
Mechanisms of Entry and Endosomal Pathway of African Swine Fever Virus

Abstract
1. Mechanisms of ASFV Entry
1.1. Cellular Receptors and Viral Proteins Involved in ASFV Entry
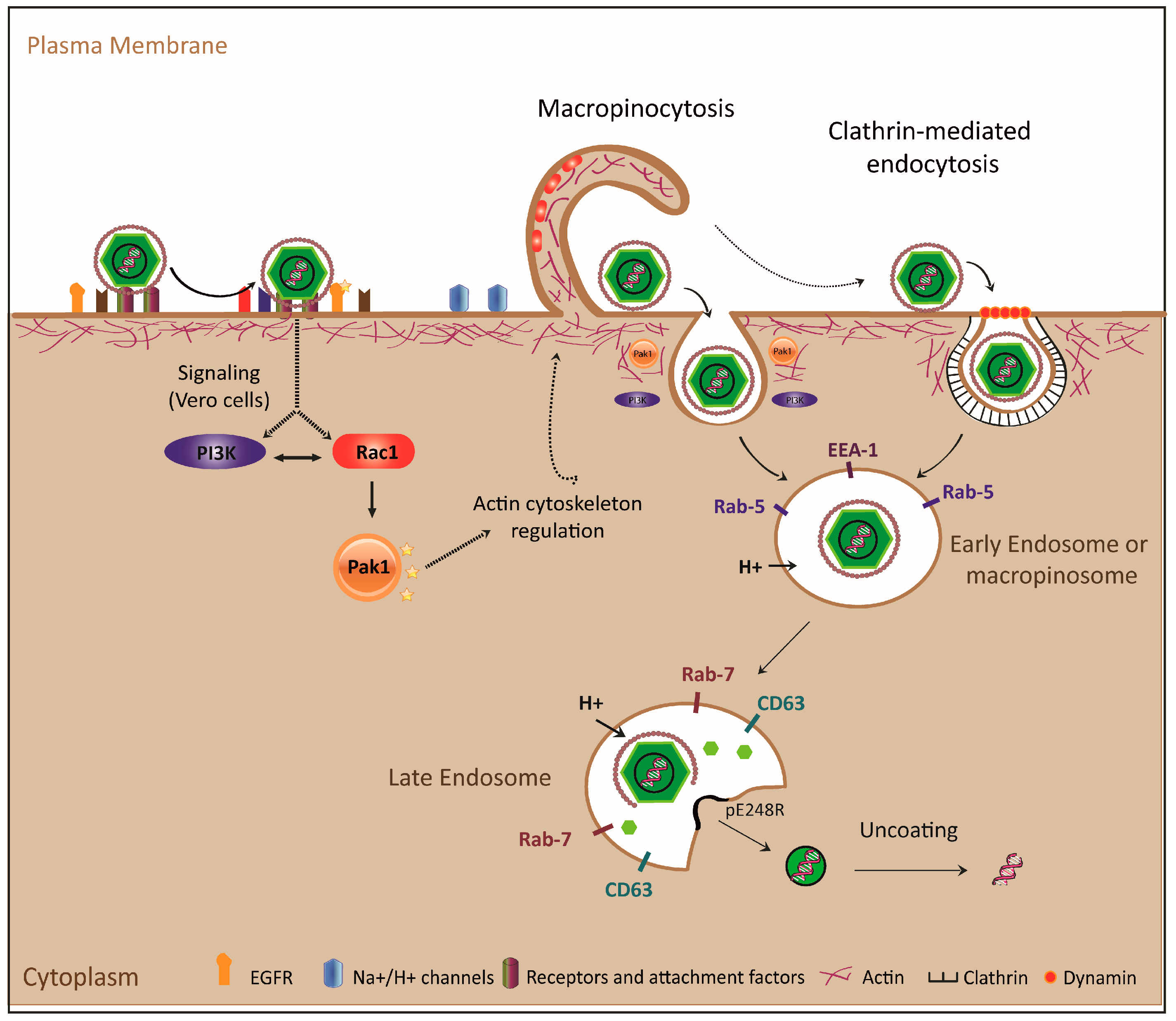
1.2. Viral Internalization: Endocytosis
1.2.1. ASFV Entry Depends on Temperature, Cholesterol, Energy and Vacuolar pH
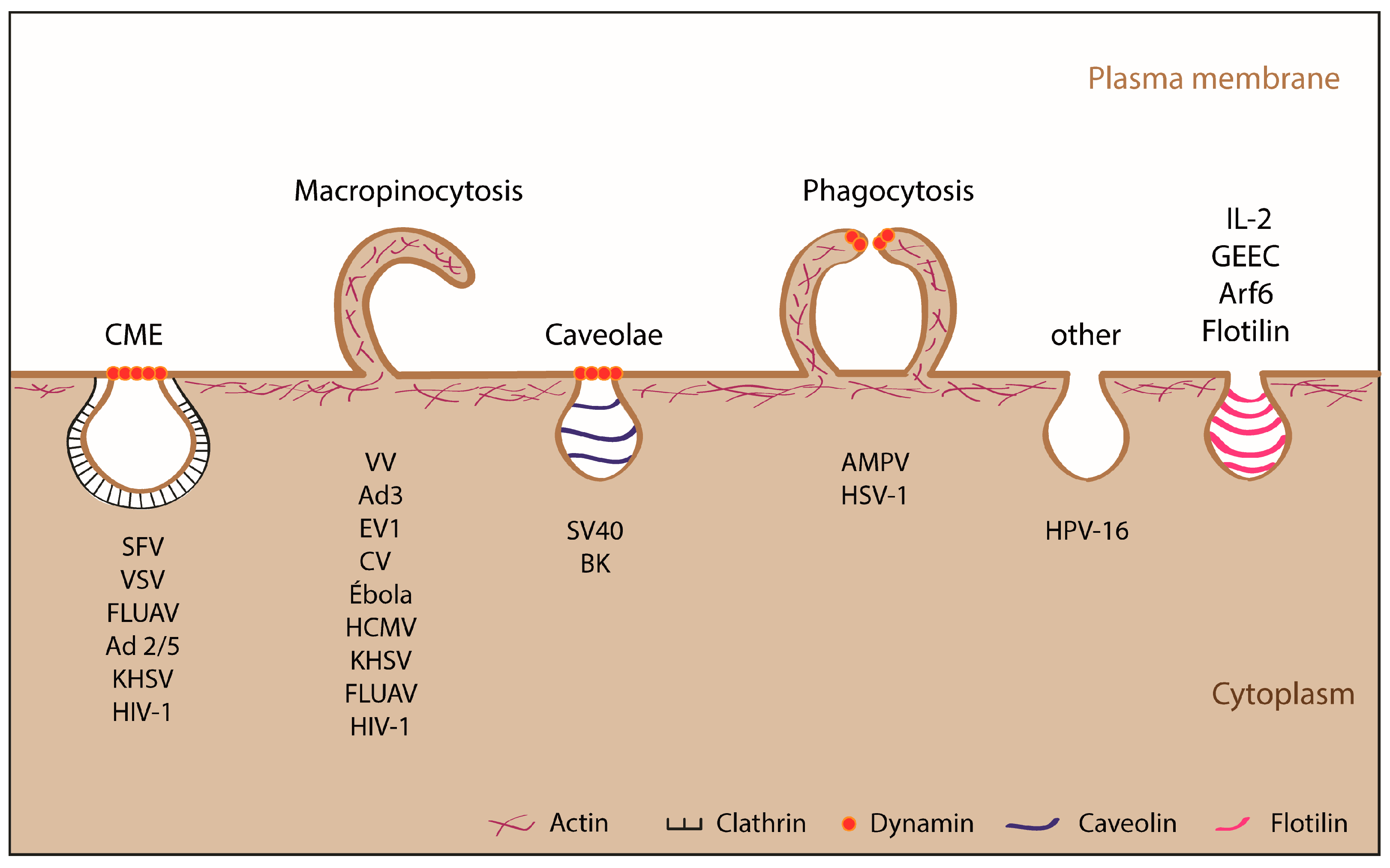
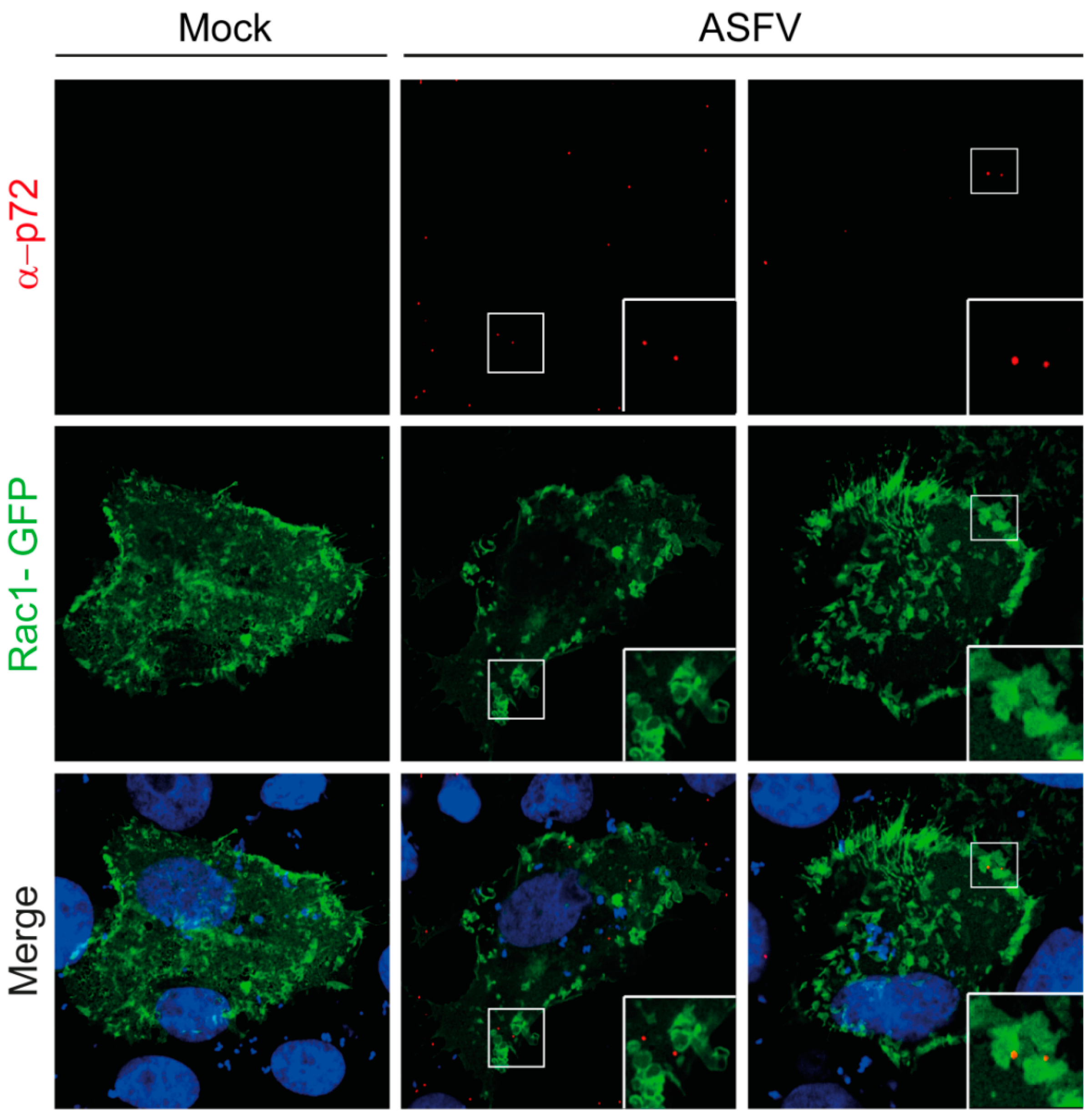
1.2.2. Endocytic Pathways Used by ASFV: CME and Macropinocytosis
2. The ASFV Endosomal Pathway
2.1. ASFV Movement through the Endolysosomal System
2.1.1. ASFV Movement across the Endolysosomal System
2.1.2. Importance of Acidification and Other Factors (i.e., Lipid Composition) in the Progression of ASFV through the Endolysosomal Route
2.2. ASFV Uncoating
3. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Alcami, A.; Carrascosa, A.L.; Vinuela, E. The entry of African swine fever virus into vero cells. Virology 1989, 171, 68–75. [Google Scholar] [CrossRef]
- Geraldes, A.; Valdeira, M.L. Effect of chloroquine on African swine fever virus infection. J. Gen. Virol. 1985, 66, 1145–1148. [Google Scholar] [CrossRef] [PubMed]
- Valdeira, M.L.; Geraldes, A. Morphological study on the entry of African swine fever virus into cells. Biol. Cell 1985, 55, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Alcami, A.; Carrascosa, A.L.; Vinuela, E. Saturable binding sites mediate the entry of African swine fever virus into vero cells. Virology 1989, 168, 393–398. [Google Scholar] [CrossRef]
- Alcami, A.; Carrascosa, A.L.; Vinuela, E. Interaction of African swine fever virus with macrophages. Virus Res. 1990, 17, 93–104. [Google Scholar] [CrossRef]
- Carrascosa, A.L.; Bustos, M.J.; Galindo, I.; Vinuela, E. Virus-specific cell receptors are necessary, but not sufficient, to confer cell susceptibility to African swine fever virus. Arch. Virol. 1999, 144, 1309–1321. [Google Scholar] [CrossRef] [PubMed]
- Alcami, A.; Vinuela, E. Fc receptors do not mediate African swine fever virus replication in macrophages. Virology 1991, 181, 756–759. [Google Scholar] [CrossRef]
- Argilaguet, J.M.; Pérez-Martín, E.; Gallardo, C.; Salguero, F.J.; Borrego, B.; Lacasta, A.; Accensi, F.; Díaz, I.; Nofrarías, M.; Pujols, J.; et al. Enhancing DNA immunization by targeting ASFV antigens to SLA-II bearing cells. Vaccine 2011, 29, 5379–5385. [Google Scholar] [CrossRef] [PubMed]
- Sunwoo, S.-Y.; Pérez-Nuñez, D.; Sánchez, E.G.; Nogal, M.; Haley, N.; Morozov, I.; Madden, D.; Mur, L.; Davis, A.S.; Trujillo, J.; et al. Development of a heterologous DNA-protein vaccination strategy against African swine fever. Manuscript in preparation. 2017. [Google Scholar]
- Sanchez-Torres, C.; Gomez-Puertas, P.; Gomez-del-Moral, M.; Alonso, F.; Escribano, J.M.; Ezquerra, A.; Dominguez, J. Expression of porcine cd163 on monocytes/macrophages correlates with permissiveness to African swine fever infection. Arch. Virol. 2003, 148, 2307–2323. [Google Scholar] [CrossRef] [PubMed]
- Lithgow, P.; Takamatsu, H.; Werling, D.; Dixon, L.; Chapman, D. Correlation of cell surface marker expression with African swine fever virus infection. Vet. Microbiol. 2014, 168, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Popescu, L.; Gaudreault, N.N.; Whitworth, K.M.; Murgia, M.V.; Nietfeld, J.C.; Mileham, A.; Samuel, M.; Wells, K.D.; Prather, R.S.; Rowland, R.R. Genetically edited pigs lacking CD163 show no resistance following infection with the African swine fever virus isolate, georgia 2007/1. Virology 2017, 501, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, E.G.; Riera, E.; Nogal, M.; Gallardo, C.; Fernandez, P.; Bello-Morales, R.; Lopez-Guerrero, J.A.; Chitko-McKown, C.G.; Richt, J.A.; Revilla, Y. Phenotyping and susceptibility of established porcine cells lines to African swine fever virus infection and viral production. Sci. Rep. 2017, 7, 10369. [Google Scholar] [CrossRef] [PubMed]
- Carrascosa, A.L.; Sastre, I.; Vinuela, E. African swine fever virus attachment protein. J. Virol. 1991, 65, 2283–2289. [Google Scholar] [PubMed]
- Angulo, A.; Vinuela, E.; Alcami, A. Inhibition of African swine fever virus binding and infectivity by purified recombinant virus attachment protein p12. J. Virol. 1993, 67, 5463–5471. [Google Scholar] [PubMed]
- Gomez-Puertas, P.; Rodriguez, F.; Oviedo, J.M.; Brun, A.; Alonso, C.; Escribano, J.M. The African swine fever virus proteins p54 and p30 are involved in two distinct steps of virus attachment and both contribute to the antibody-mediated protective immune response. Virology 1998, 243, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Puertas, P.; Rodriguez, F.; Oviedo, J.M.; Ramiro-Ibanez, F.; Ruiz-Gonzalvo, F.; Alonso, C.; Escribano, J.M. Neutralizing antibodies to different proteins of African swine fever virus inhibit both virus attachment and internalization. J. Virol. 1996, 70, 5689–5694. [Google Scholar] [PubMed]
- Carter, G.C.; Bernstone, L.; Baskaran, D.; James, W. HIV-1 infects macrophages by exploiting an endocytic route dependent on dynamin, RAC1 and PAK1. Virology 2011, 409, 234–250. [Google Scholar] [CrossRef] [PubMed]
- Stein, B.S.; Gowda, S.D.; Lifson, J.D.; Penhallow, R.C.; Bensch, K.G.; Engleman, E.G. PH-independent HIV entry into CD4-positive T cells via virus envelope fusion to the plasma membrane. Cell 1987, 49, 659–668. [Google Scholar] [CrossRef]
- Cuesta-Geijo, M.A.; Galindo, I.; Hernaez, B.; Quetglas, J.I.; Dalmau-Mena, I.; Alonso, C. Endosomal maturation, Rab7 gtpase and phosphoinositides in African swine fever virus entry. PLoS ONE 2012, 7, e48853. [Google Scholar] [CrossRef] [PubMed]
- Valdeira, M.L.; Bernardes, C.; Cruz, B.; Geraldes, A. Entry of African swine fever virus into vero cells and uncoating. Vet. Microbiol. 1998, 60, 131–140. [Google Scholar] [CrossRef]
- Bernardes, C.; Antonio, A.; Pedroso de Lima, M.C.; Valdeira, M.L. Cholesterol affects African swine fever virus infection. Biochim. Biophys. Acta 1998, 1393, 19–25. [Google Scholar] [CrossRef]
- Amstutz, B.; Gastaldelli, M.; Kalin, S.; Imelli, N.; Boucke, K.; Wandeler, E.; Mercer, J.; Hemmi, S.; Greber, U.F. Subversion of CTBP1-controlled macropinocytosis by human adenovirus serotype 3. EMBO J. 2008, 27, 956–969. [Google Scholar] [CrossRef] [PubMed]
- Clement, C.; Tiwari, V.; Scanlan, P.M.; Valyi-Nagy, T.; Yue, B.Y.; Shukla, D. A novel role for phagocytosis-like uptake in herpes simplex virus entry. J. Cell Biol. 2006, 174, 1009–1021. [Google Scholar] [CrossRef] [PubMed]
- De Vries, E.; Tscherne, D.M.; Wienholts, M.J.; Cobos-Jimenez, V.; Scholte, F.; Garcia-Sastre, A.; Rottier, P.J.; de Haan, C.A. Dissection of the influenza a virus endocytic routes reveals macropinocytosis as an alternative entry pathway. PLoS Pathog. 2011, 7, e1001329. [Google Scholar] [CrossRef] [PubMed]
- Meier, O.; Boucke, K.; Hammer, S.V.; Keller, S.; Stidwill, R.P.; Hemmi, S.; Greber, U.F. Adenovirus triggers macropinocytosis and endosomal leakage together with its clathrin-mediated uptake. J. Cell Biol. 2002, 158, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Mousavi, S.A.; Malerod, L.; Berg, T.; Kjeken, R. Clathrin-dependent endocytosis. Biochem. J. 2004, 377, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Mellman, I. Endocytosis and molecular sorting. Annu. Rev. Cell Dev. Biol. 1996, 12, 575–625. [Google Scholar] [CrossRef] [PubMed]
- Brodsky, F.M.; Chen, C.Y.; Knuehl, C.; Towler, M.C.; Wakeham, D.E. Biological basket weaving: Formation and function of clathrin-coated vesicles. Annu. Rev. Cell Dev. Biol. 2001, 17, 517–568. [Google Scholar] [CrossRef] [PubMed]
- Pearse, B.M. Clathrin: A unique protein associated with intracellular transfer of membrane by coated vesicles. Proc. Natl. Acad. Sci. USA 1976, 73, 1255–1259. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.J.; Pearse, B.M. Clathrin: Anatomy of a coat protein. Trends Cell Biol. 1999, 9, 335–338. [Google Scholar] [CrossRef]
- Takei, K.; Haucke, V. Clathrin-mediated endocytosis: Membrane factors pull the trigger. Trends Cell Biol. 2001, 11, 385–391. [Google Scholar] [CrossRef]
- Hinshaw, J.E. Dynamin and its role in membrane fission. Annu. Rev. Cell Dev. Boil. 2000, 16, 483–519. [Google Scholar] [CrossRef] [PubMed]
- Mettlen, M.; Pucadyil, T.; Ramachandran, R.; Schmid, S.L. Dissecting dynamin’s role in clathrin-mediated endocytosis. Biochem. Soc. Trans. 2009, 37, 1022–1026. [Google Scholar] [CrossRef] [PubMed]
- Sever, S.; Damke, H.; Schmid, S.L. Dynamin:Gtp controls the formation of constricted coated pits, the rate limiting step in clathrin-mediated endocytosis. J. Cell Biol. 2000, 150, 1137–1148. [Google Scholar] [CrossRef] [PubMed]
- Mercer, J.; Schelhaas, M.; Helenius, A. Virus entry by endocytosis. Annu. Rev. Biochem. 2010, 79, 803–833. [Google Scholar] [CrossRef] [PubMed]
- Meier, O.; Greber, U.F. Adenovirus endocytosis. J. Gene Med. 2004, 6, S152–S163. [Google Scholar] [CrossRef] [PubMed]
- Helenius, A.; Kartenbeck, J.; Simons, K.; Fries, E. On the entry of semliki forest virus into BHK-21 cells. J. Cell Biol. 1980, 84, 404–420. [Google Scholar] [CrossRef] [PubMed]
- Hernaez, B.; Guerra, M.; Salas, M.L.; Andres, G. African swine fever virus undergoes outer envelope disruption, capsid disassembly and inner envelope fusion before core release from multivesicular endosomes. PLoS Pathog. 2016, 12, e1005595. [Google Scholar] [CrossRef] [PubMed]
- Hernaez, B.; Alonso, C. Dynamin- and clathrin-dependent endocytosis in African swine fever virus entry. J. Virol. 2010, 84, 2100–2109. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, E.G.; Quintas, A.; Perez-Nunez, D.; Nogal, M.; Barroso, S.; Carrascosa, A.L.; Revilla, Y. African swine fever virus uses macropinocytosis to enter host cells. PLoS Pathog. 2012, 8, e1002754. [Google Scholar] [CrossRef] [PubMed]
- Galindo, I.; Cuesta-Geijo, M.A.; Hlavova, K.; Munoz-Moreno, R.; Barrado-Gil, L.; Dominguez, J.; Alonso, C. African swine fever virus infects macrophages, the natural host cells, via clathrin- and cholesterol-dependent endocytosis. Virus Res. 2015, 200, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Mercer, J.; Helenius, A. Virus entry by macropinocytosis. Nat. Cell Biol. 2009, 11, 510–520. [Google Scholar] [CrossRef] [PubMed]
- Swanson, J.A.; Watts, C. Macropinocytosis. Trends Cell Biol. 1995, 5, 424–428. [Google Scholar] [CrossRef]
- Bryant, D.M.; Kerr, M.C.; Hammond, L.A.; Joseph, S.R.; Mostov, K.E.; Teasdale, R.D.; Stow, J.L. EGF induces macropinocytosis and SNX1-modulated recycling of E-cadherin. J. Cell Sci. 2007, 120, 1818–1828. [Google Scholar] [CrossRef] [PubMed]
- Racoosin, E.L.; Swanson, J.A. Macrophage colony-stimulating factor (RM-CSF) stimulates pinocytosis in bone marrow-derived macrophages. J. Exp. Med. 1989, 170, 1635–1648. [Google Scholar] [CrossRef] [PubMed]
- Swanson, J.A. Phorbol esters stimulate macropinocytosis and solute flow through macrophages. J. Cell Sci. 1989, 94, 135–142. [Google Scholar] [PubMed]
- West, M.A.; Bretscher, M.S.; Watts, C. Distinct endocytotic pathways in epidermal growth factor-stimulated human carcinoma A431 cells. J. Cell Biol. 1989, 109, 2731–2739. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, M.; Nasser, H.; Chihara, T.; Suzu, S. Macropinocytosis and TAK1 mediate anti-inflammatory to pro-inflammatory macrophage differentiation by HIV-1 NEF. Cell Death Dis. 2014, 5, e1267. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Verreck, F.A.; de Boer, T.; Langenberg, D.M.; Hoeve, M.A.; Kramer, M.; Vaisberg, E.; Kastelein, R.; Kolk, A.; de Waal-Malefyt, R.; Ottenhoff, T.H. Human IL-23-producing type 1 macrophages promote but IL-10-producing type 2 macrophages subvert immunity to (Myco)bacteria. Proc. Natl. Acad. Sci. USA 2004, 101, 4560–4565. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Liang, H.; Zen, K. Molecular mechanisms that influence the macrophage m1-m2 polarization balance. Front. Immunol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Franzoni, G.; Graham, S.P.; Giudici, S.D.; Bonelli, P.; Pilo, G.; Anfossi, A.G.; Pittau, M.; Nicolussi, P.S.; Laddomada, A.; Oggiano, A. Characterization of the interaction of African swine fever virus with monocytes and derived macrophage subsets. Vet. Microbiol. 2017, 198, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, E.G.; Riera, E.; Revilla, Y. Study of monocyte origin porcine cell lines and M2 macrophages in susceptibility to African swine fever virus infection. CBMSO; Unpublished data. 2015. [Google Scholar]
- Quetglas, J.I.; Hernáez, B.; Galindo, I.; Muñoz-Moreno, R.; Cuesta-Geijo, M.A.; Alonso, C. Small rho GTPases and cholesterol biosynthetic pathway intermediates in African swine fever virus infection. J. Virol. 2012, 86, 1758–1767. [Google Scholar] [CrossRef] [PubMed]
- Mercer, J.; Helenius, A. Vaccinia virus uses macropinocytosis and apoptotic mimicry to enter host cells. Science 2008, 320, 531–535. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, F.I.; Bleck, C.K.; Helenius, A.; Mercer, J. Vaccinia extracellular virions enter cells by macropinocytosis and acid-activated membrane rupture. EMBO J. 2011, 30, 3647–3661. [Google Scholar] [CrossRef] [PubMed]
- Basta, S.; Gerber, H.; Schaub, A.; Summerfield, A.; McCullough, K.C. Cellular processes essential for african swine fever virus to infect and replicate in primary macrophages. Vete. Microbiol. 2010, 140, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Alonso, C.; Galindo, I.; Cuesta-Geijo, M.A.; Cabezas, M.; Hernaez, B.; Munoz-Moreno, R. African swine fever virus-cell interactions: From virus entry to cell survival. Virus Res. 2013, 173, 42–57. [Google Scholar] [CrossRef] [PubMed]
- Heath, C.M.; Windsor, M.; Wileman, T. Aggresomes resemble sites specialized for virus assembly. J. Cell Biol. 2001, 153, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Andres, G.; Garcia-Escudero, R.; Salas, M.L.; Rodriguez, J.M. Repression of African swine fever virus polyprotein pp220-encoding gene leads to the assembly of icosahedral core-less particles. J. Virol. 2002, 76, 2654–2666. [Google Scholar] [CrossRef] [PubMed]
- Alejo, A.; Andres, G.; Salas, M.L. African swine fever virus proteinase is essential for core maturation and infectivity. J. Virol. 2003, 77, 5571–5577. [Google Scholar] [CrossRef] [PubMed]
- Moss, B. Poxvirus cell entry: How many proteins does it take? Viruses 2012, 4, 688–707. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, I.; Nogal, M.L.; Redrejo-Rodriguez, M.; Bustos, M.J.; Salas, M.L. The African swine fever virus virion membrane protein pe248r is required for virus infectivity and an early postentry event. J. Virol. 2009, 83, 12290–12300. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Cellular Factors | Inhibitors | Internalization (Viral Uptake) | Post-Internalization (Endosome Transport) |
|---|---|---|---|
| Na+/H+ channels | EIPA | Yes [39,41,42] | Yes [42]/No [40,41] |
| Actin | Cytochalasin D and B Latrunculin A | Yes [39,41,42]/No [21] | Yes [41,42,57] |
| Myosin II | Blebbistatin | Yes [41] | Yes [41] |
| EGFR | 324674 | Yes [41] | Unknown |
| PI3K | LY294002Worthmanin | Yes [41,42] | Yes [20,39,40,41] |
| Rac1 | NSC23766, Rac1-N17 | Yes [41]/No [40] | Yes [40,41] |
| Pak1 | IPA-3, Pak1-AID | Yes [39,41] | No [41] |
| Tyrosin kinases | Genistein | Yes [41] | Unknown |
| Dynamin-2 | Dynasore | Yes [39,41,42] | Yes [40,41,42] |
| Clathrin | Clorpromazine | No [41] | Yes [40,41] |
| Microtubules | Nocodazol | No [41] | Yes [39,41,59] |
| Vacuolar acidification | Cloroquine, NH4Cl, Bafilomycin A | No [2,3]/Yes [42] | Yes [20,39,41,57] |
| Cholesterol | MβCD | No [22] | Yes [20,22] |
| Rab-7 | Rab-7-T22N Rab-7 siRNA | Unknown | Yes [10,11] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez, E.G.; Pérez-Núñez, D.; Revilla, Y. Mechanisms of Entry and Endosomal Pathway of African Swine Fever Virus. Vaccines 2017, 5, 42. https://doi.org/10.3390/vaccines5040042
Sánchez EG, Pérez-Núñez D, Revilla Y. Mechanisms of Entry and Endosomal Pathway of African Swine Fever Virus. Vaccines. 2017; 5(4):42. https://doi.org/10.3390/vaccines5040042
Chicago/Turabian StyleSánchez, Elena G., Daniel Pérez-Núñez, and Yolanda Revilla. 2017. "Mechanisms of Entry and Endosomal Pathway of African Swine Fever Virus" Vaccines 5, no. 4: 42. https://doi.org/10.3390/vaccines5040042
APA StyleSánchez, E. G., Pérez-Núñez, D., & Revilla, Y. (2017). Mechanisms of Entry and Endosomal Pathway of African Swine Fever Virus. Vaccines, 5(4), 42. https://doi.org/10.3390/vaccines5040042