Tumor Membrane Vesicle Vaccine Augments the Efficacy of Anti-PD1 Antibody in Immune Checkpoint Inhibitor-Resistant Squamous Cell Carcinoma Models of Head and Neck Cancer

 ,
,  ,
,  , and
, and 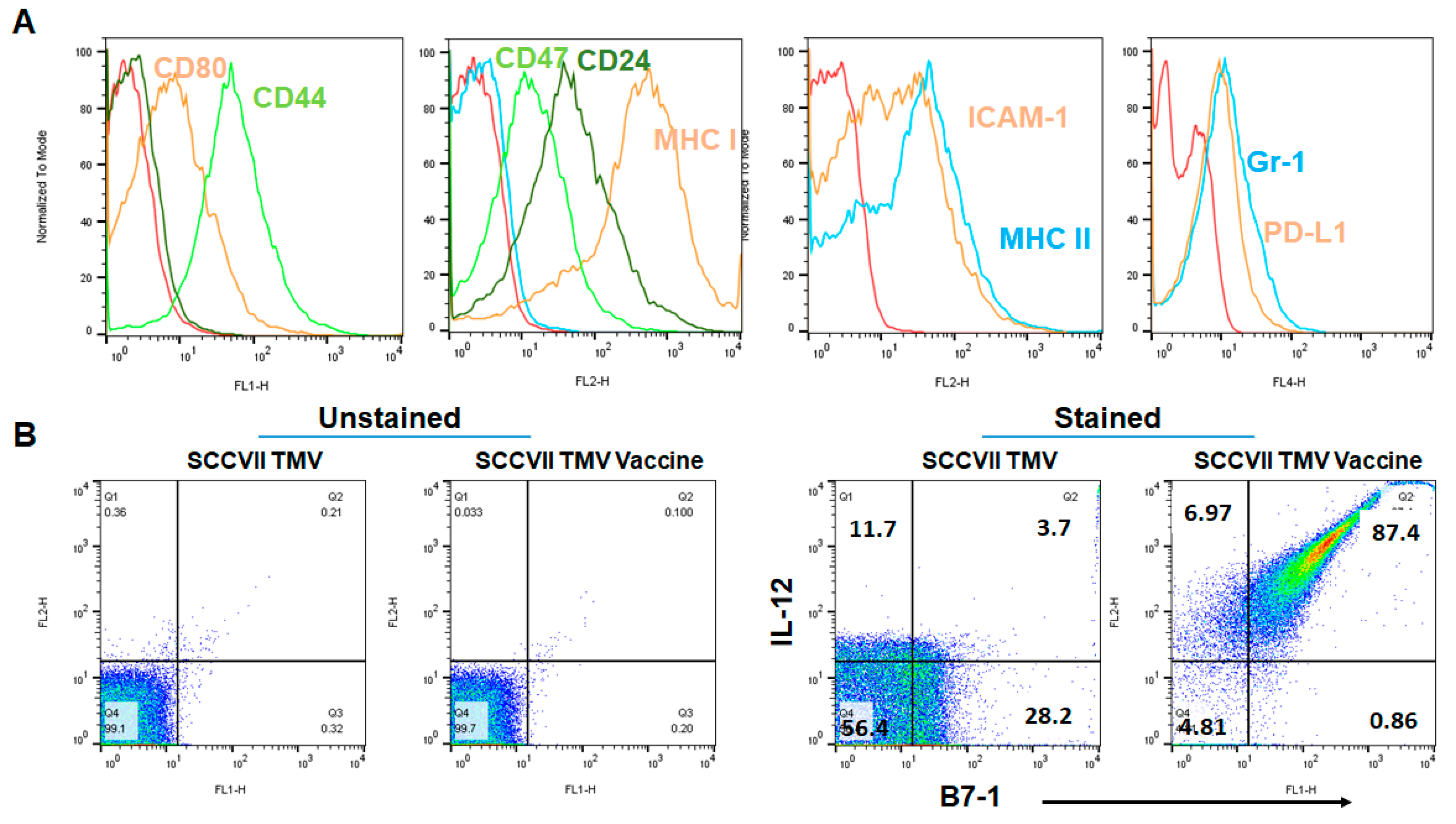{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Antibodies
2.2. Mice
2.3. Cell Lines
2.4. In Vitro IFN-γ Treatment of Murine HNSCC Cells
2.5. TMV Vaccine Preparation
2.6. TMV Vaccine Immunotherapy
2.7. Cellular Phenotyping of Immune Infiltrates
2.8. Flow Cytometry
2.9. Statistical Analysis
3. Results
3.1. GPI-B7-1 and GPI-IL-12 Are Incorporated into TMV by Protein Transfer
3.2. Efficacy of Anti-PD1 Antibody Therapy in SCC VII Tumor Model Depends on the Timing of the Administration
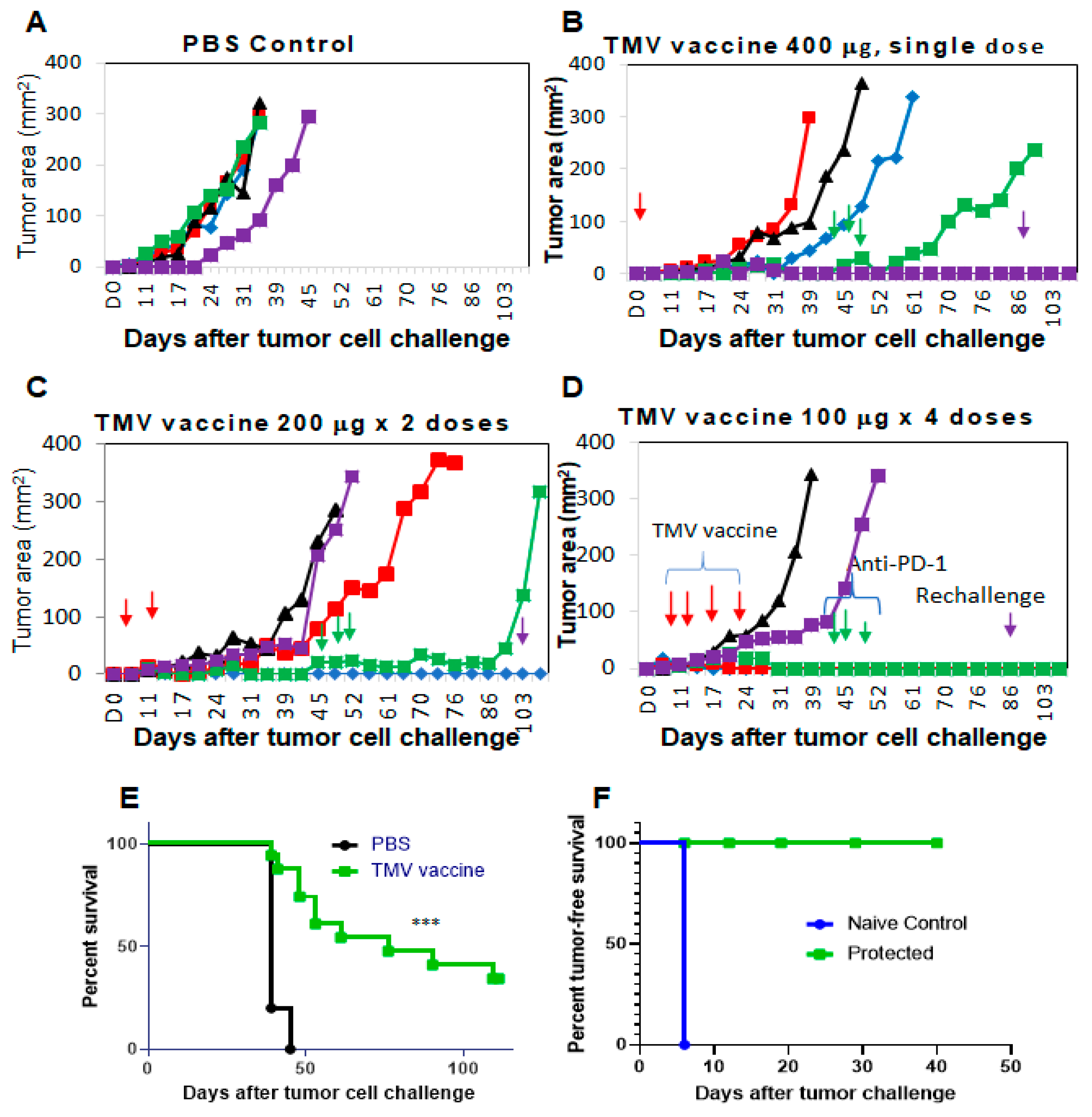
3.3. TMV Vaccine Inhibits Syngeneic SCC VII Squamous Cell Carcinoma Tumor Growth
3.4. Multiple Doses of TMV Vaccine Are More Effective than a Single Dose of Bulk TMV Vaccine in Inhibiting SCC VII Tumor Growth
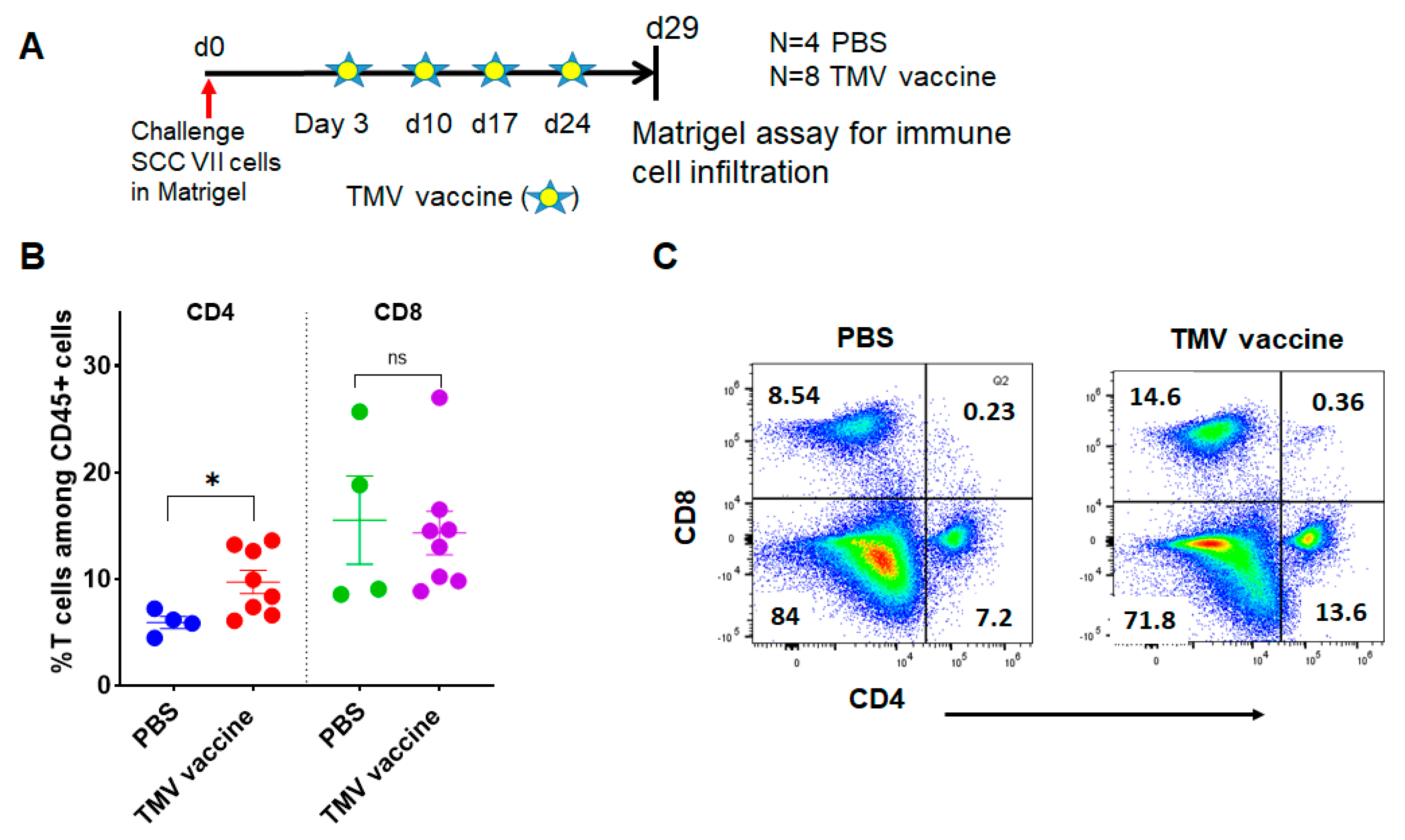
3.5. TMV Vaccine Induces T Cell Infiltration into SCC VII Squamous Cell Carcinoma Tumors
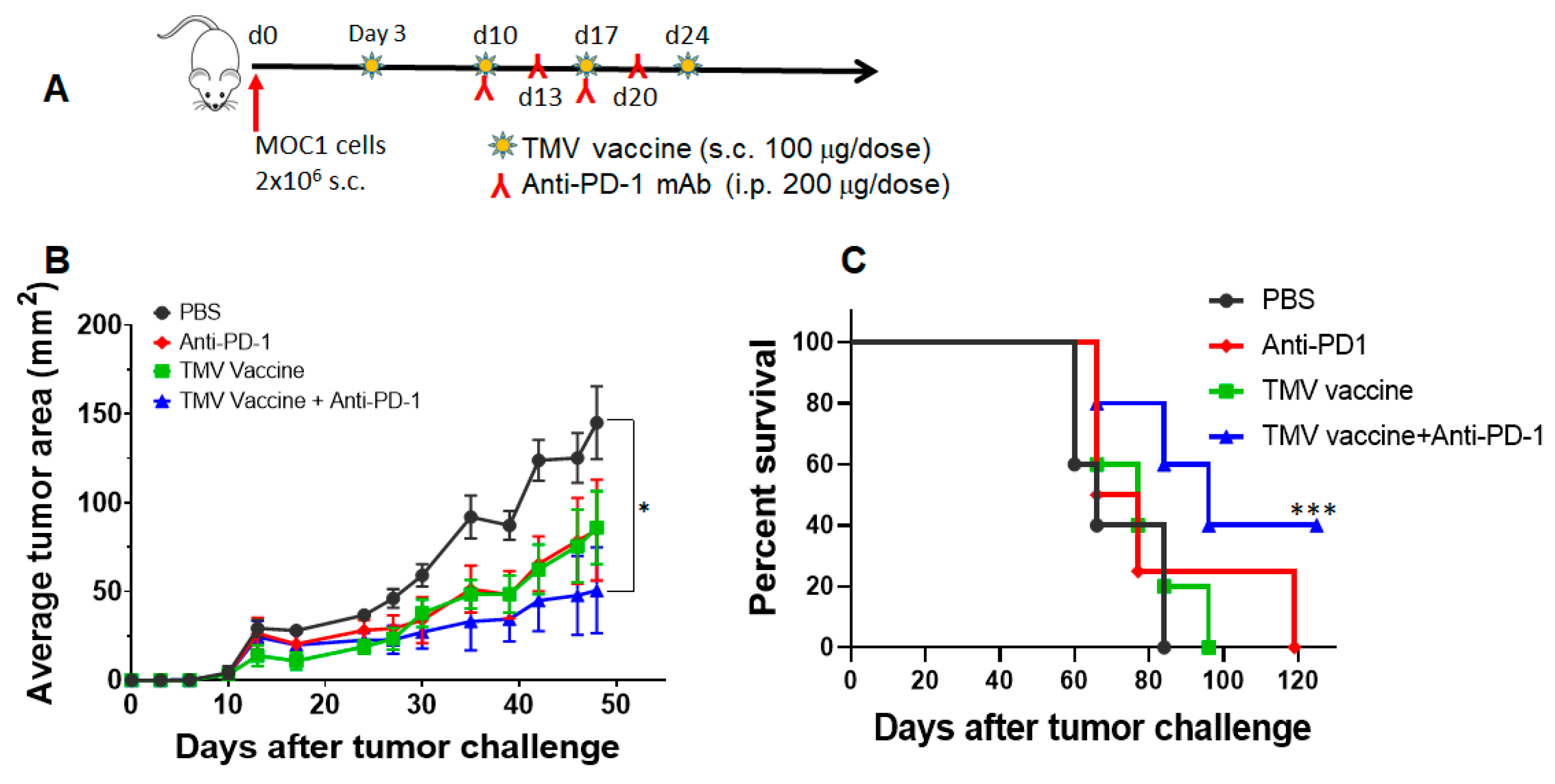
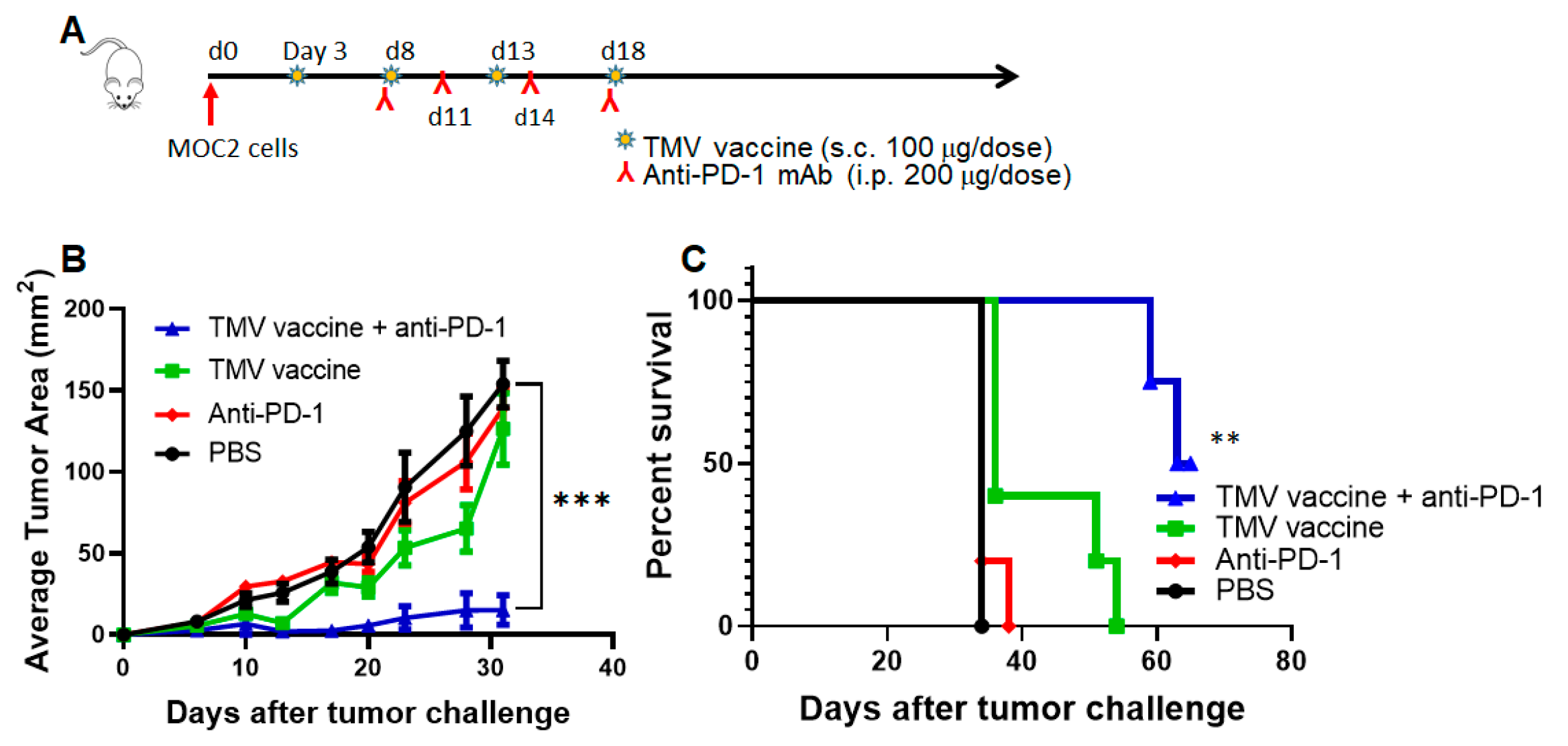
3.6. TMV Vaccine Enhances Immune Checkpoint Inhibitor Efficacy against MOC Tumors
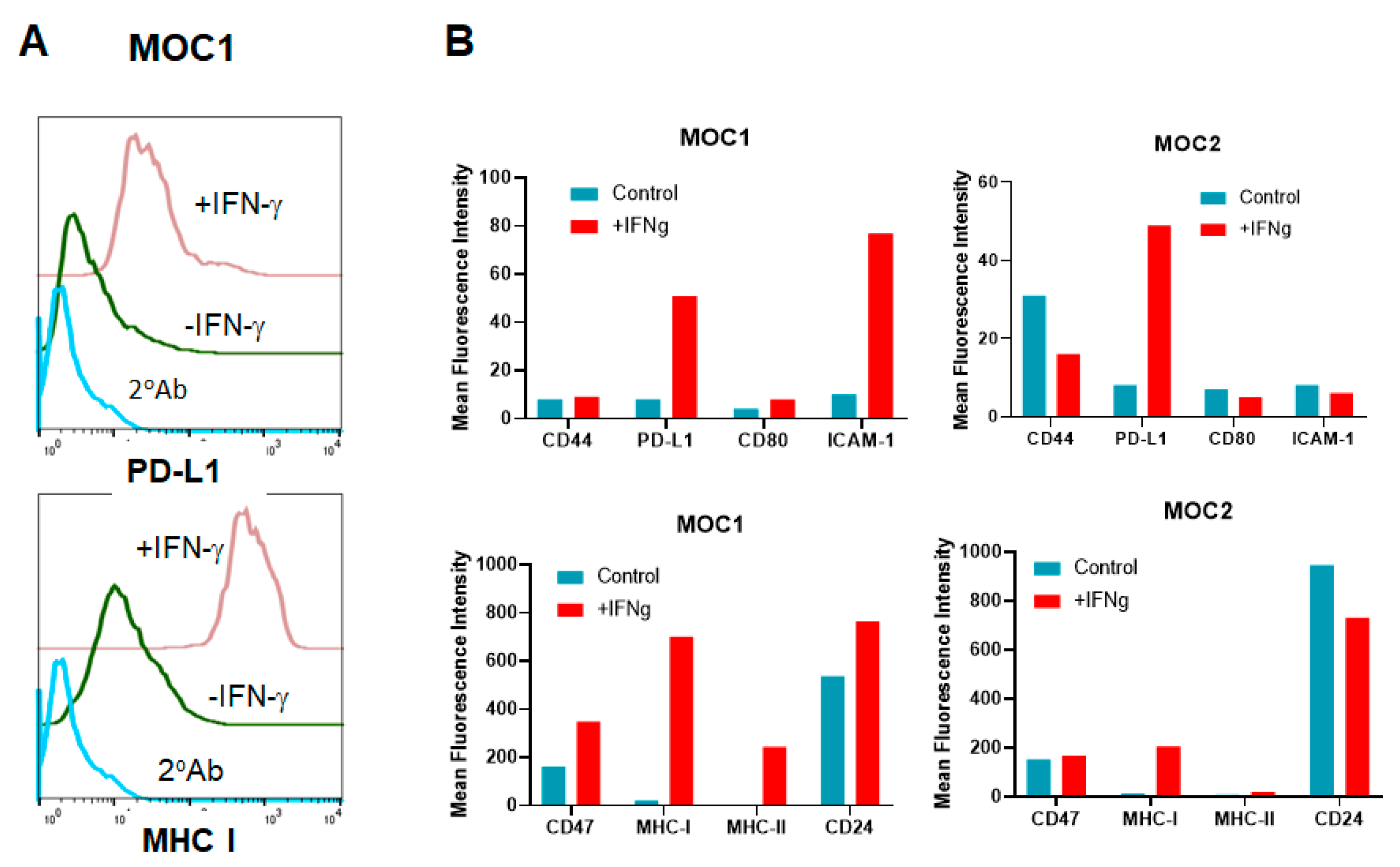
3.7. HNSCC Tumor Cells Respond to IFN-γ Treatment In Vitro
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Saba, N.F.; Blumenschein, G., Jr.; Guigay, J.; Licitra, L.; Fayette, J.; Harrington, K.J.; Kiyota, N.; Gillison, M.L.; Ferris, R.L.; Jayaprakash, V.; et al. Nivolumab versus investigator’s choice in patients with recurrent or metastatic squamous cell carcinoma of the head and neck: Efficacy and safety in CheckMate 141 by age. Oral Oncol. 2019, 96, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Moskovitz, J.; Moy, J.; Ferris, R.L. Immunotherapy for Head and Neck Squamous Cell Carcinoma. Curr. Oncol. Rep. 2018, 20, 22. [Google Scholar] [CrossRef]
- Cohen, E.E.W.; Soulieres, D.; Le Tourneau, C.; Dinis, J.; Licitra, L.; Ahn, M.J.; Soria, A.; Machiels, J.P.; Mach, N.; Mehra, R.; et al. Pembrolizumab versus methotrexate, docetaxel, or cetuximab for recurrent or metastatic head-and-neck squamous cell carcinoma (KEYNOTE-040): A randomised, open-label, phase 3 study. Lancet (London, England) 2019, 393, 156–167. [Google Scholar] [CrossRef]
- Harrington, K.J.; Ferris, R.L.; Blumenschein, G., Jr.; Colevas, A.D.; Fayette, J.; Licitra, L.; Kasper, S.; Even, C.; Vokes, E.E.; Worden, F.; et al. Nivolumab versus standard, single-agent therapy of investigator’s choice in recurrent or metastatic squamous cell carcinoma of the head and neck (CheckMate 141): Health-related quality-of-life results from a randomised, phase 3 trial. Lancet Oncol. 2017, 18, 1104–1115. [Google Scholar] [CrossRef]
- Bauml, J.; Seiwert, T.Y.; Pfister, D.G.; Worden, F.; Liu, S.V.; Gilbert, J.; Saba, N.F.; Weiss, J.; Wirth, L.; Sukari, A.; et al. Pembrolizumab for Platinum- and Cetuximab-Refractory Head and Neck Cancer: Results From a Single-Arm, Phase II Study. J. Clin. Oncol. 2017, 35, 1542–1549. [Google Scholar] [CrossRef]
- Ferris, R.L.; Blumenschein, G., Jr.; Fayette, J.; Guigay, J.; Colevas, A.D.; Licitra, L.; Harrington, K.; Kasper, S.; Vokes, E.E.; Even, C.; et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N. Engl. J. Med. 2016, 375, 1856–1867. [Google Scholar] [CrossRef] [PubMed]
- Hanna, G.J.; Liu, H.; Jones, R.E.; Bacay, A.F.; Lizotte, P.H.; Ivanova, E.V.; Bittinger, M.A.; Cavanaugh, M.E.; Rode, A.J.; Schoenfeld, J.D.; et al. Defining an inflamed tumor immunophenotype in recurrent, metastatic squamous cell carcinoma of the head and neck. Oral Oncol. 2017, 67, 61–69. [Google Scholar] [CrossRef]
- Schuler, P.J.; Harasymczuk, M.; Visus, C.; Deleo, A.; Trivedi, S.; Lei, Y.; Argiris, A.; Gooding, W.; Butterfield, L.H.; Whiteside, T.L.; et al. Phase I dendritic cell p53 peptide vaccine for head and neck cancer. Clin. Cancer. Res. 2014, 20, 2433–2444. [Google Scholar] [CrossRef]
- Schutt, C.A.; Mirandola, L.; Figueroa, J.A.; Nguyen, D.D.; Cordero, J.; Bumm, K.; Judson, B.L.; Chiriva-Internati, M. The cancer-testis antigen, sperm protein 17, a new biomarker and immunological target in head and neck squamous cell carcinoma. Oncotarget 2017, 8, 100280–100287. [Google Scholar] [CrossRef]
- Zolkind, P.; Dunn, G.P.; Lin, T.; Griffith, M.; Griffith, O.L.; Uppaluri, R. Neoantigens in immunotherapy and personalized vaccines: Implications for head and neck squamous cell carcinoma. Oral Oncol. 2017, 71, 169–176. [Google Scholar] [CrossRef]
- Srivatsan, S.; Patel, J.M.; Bozeman, E.N.; Imasuen, I.E.; He, S.; Daniels, D.; Selvaraj, P. Allogeneic tumor cell vaccines: The promise and limitations in clinical trials. Hum. Vaccin. Immunother. 2014, 10, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Mirani, N.; Baisre, A.; Fernandes, H. Molecular heterogeneity of head and neck squamous cell carcinoma defined by next-generation sequencing. Am. J. Pathol. 2014, 184, 1323–1330. [Google Scholar] [CrossRef] [PubMed]
- Peyser, N.D.; Grandis, J.R. Cancer genomics: Spot the difference. Nature 2017, 541, 162–163. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas, N. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef]
- Loyo, M.; Li, R.J.; Bettegowda, C.; Pickering, C.R.; Frederick, M.J.; Myers, J.N.; Agrawal, N. Lessons learned from next-generation sequencing in head and neck cancer. Head Neck 2013, 35, 454–463. [Google Scholar] [CrossRef]
- Agrawal, N.; Frederick, M.J.; Pickering, C.R.; Bettegowda, C.; Chang, K.; Li, R.J.; Fakhry, C.; Xie, T.X.; Zhang, J.; Wang, J.; et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science 2011, 333, 1154–1157. [Google Scholar] [CrossRef]
- Stransky, N.; Egloff, A.M.; Tward, A.D.; Kostic, A.D.; Cibulskis, K.; Sivachenko, A.; Kryukov, G.V.; Lawrence, M.S.; Sougnez, C.; McKenna, A.; et al. The mutational landscape of head and neck squamous cell carcinoma. Science 2011, 333, 1157–1160. [Google Scholar] [CrossRef]
- Gaykalova, D.A.; Mambo, E.; Choudhary, A.; Houghton, J.; Buddavarapu, K.; Sanford, T.; Darden, W.; Adai, A.; Hadd, A.; Latham, G.; et al. Novel insight into mutational landscape of head and neck squamous cell carcinoma. PLoS ONE 2014, 9, e93102. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Snyder, A. Making It Personal: Neoantigen Vaccines in Metastatic Melanoma. Immunity 2017, 47, 221–223. [Google Scholar] [CrossRef]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.P.; Simon, P.; Lower, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrors, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef]
- Tanyi, J.L.; Bobisse, S.; Ophir, E.; Tuyaerts, S.; Roberti, A.; Genolet, R.; Baumgartner, P.; Stevenson, B.J.; Iseli, C.; Dangaj, D.; et al. Personalized cancer vaccine effectively mobilizes antitumor T cell immunity in ovarian cancer. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Carreno, B.M.; Magrini, V.; Becker-Hapak, M.; Kaabinejadian, S.; Hundal, J.; Petti, A.A.; Ly, A.; Lie, W.R.; Hildebrand, W.H.; Mardis, E.R.; et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 2015, 348, 803–808. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, P.; Yerra, A.; Tein, L.; Shashidharamurthy, R. Custom designing therapeutic cancer vaccines: Delivery of immunostimulatory molecule adjuvants by protein transfer. Human Vaccines 2008, 5, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Cimino, A.M.; Palaniswami, P.; Kim, A.C.; Selvaraj, P. Cancer Vaccine Development: Protein Transfer of Membrane-anchored Cytokines and Immunostimulatory Molecules. Immunol. Res. 2004, 29, 231–240. [Google Scholar] [CrossRef]
- Patel, J.M.; Vartabedian, V.F.; Bozeman, E.N.; Caoyonan, B.E.; Srivatsan, S.; Pack, C.D.; Dey, P.; D’Souza, M.J.; Yang, L.; Selvaraj, P. Plasma membrane vesicles decorated with glycolipid-anchored antigens and adjuvants via protein transfer as an antigen delivery platform for inhibition of tumor growth. Biomaterials 2015, 74, 231–244. [Google Scholar] [CrossRef]
- McHugh, R.S.; Nagarajan, S.; Wang, Y.C.; Sell, K.W.; Selvaraj, P. Protein transfer of glycosylphosphatidylinositol-B7-1 into tumor cell membranes: A novel approach to tumor immunotherapy. Cancer Res. 1999, 59, 2433–2437. [Google Scholar]
- Li, M.; Ye, C.; Feng, C.; Riedel, F.; Liu, X.; Zeng, Q.; Grandis, J.R. Enhanced antiangiogenic therapy of squamous cell carcinoma by combined endostatin and epidermal growth factor receptor-antisense therapy. Clin. Cancer Res. 2002, 8, 3570–3578. [Google Scholar]
- Khurana, D.; Martin, E.A.; Kasperbauer, J.L.; O’Malley, B.W., Jr.; Salomao, D.R.; Chen, L.; Strome, S.E. Characterization of a spontaneously arising murine squamous cell carcinoma (SCC VII) as a prerequisite for head and neck cancer immunotherapy. Head Neck 2001, 23, 899–906. [Google Scholar] [CrossRef]
- Suit, H.D.; Sedlacek, R.; Silver, G.; Hsieh, C.C.; Epp, E.R.; Ngo, F.Q.; Roberts, W.K.; Verhey, L. Therapeutic gain factors for fractionated radiation treatment of spontaneous murine tumors using fast neutrons, photons plus O2(1) or 3 ATA, or photons plus misonidazole. Radiat. Res. 1988, 116, 482–502. [Google Scholar] [CrossRef]
- Judd, N.P.; Allen, C.T.; Winkler, A.E.; Uppaluri, R. Comparative analysis of tumor-infiltrating lymphocytes in a syngeneic mouse model of oral cancer. Otolaryngol. Head Neck Surg. Off. J. Am. Acad. Otolaryngol. Head Neck Surg. 2012, 147, 493–500. [Google Scholar] [CrossRef]
- Moore, E.; Clavijo, P.E.; Davis, R.; Cash, H.; Van Waes, C.; Kim, Y.; Allen, C. Established T Cell-Inflamed Tumors Rejected after Adaptive Resistance Was Reversed by Combination STING Activation and PD-1 Pathway Blockade. Cancer Immunol. Res. 2016, 4, 1061–1071. [Google Scholar] [CrossRef] [PubMed]
- Onken, M.D.; Winkler, A.E.; Kanchi, K.L.; Chalivendra, V.; Law, J.H.; Rickert, C.G.; Kallogjeri, D.; Judd, N.P.; Dunn, G.P.; Piccirillo, J.F.; et al. A surprising cross-species conservation in the genomic landscape of mouse and human oral cancer identifies a transcriptional signature predicting metastatic disease. Clin. Cancer Res. 2014, 20, 2873–2884. [Google Scholar] [CrossRef] [PubMed]
- Bozeman, E.N.; Cimino-Mathews, A.; Machiah, D.K.; Patel, J.M.; Krishnamoorthy, A.; Tien, L.; Shashidharamurthy, R.; Selvaraj, P. Expression of membrane anchored cytokines and B7-1 alters tumor microenvironment and induces protective antitumor immunity in a murine breast cancer model. Vaccine 2013, 31, 2449–2456. [Google Scholar] [CrossRef]
- Kang, S.H.; Keam, B.; Ahn, Y.O.; Park, H.R.; Kim, M.; Kim, T.M.; Kim, D.W.; Heo, D.S. Inhibition of MEK with trametinib enhances the efficacy of anti-PD-L1 inhibitor by regulating anti-tumor immunity in head and neck squamous cell carcinoma. Oncoimmunology 2019, 8, e1515057. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.J.; Moore, E.C.; Clavijo, P.E.; Friedman, J.; Cash, H.; Chen, Z.; Silvin, C.; Van Waes, C.; Allen, C. Anti-PD-L1 Efficacy Can Be Enhanced by Inhibition of Myeloid-Derived Suppressor Cells with a Selective Inhibitor of PI3Kdelta/gamma. Cancer Res. 2017, 77, 2607–2619. [Google Scholar] [CrossRef] [PubMed]
- Moore, E.C.; Cash, H.A.; Caruso, A.M.; Uppaluri, R.; Hodge, J.W.; Van Waes, C.; Allen, C.T. Enhanced Tumor Control with Combination mTOR and PD-L1 Inhibition in Syngeneic Oral Cavity Cancers. Cancer Immunol. Res. 2016, 4, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Zolkind, P.; Przybylski, D.; Marjanovic, N.; Nguyen, L.; Lin, T.; Johanns, T.; Alexandrov, A.; Zhou, L.; Allen, C.T.; Miceli, A.P.; et al. Cancer immunogenomic approach to neoantigen discovery in a checkpoint blockade responsive murine model of oral cavity squamous cell carcinoma. Oncotarget 2018, 9, 4109–4119. [Google Scholar] [CrossRef]
- Gao, J.; Shi, L.Z.; Zhao, H.; Chen, J.; Xiong, L.; He, Q.; Chen, T.; Roszik, J.; Bernatchez, C.; Woodman, S.E.; et al. Loss of IFN-gamma Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell 2016, 167, 397–404.e399. [Google Scholar] [CrossRef]
- Bullock, B.L.; Kimball, A.K.; Poczobutt, J.M.; Neuwelt, A.J.; Li, H.Y.; Johnson, A.M.; Kwak, J.W.; Kleczko, E.K.; Kaspar, R.E.; Wagner, E.K.; et al. Tumor-intrinsic response to IFNgamma shapes the tumor microenvironment and anti-PD-1 response in NSCLC. Life Sci. Alliance 2019, 2. [Google Scholar] [CrossRef]
- Grohmann, U.; Belladonna, M.L.; Bianchi, R.; Orabona, C.; Ayroldi, E.; Fioretti, M.C.; Puccetti, P. IL-12 acts directly on DC to promote nuclear localization of NF-kappaB and primes DC for IL-12 production. Immunity 1998, 9, 315–323. [Google Scholar] [CrossRef]
- Cavallo, F.; Martin-Fontecha, A.; Bellone, M.; Heltai, S.; Gatti, E.; Tornaghi, P.; Freschi, M.; Forni, G.; Dellabona, P.; Casorati, G. Co-expression of B7-1 and ICAM-1 on tumors is required for rejection and the establishment of a memory response. Eur. J. Immunol. 1995, 25, 1154–1162. [Google Scholar] [CrossRef] [PubMed]
- Thomson, A.J.; Greer, M.R.; Young, A.; Boswell, F.; Telfer, J.F.; Cameron, I.T.; Norman, J.E.; Campbell, S. Expression of intercellular adhesion molecules ICAM-1 and ICAM-2 in human endometrium: Regulation by interferon-gamma. Mol. Hum. Reprod. 1999, 5, 64–70. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Komita, H.; Homma, S.; Saotome, H.; Zeniya, M.; Ohno, T.; Toda, G. Interferon-gamma produced by interleukin-12-activated tumor infiltrating CD8+T cells directly induces apoptosis of mouse hepatocellular carcinoma. J. Hepatol. 2006, 45, 662–672. [Google Scholar] [CrossRef] [PubMed]
- Iway, Y.; Ishida, M.; Tanaka, Y.; Okazaki, T.; Honjo, T.; Minato, N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl. Acad. Sci. USA 2002, 99, 12293–12297. [Google Scholar] [CrossRef] [PubMed]
- Juneja, V.R.; McGuire, K.A.; Manguso, R.T.; LaFleur, M.W.; Collins, N.; Haining, W.N.; Freeman, G.J.; Sharpe, A.H. PD-L1 on tumor cells is sufficient for immune evasion in immunogenic tumors and inhibits CD8 T cell cytotoxicity. J. Exp. Med. 2017, 214, 895–904. [Google Scholar] [CrossRef]
- Soria, J.C.; Marabelle, A.; Brahmer, J.R.; Gettinger, S. Immune checkpoint modulation for non-small cell lung cancer. Clin. Cancer. Res. 2015, 21, 2256–2262. [Google Scholar] [CrossRef]
- Bozeman, E.N.; He, S.; Shafizadeh, Y.; Selvaraj, P. Therapeutic efficacy of PD-L1 blockade in a breast cancer model is enhanced by cellular vaccines expressing B7-1 and glycolipid-anchored IL-12. Human Vaccine. Immunother. 2016, 12, 421–430. [Google Scholar] [CrossRef]
- Hamanishi, J.; Mandai, M.; Matsumura, N.; Abiko, K.; Baba, T.; Konishi, I. PD-1/PD-L1 blockade in cancer treatment: Perspectives and issues. Int. J. Clin. Oncol. 2016, 21, 462–473. [Google Scholar] [CrossRef]
- Suit, H.D.; Sedlacek, R.S.; Silver, G.; Dosoretz, D. Pentobarbital anesthesia and the response of tumor and normal tissue in the C3Hf/sed mouse to radiation. Radiat. Res. 1985, 104, 47–65. [Google Scholar] [CrossRef]
- Hirst, D.G.; Brown, J.M.; Hazlehurst, J.L. Enhancement of CCNU cytotoxicity by misonidazole: Possible therapeutic gain. British J. Cancer 1982, 46, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Esaki, S.; Goshima, F.; Ozaki, H.; Takano, G.; Hatano, Y.; Kawakita, D.; Ijichi, K.; Watanabe, T.; Sato, Y.; Murata, T.; et al. Oncolytic activity of HF10 in head and neck squamous cell carcinomas. Cancer Gene Ther. 2019. [Google Scholar] [CrossRef] [PubMed]
- Grandis, J.R.; Chang, M.J.; Yu, W.D.; Johnson, C.S. Antitumor activity of interleukin-1 alpha and cisplatin in a murine model system. Arch. Otolaryngol. Head Neck Surg. 1995, 121, 197–200. [Google Scholar] [CrossRef] [PubMed]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bommireddy, R.; Munoz, L.E.; Kumari, A.; Huang, L.; Fan, Y.; Monterroza, L.; Pack, C.D.; Ramachandiran, S.; Reddy, S.J.C.; Kim, J.; et al. Tumor Membrane Vesicle Vaccine Augments the Efficacy of Anti-PD1 Antibody in Immune Checkpoint Inhibitor-Resistant Squamous Cell Carcinoma Models of Head and Neck Cancer. Vaccines 2020, 8, 182. https://doi.org/10.3390/vaccines8020182
Bommireddy R, Munoz LE, Kumari A, Huang L, Fan Y, Monterroza L, Pack CD, Ramachandiran S, Reddy SJC, Kim J, et al. Tumor Membrane Vesicle Vaccine Augments the Efficacy of Anti-PD1 Antibody in Immune Checkpoint Inhibitor-Resistant Squamous Cell Carcinoma Models of Head and Neck Cancer. Vaccines. 2020; 8(2):182. https://doi.org/10.3390/vaccines8020182
Chicago/Turabian StyleBommireddy, Ramireddy, Luis E. Munoz, Anita Kumari, Lei Huang, Yijian Fan, Lenore Monterroza, Christopher D. Pack, Sampath Ramachandiran, Shaker J.C. Reddy, Janet Kim, and et al. 2020. "Tumor Membrane Vesicle Vaccine Augments the Efficacy of Anti-PD1 Antibody in Immune Checkpoint Inhibitor-Resistant Squamous Cell Carcinoma Models of Head and Neck Cancer" Vaccines 8, no. 2: 182. https://doi.org/10.3390/vaccines8020182
APA StyleBommireddy, R., Munoz, L. E., Kumari, A., Huang, L., Fan, Y., Monterroza, L., Pack, C. D., Ramachandiran, S., Reddy, S. J. C., Kim, J., Chen, Z. G., Saba, N. F., Shin, D. M., & Selvaraj, P. (2020). Tumor Membrane Vesicle Vaccine Augments the Efficacy of Anti-PD1 Antibody in Immune Checkpoint Inhibitor-Resistant Squamous Cell Carcinoma Models of Head and Neck Cancer. Vaccines, 8(2), 182. https://doi.org/10.3390/vaccines8020182