Recent Advances in the Development of Peptide Vaccines and Their Delivery Systems against Group A Streptococcus

 , ,
, , 


Abstract
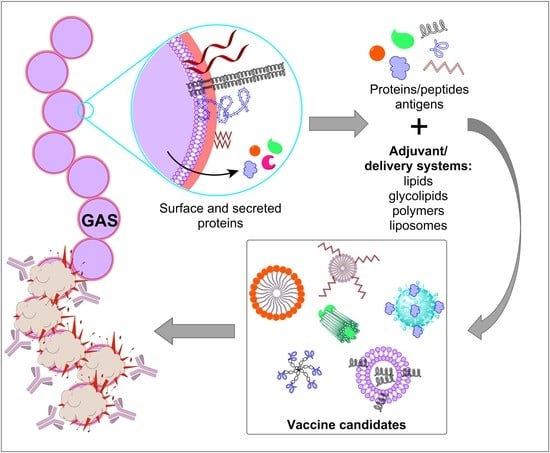
1. Introduction
2. Vaccine Development against GAS
2.1. Vaccination
Peptide-Based Subunit Vaccines
2.2. GAS Vaccine Development
3. Major Antigen Targets for GAS Vaccines
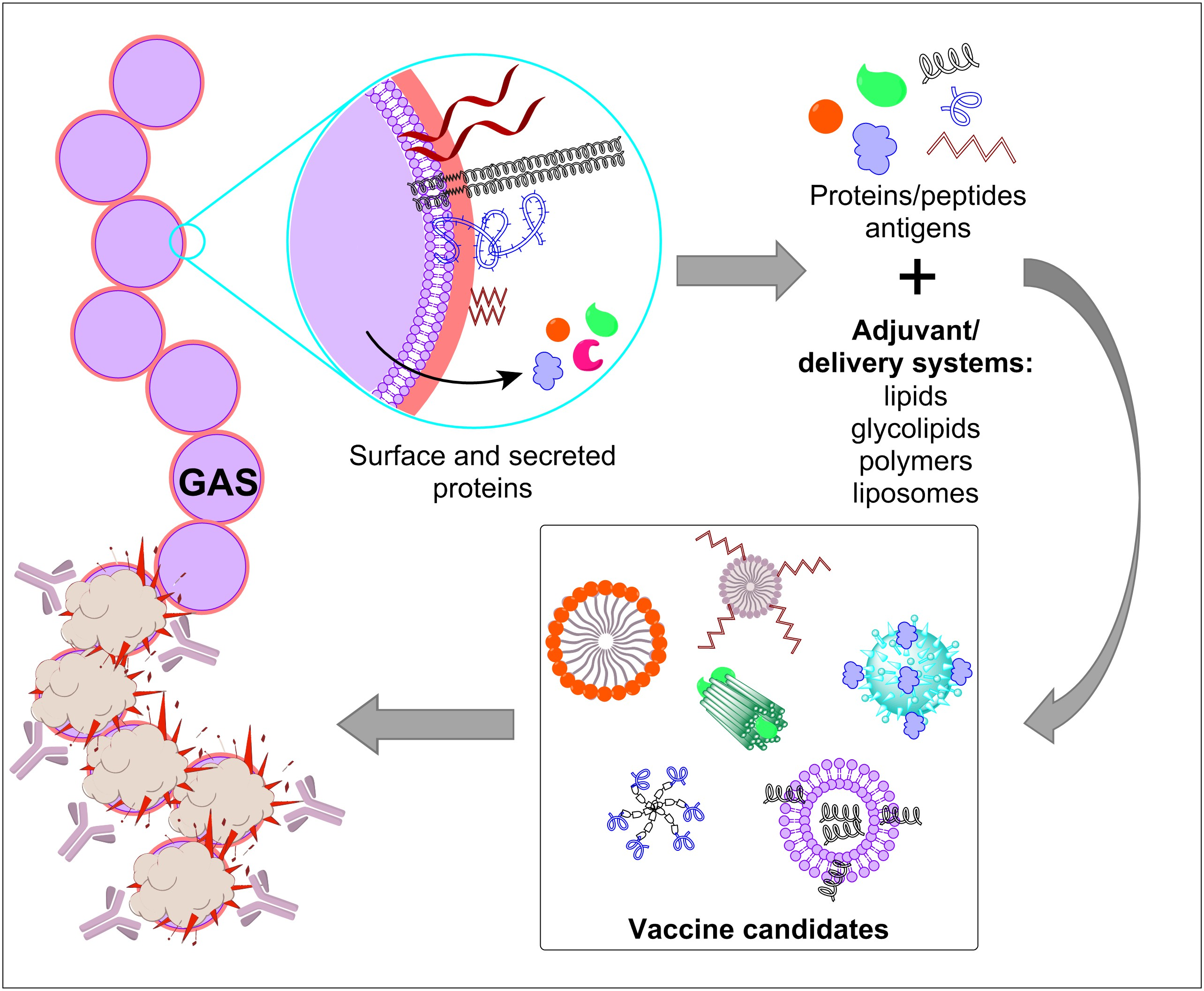
3.1. GAS M Protein
3.1.1. Variable Region N-Terminal GAS Epitopes
Advanced Preclinical and Clinical Trials
3.1.2. Conserved Region M Protein Epitopes
Clinical Trial
3.1.3. Combined Epitopes
3.2. Non-M Proteins
3.2.1. Fibronectin-Binding Proteins
3.2.2. Interleukine-8 (IL-8) Protease (SpyCEP)
3.2.3. Surface-Bound C5a Peptidase (SCPA)
3.2.4. Other Potential Antigens
3.3. GAS Carbohydrate (GAC)
4. Development of Adjuvants and Delivery Systems for GAS Peptide
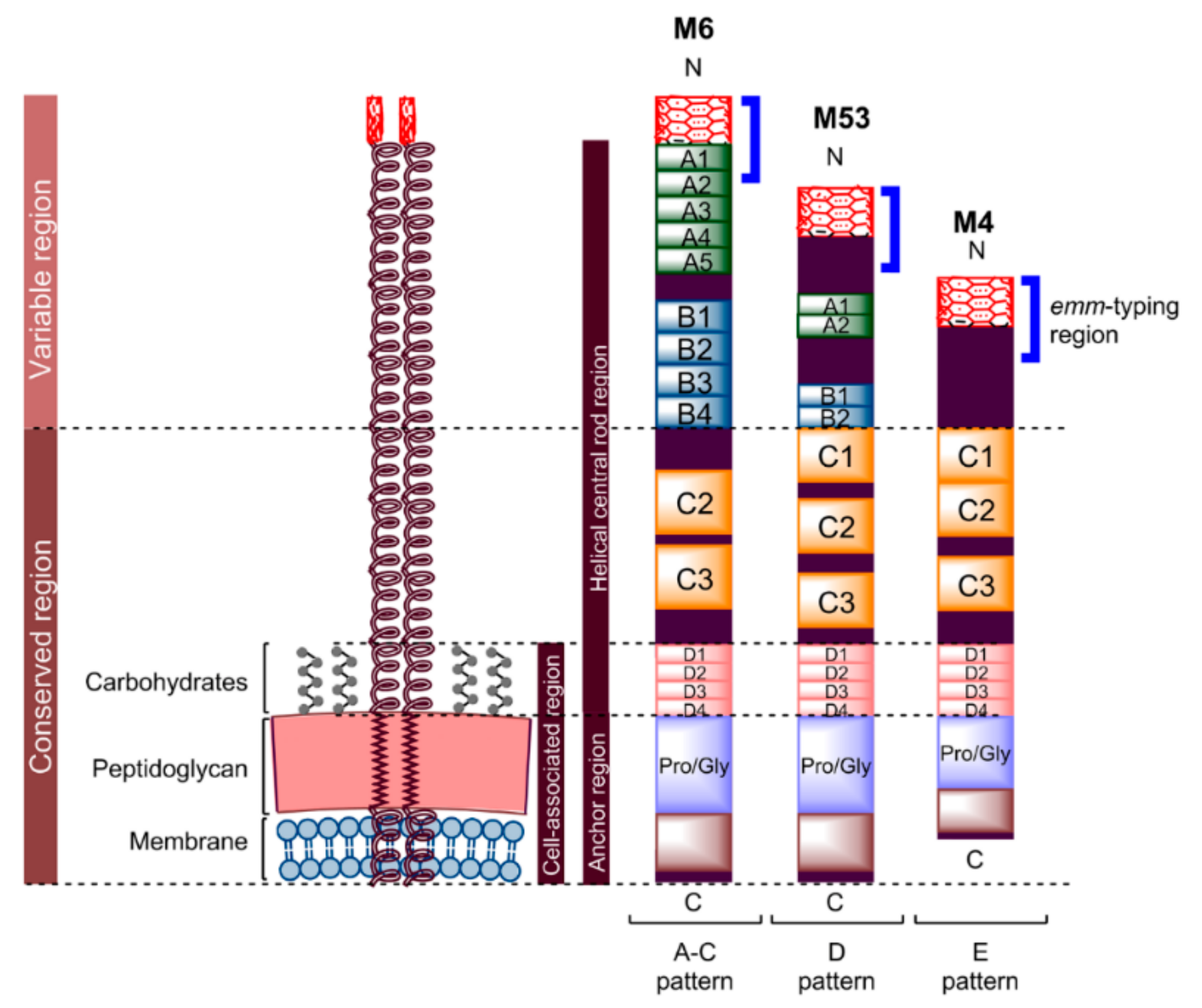
4.1. Peptide Lipidation
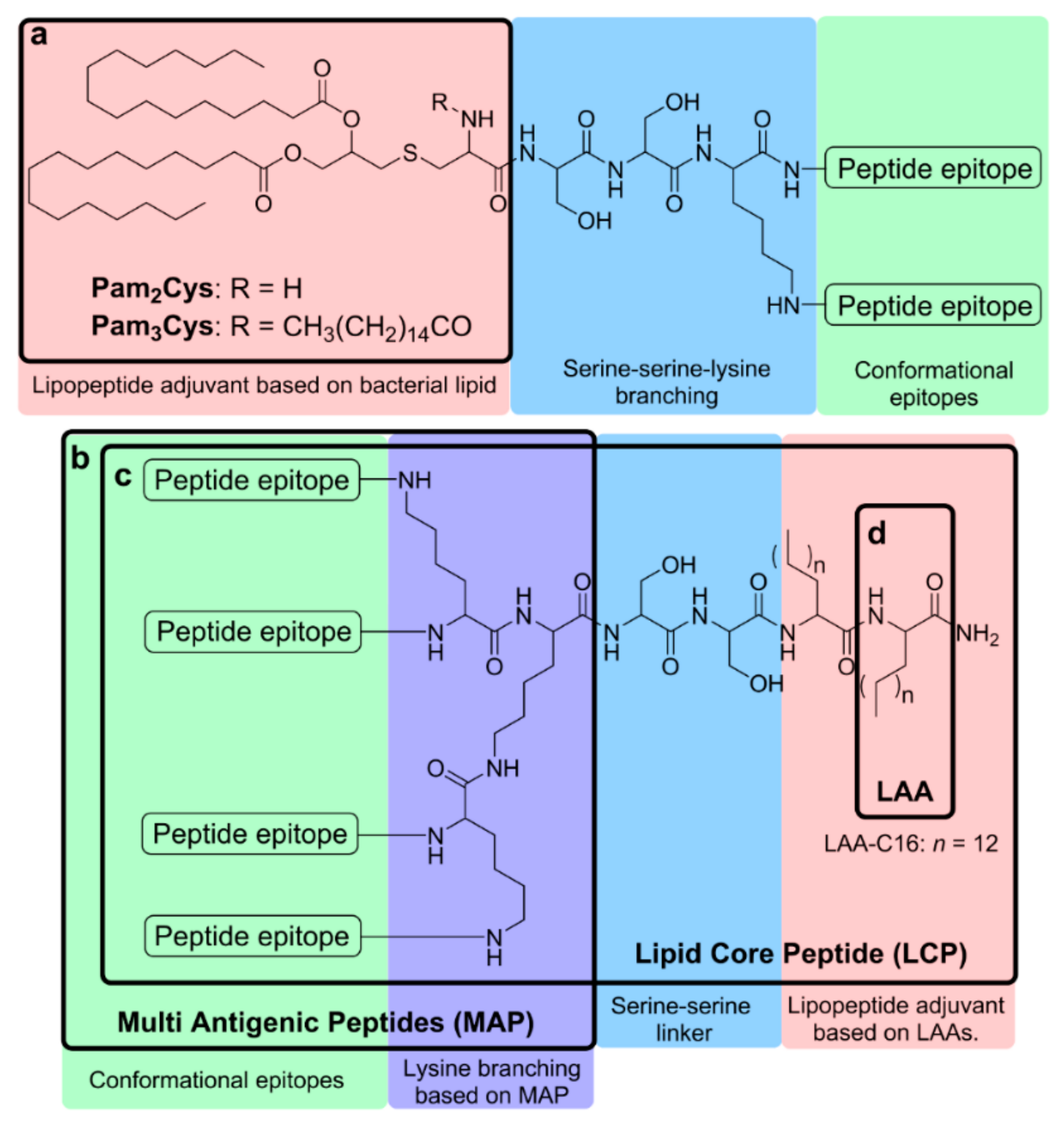
4.1.1. Pam3Cys and Pam2Cys
4.1.2. Lipoamino Acid (LAA) and Lipid Core Peptide (LCP)
4.2. Glycolipid
4.3. Polymers
4.4. Liposomes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cole, J.N.; Barnett, T.C.; Nizet, V.; Walker, M.J. Molecular insight into invasive Group A Streptococcal disease. Nat. Rev. Microbiol. 2011, 9, 724–736. [Google Scholar] [CrossRef] [PubMed]
- Carapetis, J.R.; Steer, A.C.; Mulholland, E.K.; Weber, M. The global burden of Group A Streptococcal diseases. Lancet Infect. Dis. 2005, 5, 685–694. [Google Scholar] [CrossRef]
- Snelling, T.L.; Carapetis, J.R. Group A Streptococcus In Hunter’s Tropical Medicine and Emerging Infectious Disease, 9th ed.; Hill, D.R., Solomon, T., Ryan, E.T., Eds.; W.B. Saunders: London, UK, 2013; pp. 391–401. [Google Scholar]
- Ellis, N.M.J.; Li, Y.; Hildebrand, W.; Fischetti, V.A.; Cunningham, M.W. T cell mimicry and epitope specificity of cross-reactive T cell clones from rheumatic heart disease. J. Immunol. 2005, 175, 5448. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, M.W. Pathogenesis of Group A Streptococcal infections. Clin. Microbiol. Rev. 2000, 13, 470–511. [Google Scholar] [CrossRef] [PubMed]
- Root-Bernstein, R. Rethinking molecular mimicry in rheumatic heart disease and autoimmune myocarditis: Laminin, collagen IV, CAR, and B1AR as initial targets of disease. Front. Pediatr. 2014, 2. [Google Scholar] [CrossRef] [PubMed]
- Dinkla, K.; Nitsche-Schmitz, D.P.; Barroso, V.; Reissmann, S.; Johansson, H.M.; Frick, I.M.; Rohde, M.; Chhatwal, G.S. Identification of a streptococcal octapeptide motif involved in acute rheumatic fever. J. Biol. Chem. 2007, 282, 18686–18693. [Google Scholar] [CrossRef] [PubMed]
- Dinkla, K.; Rohde, M.; Jansen, W.T.M.; Kaplan, E.L.; Chhatwal, G.S.; Talay, S.R. Rheumatic fever-associated Streptococcus pyogenes isolates aggregate collagen. J. Clin. Investig. 2003, 111, 1905–1912. [Google Scholar] [CrossRef]
- Olive, C. Development of subunit vaccines for Group A Streptococcus. In Molecular Vaccines: From Prophylaxis to Therapy—Volume 1; Giese, M., Ed.; Springer: Vienna, Austria, 2013; pp. 207–216. [Google Scholar]
- Kotloff, K.L. Streptococcus Group A vaccines. In Vaccines, 6th ed.; Plotkin, S.A., Orenstein, W.A., Offit, P.A., Eds.; W.B. Saunders: London, UK, 2013; pp. 1169–1175. [Google Scholar]
- Fischetti, V.A. Streptococcal M protein. Sci. Am. 1991, 264, 58–65. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. The Current Evidence for the Burden of Group A Streptococcal Diseases; WHO: Geneva, Switzerland, 2005; pp. 1–52. [Google Scholar]
- Musser, J.M.; Shelburne, S.A., III. A decade of molecular pathogenomic analysis of Group A Streptococcus. J. Clin. Investig. 2009, 119, 2455–2463. [Google Scholar] [CrossRef]
- Watkins, D.A.; Johnson, C.O.; Colquhoun, S.M.; Karthikeyan, G.; Beaton, A.; Bukhman, G.; Forouzanfar, M.H.; Longenecker, C.T.; Mayosi, B.M.; Mensah, G.A.; et al. Global, regional, and national burden of rheumatic heart disease, 1990–2015. N. Engl. J. Med. 2017, 377, 713–722. [Google Scholar] [CrossRef]
- Paar, J.A.; Berrios, N.M.; Rose, J.D.; Caceres, M.; Pena, R.; Perez, W.; Chen-Mok, M.; Jolles, E.; Dale, J.B. Prevalence of rheumatic heart disease in children and young adults in Nicaragua. Am. J. Cardiol. 2010, 105, 1809–1814. [Google Scholar] [CrossRef] [PubMed]
- Sanyahumbi, A.S.; Colquhoun, S.; Wyber, R.; Carapetis, J.R. Global disease burden of Group A Streptococcus. In Streptococcus pyogenes: Basic Biology to Clinical Manifestations; Ferretti, J.J., Stevens, D.L., Fischetti, V.A., Eds.; University of Oklahoma Health Sciences Center: Oklahoma City, OK, USA, 2016; pp. 510–539. [Google Scholar]
- Efstratiou, A.; Lamagni, T. Epidemiology of Streptococcus pyogenes. In Streptococcus pyogenes: Basic Biology to Clinical Manifestations; Ferretti, J.J., Stevens, D.L., Fischetti, V.A., Eds.; University of Oklahoma Health Sciences Center: Oklahoma City, OK, USA, 2016; pp. 465–485. [Google Scholar]
- Stevens, D.L. Invasive Group A Streptococcus infections. Clin. Infect. Dis. 1992, 14, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Zachariadou, L.; Papaparaskevas, J.; Paraskakis, I.; Efstratiou, A.; Pangalis, A.; Legakis, N.J.; Tassios, P.T. Predominance of two M-types among erythromycin-resistant Group A Streptococci from Greek children. Clin. Microbiol. Infect. 2003, 9, 310–314. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cornaglia, G.; Ligozzi, M.; Mazzariol, A.; Valentini, M.; Orefici, G.; Fontana, R. Rapid increase of resistance to erythromycin and clindamycin in Streptococcus pyogenes in Italy, 1993–1995. The Italian surveillance group for antimicrobial resistance. Emerg. Infect. Dis. 1996, 2, 339–342. [Google Scholar] [CrossRef] [PubMed]
- Desjardins, M.; Delgaty, K.L.; Ramotar, K.; Seetaram, C.; Toye, B. Prevalence and mechanisms of erythromycin resistance in Group A and Group B Streptococcus: Implications for reporting susceptibility results. J. Clin. Microbiol. 2004, 42, 5620–5623. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Group A Streptococcal Vaccine Development: Current Status and Issues of Relevance to Less Developed Countries; WHO: Geneva, Switzerland, 2005; pp. 1–18. [Google Scholar]
- Greenwood, B. The contribution of vaccination to global health: Past, present and future. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130433. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, S. History of vaccination. Proc. Natl. Acad. Sci. USA 2014, 111, 12283. [Google Scholar] [CrossRef]
- Nevagi, R.J.; Toth, I.; Skwarczynski, M. Peptide-based vaccines. In Peptide Applications in Biomedicine, Biotechnology and Bioengineering; Koutsopoulos, S., Ed.; Woodhead Publishing: Sawston, UK, 2018; pp. 327–358. [Google Scholar]
- Skwarczynski, M.; Toth, I. Peptide-based synthetic vaccines. Chem. Sci. 2016, 7, 842–854. [Google Scholar] [CrossRef]
- Azmi, F.; Ahmad Fuaad, A.A.H.; Skwarczynski, M.; Toth, I. Recent progress in adjuvant discovery for peptide-based subunit vaccines. Hum. Vaccines Immunother. 2014, 10, 778–796. [Google Scholar] [CrossRef]
- Reed, S.G.; Orr, M.T.; Fox, C.B. Key roles of adjuvants in modern vaccines. Nat. Med. 2013, 19, 1597–1608. [Google Scholar] [CrossRef]
- Skwarczynski, M.; Zaman, M.; Toth, I. Lipo-peptides/saccharides for peptide vaccine delivery. In Handbook of the Biologically Active Peptides, 2nd ed.; Kastin, A., Ed.; Elsevier Inc.: Burlington, NJ, USA, 2013; pp. 571–579. [Google Scholar]
- Dale, J.B.; Batzloff, M.R.; Cleary, P.P.; Courtney, H.S.; Good, M.F.; Grandi, G.; Halperin, S.; Margarit, I.Y.; McNeil, S.; Pandey, M. Current approaches to Group A Streptococcal vaccine development. In Streptococcus pyogenes: Basic Biology to Clinical Manifestations; Ferretti, J.J., Stevens, D.L., Fischetti, V.A., Eds.; University of Oklahoma Health Sciences Center: Oklahoma City, OK, USA, 2016. [Google Scholar]
- Good, M.F.; Pandey, M.; Batzloff, M.R.; Tyrrell, G.J. Strategic development of the conserved region of the M protein and other candidates as vaccines to prevent infection with Group A Streptococci. Expert Rev. Vaccines 2015, 14, 1459–1470. [Google Scholar] [CrossRef] [PubMed]
- Smeesters, P.R.; Mardulyn, P.; Vergison, A.; Leplae, R.; Van Melderen, L. Genetic diversity of Group A Streptococcus m protein: Implications for typing and vaccine development. Vaccine 2008, 26, 5835–5842. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.J.; Georgousakis, M.M.; Vu, T.; Henningham, A.; Hofmann, A.; Rettel, M.; Hafner, L.M.; Sriprakash, K.S.; McMillan, D.J. Evaluation of novel Streptococcus pyogenes vaccine candidates incorporating multiple conserved sequences from the C-repeat region of the M-protein. Vaccine 2012, 30, 2197–2205. [Google Scholar] [CrossRef] [PubMed]
- Dale, J.B.; Fischetti, V.A.; Carapetis, J.R.; Steer, A.C.; Sow, S.; Kumar, R.; Mayosi, B.M.; Rubin, F.A.; Mulholland, K.; Hombach, J.M.; et al. Group A Streptococcal vaccines: Paving a path for accelerated development. Vaccine 2013, 31, B216–B222. [Google Scholar] [CrossRef]
- Young, D.C. Failure of type specific Streptococcus pyogenes vaccine to prevent respiratory infections. U.S. Navy Med. Bull. 1946, 46, 709–718. [Google Scholar]
- Massell, B.F.; Michael, J.G.; Amezcua, J.; Siner, M. Secondary and apparent primary antibody responses after Group A Streptococcal vaccination of 21 children. Appl. Microbiol. 1968, 16, 509–518. [Google Scholar]
- Massell, B.F.; Honikman, L.H.; Amezcua, J. Rheumatic fever following Streptococcal vaccination: Report of three cases. J. Am. Med. Assoc. 1969, 207, 1115–1119. [Google Scholar] [CrossRef]
- Food and Drug Administration. Revocation of status of specific products; Group A streptococcus. Direct final rule. Fed. Regist. 2005, 70, 72197–72199. [Google Scholar]
- Fox, E.N.; Wittner, M.K.; Dorfman, A. Antigenicity of the M proteins of group A hemolytic streptococci. III. Antibody responses and cutaneous hypersensitivity in humans. J. Exp. Med. 1966, 124, 1135–1151. [Google Scholar] [CrossRef]
- Fox, E.N.; Pachman, L.M.; Wittner, M.K.; Dorfman, A. Primary immunization of infants and children with Group A Streptococcal M protein. J. Infect. Dis. 1969, 120, 598–604. [Google Scholar] [CrossRef]
- Fox, E.N.; Waldman, R.H.; Wittner, M.K.; Mauceri, A.A.; Dorfman, A. Protective study with a Group A Streptococcal M protein vaccine. Infectivity challenge of human volunteers. J. Clin. Investig. 1973, 52, 1885–1892. [Google Scholar] [CrossRef] [PubMed]
- Polly, S.M.; Waldman, R.H.; High, P.; Wittner, M.K.; Dorfman, A.; Fox, E.N. Protective studies with a Group A Streptococcal M Protein Vaccine. II. Challenge of volunteers after local immunization in the upper respiratory tract. J. Infect. Dis. 1975, 131, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, M.W.; McCormack, J.M.; Fenderson, P.G.; Ho, M.K.; Beachey, E.H.; Dale, J.B. Human and murine antibodies cross-reactive with streptococcal M protein and myosin recognize the sequence Gln-Lys-Ser-Lys-Gln in M protein. J. Immunol. 1989, 143, 2677–2683. [Google Scholar] [PubMed]
- Dale, J.B.; Beachey, E.H. Multiple, heart-cross-reactive epitopes of streptococcal M proteins. J. Exp. Med. 1985, 161, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Dale, J.B.; Beachey, E.H. Sequence of myosin-crossreactive epitopes of streptococcal M protein. J. Exp. Med. 1986, 164, 1785–1790. [Google Scholar] [CrossRef] [PubMed]
- Bronze, M.S.; Beachey, E.H.; Dale, J.B. Protective and heart-crossreactive epitopes located within the NH2 terminus of type 19 streptococcal M protein. J. Exp. Med. 1988, 167, 1849–1859. [Google Scholar] [CrossRef]
- Pruksakorn, S.; Currie, B.; Brandt, E.; Phornphutkul, C.; Hunsakunachal, S.; Manmontri, A.; Robinson, J.H.; Kehoe, M.A.; Galbraith, A.; Good, M.F. Identification of T cell autoepitopes that cross-react with the C-terminal segment of the M protein of Group A Streptococci. Int. Immunol. 1994, 6, 1235–1244. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, M.W.; Antone, S.M.; Smart, M.; Liu, R.; Kosanke, S. Molecular analysis of human cardiac myosin-cross-reactive B- and T-cell epitopes of the Group A Streptococcal M5 protein. Infect. Immun. 1997, 65, 3913–3923. [Google Scholar]
- Hayman, W.A.; Brandt, E.R.; Relf, W.A.; Cooper, J.; Saul, A.; Good, M.F. Mapping the minimal murine T cell and B cell epitopes within a peptide vaccine candidate from the conserved region of the M protein of Group A Streptococcus. Int. Immunol. 1997, 9, 1723–1733. [Google Scholar] [CrossRef]
- Quinn, A.; Ward, K.; Fischetti, V.A.; Hemric, M.; Cunningham, M.W. Immunological relationship between the class I epitope of streptococcal M protein and myosin. Infect. Immun. 1998, 66, 4418–4424. [Google Scholar]
- Smeesters, P.R.; McMillan, D.J.; Sriprakash, K.S. The streptococcal m protein: A highly versatile molecule. Trend Microbiol. 2010, 18, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Olive, C.; Ho, M.F.; Dyer, J.; Lincoln, D.; Barozzi, N.; Toth, I.; Good, M.F. Immunization with a tetraepitopic lipid core peptide vaccine construct induces broadly protective immune responses against Group A Streptococcus. J. Infect. Dis. 2006, 193, 1666–1676. [Google Scholar] [CrossRef][Green Version]
- Olive, C.; Batzloff, M.; Horvath, A.; Clair, T.; Yarwood, P.; Toth, I.; Good, M.F. Potential of lipid core peptide technology as a novel self-adjuvanting vaccine delivery system for multiple different synthetic peptide immunogens. Infect. Immun. 2003, 71, 2373–2383. [Google Scholar] [CrossRef] [PubMed]
- McMillan, D.J.; Batzloff, M.R.; Browning, C.L.; Davies, M.R.; Good, M.F.; Sriprakash, K.S.; Janulczyk, R.; Rasmussen, M. Identification and assessment of new vaccine candidates for Group A Streptococcal infections. Vaccine 2004, 22, 2783–2790. [Google Scholar] [CrossRef]
- Steer, A.C.; Batzloff, M.R.; Mulholland, K.; Carapetis, J.R. Group A Streptococcal vaccines: Facts versus fantasy. Curr. Opin. Infect. Dis. 2009, 22, 544–552. [Google Scholar] [CrossRef]
- Lynskey, N.N.; Lawrenson, R.A.; Sriskandan, S. New understandings in Streptococcus pyogenes. Curr. Opin. Infect. Dis. 2011, 24, 196. [Google Scholar] [CrossRef]
- McMillan, D.J.; Davies, M.R.; Browning, C.L.; Good, M.F.; Sriprakash, K.S. Prospecting for new Group A Streptococcal vaccine candidates. Indian J. Med. Res. 2004, 119 Suppl, 121–125. [Google Scholar]
- Rivera-Hernandez, T.; Pandey, M.; Henningham, A.; Cole, J.; Choudhury, B.; Cork, A.J.; Gillen, C.M.; Ghaffar, K.A.; West, N.P.; Silvestri, G.; et al. Differing efficacies of lead Group A Streptococcal vaccine candidates and full-length m protein in cutaneous and invasive disease models. mBio 2016, 7, e00618–e00616. [Google Scholar] [CrossRef] [PubMed]
- Henningham, A.; Chiarot, E.; Gillen, C.M.; Cole, J.; Rohde, M.; Fulde, M.; Ramachandran, V.; Cork, A.; Hartas, J.; Magor, G.; et al. Conserved anchorless surface proteins as Group A Streptococcal vaccine candidates. J. Mol. Med. 2012, 90, 1197–1207. [Google Scholar] [CrossRef]
- Johnson, D.R.; Kaplan, E.L.; VanGheem, A.; Facklam, R.R.; Beall, B. Characterization of Group A Streptococci (Streptococcus pyogenes): Correlation of m-protein and emm-gene type with T-protein agglutination pattern and serum opacity factor. J. Med. Microbiol. 2006, 55, 157–164. [Google Scholar] [CrossRef]
- Fischetti, V.A. M protein and other surface proteins on Streptococci. In Streptococcus pyogenes: Basic Biology to Clinical Manifestations; Ferretti, J.J., Stevens, D.L., Fischetti, V.A., Eds.; University of Oklahoma Health Sciences Center: Oklahoma City, OK, USA, 2016; pp. 23–43. [Google Scholar]
- Olive, C.; Moyle, P.M.; Toth, I. Towards the development of a broadly protective Group A Streptococcal vaccine based on the lipid-core peptide system. Curr. Med. Chem. 2007, 14, 2976–2988. [Google Scholar] [CrossRef] [PubMed]
- Beres, S.B.; Musser, J.M. Contribution of exogenous genetic elements to the Group A Streptococcus metagenome. PLoS ONE 2007, 2, e800. [Google Scholar] [CrossRef] [PubMed]
- Olive, C.; Schulze, K.; Sun, H.K.; Ebensen, T.; Horvath, A.; Toth, I.; Guzman, C.A. Enhanced protection against Streptococcus pyogenes infection by intranasal vaccination with a dual antigen component M protein/SfbI lipid core peptide vaccine formulation. Vaccine 2007, 25, 1789–1797. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.A.; Hayman, W.; Reed, C.; Kagawa, H.; Good, M.F.; Saul, A. Mapping of conformational B cell epitopes within alpha-helical coiled coil proteins. Mol. Immunol. 1997, 34, 433–440. [Google Scholar] [CrossRef]
- Bruner, M.; James, A.; Beall, B.; Carlone, G.M.; Ades, E.; Johnson, S.; Guarner, J.; Sampson, J. Evaluation of synthetic, M type-specific peptides as antigens in a multivalent Group A Streptococcal vaccine. Vaccine 2003, 21, 2698–2703. [Google Scholar] [CrossRef]
- Hu, M.C.; Walls, M.A.; Stroop, S.D.; Reddish, M.A.; Beall, B.; Dale, J.B. Immunogenicity of a 26-valent Group A Streptococcal vaccine. Infect. Immun. 2002, 70, 2171–2177. [Google Scholar] [CrossRef]
- Dale, J.B.; Penfound, T.A.; Chiang, E.Y.; Walton, W.J. New 30-valent M protein-based vaccine evokes cross-opsonic antibodies against non-vaccine serotypes of Group A Streptococci. Vaccine 2011, 29, 8175–8178. [Google Scholar] [CrossRef]
- Dey, N.; McMillan, D.J.; Yarwood, P.J.; Joshi, R.M.; Kumar, R.; Good, M.F.; Sriprakash, K.S.; Vohra, H. High diversity of Group A Streptococcal emm types in an Indian community: The need to tailor multivalent vaccines. Clin. Infect. Dis. 2005, 40, 46–51. [Google Scholar] [CrossRef]
- Dale, J.B. Multivalent Group A Streptococcal vaccine designed to optimize the immunogenicity of six tandem M protein fragments. Vaccine 1999, 17, 193–200. [Google Scholar] [CrossRef]
- Kotloff, K.L.; Corretti, M.; Palmer, K.; Campbell, J.D.; Reddish, M.A.; Hu, M.C.; Wasserman, S.S.; Dale, J.B. Safety and immunogenicity of a recombinant multivalent Group A Streptococcal vaccine in healthy adults: Phase 1 trial. J. Am. Med. Assoc. 2004, 292, 709–715. [Google Scholar] [CrossRef]
- McNeil, S.A.; Halperin, S.A.; Langley, J.M.; Smith, B.; Warren, A.; Sharratt, G.P.; Baxendale, D.M.; Reddish, M.A.; Hu, M.C.; Stroop, S.D.; et al. Safety and immunogenicity of 26-valent group a streptococcus vaccine in healthy adult volunteers. Clin. Infect. Dis. 2005, 41, 1114–1122. [Google Scholar] [CrossRef] [PubMed]
- McNeil, S.A.; Halperin, S.A.; Langley, J.M.; Smith, B.; Baxendale, D.M.; Warren, A.; Sharratt, G.P.; Reddish, M.A.; Fries, L.F.; Vink, P.E.; et al. A double-blind, randomized Phase II trial of the safety and immunogenicity of 26-valent Group A Streptococcus vaccine in healthy adults. Int. Congr. Ser. 2006, 1289, 303–306. [Google Scholar] [CrossRef]
- Dale, J.B.; Penfound, T.A.; Tamboura, B.; Sow, S.O.; Nataro, J.P.; Tapia, M.; Kotloff, K.L. Potential coverage of a multivalent M protein-based Group A Streptococcal vaccine. Vaccine 2013, 31, 1576–1581. [Google Scholar] [CrossRef] [PubMed]
- Dale, J.B.; Smeesters, P.R.; Courtney, H.S.; Penfound, T.A.; Hohn, C.M.; Smith, J.C.; Baudry, J.Y. Structure-based design of broadly protective Group A Streptococcal M protein-based vaccines. Vaccine 2017, 35, 19–26. [Google Scholar] [CrossRef] [PubMed]
- O’Loughlin, R.E.; Roberson, A.; Cieslak, P.R.; Lynfield, R.; Gershman, K.; Craig, A.; Albanese, B.A.; Farley, M.M.; Barrett, N.L.; Spina, N.L.; et al. The epidemiology of invasive Group A Streptococcal infection and potential vaccine implications: United States, 2000–2004. Clin. Infect. Dis. 2007, 45, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Shulman, S.T.; Tanz, R.R.; Dale, J.B.; Beall, B.; Kabat, W.; Kabat, K.; Cederlund, E.; Patel, D.; Rippe, J.; Li, Z.; et al. Seven-year surveillance of north american pediatric Group A Streptococcal pharyngitis isolates. Clin. Infect. Dis. 2009, 49, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Luca-Harari, B.; Darenberg, J.; Neal, S.; Siljander, T.; Strakova, L.; Tanna, A.; Creti, R.; Ekelund, K.; Koliou, M.; Tassios, P.T.; et al. Clinical and microbiological characteristics of severe Streptococcus pyogenes disease in Europe. J. Clin. Microbiol. 2009, 47, 1155. [Google Scholar] [CrossRef] [PubMed]
- Steer, A.C.; Carapetis, J.R.; Dale, J.B.; Fraser, J.D.; Good, M.F.; Guilherme, L.; Moreland, N.J.; Mulholland, E.K.; Schodel, F.; Smeesters, P.R. Status of research and development of vaccines for Streptococcus pyogenes. Vaccine 2016, 34, 2953–2958. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, D.M.P.; Sanderson-Smith, M.; Bessen, D.E.; Dale, J.B.; Beall, B.W.; Guglielmini, J.; Carapetis, J.R.; Sriprakash, K.S.; McMillan, D.J.; Vu, T.; et al. A systematic and functional classification of Streptococcus pyogenes that serves as a new tool for molecular typing and vaccine development. J. Infect. Dis. 2014, 210, 1325–1338. [Google Scholar] [CrossRef]
- Ozberk, V.; Pandey, M.; Good, M.F. Contribution of cryptic epitopes in designing a Group A Streptococcal vaccine. Hum. Vaccines Immunother. 2018, 14, 2034–2052. [Google Scholar] [CrossRef]
- Olive, C.; Clair, T.; Yarwood, P.; Good, M.R. Protection of mice from Group A Streptococcal infection by intranasal immunisation with a peptide vaccine that contains a conserved M protein B cell epitope and lacks a T cell autoepitope. Vaccine 2002, 20, 2816–2825. [Google Scholar] [CrossRef]
- Hayman, W.A.; Toth, I.; Flinn, N.; Scanlon, M.; Good, M.F. Enhancing the immunogenicity and modulating the fine epitope recognition of antisera to a helical Group A Streptococcal peptide vaccine candidate from the M protein using lipid-core peptide technology. Immunol. Cell Biol. 2002, 80, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Zaman, M.; Abdel-Aal, A.B.; Fujita, Y.; Ziora, Z.M.; Batzloff, M.R.; Good, M.F.; Toth, I. Structure-activity relationship for the development of a self-adjuvanting mucosally active lipopeptide vaccine against Streptococcus pyogenes. J. Med. Chem. 2012, 55, 8515–8523. [Google Scholar] [CrossRef] [PubMed]
- Brandt, E.R.; Sriprakash, K.S.; Hobb, R.I.; Hayman, W.A.; Zeng, W.; Batzloff, M.R.; Jackson, D.C.; Good, M.F. New multi-determinant strategy for a Group A Streptococcal vaccine designed for the Australian Aboriginal population. Nat. Med. 2000, 6, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Shaila, M.S.; Nayak, R.; Prakash, S.S.; Georgousakis, M.; Brandt, E.; McMillan, D.J.; Batzloff, M.R.; Pruksakorn, S.; Good, M.F.; Sriprakash, K.S. Comparative in silico analysis of two vaccine candidates for Group A Streptococcus predicts that they both may have similar safety profiles. Vaccine 2007, 25, 3567–3573. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Chandrudu, S.; Skwarczynski, M.; Toth, I. The application of self-assembled nanostructures in peptide-based subunit vaccine development. Eur. Polym. J. 2017, 93, 670–681. [Google Scholar] [CrossRef]
- Skwarczynski, M.; Toth, I. Recent advances in peptide-based subunit nanovaccines. Nanomedicine (Lond.) 2014, 9, 2657–2669. [Google Scholar] [CrossRef]
- Skwarczynski, M.; Parhiz, B.H.; Soltani, F.; Srinivasan, S.; Kamaruzaman, K.A.; Lin, I.-C.; Toth, I. Lipid peptide core nanoparticles as multivalent vaccine candidates against Streptococcus pyogenes. Aust. J. Chem. 2012, 65, 35–39. [Google Scholar] [CrossRef]
- Skwarczynski, M.; Toth, I. Lipid-core-peptide system for self-adjuvanting synthetic vaccine delivery. In Bioconjugation Protocols; Mark, S.S., Ed.; Humana Press: New York, NY, USA, 2011; Volume 751, pp. 297–308. [Google Scholar]
- Zhong, W.; Skwarczynski, M.; Toth, I. Lipid core peptide system for gene, drug, and vaccine delivery. Aust. J. Chem. 2009, 62, 956–967. [Google Scholar] [CrossRef]
- Batzloff, M.R.; Hayman, W.A.; Davies, M.R.; Zeng, M.; Pruksakorn, S.; Brandt, E.R.; Good, M.F. Protection against Group A Streptococcus by immunization with J8-diphtheria toxoid: Contribution of J8-and diphtheria toxoid-specific antibodies to protection. J. Infect. Dis 2003, 187, 1598–1608. [Google Scholar] [CrossRef]
- Pandey, M.; Wykes, M.N.; Hartas, J.; Good, M.F.; Batzloff, M.R. Long-term antibody memory induced by synthetic peptide vaccination is protective against Streptococcus pyogenes infection and is independent of memory T cell help. J. Immunol. 2013, 190, 2692–2701. [Google Scholar] [CrossRef] [PubMed]
- Sekuloski, S.; Batzloff, M.R.; Griffin, P.; Parsonage, W.; Elliott, S.; Hartas, J.; O’Rourke, P.; Marquart, L.; Pandey, M.; Rubin, F.A.; et al. Evaluation of safety and immunogenicity of a Group A Streptococcus vaccine candidate (MJ8VAX) in a randomized clinical trial. PLoS ONE 2018, 13, e0198658. [Google Scholar] [CrossRef] [PubMed]
- Good, M.F.; Xu, H.; Batzloff, M. Adapting immunity with subunit vaccines: Case studies with Group A Streptococcus and malaria. Int. J. Parasitol. 2002, 32, 575–580. [Google Scholar] [CrossRef]
- Moyle, P.M.; Hartas, J.; Henningham, A.; Batzloff, M.R.; Good, M.F.; Toth, I. An efficient, chemically-defined semisynthetic lipid-adjuvanted nanoparticulate vaccine development system. Nanomed. Nanotechnol. Biol. Med. 2013, 9, 935–944. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Ortega, M.J.; Norais, N.; Bensi, G.; Liberatori, S.; Capo, S.; Mora, M.; Scarselli, M.; Doro, F.; Ferrari, G.; Garaguso, I.; et al. Characterization and identification of vaccine candidate proteins through analysis of the Group A Streptococcus surface proteome. Nat. Biotechnol. 2006, 24, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Lei, B.; Liu, M.; Chesney, G.L.; Musser, J.M. Identification of new candidate vaccine antigens made by Streptococcus pyogenes: Purification and characterization of 16 putative extracellular lipoproteins. J. Infect. Dis. 2004, 189, 79–89. [Google Scholar] [CrossRef]
- Bensi, G.; Mora, M.; Tuscano, G.; Biagini, M.; Chiarot, E.; Bombaci, M.; Capo, S.; Falugi, F.; Manetti, A.G.; Donato, P.; et al. Multi high-throughput approach for highly selective identification of vaccine candidates: the Group A Streptococcus case. Mol. Cell. Proteom. 2012, 11, M111–015693. [Google Scholar] [CrossRef] [PubMed]
- Fritzer, A.; Senn, B.M.; Minh, D.B.; Hanner, M.; Gelbmann, D.; Noiges, B.; Henics, T.; Schulze, K.; Guzman, C.A.; Goodacre, J.; et al. Novel conserved Group A Streptococcal proteins identified by the antigenome technology as vaccine candidates for a non-M protein-based vaccine. Infect. Immun. 2010, 78, 4051–4067. [Google Scholar] [CrossRef]
- Pandey, M.; Good, M.F. The quest for GAS vaccine. Oncotarget 2015, 6, 34063–34064. [Google Scholar] [CrossRef]
- Pandey, M.; Mortensen, R.; Calcutt, A.; Powell, J.; Batzloff, M.R.; Dietrich, J.; Good, M.F. Combinatorial synthetic peptide vaccine strategy protects against hypervirulent CovR/S mutant Streptococci. J. Immunol. 2016, 196, 3364–3374. [Google Scholar] [CrossRef]
- Kreikemeyer, B.; Talay, S.R.; Chhatwal, G.S. Characterization of a novel fibronectin-binding surface protein in Group A Streptococci. Mol. Microbiol. 1995, 17, 137–145. [Google Scholar] [CrossRef] [PubMed]
- McArthur, J.; Medina, E.; Mueller, A.; Chin, J.; Currie, B.J.; Sriprakash, K.S.; Talay, S.R.; Chhatwal, G.S.; Walker, M.J. Intranasal vaccination with streptococcal fibronectin binding protein Sfb1 fails to prevent growth and dissemination of Streptococcus pyogenes in a murine skin infection model. Infect. Immun. 2004, 72, 7342–7345. [Google Scholar] [CrossRef] [PubMed]
- Guzman, C.A.; Talay, S.R.; Molinari, G.; Medina, E.; Chhatwal, G.S. Protective immune response against Streptococcus pyogenes in mice after intranasal vaccination with the fibronectin-binding protein SfbI. J. Infect. Dis 1999, 179, 901–906. [Google Scholar] [CrossRef] [PubMed]
- Schulze, K.; Medina, E.; Chhatwal, G.S.; Guzmán, C.A. Identification of B- and T-cell epitopes within the fibronectin-binding domain of the SfbI protein of Streptococcus pyogenes. Infect. Immun. 2003, 71, 7197–7201. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Molinari, G.; Talay, S.R.; Valentin-Weigand, P.; Rohde, M.; Chhatwal, G.S. The fibronectin-binding protein of Streptococcus pyogenes, SfbI, is involved in the internalization of Group A Streptococci by epithelial cells. Infect. Immun. 1997, 65, 1357–1363. [Google Scholar] [PubMed]
- Talay, S.R.; Valentin-Weigand, P.; Jerlström, P.G.; Timmis, K.N.; Chhatwal, G.S. Fibronectin-binding protein of Streptococcus pyogenes: Sequence of the binding domain involved in adherence of streptococci to epithelial cells. Infect. Immun. 1992, 60, 3837–3844. [Google Scholar]
- Medina, E.; Molinari, G.; Rohde, M.; Haase, B.; Chhatwal, G.S.; Guzmán, C.A. Fc-mediated nonspecific binding between fibronectin-binding protein of Streptococcus pyogenes and human immunoglobulins. J. Immunol. 1999, 163, 3396. [Google Scholar] [PubMed]
- Ma, C.Q.; Li, C.H.; Wang, X.R.; Zeng, R.H.; Yin, X.L.; Feng, H.D.; Wei, L. Similar ability of FbaA with M protein to elicit protective immunity against Group A Streptococcus challenge in mice. Cell. Mol. Immunol. 2009, 6, 73–77. [Google Scholar] [CrossRef]
- Kawabata, S.; Kunitomo, E.; Terao, Y.; Nakagawa, I.; Kikuchi, K.; Totsuka, K.; Hamada, S. Systemic and mucosal immunizations with fibronectin-binding protein FBP54 induce protective immune responses against Streptococcus pyogenes challenge in mice. Infect. Immun. 2001, 69, 924–930. [Google Scholar] [CrossRef]
- Schulze, K.; Ebensen, T.; Chandrudu, S.; Skwarczynski, M.; Toth, I.; Olive, C.; Guzman, C.A. Bivalent mucosal peptide vaccines administered using the LCP carrier system stimulate protective immune responses against Streptococcus pyogenes infection. Nanomed. Nanotechnol. Biol. Med. 2017, 13, 2463–2474. [Google Scholar] [CrossRef]
- Turner, C.E.; Kurupati, P.; Jones, M.D.; Edwards, R.J.; Sriskandan, S. Emerging role of the interleukin-8 cleaving enzyme SpyCEP in clinical Streptococcus pyogenes infection. J. Infect. Dis. 2009, 200, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Zinkernagel, A.S.; Timmer, A.M.; Pence, M.A.; Locke, J.B.; Buchanan, J.T.; Turner, C.E.; Mishalian, I.; Sriskandan, S.; Hanski, E.; Nizet, V. The IL-8 protease SpyCEP/ScpC of Group A Streptococcus promotes resistance to neutrophil killing. Cell Host Microbe 2008, 4, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Turner, C.E.; Kurupati, P.; Wiles, S.; Edwards, R.J.; Sriskandan, S. Impact of immunization against SpyCEP during invasive disease with two Streptococcal species: Streptococcus pyogenes and Streptococcus equi. Vaccine 2009, 27, 4923–4929. [Google Scholar] [CrossRef]
- Ji, Y.; Carlson, B.; Kondagunta, A.; Cleary, P.P. Intranasal immunization with C5a peptidase prevents nasopharyngeal colonization of mice by the Group A Streptococcus. Infect. Immun. 1997, 65, 2080–2087. [Google Scholar]
- Shet, A.; Kaplan, E.L.; Johnson, D.R.; Cleary, P.P. Immune response to Group A Streptococcal C5a peptidase in children: implications for vaccine development. J. Infect. Dis 2003, 188, 809–817. [Google Scholar] [CrossRef]
- Park, H.S.; Cleary, P.P. Active and passive intranasal immunizations with streptococcal surface protein C5a peptidase prevent infection of murine nasal mucosa-associated lymphoid tissue, a functional homologue of human tonsils. Infect. Immun. 2005, 73, 7878–7886. [Google Scholar] [CrossRef] [PubMed]
- Cleary, P.P.; Matsuka, Y.V.; Huynh, T.; Lam, H.; Olmsted, S.B. Immunization with C5a peptidase from either Group A or B Streptococci enhances clearance of Group A Streptococci from intranasally infected mice. Vaccine 2004, 22, 4332–4341. [Google Scholar] [CrossRef]
- Rasmussen, M.; Muller, H.P.; Bjorck, L. Protein GRAB of streptococcus pyogenes regulates proteolysis at the bacterial surface by binding alpha2-macroglobulin. J. Biol. Chem. 1999, 274, 15336–15344. [Google Scholar] [CrossRef]
- Zhang, Q.; Choo, S.; Finn, A. Immune responses to novel pneumococcal proteins pneumolysin, PspA, PsaA, and CbpA in adenoidal B cells from children. Infect. Immun. 2002, 70, 5363–5369. [Google Scholar] [CrossRef] [PubMed]
- Gibson, C.M.; Caparon, M.G. Insertional inactivation of Streptococcus pyogenes SOD suggests that prtF is regulated in response to a superoxide signal. J. Bacteriol. 1996, 178, 4688–4695. [Google Scholar] [CrossRef]
- McMillan, D.J.; Davies, M.R.; Good, M.F.; Sriprakash, K.S. Immune response to superoxide dismutase in Group A Streptococcal infection. FEMS Immunol. Med. Microbiol. 2004, 40, 249–256. [Google Scholar] [CrossRef]
- Lancefield, R.C. The antigenic complex of Streptococcus haemolyticus: I. Demonstration of a type-specific substance in extracts of Streptococcus haemolyticus. J. Exp. Med. 1928, 47, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Caliot, E.; Dramsi, S.; Chapot-Chartier, M.P.; Courtin, P.; Kulakauskas, S.; Pechoux, C.; Trieu-Cuot, P.; Mistou, M.Y. Role of the Group B antigen of Streptococcus agalactiae: a peptidoglycan-anchored polysaccharide involved in cell wall biogenesis. PLoS Pathog. 2012, 8, e1002756. [Google Scholar] [CrossRef] [PubMed]
- McCarty, M. The lysis of Group A Hemolytic Streptococci by extracellular enzymes of Streptomyces albus. Nature of the cellular substrate attacked by the lytic enzymes. J. Exp. Med. 1952, 96, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Van Sorge, N.M.; Cole, J.N.; Kuipers, K.; Henningham, A.; Aziz, R.K.; Kasirer-Friede, A.; Lin, L.; Berends, E.T.M.; Davies, M.R.; Dougan, G.; et al. The classical Lancefield antigen of Group A Streptococcus is a virulence determinant with implications for vaccine design. Cell Host Microbe 2014, 15, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, I.; Halpern, B.; Robert, L. Immunological relationship between Streptococcus A polysaccharide and the structural glycoproteins of heart valve. Nature 1967, 213, 44–47. [Google Scholar] [CrossRef]
- Goldstein, I.; Rebeyrotte, P.; Parlebas, J.; Halpern, B. Isolation from heart valves of glycopeptides which share immunological properties with Streptococcus haemolyticus group A polysaccharides. Nature 1968, 219, 866–868. [Google Scholar] [CrossRef]
- Kirvan, C.A.; Swedo, S.E.; Heuser, J.S.; Cunningham, M.W. Mimicry and autoantibody-mediated neuronal cell signaling in Sydenham chorea. Nat. Med. 2003, 9, 914–920. [Google Scholar] [CrossRef]
- Salvadori, L.G.; Blake, M.S.; McCarty, M.; Tai, J.Y.; Zabriskie, J.B. Group A Streptococcus-liposome ELISA antibody titers to group A polysaccharide and opsonophagocytic capabilities of the antibodies. J. Infect. Dis 1995, 171, 593–600. [Google Scholar] [CrossRef]
- Sabharwal, H.; Michon, F.; Nelson, D.; Dong, W.; Fuchs, K.; Manjarrez, R.C.; Sarkar, A.; Uitz, C.; Viteri-Jackson, A.; Suarez, R.S.; et al. Group A Streptococcus (GAS) carbohydrate as an immunogen for protection against GAS infection. J. Infect. Dis. 2006, 193, 129–135. [Google Scholar] [CrossRef]
- Henningham, A.; Davies, M.R.; Uchiyama, S.; van Sorge, N.M.; Lund, S.; Chen, K.T.; Walker, M.J.; Cole, J.N.; Nizet, V. Virulence role of the GlcNAc side chain of the Lancefield cell wall carbohydrate antigen in non-M1-serotype Group A Streptococcus. mBio 2018, 9, e02217–e02294. [Google Scholar] [CrossRef] [PubMed]
- Kabanova, A.; Margarit, I.; Berti, F.; Romano, M.R.; Grandi, G.; Bensi, G.; Chiarot, E.; Proietti, D.; Swennen, E.; Cappelletti, E.; et al. Evaluation of a Group A Streptococcus synthetic oligosaccharide as vaccine candidate. Vaccine 2010, 29, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.T.; Gotz, F. Lipoproteins of gram-positive bacteria: Key players in the immune response and virulence. Microbiol. Mol. Biol. R. 2016, 80, 891–903. [Google Scholar] [CrossRef] [PubMed]
- Zaman, M.; Abdel-Aal, A.B.; Fujita, Y.; Phillipps, K.S.; Batzloff, M.R.; Good, M.F.; Toth, I. Immunological evaluation of lipopeptide Group A Streptococcus (GAS) vaccine: structure-activity relationship. PLoS ONE 2012, 7, e30146. [Google Scholar] [CrossRef] [PubMed]
- Skwarczynski, M.; Dougall, A.M.; Khoshnejad, M.; Chandrudu, S.; Pearson, M.S.; Loukas, A.; Toth, I. Peptide-based subunit vaccine against hookworm infection. PLoS ONE 2012, 7, e46870. [Google Scholar] [CrossRef] [PubMed]
- Dougall, A.M.; Skwarczynski, M.; Khoshnejad, M.; Chandrudu, S.; Daly, N.L.; Toth, I.; Loukas, A. Lipid core peptide targeting the cathepsin D hemoglobinase of Schistosoma mansoni as a component of a schistosomiasis vaccine. Hum. Vaccines Immunother. 2014, 10, 399–409. [Google Scholar] [CrossRef]
- Hussein, W.M.; Choi, P.; Zhang, C.; Su, M.; Sierecki, E.; Johnston, W.; Fagan, V.; Alexandrov, K.; Skwarczynski, M.; Gambin, Y.; et al. Evaluation of lipopeptides as toll-like receptor 2 ligands. Curr. Drug Deliv. 2016. [Google Scholar] [CrossRef]
- Zeng, W.; Eriksson, E.; Chua, B.; Grollo, L.; Jackson, D.C. Structural requirement for the agonist activity of the TLR2 ligand Pam2Cys. Amino Acids 2010, 39, 471–480. [Google Scholar] [CrossRef]
- Hussein, W.M.; Liu, T.Y.; Skwarczynski, M.; Toth, I. Toll-like receptor agonists: A patent review (2011 - 2013). Expert Opin. Ther. Pat. 2014, 24, 453–470. [Google Scholar] [CrossRef]
- Zaman, M.; Toth, I. Immunostimulation by synthetic lipopeptide-based vaccine candidates: Structure-activity relationships. Front. Immunol. 2013, 4, 318. [Google Scholar] [CrossRef]
- Ignacio, B.J.; Albin, T.J.; Esser-Kahn, A.P.; Verdoes, M. Toll-like Receptor Agonist Conjugation: A Chemical Perspective. Bioconjug. Chem. 2018, 29, 587–603. [Google Scholar] [CrossRef] [PubMed]
- Horvath, A.; Olive, C.; Wong, A.; Clair, T.; Yarwood, P.; Good, M.; Toth, I. A lipophilic adjuvant carrier system for antigenic peptides. Lett. Pept. Sci. 2001, 8, 285–288. [Google Scholar] [CrossRef]
- Arai, Y.; Inuki, S.; Fujimoto, Y. Site-specific effect of polar functional group-modification in lipids of TLR2 ligands for modulating the ligand immunostimulatory activity. Bioorg. Med. Chem. Lett. 2018, 28, 1638–1641. [Google Scholar] [CrossRef] [PubMed]
- Moyle, P.M.; Dai, W.; Zhang, Y.; Batzloff, M.R.; Good, M.F.; Toth, I. Site-specific incorporation of three toll-like receptor 2 targeting adjuvants into semisynthetic, molecularly defined nanoparticles: Application to Group A Streptococcal vaccines. Bioconjug. Chem. 2014, 25, 965–978. [Google Scholar] [CrossRef] [PubMed]
- Batzloff, M.R.; Hartas, J.; Zeng, W.; Jackson, D.C.; Good, M.F. Intranasal vaccination with a lipopeptide containing a conformationally constrained conserved minimal peptide, a universal T cell epitope, and a self-adjuvanting lipid protects mice from Group A Streptococcus challenge and reduces throat colonization. J. Infect. Dis 2006, 194, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Barrett, J.C.; Ulery, B.D.; Trent, A.; Liang, S.; David, N.A.; Tirrell, M.V. Modular peptide amphiphile micelles improving an antibody-mediated immune response to Group A Streptococcus. ACS Biomater. Sci. Eng. 2017, 3, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Fagan, V.; Hussein, W.M.; Su, M.; Giddam, A.K.; Batzloff, M.R.; Good, M.F.; Toth, I.; Simerska, P. Synthesis, characterization and immunological evaluation of self-adjuvanting Group A Streptococcal vaccine candidates bearing various lipidic adjuvanting moieties. ChemBioChem 2017, 18, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Eskandari, S.; Pattinson, D.J.; Stephenson, R.J.; Groves, P.L.; Apte, S.H.; Sedaghat, B.; Chandurudu, S.; Doolan, D.L.; Toth, I. Influence of physicochemical properties of lipopeptide adjuvants on the immune response: A rationale for engineering a potent vaccine. Chem. Eur. J. 2018, 24, 9892–9902. [Google Scholar] [CrossRef]
- Eskandari, S.; Stephenson, R.J.; Fuaad, A.A.; Apte, S.H.; Doolan, D.L.; Toth, I. Synthesis and characterisation of self-assembled and self-adjuvanting asymmetric multi-epitope lipopeptides of ovalbumin. Chem. Eur. J. 2015, 21, 1251–1261. [Google Scholar] [CrossRef]
- Chan, A.; Hussein, W.M.; Ghaffar, K.A.; Marasini, N.; Mostafa, A.; Eskandari, S.; Batzloff, M.R.; Good, M.F.; Skwarczynski, M.; Toth, I. Structure-activity relationship of lipid core peptide-based Group A Streptococcus vaccine candidates. Bioorg. Med. Chem. 2016, 24, 3095–3101. [Google Scholar] [CrossRef]
- Tam, J.P. Synthetic peptide vaccine design: Synthesis and properties of a high-density multiple antigenic peptide system. Proc. Natl. Acad. Sci. USA 1988, 85, 5409–5413. [Google Scholar] [CrossRef] [PubMed]
- Fujita, Y.; Taguchi, H. Nanoparticle-based peptide vaccines. In Micro and Nanotechnology in Vaccine Development; Skwarczynski, M., Toth, I., Eds.; William Andrew Publishing: Norwich, NY, USA, 2017; pp. 149–170. [Google Scholar]
- Ketchum, C.; Miller, H.; Song, W.; Upadhyaya, A. Ligand mobility regulates B cell receptor clustering and signaling activation. Biophys. J. 2014, 106, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Olive, C.; Batzloff, M.R.; Horvath, A.; Wong, A.; Clair, T.; Yarwood, P.; Toth, I.; Good, M.F. A lipid core peptide construct containing a conserved region determinant of the Group A Streptococcal M protein elicits heterologous opsonic antibodies. Infect. Immun. 2002, 70, 2734–2738. [Google Scholar] [CrossRef] [PubMed]
- Olive, C.; Hsien, K.; Horvath, A.; Clair, T.; Yarwood, P.; Toth, I.; Good, M.F. Protection against Group A Streptococcal infection by vaccination with self-adjuvanting lipid core M protein peptides. Vaccine 2005, 23, 2298–2303. [Google Scholar] [CrossRef] [PubMed]
- Zaman, M.; Abdel-Aal, A.B.; Phillipps, K.S.; Fujita, Y.; Good, M.F.; Toth, I. Structure-activity relationship of lipopeptide Group A Streptococcus (GAS) vaccine candidates on toll-like receptor 2. Vaccine 2010, 28, 2243–2248. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Aal, A.B.; Batzloff, M.R.; Fujita, Y.; Barozzi, N.; Faria, A.; Simerska, P.; Moyle, P.M.; Good, M.F.; Toth, I. Structure-activity relationship of a series of synthetic lipopeptide self-adjuvanting Group A Streptococcal vaccine candidates. J. Med. Chem. 2008, 51, 167–172. [Google Scholar] [CrossRef]
- Zaman, M.; Chandrudu, S.; Giddam, A.K.; Reiman, J.; Skwarczynski, M.; McPhun, V.; Moyle, P.M.; Batzloff, M.R.; Good, M.F.; Toth, I. Group A Streptococcal vaccine candidate: Contribution of epitope to size, antigen presenting cell interaction and immunogenicity. Nanomedicine 2014, 9, 2613–2624. [Google Scholar] [CrossRef]
- Marasini, N.; Abdul Ghaffar, K.; Kumar Giddam, A.R.; Batzloff, M.F.; Good, M.; Skwarczynski, M.; Toth, I. Highly immunogenic trimethyl chitosan-based delivery system for intranasal lipopeptide vaccines against Group A Streptococcus. Curr. Drug Deliv. 2017, 14, 701–708. [Google Scholar] [CrossRef]
- Ghaffar, K.A.; Marasini, N.; Giddam, A.K.; Batzloff, M.R.; Good, M.F.; Skwarczynski, M.; Toth, I. Liposome-based intranasal delivery of lipopeptide vaccine candidates against Group A Streptococcus. Acta Biomater. 2016, 41, 161–168. [Google Scholar] [CrossRef]
- Zhong, W.; Skwarczynski, M.; Simerska, P.; Good, M.F.; Toth, I. Development of highly pure alpha-helical lipoglycopeptides as self-adjuvanting vaccines. Tetrahedron 2009, 65, 3459–3464. [Google Scholar] [CrossRef]
- Zhong, W.; Skwarczynski, M.; Fujita, Y.; Simerska, P.; Good, M.F.; Toth, I. Design and synthesis of lipopeptide-carbohydrate assembled multivalent vaccine candidates using native chemical ligation. Aust. J. Chem. 2009, 62, 993–999. [Google Scholar] [CrossRef]
- Simerska, P.; Abdel-Aal, A.B.M.; Fujita, Y.; Moyle, P.M.; McGeary, R.P.; Batzloff, M.R.; Olive, C.; Good, M.F.; Toth, I. Development of a liposaccharide-based delivery system and its application to the design of Group A Streptococcal vaccines. J. Med. Chem. 2008, 51, 1447–1452. [Google Scholar] [CrossRef] [PubMed]
- Simerska, P.; Moyle, P.M.; Toth, I. Modern lipid-, carbohydrate-, and peptide-based delivery systems for peptide, vaccine, and gene products. Med. Res. Rev. 2011, 31, 520–547. [Google Scholar] [CrossRef] [PubMed]
- Simerska, P.; Abdel-Aal, A.B.; Fujita, Y.; Batzloff, M.R.; Good, M.F.; Toth, I. Synthesis and in vivo studies of carbohydrate-based vaccines against Group A Streptococcus. Biopolymers 2008, 90, 611–616. [Google Scholar] [CrossRef] [PubMed]
- Fujita, Y.; Abdel-Aal, A.B.; Wimmer, N.; Batzloff, M.R.; Good, M.F.; Toth, I. Synthesis and immunological evaluation of self-adjuvanting glycolipopeptide vaccine candidates. Bioorg. Med. Chem. 2008, 16, 8907–8913. [Google Scholar] [CrossRef]
- Shae, D.; Postma, A.; Wilson, J.T. Vaccine delivery: where polymer chemistry meets immunology. Ther. Deliv. 2016, 7, 193–196. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.M.; Videira, M.; Gaspar, R.; Preat, V.; Florindo, H.F. Immune system targeting by biodegradable nanoparticles for cancer vaccines. J. Control. Release 2013, 168, 179–199. [Google Scholar] [CrossRef]
- Zolnik, B.S.; Burgess, D.J. Effect of acidic pH on PLGA microsphere degradation and release. J. Control. Release 2007, 122, 338–344. [Google Scholar] [CrossRef]
- Danhier, F.; Ansorena, E.; Silva, J.M.; Coco, R.; Le Breton, A.; Preat, V. PLGA-based nanoparticles: an overview of biomedical applications. J. Control. Release 2012, 161, 505–522. [Google Scholar] [CrossRef]
- Shive, M.S.; Anderson, J.M. Biodegradation and biocompatibility of PLA and PLGA microspheres. Adv. Drug Deliv. Rev. 1997, 28, 5–24. [Google Scholar] [CrossRef]
- Li, X.; Min, M.; Du, N.; Gu, Y.; Hode, T.; Naylor, M.; Chen, D.; Nordquist, R.E.; Chen, W.R. Chitin, chitosan, and glycated chitosan regulate immune responses: The novel adjuvants for cancer vaccine. Clin. Dev. Immunol. 2013, 2013, 387023. [Google Scholar] [CrossRef] [PubMed]
- Lambricht, L.; Peres, C.; Florindo, H.; Préat, V.; Vandermeulen, G. Polymer-based nanoparticles as modern vaccine delivery systems. In Micro and Nanotechnology in Vaccine Development; Skwarczynski, M., Toth, I., Eds.; William Andrew Publishing: Norwich, NY, USA, 2017; pp. 185–203. [Google Scholar]
- Marasini, N.; Giddam, A.K.; Khalil, Z.G.; Hussein, W.M.; Capon, R.J.; Batzloff, M.R.; Good, M.F.; Toth, I.; Skwarczynski, M. Double adjuvanting strategy for peptide-based vaccines: Trimethyl chitosan nanoparticles for lipopeptide delivery. Nanomedicine 2016, 11, 3223–3235. [Google Scholar] [CrossRef] [PubMed]
- Marasini, N.; Khalil, Z.G.; Giddam, A.K.; Ghaffar, K.A.; Hussein, W.M.; Capon, R.J.; Batzloff, M.R.; Good, M.F.; Skwarczynski, M.; Toth, I. Lipid core peptide/poly(lactic-co-glycolic acid) as a highly potent intranasal vaccine delivery system against Group A Streptococcus. Int. J. Pharm. 2016, 513, 410–420. [Google Scholar] [CrossRef] [PubMed]
- Nevagi, R.J.; Khalil, Z.G.; Hussein, W.M.; Powell, J.; Batzloff, M.R.; Capon, R.J.; Good, M.F.; Skwarczynski, M.; Toth, I. Polyglutamic acid-trimethyl chitosan-based intranasal peptide nano-vaccine induces potent immune responses against Group A Streptococcus. Acta Biomater. 2018, 80, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Skwarczynski, M.; Zaman, M.; Urbani, C.N.; Lin, I.C.; Jia, Z.; Batzloff, M.R.; Good, M.F.; Monteiro, M.J.; Toth, I. Polyacrylate dendrimer nanoparticles: a self-adjuvanting vaccine delivery system. Angew. Chem. Int. Ed. Engl. 2010, 49, 5742–5745. [Google Scholar] [CrossRef] [PubMed]
- Zaman, M.; Skwarczynski, M.; Malcolm, J.M.; Urbani, C.N.; Jia, Z.F.; Batzloff, M.R.; Good, M.F.; Monteiro, M.J.; Toth, I. Self-adjuvanting polyacrylic nanoparticulate delivery system for Group A Streptococcus (GAS) vaccine. Nanomed. Nanotechnol. Biol. Med. 2011, 7, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Ahmad Fuaad, A.A.; Jia, Z.; Zaman, M.; Hartas, J.; Ziora, Z.M.; Lin, I.C.; Moyle, P.M.; Batzloff, M.R.; Good, M.F.; Monteiro, M.J.; et al. Polymer-peptide hybrids as a highly immunogenic single-dose nanovaccine. Nanomedicine 2014, 9, 35–43. [Google Scholar] [CrossRef]
- Chandrudu, S.; Bartlett, S.; Khalil, Z.G.; Jia, Z.; Hussein, W.M.; Capon, R.J.; Batzloff, M.R.; Good, M.F.; Monteiro, M.J.; Skwarczynski, M.; et al. Linear and branched polyacrylates as a delivery platform for peptide-based vaccines. Ther. Deliv. 2016, 7, 601–609. [Google Scholar] [CrossRef]
- Torchilin, V.P. Recent advances with liposomes as pharmaceutical carriers. Nat. Rev. Drug Discov. 2005, 4, 145–160. [Google Scholar] [CrossRef]
- Henriksen-Lacey, M.; Korsholm, K.S.; Andersen, P.; Perrie, Y.; Christensen, D. Liposomal vaccine delivery systems. Expert Opin. Drug Deliv. 2011, 8, 505–519. [Google Scholar] [CrossRef]
- Gregoriadis, G. Engineering liposomes for drug delivery: Progress and problems. Trends Biotechnol. 1995, 13, 527–537. [Google Scholar] [CrossRef]
- Allison, A.C.; Gregoriadis, G. Liposomes as immunological adjuvants. In Recent Results in Cancer Research, 1976/01/01 ed.; Mathé, G., Florentin, I., Simmler, M.C., Eds.; Springer: Berlin, Germany; Heidelberg, Germany, 1976; pp. 58–64. [Google Scholar]
- Allison, A.G.; Gregoriadis, G. Liposomes as immunological adjuvants. Nature 1974, 252, 252. [Google Scholar] [CrossRef] [PubMed]
- Ghaffar, K.A.; Giddam, A.K.; Zaman, M.; Skwarczynski, M.; Toth, I. Liposomes as nanovaccine delivery systems. Curr. Top. Med. Chem. 2014, 14, 1194–1208. [Google Scholar] [CrossRef] [PubMed]
- Marasini, N.; Ghaffar, K.A.; Skwarczynski, M.; Toth, I. Liposomes as a Vaccine Delivery System; William Andrew Inc.: Norwich, UK, 2017; pp. 221–223. [Google Scholar]
- Giddam, A.K.; Zaman, M.; Skwarczynski, M.; Toth, I. Liposome-based delivery system for vaccine candidates: Constructing an effective formulation. Nanomedicine 2012, 7, 1877–1893. [Google Scholar] [CrossRef] [PubMed]
- Black, M.; Trent, A.; Tirrell, M.; Olive, C. Advances in the design and delivery of peptide subunit vaccines with a focus on toll-like receptor agonists. Expert Rev. Vaccines 2010, 9, 157–173. [Google Scholar] [CrossRef] [PubMed]
- Ghaffar, K.A.; Marasini, N.; Giddam, A.K.; Batzloff, M.R.; Good, M.F.; Skwarczynski, M.; Toth, I. The role of size in development of mucosal liposome-lipopeptide vaccine candidates against Group A Streptococcus. Med. Chem. 2016, 13, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Zaman, M.; Ozberk, V.; Langshaw, E.L.; McPhun, V.; Powell, J.L.; Phillips, Z.N.; Ho, M.F.; Calcutt, A.; Batzloff, M.R.; Toth, I.; et al. Novel platform technology for modular mucosal vaccine that protects against Streptococcus. Sci. Rep. UK 2016, 6. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Non-Invasive Diseases | ||
| Diseases | Description | Symptoms |
| Pharyngitis | Benign local throat infection. Common manifestation of GAS infection. Arises from complex host-pathogen interaction. | Sore throat, high fever, cervical lymphadenopathy, tonsil exudates, raised peripheral white cell count. |
| Tonsillitis | Benign local tonsil infection. | White/yellow spots on tonsils, sore throat, swollen jaw lymph glands, fever, bad breath. |
| Impetigo/Pyoderma | Benign local skin infection. Common in childhood. Arises from complex host-pathogen interaction. | Superficial, non-follicular, crusted lesion on the face and other exposed body parts. |
| Scarlet fever | Disease that can follow an episode of GAS-mediated pharyngitis. Commonly occurs in children. | Diffuse blanching rash on chest to abdomen, sandpaper-like texture to the skin. |
| Otitis media | Infection in the middle ear. | Otalgia, otorrhea, headache, fever, appetite loss, vomiting, diarrhoea, hearing loss, tinnitus, vertigo. |
| Invasive Diseases | ||
| Diseases | Description | Symptoms |
| Cellulitis | Painful skin infection on deeper subcutaneous tissue. Frequent manifestation of invasive GAS. Incidence increases with age. | Fever, systemic toxicity, inflammation of the skin. |
| Pneumonia | Infection of the lung. | Difficulty in breathing, fever, appetite loss, abdominal pain, headache, chest pain, cough, cyanosis. |
| Necrotising fasciitis (flesh eating bacteria) | Rapidly progressing skin infection that causes a destruction of subcutaneous fat, tissue and muscle. | Fever, spared overlying skin, severe pain, violaceous, bullae and slough skin, shock, multi-organ failure. |
| Streptococcal toxic shock syndrome (STSS) | Associated with GAS necrotising fasciitis. | High fever, hypotension, rash, coagulopathy, respiratory distress syndrome, renal failure, hepatic impairment, multi-organ failure. |
| Erysipelas | Painful skin infection. Frequent manifestation of invasive GAS. Incidence increases with age. | Fever, systemic toxicity, clear demarcated red inflammation, formation of superficial bullae. |
| Meningitis | Inflammation of the meninges. GAS is an uncommon cause of meningitis. | Fever, headache, stiff/sore neck, vomiting, appetite loss, tiredness, drowsiness, irritability. |
| Bacteraemia/septicaemia | Blood poisoning due to the presence of bacteria/toxin in the blood. | Sudden high fever with chills, nausea, vomiting, diarrhoea, abdominal pain, confusion, anxiety, tachycardia. |
| Lymphangitis | Infection of draining lymphatic tracts. | Tender linear streak extending from the site of infection. |
| Septic arthritis | Painful infection of the joint following episode of GAS-mediated pharyngitis. | Fever, enlarged joints. |
| Puerperal sepsis | Infection resulting from the birthing process. Frequent cause of death in the pre-antibiotic era. | Postpartum endometritis, peritonitis, septic, thrombophlebitis/bacteraemia without focus, fever for 24 h or recurring after childbirth/abortion. |
| Post-Infectious Diseases | ||
| Diseases | Description | Symptoms |
| Rheumatic fever (RF) | Inflammatory disease caused by cross-reactive antibodies induced after GAS infection. | The combination of fever, polyarthritis/arthralgia, carditis, erythema marginatum, chorea, subcutaneous nodules, and mitral/aortic valve damage. Can turn into RHD. |
| Rheumatic heart disease (RHD) | Inflammation caused by cross-reactive antibodies induced after GAS infection leading to permanent damage to heart tissue and valves. | Tissue inflammation that results in carditis, valvulitis, arthritis, chorea, erythema marginatum and/or subcutaneous nodules. |
| Post-streptococcal glomerulonephritis (PSGN) | Inflammation of the glomeruli in the kidney caused by a build-up of immune complexes induced by GAS infection. Follows an episode of GAS-mediated pharyngitis/pyoderma. | Rapid onset of gross/microscopic haematuria oedema, hypertension, and encephalopathy. |
| Post-streptococcal reactive arthritis | Syndrome of polyarthritis that differs from acute RF/carditis. | Range of smaller joints unreactive to anti-inflammatory treatment. |
| Paediatric autoimmune neuropsychiatric disorder associated with GAS infection (PANDAS) | Cross-reactive antibodies induced after GAS infection that interferes with basal ganglia function causing symptoms of exacerbation in children. The existence of this disease is controversial. | Children with tie/obsessive compulsive disorder (OCD) worsened/developed after GAS infection. |
| Name | Stage of Development | Comments | Ref. | ||
|---|---|---|---|---|---|
| Preclinical | Phase 1 | Phase 2 | |||
| 6-valent vaccine | 9 white rabbits IM injection | 28 healthy adults | NI | Tolerable. No human tissue cross-reactivity. No clinical complications. Limited by small-scale trials. | [70,71] |
| 26-valent vaccine | 3 white rabbits IM injection | 30 healthy adults IM injection | 90 healthy adults IM injection | No evidence of RF or human tissue cross-reactivity. Highly immunogenic No control group was set. | [67,72,73] |
| 30-valent vaccine | 12 white rabbits IM injection | NI | NI | Highly immunogenic. Potential efficacy against non-vaccine-targeted GAS serotypes | [68,74] |
| 5-valent E4 vaccine | 3 white rabbits IM injection | NI | NI | Highly immunogenic. Potential to provide broad protection against GAS | [75] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azuar, A.; Jin, W.; Mukaida, S.; Hussein, W.M.; Toth, I.; Skwarczynski, M. Recent Advances in the Development of Peptide Vaccines and Their Delivery Systems against Group A Streptococcus. Vaccines 2019, 7, 58. https://doi.org/10.3390/vaccines7030058
Azuar A, Jin W, Mukaida S, Hussein WM, Toth I, Skwarczynski M. Recent Advances in the Development of Peptide Vaccines and Their Delivery Systems against Group A Streptococcus. Vaccines. 2019; 7(3):58. https://doi.org/10.3390/vaccines7030058
Chicago/Turabian StyleAzuar, Armira, Wanli Jin, Saori Mukaida, Waleed M. Hussein, Istvan Toth, and Mariusz Skwarczynski. 2019. "Recent Advances in the Development of Peptide Vaccines and Their Delivery Systems against Group A Streptococcus" Vaccines 7, no. 3: 58. https://doi.org/10.3390/vaccines7030058
APA StyleAzuar, A., Jin, W., Mukaida, S., Hussein, W. M., Toth, I., & Skwarczynski, M. (2019). Recent Advances in the Development of Peptide Vaccines and Their Delivery Systems against Group A Streptococcus. Vaccines, 7(3), 58. https://doi.org/10.3390/vaccines7030058