
Human T Cell Memory: A Dynamic View

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Part I: Thinking Kinetically—How Long Is a Memory?
2.1. The Longevity of Immune Responses
2.2. The Paradox of Long-Lived Memory and Short-Lived Cells
2.3. Resolving the Paradox—Short-Lived Cells Conferring Long Lived-Memory
2.4. Are There Really No Long-Lived Memory-Cells?
2.5. Circulating Memory Cells
2.6. Tissue-Resident Memory
3. Part II—The Vaccine Response—A Kinetic Model
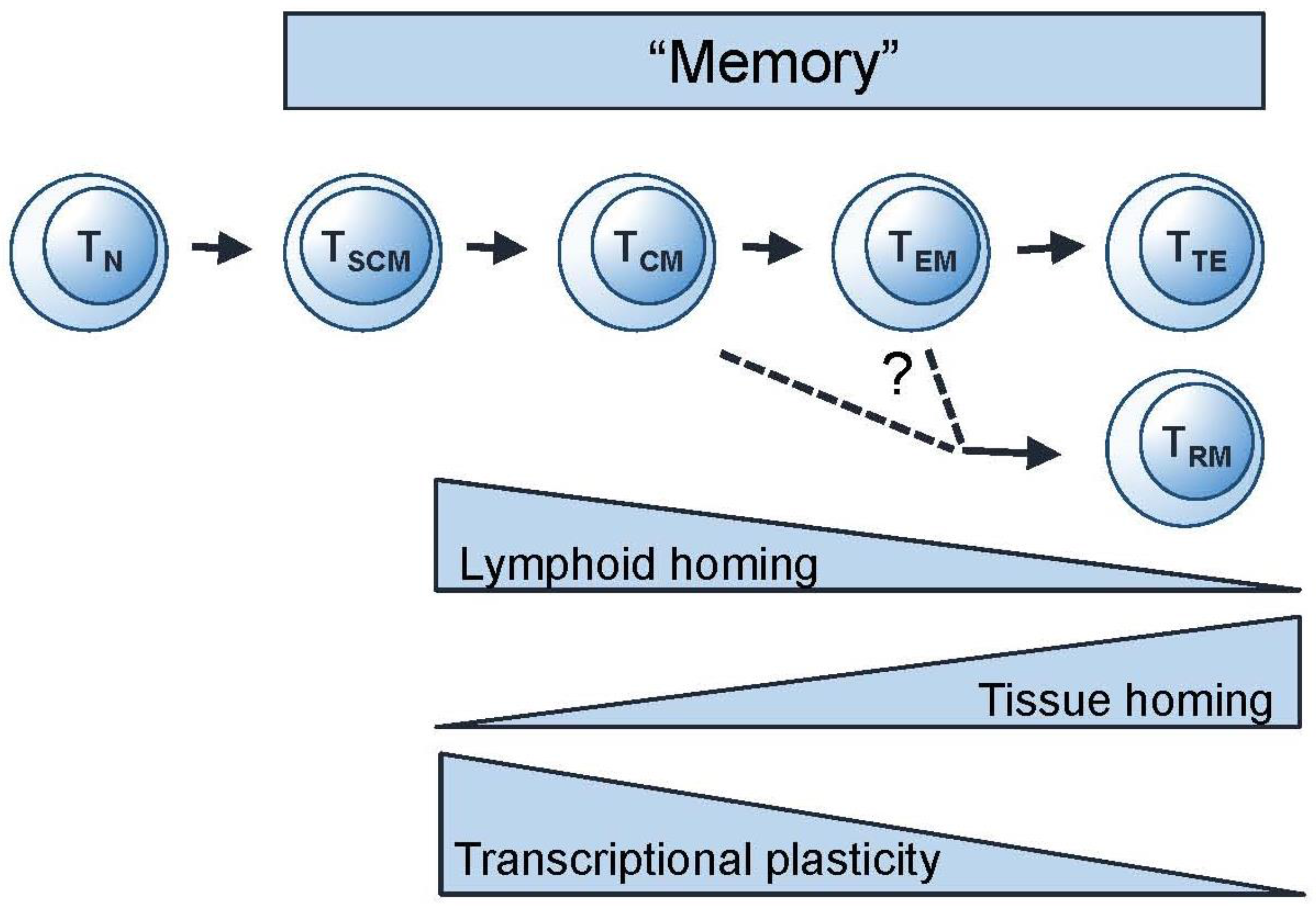
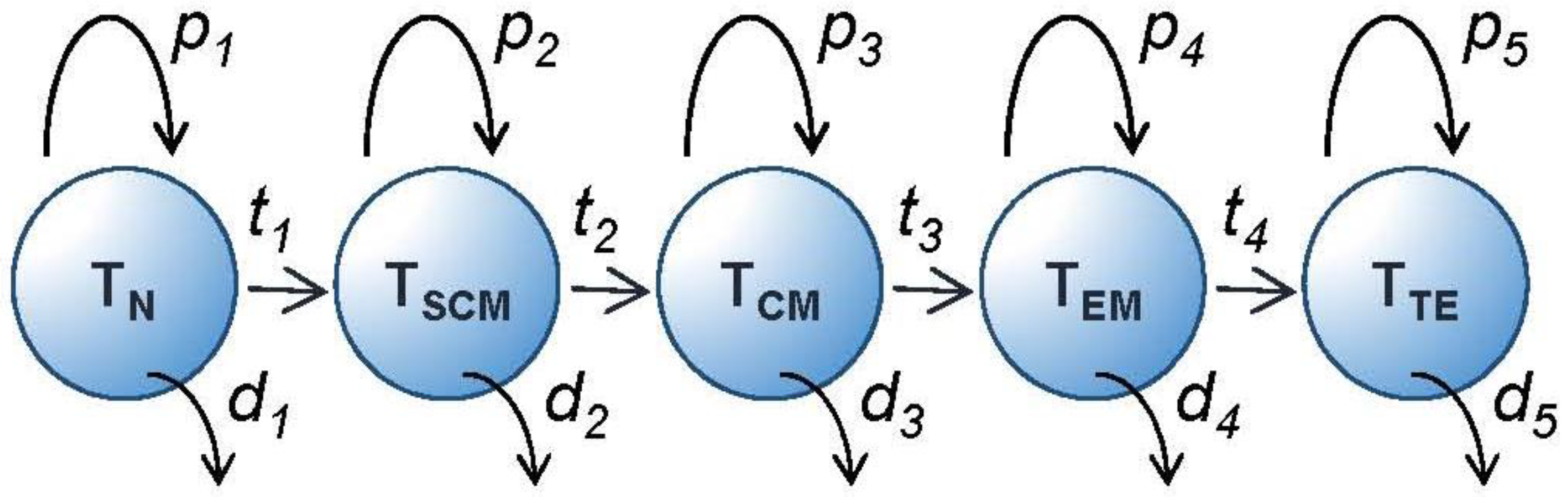
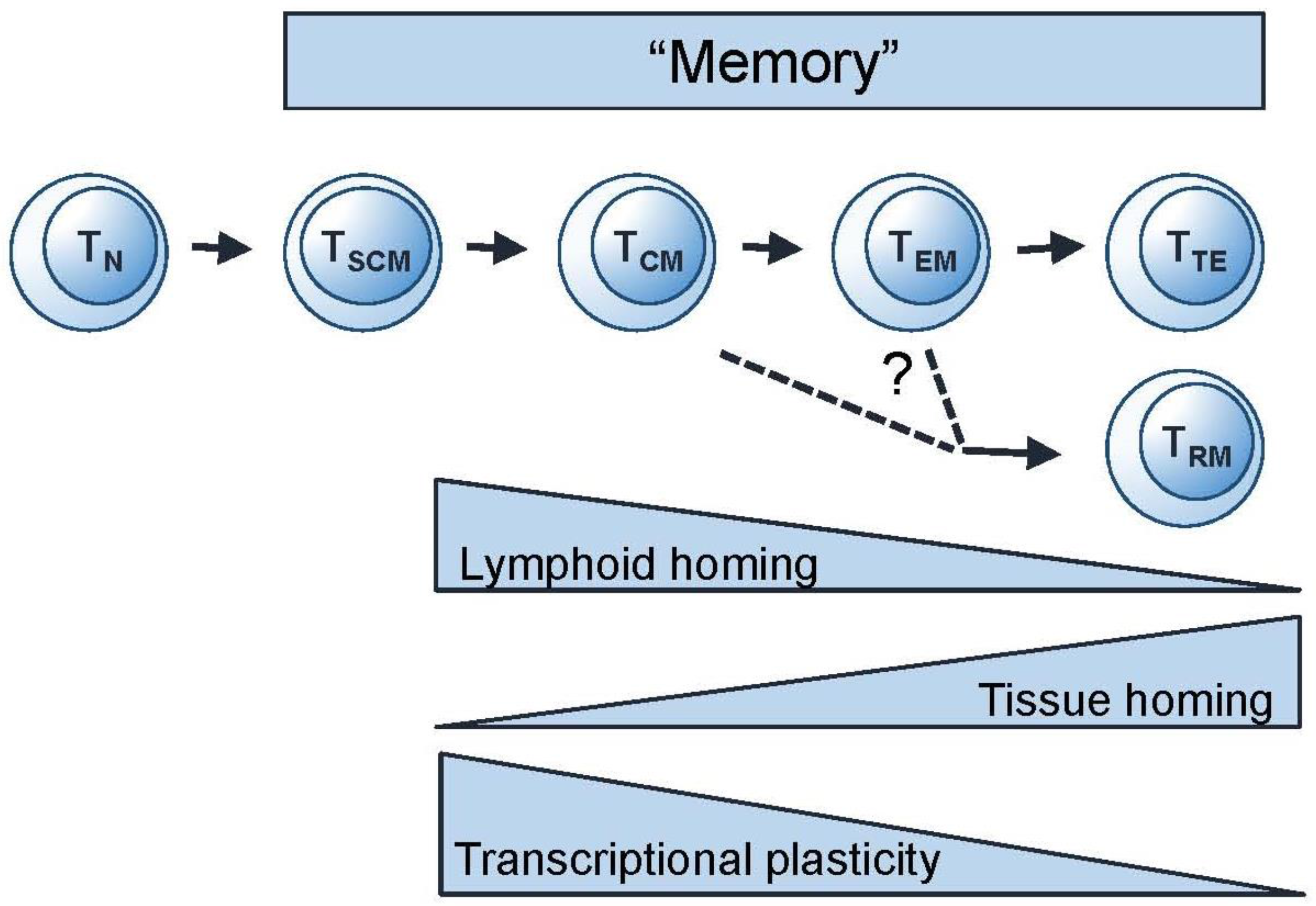
3.1. Thinking Phenotypically—Differentiation Pathways for Memory T Cells
3.2. Thinking Clonotypically—Remodelling and Focussing
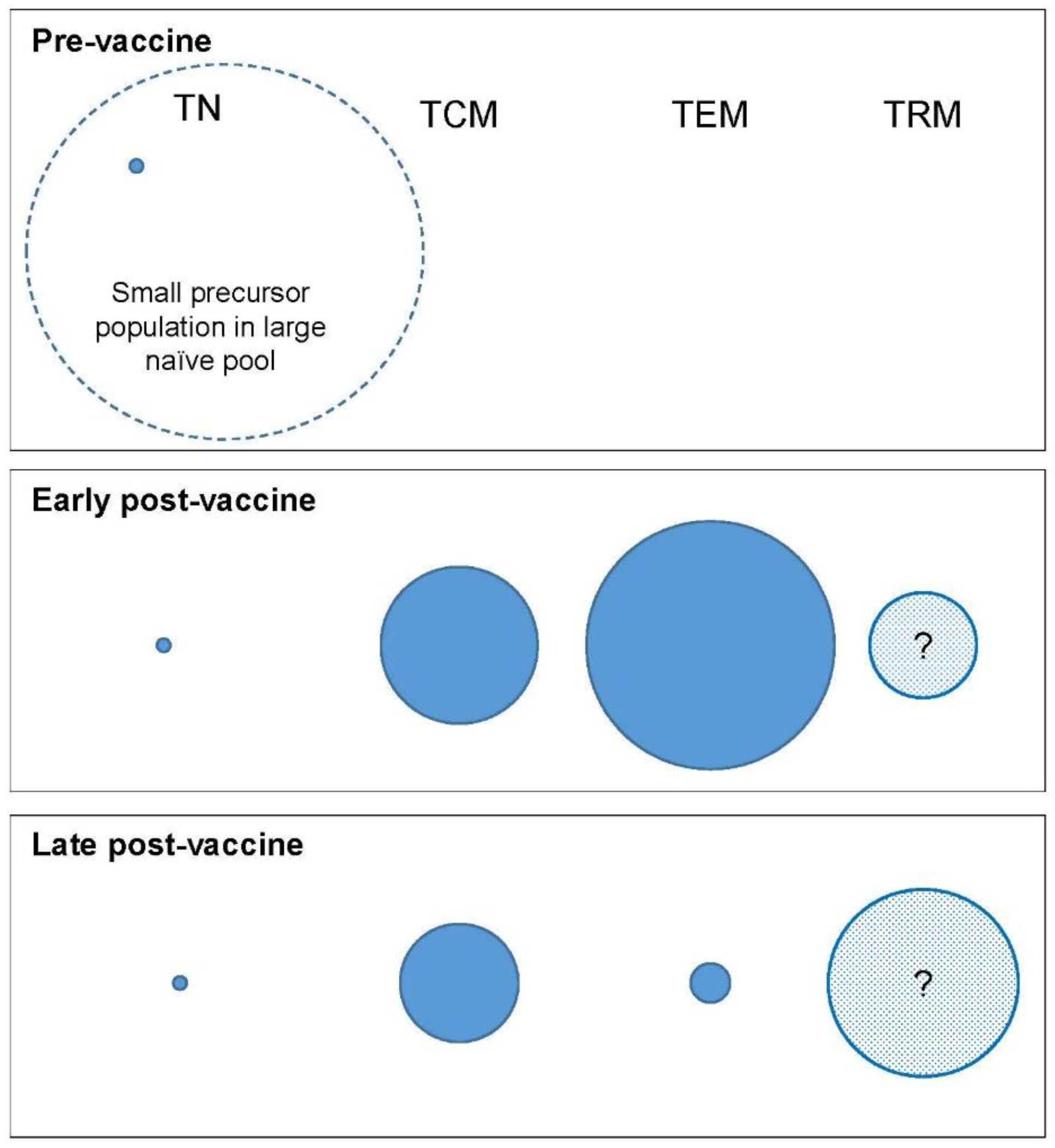
3.3. Preservation of a Memory Population—Stochastic or Deterministic?
4. Part III—Consequences of Thinking Kinetically about Vaccines
4.1. Rethinking the Target—Beyond ‘Bigger Is Better’
4.2. Focussing on Phenotype—and Beyond
4.3. Room for Change
4.4. Dynamic Systems Offer Opportunities for Modulation
4.5. Thinking Locally—Directing the Immune Response
4.6. Implications for Viral Latency
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Gotuzzo, E.; Yactayo, S.; Cordova, E. Efficacy and duration of immunity after yellow fever vaccination: Systematic review on the need for a booster every 10 years. Am. J. Trop. Med. Hyg. 2013, 89, 434–444. [Google Scholar] [CrossRef] [PubMed]
- Fisman, D.N.; Savage, R.; Gubbay, J.; Achonu, C.; Akwar, H.; Farrell, D.J.; Crowcroft, N.S.; Jackson, P. Older age and a reduced likelihood of 2009 H1N1 virus infection. N. Engl. J. Med. 2009, 361, 2000–2001. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Ekiert, D.C.; Krause, J.C.; Hai, R.; Crowe, J.E., Jr.; Wilson, I.A. Structural basis of preexisting immunity to the 2009 H1N1 pandemic influenza virus. Science 2010, 328, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Hammarlund, E.; Lewis, M.W.; Hansen, S.G.; Strelow, L.I.; Nelson, J.A.; Sexton, G.J.; Hanifin, J.M.; Slifka, M.K. Duration of antiviral immunity after smallpox vaccination. Nat. Med. 2003, 9, 1131–1137. [Google Scholar] [CrossRef] [PubMed]
- Crotty, S.; Felgner, P.; Davies, H.; Glidewell, J.; Villarreal, L.; Ahmed, R. Cutting edge: Long-term B cell memory in humans after smallpox vaccination. J. Immunol. 2003, 171, 4969–4973. [Google Scholar] [CrossRef] [PubMed]
- Tough, D.F.; Sprent, J. Life span of naive and memory T cells. Stem Cells 1995, 13, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Macallan, D.C.; Wallace, D.L.; Zhang, Y.; Ghattas, H.; Asquith, B.; de Lara, C.; Worth, A.; Panayiotakopoulos, G.; Griffin, G.E.; Tough, D.F.; et al. B cell kinetics in humans: Rapid turnover of peripheral blood memory cells. Blood 2005, 105, 3633–3640. [Google Scholar] [CrossRef] [PubMed]
- McLean, A.; Michie, C.A. In vivo estimates of division and death rates of human T lymphocytes. Proc. Natl. Acad. Sci. USA 1995, 92, 3707–3711. [Google Scholar] [CrossRef] [PubMed]
- Sachsenberg, N.; Perelson, A.S.; Yerly, S.; Schockmel, G.A.; Leduc, D.; Hirschel, B.; Perrin, L. Turnover of CD4+ and CD8+ T lymphocytes in HIV-1 infection as measured by Ki-67 antigen. J. Exp. Med. 1998, 187, 1295–1303. [Google Scholar] [CrossRef] [PubMed]
- Hazenberg, M.D.; Stuart, J.W.; Otto, S.A.; Borleffs, J.C.; Boucher, C.A.; De Boer, R.J.; Miedema, F.; Hamann, D. T-cell division in human immunodeficiency virus (HIV)-1 infection is mainly due to immune activation: A longitudinal analysis in patients before and during highly active antiretroviral therapy (HAART). Blood 2000, 95, 249–255. [Google Scholar] [PubMed]
- Vrisekoop, N.; den, B.I.; de Boer, A.B.; Ruiter, A.F.; Ackermans, M.T.; van der Crabben, S.N.; Schrijver, E.H.; Spierenburg, G.; Sauerwein, H.P.; Hazenberg, M.D.; et al. Sparse production but preferential incorporation of recently produced naive T cells in the human peripheral pool. Proc. Natl. Acad. Sci. USA 2008, 105, 6115–6120. [Google Scholar] [CrossRef] [PubMed]
- Macallan, D.C.; Asquith, B.; Irvine, A.; Wallace, D.; Worth, A.; Ghattas, H.; Zhang, Y.; Griffin, G.E.; Tough, D.; Beverley, P.C. Measurement and Modeling of Human T cell kinetics. Eur. J. Immunol. 2003, 33, 2316–2326. [Google Scholar] [CrossRef] [PubMed]
- Macallan, D.C.; Wallace, D.; Zhang, Y.; de Lara, C.; Worth, A.T.; Ghattas, H.; Griffin, G.E.; Beverley, P.C.; Tough, D.F. Rapid turnover of effector-memory CD4(+) T cells in healthy humans. J. Exp. Med. 2004, 200, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Westera, L.; van Hoeven, V.; Drylewicz, J.; Spierenburg, G.; van Velzen, J.F.; de Boer, R.J.; Tesselaar, K.; Borghans, J.A. Lymphocyte maintenance during healthy aging requires no substantial alterations in cellular turnover. Aging Cell 2015, 14, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Westera, L.; Drylewicz, J.; den Braber, I.; Mugwagwa, T.; van der Maas, I.; Kwast, L.; Volman, T.; van de Weg-Schrijver, E.H.; Bartha, I.; Spierenburg, G.; et al. Closing the gap between T-cell life span estimates from stable isotope-labeling studies in mice and humans. Blood 2013, 122, 2205–2212. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, R.; Westera, L.; Drylewicz, J.; Elemans, M.; Zhang, Y.; Kelly, E.; Reljic, R.; Tesselaar, K.; de Boer, R.J.; Macallan, D.C.; et al. Reconciling Estimates of Cell Proliferation from Stable Isotope Labeling Experiments. PLoS Comput. Biol. 2015, 11, e1004355. [Google Scholar] [CrossRef] [PubMed]
- Michie, C.A.; McLean, A.; Alcock, C.; Beverley, P.C. Lifespan of human lymphocyte subsets defined by CD45 isoforms. Nature 1992, 360, 264–265. [Google Scholar] [CrossRef] [PubMed]
- Youngblood, B.; Hale, J.S.; Ahmed, R. T-cell memory differentiation: Insights from transcriptional signatures and epigenetics. Immunology 2013, 139, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Young, A.J. The physiology of lymphocyte migration through the single lymph node in vivo. Semin. Immunol. 1999, 11, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Mackay, L.K.; Stock, A.T.; Ma, J.Z.; Jones, C.M.; Kent, S.J.; Mueller, S.N.; Heath, W.R.; Carbone, F.R.; Gebhardt, T. Long-lived epithelial immunity by tissue-resident memory T (TRM) cells in the absence of persisting local antigen presentation. Proc. Natl. Acad. Sci. USA 2012, 109, 7037–7042. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Clark, R.A.; Liu, L.; Wagers, A.J.; Fuhlbrigge, R.C.; Kupper, T.S. Skin infection generates non-migratory memory CD8+ T(RM) cells providing global skin immunity. Nature 2012, 483, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Gebhardt, T.; Wakim, L.M.; Eidsmo, L.; Reading, P.C.; Heath, W.R.; Carbone, F.R. Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat. Immunol. 2009, 10, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Cuburu, N.; Graham, B.S.; Buck, C.B.; Kines, R.C.; Pang, Y.Y.; Day, P.M.; Lowy, D.R.; Schiller, J.T. Intravaginal immunization with HPV vectors induces tissue-resident CD8+ T cell responses. J. Clin. Investig. 2012, 122, 4606–4620. [Google Scholar] [CrossRef] [PubMed]
- Di Rosa, F. Maintenance of memory T cells in the bone marrow: Survival or homeostatic proliferation? Nat. Rev. Immunol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Okhrimenko, A.; Grun, J.R.; Westendorf, K.; Fang, Z.; Reinke, S.; von Roth, P.; Wassilew, G.; Kuhl, A.A.; Kudernatsch, R.; Demski, S.; et al. Human memory T cells from the bone marrow are resting and maintain long-lasting systemic memory. Proc. Natl. Acad. Sci. USA 2014, 111, 9229–9234. [Google Scholar] [CrossRef] [PubMed]
- Di Rosa, F. Commentary: Memory CD8+ T cells colocalize with IL-7+ stromal cells in bone marrow and rest in terms of proliferation and transcription. Front. Immunol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Sercan Alp, O.; Durlanik, S.; Schulz, D.; McGrath, M.; Grun, J.R.; Bardua, M.; Ikuta, K.; Sgouroudis, E.; Riedel, R.; Zehentmeier, S.; et al. Memory CD8+ T cells colocalize with IL-7+ stromal cells in bone marrow and rest in terms of proliferation and transcription. Eur. J. Immunol. 2015, 45, 975–987. [Google Scholar] [CrossRef] [PubMed]
- Cavanagh, L.L.; Bonasio, R.; Mazo, I.B.; Halin, C.; Cheng, G.; van der Velden, A.W.; Cariappa, A.; Chase, C.; Russell, P.; Starnbach, M.N.; et al. Activation of bone marrow-resident memory T cells by circulating, antigen-bearing dendritic cells. Nat. Immunol. 2005, 6, 1029–1037. [Google Scholar] [CrossRef] [PubMed]
- Schenkel, J.M.; Masopust, D. Tissue-resident memory T cells. Immunity 2014, 41, 886–897. [Google Scholar] [CrossRef] [PubMed]
- Mahnke, Y.D.; Brodie, T.M.; Sallusto, F.; Roederer, M.; Lugli, E. The who’s who of T-cell differentiation: Human memory T-cell subsets. Eur. J. Immunol. 2013, 43, 2797–2809. [Google Scholar] [CrossRef] [PubMed]
- Youngblood, B.; Hale, J.S.; Ahmed, R. Memory CD8 T cell transcriptional plasticity. F1000Prime Rep. 2015. [Google Scholar] [CrossRef] [PubMed]
- Appay, V.; Van Lier, R.A.; Sallusto, F.; Roederer, M. Phenotype and function of human T lymphocyte subsets: Consensus and issues. Cytometry A 2008, 73, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Teichgraber, V.; Becker, T.C.; Masopust, D.; Kaech, S.M.; Antia, R.; von Andrian, U.H.; Ahmed, R. Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat. Immunol. 2003, 4, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Restifo, N.P.; Gattinoni, L. Lineage relationship of effector and memory T cells. Curr. Opin. Immunol. 2013, 25, 556–563. [Google Scholar] [CrossRef] [PubMed]
- Hale, J.S.; Ahmed, R. Memory T follicular helper CD4 T cells. Front. Immunol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, C.; Rohr, J.C.; Perie, L.; van Rooij, N.; van Heijst, J.W.; Velds, A.; Urbanus, J.; Naik, S.H.; Jacobs, H.; Beltman, J.B.; et al. Heterogeneous differentiation patterns of individual CD8+ T cells. Science 2013, 340, 635–639. [Google Scholar] [CrossRef] [PubMed]
- Day, E.K.; Carmichael, A.J.; ten Berge, I.J.; Waller, E.C.; Sissons, J.G.; Wills, M.R. Rapid CD8+ T cell repertoire focusing and selection of high-affinity clones into memory following primary infection with a persistent human virus: Human cytomegalovirus. J. Immunol. 2007, 179, 3203–3213. [Google Scholar] [CrossRef] [PubMed]
- Kemball, C.C.; Lee, E.D.; Vezys, V.; Pearson, T.C.; Larsen, C.P.; Lukacher, A.E. Late priming and variability of epitope-specific CD8+ T cell responses during a persistent virus infection. J. Immunol. 2005, 174, 7950–7960. [Google Scholar] [CrossRef] [PubMed]
- Sen, P.; Charini, W.A.; Subbramanian, R.A.; Manuel, E.R.; Kuroda, M.J.; Autissier, P.A.; Letvin, N.L. Clonal focusing of epitope-specific CD8+ T lymphocytes in rhesus monkeys following vaccination and simian-human immunodeficiency virus challenge. J. Virol. 2008, 82, 805–816. [Google Scholar] [CrossRef] [PubMed]
- Akondy, R.S.; Johnson, P.L.; Nakaya, H.I.; Edupuganti, S.; Mulligan, M.J.; Lawson, B.; Miller, J.D.; Pulendran, B.; Antia, R.; Ahmed, R. Initial viral load determines the magnitude of the human CD8 T cell response to yellow fever vaccination. Proc. Natl. Acad. Sci. USA 2015, 112, 3050–3055. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.J.; Chu, H.H.; Pepper, M.; McSorley, S.J.; Jameson, S.C.; Kedl, R.M.; Jenkins, M.K. Naive CD4+ T Cell Frequency Varies for Different Epitopes and Predicts Repertoire Diversity and Response Magnitude. Immunity 2007, 27, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Picker, L.J. Are effector memory T cells the key to an effective HIV/AIDS vaccine? EMBO Rep. 2014, 15, 820–821. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.G.; Ford, J.C.; Lewis, M.S.; Ventura, A.B.; Hughes, C.M.; Coyne-Johnson, L.; Whizin, N.; Oswald, K.; Shoemaker, R.; Swanson, T.; et al. Profound early control of highly pathogenic SIV by an effector memory T-cell vaccine. Nature 2011, 473, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Angelosanto, J.M.; Blackburn, S.D.; Crawford, A.; Wherry, E.J. Progressive loss of memory T cell potential and commitment to exhaustion during chronic viral infection. J. Virol. 2012, 86, 8161–8170. [Google Scholar] [CrossRef] [PubMed]
- Wirth, T.C.; Xue, H.H.; Rai, D.; Sabel, J.T.; Bair, T.; Harty, J.T.; Badovinac, V.P. Repetitive antigen stimulation induces stepwise transcriptome diversification but preserves a core signature of memory CD8(+) T cell differentiation. Immunity 2010, 33, 128–140. [Google Scholar] [CrossRef] [PubMed]
- Vezys, V.; Yates, A.; Casey, K.A.; Lanier, G.; Ahmed, R.; Antia, R.; Masopust, D. Memory CD8 T-cell compartment grows in size with immunological experience. Nature 2009, 457, 196–199. [Google Scholar] [CrossRef] [PubMed]
- Odumade, O.A.; Knight, J.A.; Schmeling, D.O.; Masopust, D.; Balfour, H.H., Jr.; Hogquist, K.A. Primary Epstein-Barr virus infection does not erode preexisting CD8(+) T cell memory in humans. J. Exp. Med. 2012, 209, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Schenkel, J.M.; Fraser, K.A.; Casey, K.A.; Beura, L.K.; Pauken, K.E.; Vezys, V.; Masopust, D. IL-15-Independent Maintenance of Tissue-Resident and Boosted Effector Memory CD8 T Cells. J. Immunol. 2016, 196, 3920–3926. [Google Scholar] [CrossRef] [PubMed]
- Rosenblum, M.D.; Gratz, I.K.; Paw, J.S.; Abbas, A.K. Treating human autoimmunity: Current practice and future prospects. Sci. Transl. Med. 2012. [Google Scholar] [CrossRef] [PubMed]
- Buchbinder, E.; Hodi, F.S. Cytotoxic T lymphocyte antigen-4 and immune checkpoint blockade. J. Clin. Investig. 2015, 125, 3377–3383. [Google Scholar] [CrossRef] [PubMed]
- Kamphorst, A.O.; Araki, K.; Ahmed, R. Beyond adjuvants: Immunomodulation strategies to enhance T cell immunity. Vaccine 2015, 33 (Suppl. S2), B21–B28. [Google Scholar] [CrossRef] [PubMed]
- Richer, M.J.; Pewe, L.L.; Hancox, L.S.; Hartwig, S.M.; Varga, S.M.; Harty, J.T. Inflammatory IL-15 is required for optimal memory T cell responses. J. Clin. Investig. 2015, 125, 3477–3490. [Google Scholar] [CrossRef] [PubMed]
- Henson, S.M.; Macaulay, R.; Riddell, N.E.; Nunn, C.J.; Akbar, A.N. Blockade of PD-1 or p38 MAP kinase signaling enhances senescent human CD8(+) T-cell proliferation by distinct pathways. Eur. J. Immunol. 2015, 45, 1441–1451. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, A. Exploiting Mucosal Immunity for Antiviral Vaccines. Annu. Rev. Immunol. 2016, 34, 575–608. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.; Iwasaki, A. A vaccine strategy that protects against genital herpes by establishing local memory T cells. Nature 2012, 491, 463–467. [Google Scholar] [CrossRef] [PubMed]
- Ariotti, S.; Hogenbirk, M.A.; Dijkgraaf, F.E.; Visser, L.L.; Hoekstra, M.E.; Song, J.Y.; Jacobs, H.; Haanen, J.B.; Schumacher, T.N. T cell memory. Skin-resident memory CD8+ T cells trigger a state of tissue-wide pathogen alert. Science 2014, 346, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Murray, A.J.; Kwon, K.J.; Farber, D.L.; Siliciano, R.F. The Latent Reservoir for HIV-1: How Immunologic Memory and Clonal Expansion Contribute to HIV-1 Persistence. J. Immunol. 2016, 197, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Den Braber, I.; Mugwagwa, T.; Vrisekoop, N.; Westera, L.; Mogling, R.; de Boer, A.B.; Willems, N.; Schrijver, E.H.; Spierenburg, G.; Gaiser, K.; et al. Maintenance of peripheral naive T cells is sustained by thymus output in mice but not humans. Immunity 2012, 36, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Beura, L.K.; Hamilton, S.E.; Bi, K.; Schenkel, J.M.; Odumade, O.A.; Casey, K.A.; Thompson, E.A.; Fraser, K.A.; Rosato, P.C.; Filali-Mouhim, A.; et al. Normalizing the environment recapitulates adult human immune traits in laboratory mice. Nature 2016, 532, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Reese, T.A.; Bi, K.; Kambal, A.; Filali-Mouhim, A.; Beura, L.K.; Burger, M.C.; Pulendran, B.; Sekaly, R.P.; Jameson, S.C.; Masopust, D.; et al. Sequential Infection with Common Pathogens Promotes Human-like Immune Gene Expression and Altered Vaccine Response. Cell Host Microbe 2016, 19, 713–719. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Macallan, D.C.; Borghans, J.A.M.; Asquith, B. Human T Cell Memory: A Dynamic View. Vaccines 2017, 5, 5. https://doi.org/10.3390/vaccines5010005
Macallan DC, Borghans JAM, Asquith B. Human T Cell Memory: A Dynamic View. Vaccines. 2017; 5(1):5. https://doi.org/10.3390/vaccines5010005
Chicago/Turabian StyleMacallan, Derek C., José A. M. Borghans, and Becca Asquith. 2017. "Human T Cell Memory: A Dynamic View" Vaccines 5, no. 1: 5. https://doi.org/10.3390/vaccines5010005
APA StyleMacallan, D. C., Borghans, J. A. M., & Asquith, B. (2017). Human T Cell Memory: A Dynamic View. Vaccines, 5(1), 5. https://doi.org/10.3390/vaccines5010005