e0168362. [Google Scholar] [CrossRef]
- Marin-Mogollon, C.; Salman, A.M.; Koolen, K.M.J.; Bolscher, J.M.; van Pul, F.J.A.; Miyazaki, S.; Imai, T.; Othman, A.S.; Ramesar, J.; van Gemert, G.J.; et al. A p. Falciparum nf54 reporter line expressing mcherry-luciferase in gametocytes, sporozoites, and liver-stages. Front. Cell Infect. Microbiol. 2019, 9, 96. [Google Scholar] [CrossRef] [PubMed]
- Butcher, G.A. The role of the spleen and immunization against malaria. Trends Parasitol. 2005, 21, 356–357. [Google Scholar] [CrossRef]
- Karrer, U.; Althage, A.; Odermatt, B.; Hengartner, H.; Zinkernagel, R.M. Immunodeficiency of alymphoplasia mice (aly/aly) in vivo: Structural defect of secondary lymphoid organs and functional b cell defect. Eur. J. Immunol. 2000, 30, 2799–2807. [Google Scholar] [CrossRef]
- Obeid, M.; Franetich, J.F.; Lorthiois, A.; Gego, A.; Gruner, A.C.; Tefit, M.; Boucheix, C.; Snounou, G.; Mazier, D. Skin-draining lymph node priming is sufficient to induce sterile immunity against pre-erythrocytic malaria. EMBO Mol. Med. 2013, 5, 250–263. [Google Scholar] [CrossRef]
- Olivier, M.; Van Den Ham, K.; Shio, M.T.; Kassa, F.A.; Fougeray, S. Malarial pigment hemozoin and the innate inflammatory response. Front. Immunol. 2014, 5, 25. [Google Scholar] [CrossRef]
- Frita, R.; Carapau, D.; Mota, M.M.; Hanscheid, T. In vivo hemozoin kinetics after clearance of plasmodium berghei infection in mice. Malar. Res. Treat 2012, 2012, 373086. [Google Scholar]
- Lin, J.W.; Spaccapelo, R.; Schwarzer, E.; Sajid, M.; Annoura, T.; Deroost, K.; Ravelli, R.B.; Aime, E.; Capuccini, B.; Mommaas-Kienhuis, A.M.; et al. Replication of plasmodium in reticulocytes can occur without hemozoin formation, resulting in chloroquine resistance. J. Exp. Med. 2015, 212, 893–903. [Google Scholar] [CrossRef] [PubMed]
- Duffy, S.; Avery, V.M. Routine in vitro culture of plasmodium falciparum: Experimental consequences? Trends Parasitol. 2018, 34, 564–575. [Google Scholar] [CrossRef] [PubMed]
- Stanisic, D.I.; Fink, J.; Mayer, J.; Coghill, S.; Gore, L.; Liu, X.Q.; El-Deeb, I.; Rodriguez, I.B.; Powell, J.; Willemsen, N.M.; et al. Vaccination with chemically attenuated plasmodium falciparum asexual blood-stage parasites induces parasite-specific cellular immune responses in malaria-naive volunteers: A pilot study. BMC Med. 2018, 16, 184. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mouse | N (7–60) | Cure rate of PyNL Infection | Result of PyL Challenge to PyNL Immunized |
|---|---|---|---|
| C57BL/6 (WT) | 60 | 97% | Complete protection |
| CD4 depletion | 20 | 50% | Complete protection |
| CD8 depletion | 56 | 50% | Complete protection |
| Macrophage depletion | 10 | All died. | Not tested. |
| IL-17A KO | 9 | 100% | Complete protection |
| LMP7 (immuno proteasome subunit) KO | 16 | 100% | Complete protection |
| MFGE-8 KO | 7 | 100% | Complete protection |
| Lpr (Fas mutant) | 20 | 85% | Complete protection |
| Perforin KO | 12 | 50% | Complete protection |
| Gld (Fas L mutant) | 25 | 48% | Complete protection |
| IFN-gamma Receptor KO | 7 | All died. | Not tested. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Imai, T.; Suzue, K.; Ngo-Thanh, H.; Shimokawa, C.; Hisaeda, H. Potential and Limitations of Cross-Protective Vaccine against Malaria by Blood-Stage Naturally Attenuated Parasite. Vaccines 2020, 8, 375. https://doi.org/10.3390/vaccines8030375
Imai T, Suzue K, Ngo-Thanh H, Shimokawa C, Hisaeda H. Potential and Limitations of Cross-Protective Vaccine against Malaria by Blood-Stage Naturally Attenuated Parasite. Vaccines. 2020; 8(3):375. https://doi.org/10.3390/vaccines8030375
Chicago/Turabian StyleImai, Takashi, Kazutomo Suzue, Ha Ngo-Thanh, Chikako Shimokawa, and Hajime Hisaeda. 2020. "Potential and Limitations of Cross-Protective Vaccine against Malaria by Blood-Stage Naturally Attenuated Parasite" Vaccines 8, no. 3: 375. https://doi.org/10.3390/vaccines8030375
APA StyleImai, T., Suzue, K., Ngo-Thanh, H., Shimokawa, C., & Hisaeda, H. (2020). Potential and Limitations of Cross-Protective Vaccine against Malaria by Blood-Stage Naturally Attenuated Parasite. Vaccines, 8(3), 375. https://doi.org/10.3390/vaccines8030375