
Oncolytic Alphaviruses in Cancer Immunotherapy

Abstract
:1. Introduction
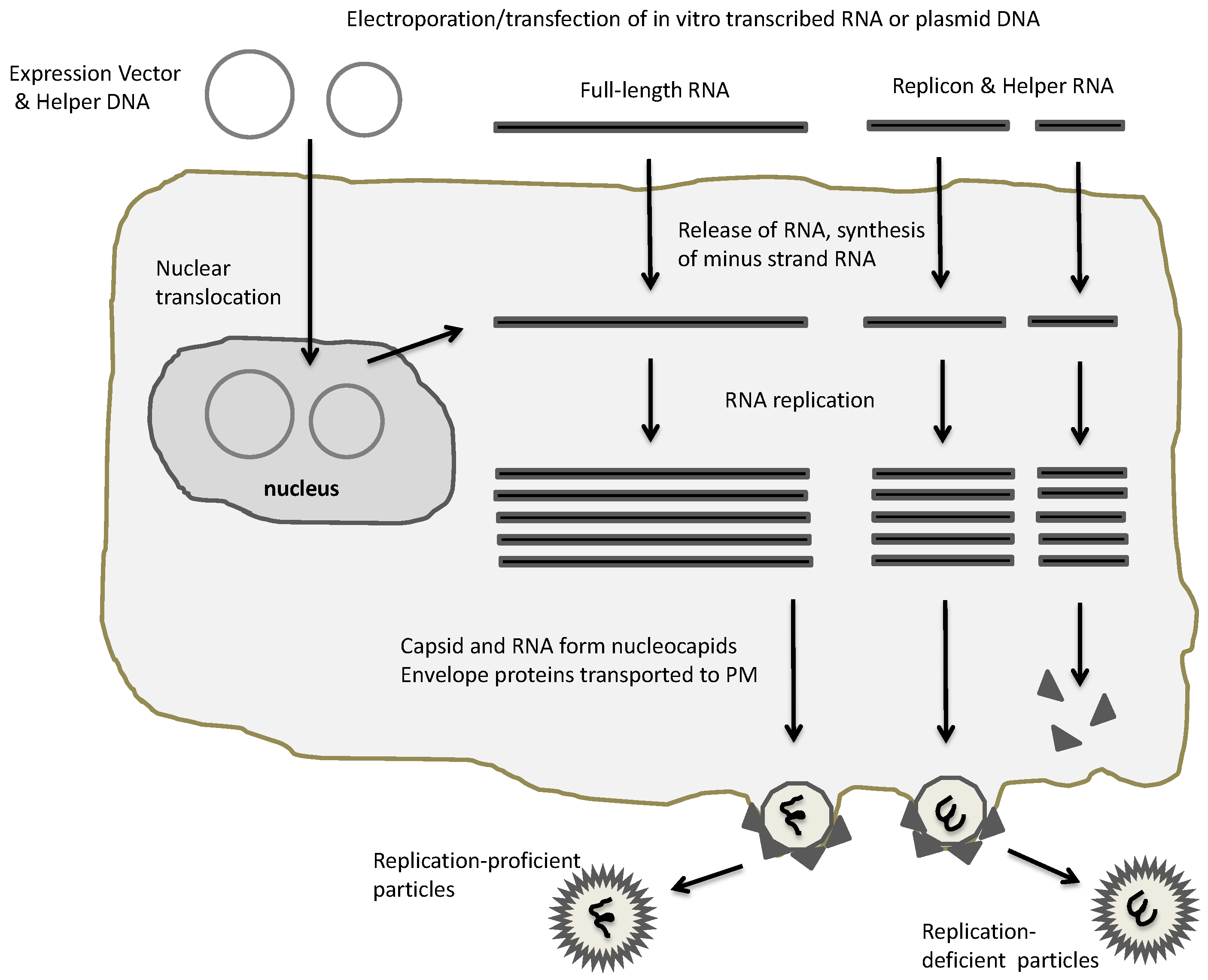
2. Expression Vectors for Alphaviruses
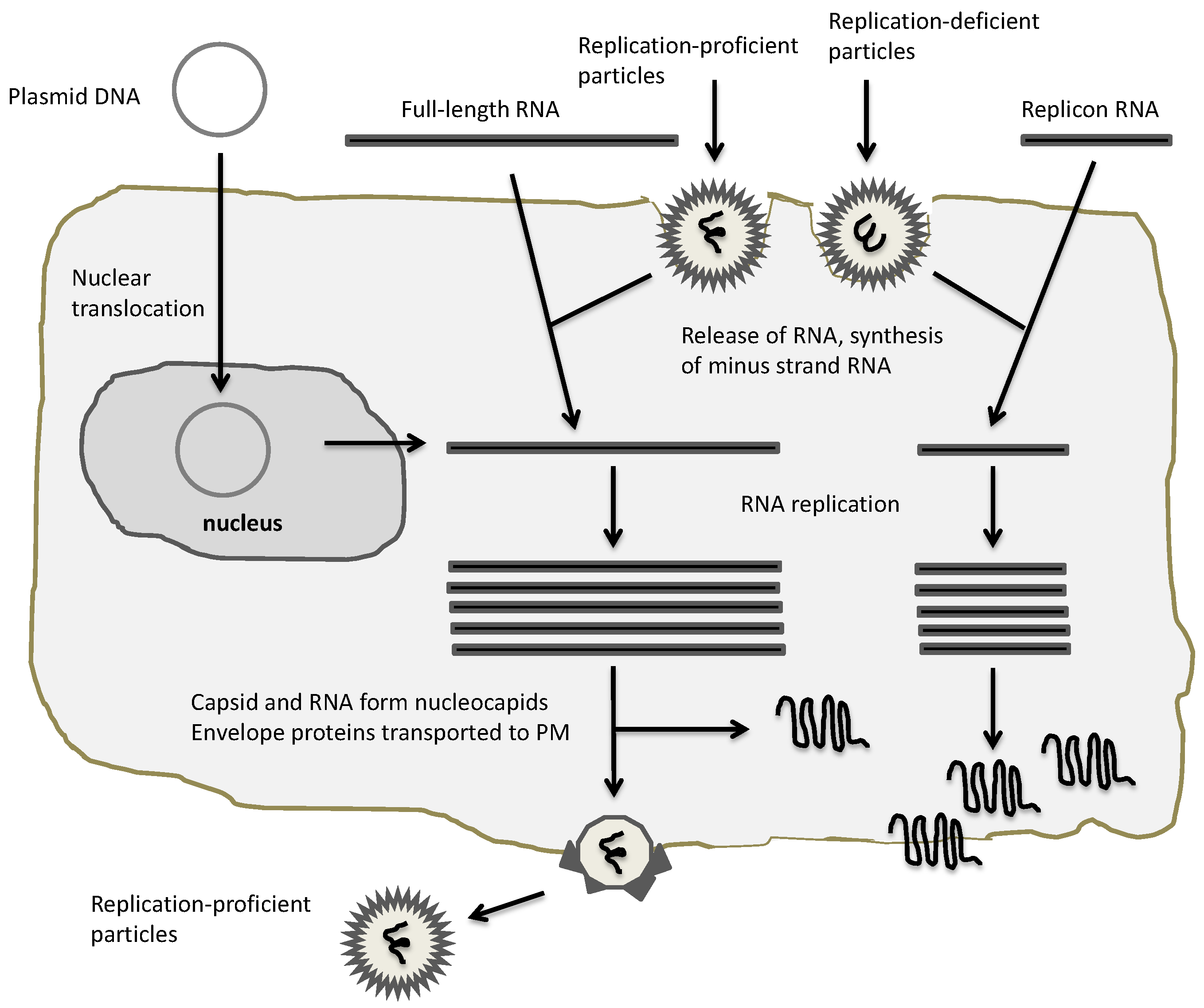
3. Oncolytic Alphavirus Approaches
4. Comparison to Other Oncolytic RNA Viruses
5. Discussion
6. Conclusions
Conflicts of Interest
References
- Strauss, J.H.; Strauss, E.G. The Alphaviruses: Gene Expression, Replication and Evolution. Micobiol. Rev. 1994, 58, 491–562. [Google Scholar]
- Garmashova, N.; Gorchakov, R.; Volkova, E.; Paessler, S.; Frolova, E.; Frolov, I. The Old and New World Alphaviruses use different virus-specific proteins for induction of transcriptional shutoff. J. Virol. 2007, 81, 2472–2484. [Google Scholar] [CrossRef] [PubMed]
- Peters, C.J.; Dalrymple, J.M. Alphaviruses. In Fields Virology, 1st ed.; Fields, B.N., Knipe, D.M., Howley, P.M., Eds.; Raven Press: New York, NY, USA, 1990; pp. 713–761. [Google Scholar]
- Phillips, D.A.; Murray, J.R.; Aaskov, J.G.; Wiemers, M.A. Clinical and subclinical Barmah Forest virus infection in Queensland. Med. J. Aust. 1990, 152, 463–466. [Google Scholar] [PubMed]
- Rennels, M.B. Arthropod-borne virus infections of the central nervous system. Neurol. Clin. 1984, 2, 241–254. [Google Scholar] [PubMed]
- Arechiga-Ceballos, N.; Aguilar-Setien, A. Alphaviral equine encephalomyelitis (Eastern, Western and Venezuelan). Rev. Sci. Tech. 2015, 34, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Mathiot, C.C.; Grimaud, G.; Garry, P.; Bouguety, J.C.; Mada, A.; Daguisy, A.M.; Georges, A.J. An outbreak of human Semliki Forest virus infection in Central African Republic. Am. J. Trop. Med. Hyg. 1990, 42, 386–393. [Google Scholar] [PubMed]
- Niklasson, B. Sindbis and Sindbis-like viruses. In The Arboviruses: Epidemiology and Ecology; Monath, T.P., Ed.; CRC Press Inc.: Boca Raton, FL, USA, 1988; pp. 167–176. [Google Scholar]
- Vilcarromero, S.; Aguilar, P.V.; Halsey, E.S.; Laguna-Torres, V.A.; Razuri, H.; Perez, J.; Valderrama, Y.; Gotuzzo, E.; Suarez, L.; Cespedes, M.; et al. Venezuelan equine encephalitis and 2 human deaths, Peru. Emerg. Infect. Dis. 2010, 16, 553–556. [Google Scholar] [CrossRef] [PubMed]
- Kelvin, A.A. Outbreak of Chikungunya in the Republic of Congo and the global picture. J. Infect. Dev. Ctries. 2011, 5, 441–444. [Google Scholar] [CrossRef]
- Jansen, K.A. The 2005–2007 Chikungunya epidemic in Reunion: Ambiguous etiologies, memories, and meaning-making. Med. Anthropol. 2013, 32, 174–189. [Google Scholar] [CrossRef] [PubMed]
- Faria, N.R.; Lourenço, J.; de Cerqueira, E.M.; de Lima, M.M.; Pybus, O.; Alcantara, L.C.J. Epidemiology of Chikungunya virus in Bahia, Brazil, 2014–2015. PLoS Curr. 2016. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, R.W. Epidemiology of arthropod-borne togaviruses: The role of arthropods as hosts and vectors and of vertebrate hosts in natural transmission cycles. In The Togaviruses: Biology, Structure, Replication; Schlesinger, R.W., Ed.; Academic Press, Inc.: New York, NY, USA, 1980; pp. 175–227. [Google Scholar]
- Helenius, A.; Morein, B.; Fries, E.; Simons, K.; Robinson, P.; Schirrmacher, V.; Terhorst, T.; Strominger, J.L. Human (HLA-A and -B) and murine (H2-K and -D) histocompatibility antigens are cell surface receptors for Semliki Forest virus. Proc. Natl. Acad. Sci. USA 1978, 75, 3846–3850. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.-S.; Kuhn, R.J.; Strauss, E.G.; Ou, S.; Strauss, J.H. High-affinity laminin receptor is a receptor for Sindbis virus in mammalian cells. J. Virol. 1992, 66, 4992–5001. [Google Scholar] [PubMed]
- Byrnes, A.P.; Griffin, D.E. Binding of Sindbis virus to cell surface heparan sulfate. J. Virol. 1998, 72, 7349–7356. [Google Scholar] [PubMed]
- Kondor-Koch, C.; Burke, B.; Garoff, H. Expression of Semliki Forest virus proteins from cloned complementary DNA. I. The fusion activity of the spike glycoprotein. J. Cell Biol. 1983, 97, 644–651. [Google Scholar] [CrossRef] [PubMed]
- Helenius, A. Semliki Forest virus penetration from endosomes. Biol. Cell 1984, 51, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Kaarianen, L.; Ranki, M. Inhibition of cell functions by RNA-virus infections. Annu. Rev. Microbiol. 1984, 38, 91–109. [Google Scholar] [CrossRef] [PubMed]
- Braakman, I.; Helenius, J.; Helenius, A. Role of ATP and disulphide bonds during protein folding in the endoplasmic reticulum. Nature 1992, 356, 260–262. [Google Scholar] [CrossRef] [PubMed]
- Lobigs, M.; Hongxing, Z.; Garoff, H. Function of Semliki Forest virus E3 peptide in virus assembly: Replacement of E3 with an artificial signal peptide abolishes spike heterodimerization and surface expression of El. J. Virol. 1990, 64, 4346–4355. [Google Scholar] [PubMed]
- Garoff, H.; Simons, K. Location of the spike glycoproteins in the Semliki Forest virus membrane. Proc. Natl. Acad. Sci. USA 1974, 71, 3988–3992. [Google Scholar] [CrossRef] [PubMed]
- Hanson, R.P.; Sulkin, S.E.; Beuscher, E.L.; Hammon, W.M.; McKinney, R.W.; Work, T.H. Arbovirus infections of laboratory workers. Science 1967, 158, 1283–1286. [Google Scholar] [CrossRef] [PubMed]
- Lundstrom, K. Viral vector-based cancer immunotherapy. Austin Immunol. 2016, 1, 1008. [Google Scholar]
- Lundstrom, K. Alphavirus-based vaccines. Viruses 2014, 6, 2392–2415. [Google Scholar] [CrossRef] [PubMed]
- Lundstrom, K. Alphavirus vectors for vaccine production and gene therapy. Expert Rev. Vaccines 2003, 2, 447–459. [Google Scholar] [CrossRef]
- Quetglas, J.I.; Ruiz-Guillen, M.; Aranda, A.; Casales, E.; Bezurnartea, J.; Smerdou, C. Alphavirus vectors for cancer therapy. Virus Res. 2010, 153, 179–196. [Google Scholar] [CrossRef] [PubMed]
- Colmenero, P.; Liljeström, P.; Jondal, M. Induction of P815 tumor immunity by recombinant Semliki Forest virus expressing the P1A gene. Gene Ther. 1999, 6, 1728–1733. [Google Scholar] [CrossRef] [PubMed]
- Daemen, T.; Riezebos-Brilman, A.; Regts, J.; Dontje, B.; van der Zee, A.; Wilschut, J. Superior therapeutic efficacy of alphavirus-mediated immunization against human papilloma virus type 16 antigens in a murine tumour model: Effects of the route of immunization. Antivir. Ther. 2004, 9, 733–742. [Google Scholar] [PubMed]
- Daemen, T.; Riezebos-Brilman, A.; Bungener, L.; Regts, J.; Dontje, B.; Wilschut, J. Eradication of established HPV-16 transformed tumours after immunisation with recombinant Semliki Forest virus expressing a fusion protein of E6 and E7. Vaccine 2003, 21, 1082–1088. [Google Scholar] [CrossRef]
- Velders, M.P.; McElhiney, S.; Cassetti, M.C.; Elben, G.L.; Higgins, T.; Kovacs, G.R.; Elmiishad, A.G.; Kast, W.M.; Smith, L.R. Eradication of established tumors by vaccination with Venezuelan equine encephalitis virus replicon particles delivering human papillomavirus 16 E7 RNA. Cancer Res. 2001, 61, 7861–7867. [Google Scholar] [PubMed]
- Ying, H.; Zaks, T.Z.; Wang, R.F.; Irvine, K.R.; Kammula, U.S.; Marincola, F.M.; Leitner, W.W.; Restifo, N.P. Cancer therapy using a self-replicating RNA vaccine. Nat. Med. 1999, 5, 823–827. [Google Scholar] [PubMed]
- Wang, X.; Wang, J.P.; Rao, X.M.; Price, J.E.; Zhou, H.S.; Lachman, L.B. Prime-boost vaccination with plasmid and adenovirus gene vaccines control HER2/neu+ metastatic breast cancer in mice. Breast Cancer Res. 2005, 7, R580–R588. [Google Scholar] [CrossRef] [PubMed]
- Chikkana-Gowda, C.P.; Sheahan, B.J.; Fleeton, M.N.; Atkins, G.J. Regression of mouse tumours and inhibition of metastases following administration of a Semliki Forest virus vector with enhanced expression of IL-12. Gene Ther. 2005, 12, 1253–1263. [Google Scholar] [CrossRef] [PubMed]
- Chikkana-Gowda, C.P.; McNally, S.; Sheahan, B.J.; Fleeton, M.N.; Atkins, G.J. Inhibition of murine K-BALB and CT26 tumour growth using a Semliki Forest virus vector with enhanced expression of IL-18. Oncol. Rep. 2006, 16, 713–719. [Google Scholar] [CrossRef]
- Lyons, J.A.; Sheahan, B.J.; Galbraith, S.E.; Mehra, R.; Atkins, G.J.; Fleeton, M.N. Inhibition of angiogenesis by a Semliki Forest virus vector expressing VEGFR-2 reduces tumour growth and metastasis in mice. Gene Ther. 2007, 14, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Morse, M.A.; Hobeika, A.C.; Osada, T.; Berglund, P.; Hubby, B.; Negri, S.; Niedzwiecki, D.; Devi, G.R.; Burnett, B.K.; Clay, T.M.; et al. An alphavirus vector overcomes the presence of neutralizing antibodies and elevated numbers of Tregs to induce immune responses in humans with advanced cancer. J. Clin. Investig. 2010, 120, 3234–3241. [Google Scholar] [CrossRef] [PubMed]
- Slovin, S.F.; Kehoe, M.; Durso, R.; Fernadez, C.; Olson, W.; Gao, J.P.; Israel, R.; Scher, H.I.; Morris, S. A phase I dose escalation trial of vaccine replicon particles (VRP) expressing prostate-specific membrane antigen (PSMA) in subjects with prostate cancer. Vaccine 2013, 31, 943–949. [Google Scholar] [CrossRef] [PubMed]
- Keller, B.A.; Bell, J.C. Oncolytic viruses-immunotherapeutics on the rise. J. Mol. Med. 2016, 94, 979–991. [Google Scholar] [CrossRef] [PubMed]
- Liljestrom, P.; Garoff, H. A new generation of animal cell expression vectors based on the Semliki Forest virus replicon. Biotechnology 1991, 9, 1356–1361. [Google Scholar] [CrossRef] [PubMed]
- Xiong, C.; Levis, R.; Shen, P.; Schlesinger, S.; Rice, C.M.; Huang, H.V. Sindbis virus: An efficient, broad host range vector for gene expression in animal cells. Science 1989, 243, 1188–1191. [Google Scholar] [CrossRef] [PubMed]
- Davis, N.L.; Willis, L.V.; Smith, J.F.; Johnston, R.E. In vitro synthesis of infectious Venezuelan equine encephalitis virus RNA from a cDNA clone: Analysis of a viable deletion mutant. Virology 1989, 171, 189–204. [Google Scholar] [CrossRef]
- Di Ciommo, D.P.; Bremner, R. Rapid, high level protein production using DNA-based Semliki Forest virus vectors. J. Biol. Chem. 1998, 273, 18060–18066. [Google Scholar] [CrossRef]
- Moffatt, P.; Salois, P.; Gaumond, M.-H.; St., Amant, N.; Godin, E.; Lanctot, C. Engineered viruses to select genes encoding secreted and membrane-bound proteins in mammalian cells. Nucleic Acid Res. 2002, 30, 4285–4294. [Google Scholar] [CrossRef] [PubMed]
- Lundstrom, K.; Rotmann, D.; Hermann, D.; Schneider, E.M.; Ehrengruber, M.U. Novel mutant Semliki Forest virus vectors: Gene expression and localization studies in neuronal cells. Histochem. Cell Biol. 2000, 115, 83–91. [Google Scholar] [CrossRef]
- Lundstrom, K.; Abenavoli, A.; Margaroli, A.; Ehrengruber, M.U. Novel Semliki Forest virus vectors with reduced cytotoxicity and temperature sensitivity for long-term enhancement of transgene expression. Mol. Ther. 2003, 7, 202–209. [Google Scholar] [CrossRef]
- Agapov, E.V.; Frolov, I.; Lindenbach, B.D.; Pragai, B.M.; Schlesinger, S.; Rice, C.M. Noncythopathic Sindbis virus RNA vectors for heterologous gene expression. Proc. Natl. Acad. Sci. USA 1998, 95, 12989–12994. [Google Scholar] [CrossRef] [PubMed]
- Dryga, S.A.; Dryga, O.A.; Schlesinger, S. Identification of mutations in a Sindbis virus variant able to establish a persistent infection in BHK cells: The importance of a mutation in the nsP2 gene. Virology 1997, 228, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Perri, S.; Driver, D.A.; Gardner, J.P.; Sherrill, S.; Belli, B.A.; Dubensky T.W., Jr.; Polo, J.M. Replicon vectors derived from Sindbis virus and Semliki Forest virus that establish persistent replication in host cells. J. Virol. 2000, 74, 9402–9407. [Google Scholar] [CrossRef]
- Ehrengruber, M.U.; Renggli, M.; Raineteau, O.; Hennou, S.; Vähä-Koskela, M.J.; Hinkkanen, A.E.; Lundstrom, K. Semliki Forest virus A7(74) transduces hippocampal neurons and glial cells in a temperature-dependent dual manner. J. Neurovirol. 2003, 9, 16–28. [Google Scholar] [CrossRef] [PubMed]
- Sjöberg, E.M.; Suomalainen, M.; Garoff, H. A significantly improved Semliki Forest virus expression system based on translation enhancer segments from the viral capsid gene. Biotechnology 1994, 12, 1127–1131. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Madoz, J.R.; Prieto, J.; Smerdou, C. Semliki forest virus vectors engineered to express higher IL-12 levels induce efficient elimination of murine colon adenocarcinomas. Mol. Ther. 2005, 12, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.Y.; Atasheva, S.; McAuley, A.J.; Plante, J.A.; Frolova, E.I.; Beasley, D.W.; Frolov, I. Enhancement of protein expression by alphavirus replicons by designing self-replicating subgenomic RNAs. Proc. Natl. Acad. Sci. USA 2014, 111, 10708–10713. [Google Scholar] [CrossRef] [PubMed]
- Nanda, K.; Vancini, R.; Ribeiro, M.; Brown, D.T.; Hernandez, R. A high capacity Alphavirus heterologous gene delivery system. Virology 2009, 390, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Berglund, P.; Sjöberg, M.; Garoff, H.; Atkins, G.J.; Sheahan, B.J.; Liljestrom, P. Semliki Forest virus expression system: Production of conditionally infectious recombinant particles. Bio/Technology 1993, 11, 916–920. [Google Scholar] [CrossRef] [PubMed]
- Smerdou, C.; Liljestrom, P. Two-helper RNA system for production of recombinant Semliki Forest virus particles. J. Virol. 1999, 73, 1092–1098. [Google Scholar] [PubMed]
- Schlesinger, S. Alphavirus vectors: Development and potential therapeutic applications. Exp. Opin. Biol. Ther. 2001, 1, 177–191. [Google Scholar] [CrossRef] [PubMed]
- Tseng, J.C.; Daniels, G.; Meruelo, D. Controlled propagation of replication-competent Sindbis viral vector using suicide gene strategy. Gene Ther. 2009, 16, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Ohno, K.; Sawai, K.; Iijima, Y.; Levin, B.; Meruelo, D. Cell-specific targeting of Sindbis virus vectors displaying IgG-binding domains of protein A. Nat. Biotechnol. 1997, 15, 763–767. [Google Scholar] [CrossRef] [PubMed]
- Tseng, J.C.; Levin, B.; Hurtado, A.; Yee, H.; Perez de Castro, I.; Jimenez, M.; Shamamian, P.; Jin, R.; Novick, R.P.; Pellicer, A.; et al. Systemic tumor targeting and killing by Sindbis viral vectors. Nat. Biotechnol. 2004, 22, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Granot, T.; Yamanashi, Y.; Meruelo, D. Sindbis virus vectors transiently deliver tumor-associated antigens to lymph nodes and elicit diversified antitumor CD8+ T-cell immunity. Mol. Ther. 2014, 22, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Madoz, J.R.; Prieto, J.; Smerdou, C. Biodistribution and tumor infectivity of Semliki Forest virus vectors in mice: Effects of re-administration. Mol. Ther. 2007, 15, 2164–2171. [Google Scholar] [CrossRef] [PubMed]
- Suzme, R.; Tseng, J.C.; Levin, B.; Ibrahim, S.; Meruelo, D.; Pellicer, A. Sindbis viral vectors target hematopoietic malignant cells. Cancer Gene Ther. 2012, 19, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.Y.; Guo, J.H.; Hwang, L.H. Oncolytic Sindbis virus targets tumors defective in interferon response and induces significant bystander antitumor immunity in vivo. Mol. Ther. 2012, 20, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Unno, Y.; Shino, Y.; Kondo, F.; Igarashi, N.; Wang, G.; Shimura, R.; Yamaguchi, T.; Asano, T.; Saisho, H.; Sekiya, S.; et al. Oncolytic viral therapy for cervical and ovarian cancer cells by Sindbis virus AR339 strain. Clin. Cancer Res. 2005, 11, 4553–4560. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Uzawa, K.; Kasamatsu, A.; Shinozuka, K.; Sakuma, K.; Yamatoji, M.; Shiiba, M.; Shino, Y.; Shirasawa, H.; Tanzawa, H. Oncolytic activity of Sindbis virus in human oral squamous carcinoma cells. Br. J. Cancer 2009, 101, 684–690. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Shirasawa, H.; Isegawa, N.; Shiiba, M.; Uzawa, K.; Tanzawa, H. Oncolytic virotherapy for oral squamous cell carcinoma using replication-competent viruses. Oral Oncol. 2009, 45, 1021–1027. [Google Scholar] [CrossRef] [PubMed]
- Takenouchi, A.; Saito, K.; Saito, E.; Saito, T.; Hishiki, T.; Matusnaga, T.; Isegawa, N.; Yoshida, H.; Ohnuma, N.; Shirasawa, H. Oncolytic viral therapy for neuroblastoma cells with Sindbis virus AR339 strain. Pediatr. Surg. Int. 2015, 31, 1151–1159. [Google Scholar] [CrossRef] [PubMed]
- Tseng, J.C.; Zanzonico, P.B.; Levin, B.; Finn, R.; Larson, S.M.; Meruelo, D. Tumor-specific in vivo transfection with HSV-1 thymidine kinase gene using a Sindbis viral vector as a basis for prodrug ganciclovir activation and PET. J. Nucl. Med. 2006, 47, 1136–1143. [Google Scholar] [PubMed]
- Vähä-Koskela, M.J.; Kallio, J.P.; Jansson, L.C.; Heikkilä, J.E.; Zakhartchenko, V.A.; Kallajoki, M.A.; Kähäri, V.M.; Hinkkanen, A.E. Oncolytic capacity of attenuated replicative semliki forest virus in human melanoma xenografts in severe combined immunodeficient mice. Cancer Res. 2006, 66, 7185–7194. [Google Scholar] [CrossRef] [PubMed]
- Määttä, A.M.; Liimatainen, T.; Wahlfors, T.; Wirth, T.; Vähä-Koskela, M.; Jansson, L.; Valonen, P.; Häkkinen, K.; Rautsi, O.; Pellinen, R.; et al. Evaluation of cancer virotherapy with attenuated replicative Semliki forest virus in different rodent tumor models. Int. J. Cancer 2007, 121, 863–870. [Google Scholar] [CrossRef] [PubMed]
- Heikkilä, J.E.; Vähä-Koskela, M.J.; Ruotsalainen, J.J.; Martikainen, M.W.; Stanford, M.M.; McCart, J.A.; Bell, J.C.; Hinkkanen, A.E. Intravenously administered alphavirus vector VA7 eradicates orthotopic human glioma xenografts in nude mice. PLoS ONE 2010, 5, e8603. [Google Scholar] [CrossRef] [PubMed]
- Wollmann, G.; Tattersall, P.; van den Pol, A.N. Targeting human glioblastoma cells: Comparison of nine viruses with oncolytic potential. J. Virol. 2005, 79, 6005–6022. [Google Scholar] [CrossRef] [PubMed]
- Abraham, R.; Mudaliar, P.; Padmanabhan, A.; Sreekumar, E. Induction of cytopathogenicity in human glioblastoma cells by chikungunya virus. PLoS ONE 2013, 8, e75854. [Google Scholar] [CrossRef] [PubMed]
- Määttä, A.M.; Mäkinen, K.; Ketola, A.; Liimatainen, T.; Yongabi, F.N.; Vähä-Koskela, M.; Pirinen, R.; Rautsi, O.; Pellinen, R.; Hinkkanen, A.; et al. Replication competent Semliki Forest virus prolongs survival in experimental lung cancer. Int. J. Cancer 2008, 123, 1704–1711. [Google Scholar] [CrossRef] [PubMed]
- Ketola, A.; Hinkkanen, A.; Yongabi, F.; Furu, P.; Määttä, A.M.; Liimatainen, T.; Pirinen, R.; Björn, M.; Hakkarainen, T.; Mäkinen, K.; et al. Oncolytic Semliki forest virus vector as a novel candidate against unresectable osteosarcoma. Cancer Res. 2008, 68, 8342–8350. [Google Scholar] [CrossRef] [PubMed]
- Roche, F.P.; Sheahan, B.J.; O’Mara, S.M.; Atkins, G.J. Semliki Forest virus-mediated gene therapy of the RG2 rat glioma. Neuropathol. Appl. Neurobiol. 2010, 36, 648–660. [Google Scholar] [CrossRef] [PubMed]
- Vähä-Koskela, M.J.; Le Boeuf, F.; Lemay, C.; De Silva, N.; Diallo, J.S.; Cox, J.; Becker, M.; Choi, Y.; Ananth, A.; Sellers, C.; et al. Resistance to two heterologous neurotropic oncolytic viruses, Semliki Forest virus and vaccinia virus, in experimental glioma. J. Virol. 2013, 87, 2363–2366. [Google Scholar] [CrossRef] [PubMed]
- Ruotsalainen, J.J.; Kaikkonen, M.U.; Niittykoski, M.; Martikainen, M.W.; Lemay, C.G.; Cox, J.; De Silva, N.S.; Kus, A.; Falls, T.J.; Diallo, J.S.; et al. Clonal variation in interferon response determines the outcome of oncolytic virotherapy in mouse CT26 colon carcinoma model. Gene Ther. 2015, 22, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Autio, K.P.; Ruotsalainen, J.J.; Anttila, M.O.; Niittykoski, M.; Waris, M.; Hemminki, A.; Vähä-Koskela, M.J.; Hinkkanen, A.E. Attenuated Semliki Forest virus for cancer treatment in dogs: Safety assessment in two laboratory Beagles. BMC Vet. Res. 2015, 11, 170. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Q.; Tsai, Y.C.; Monie, A.; Wu, T.C.; Hung, C.F. Enhancing the therapeutic effect against ovarian cancer through a combination of viral oncolysis and antigen-specific immunotherapy. Mol. Ther. 2010, 18, 692–699. [Google Scholar] [CrossRef] [PubMed]
- Quetglas, J.I.; Labiano, S.; Aznar, M.Á.; Bolaños, E.; Azpilikueta, A.; Rodriguez, I.; Casales, E.; Sánchez-Paulete, A.R.; Segura, V.; Smerdou, C.; et al. Virotherapy with a Semliki Forest Virus-Based Vector Encoding IL12 Synergizes with PD-1/PD-L1 Blockade. Cancer Immunol. Res. 2015, 3, 449–454. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Zhang, H.; Liang, J.; Li, K.; Zhu, W.; Fu, L.; Wang, F.; Zheng, X.; Shi, H.; Wu, S.; et al. Identification and characterization of alphavirus M1 as a selective oncolytic virus targeting ZAP-defective human cancers. Proc. Natl. Acad. Sci. USA 2014, 111, E4504–4512. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Zhang, H.; Qiu, J.; Lin, Y.; Liang, J.; Xiao, X.; Fu, L.; Wang, F.; Cai, J.; Tan, Y.; et al. Activation of Cyclic Adenosine Monophosphate Pathway Increases the Sensitivity of Cancer Cells to the Oncolytic Virus M1. Mol. Ther. 2016, 24, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Lin, Y.; Li, K.; Liang, J.; Xiao, X.; Cai, J.; Tan, Y.; Xing, F.; Mai, J.; Li, Y.; et al. Naturally Existing Oncolytic Virus M1 Is Nonpathogenic for the Nonhuman Primates After Multiple Rounds of Repeated Intravenous Injections. Hum. Gene Ther. 2016, 27, 700–711. [Google Scholar] [CrossRef] [PubMed]
- Tseng, J.C.; Granot, T.; DiGiacomo, V.; Levin, B.; Meruelo, D. Enhanced specific delivery and targeting of oncolytic Sindbis viral vectors by modulating vascular leakiness in tumor. Cancer Gene Ther. 2010, 17, 244–255. [Google Scholar] [CrossRef] [PubMed]
- Ylosmaki, E.; Martikainen, M.; Hinkkanen, A.; Saksela, K. Attenuation of Semliki Forest virus neurovirulence by microRNA-mediated detargeting. J. Virol. 2013, 87, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Martikainen, M.; Niittykoski, M.; von und zu Fraunberg, M.; Immonen, A.; Koponen, S.; van Geenen, M.; Vähä-Koskela, M.; Ylösmäki, E.; Jääskeläinen, J.E.; Saksela, K.; et al. MicroRNA-Attenuated Clone of Virulent Semliki Forest Virus Overcomes Antiviral Type I Interferon in Resistant Mouse CT-2A Glioma. J. Virol. 2015, 89, 10637–10647. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Tatsuya, T.; Nishiyama, Y. Oncotarget Strategies For Herpes Simplex Virus-1. Curr. Gene Ther. 2016, 16, 130–143. [Google Scholar] [CrossRef] [PubMed]
- Msaouel, P.; Iankov, I.D.; Dispenzieri, A.; Galanis, E. Attenuated oncolysis measles virus strains as cancer therapeutics. Curr. Pharm. Biotechnol. 2012, 13, 1732–1741. [Google Scholar] [CrossRef] [PubMed]
- Grote, D.; Russell, S.J.; Cornu, T.I.; Cattaneo, R.; Vile, R.; Poland, G.A.; Fielding, A.K. Live attenuated measles virus induces regression of human lymphoma xenografts in immunodeficient mice. Blood 2001, 97, 3746–3754. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, K.; Pham, L.; O’Connor, M.K.; Federspiel, M.J.; Russell, S.J.; Peng, K.W. Dual therapy of ovarian cancer using measles viruses expressing carcinoembryonic antigen and sodium iodide symporter. Clin. Cancer Res. 2006, 12, 1868–1875. [Google Scholar] [CrossRef] [PubMed]
- Paraskevakou, G.; Allen, C.; Nakamura, T.; Zollman, P.; James, C.D.; Peng, K.W.; Schroeder, M.; Russell, S.J.; Galanis, E. Epidermal growth factor receptor (EGFR)-retargeted measles virus strains effectively target EGFR- or EGFRvIII expressing gliomas. Mol. Ther. 2007, 15, 677–686. [Google Scholar] [CrossRef] [PubMed]
- McDonald, C.J.; Erlichman, C.; Ingle, J.N.; Rosales, G.A.; Allen, C.; Greiner, S.M.; Harvey, M.E.; Zollman, P.; Russell, S.J.; Galanis, E. A measles virus vaccine strain derivative as a novel oncolytic agent against breast cancer. Breast Cancer Res. Treat. 2006, 99, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Msaouel, P.; Iankov, I.D.; Allen, C.; Moris, J.C.; von Messing, V.; Cattaneo, R.; Koustilieris, M.; Russell, S.J.; Galanis, E. Engineered measles virus as a novel oncolytic therapy against prostate cancer. Prostate 2009, 69, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.; Galanis, E. Potential and clinical translation of oncolytic measles viruses. Expert Opin. Biol. Ther. 2017, 17, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Fehl, D.J.; Ahmed, M. Curcumin promotes the oncolytic capacity of vesicular stomatitis virus for the treatment of prostate cancers. Virus Res. 2017, 228, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Steele, M.B.; Jenks, N.; Grell, J.; Behrens, M.; Nace, R.; Naik, S.; Federspeil, M.J.; Russell, S.J.; Peng, K.W. Robust oncolytic virotherapy induces tumor lysis syndrome and associated toxicities in the MPC-11 plasmacytoma model. Mol. Ther. 2016, 24, 2109–2117. [Google Scholar] [CrossRef] [PubMed]
- Dold, C.; Rodriguez Urbiola, C.; Wollmann, G.; Egerer, L.; Muik, A.; Bellmann, L.; Fiegl, H.; Marth, C.; Kimpel, J.; von Laer, D. Application of interferon modulators to overcome partial resistance of human ovarian cancers to VSV-GP oncolytic viral therapy. Mol. Ther. Oncol. 2016, 3, 16021. [Google Scholar] [CrossRef] [PubMed]
- Muik, A.; Stubbert, L.J.; Jahedi, R.Z.; Geiss, Y.; Kimpel, J.; Dold, C.; Tober, R.; Volk, A.; Klein, S.; Dietrich, U.; et al. Re-engineering vesicular stomatitis virus to abrogate neurotoxicity, circumvent humor response, and enhance oncolytic potency. Cancer Res. 2014, 74, 3567–3578. [Google Scholar] [CrossRef] [PubMed]
- Wollmann, G.; Drokhlyansky, E.; Davis, J.N.; Cepko, C.; van den Pol, A.N. Lassa-Vesicular stomatitis chimeic virus safely destroys brain tumors. J. Virol. 2015, 89, 6711–6724. [Google Scholar] [CrossRef] [PubMed]
- Alkayyal, A.A.; Tai, L.H.; Kennedy, M.A.; de Souza, C.T.; Zhang, J.; Lefebvre, C.; Sahi, S.; Ananth, A.A.; Mahmoud, A.B.; Cron, G.O.; et al. NK-cell recruitment is necessary for eradication of peritoneal carcinomatosis with an IL12-expressing Maraba virus cellular vaccine. Cancer Immunol. Res. 2017, 5, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.X.; Danquah, M.K.; Sidhu, A.; Ongkudon, C.M.; Lau, S.Y. Towards targeted cancer therapy: Apatmer or oncolytic virus? Eur. J. Pharm. Sci. 2016, 96, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Bilsland, A.E.; Spiliopoulou, P.; Evans, T.R. Virotherapy: Cancer gene therapy at last? F1000 Res. 2016, 5, 2105. [Google Scholar] [CrossRef] [PubMed]
- Lundstrom, K. Biology and application of alphaviruses in gene therapy. Gene Ther. 2005, 12 (Suppl. 1), S92–S97. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Boulikas, T.; Lundstrom, K.; Söling, A.; Warnke, P.C.; Rainov, N.G. Immunogene therapy of recurrent glioblastoma multiforme with a liposomally encapsulated replication-incompetent Semliki forest virus vector carrying the human interleukin-12 gene—A phase I/II clinical protocol. J. Neurooncol. 2003, 64, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Riezebos-Brilman, A.; Walczak, M.; Regts, J.; Rots, M.G.; Kamps, G.; Dontje, B.; Haisma, H.Y.; Wilschut, J.; Daemen, T. A comparative study on the immunotherapeutic efficacy of recombinant Semliki Forest virus and adenovirus vector systems in a murine model for cervical cancer. Gene Ther. 2007, 14, 1695–1704. [Google Scholar] [CrossRef] [PubMed]
- Polo, J.M.; Belli, B.A.; Driver, D.A.; Frolov, I.; Sherrill, S.; Hariharan, M.J.; Townsend, J.; Perri, S.; Mento, S.J.; Jolly, D.J.; et al. Stable alphavirus packaging cell lines for Sindbis and Semliki Forest virus-derived vectors. Proc. Natl. Acad. Sci. USA 1999, 96, 4598–4603. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.Y.; Liang, G.D. Selection and characterization of packaging cell lines for XJ-160 virus. Intervirology 2009, 52, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Zajakina, A.; Vasilevska, J.; Kozlovska, T.; Lundstrom, K. Alphaviral vectors for cancer treatment. In Viral Nanotechnology; Khudyakov, Y., Pumpens, P., Eds.; CRC Press: Boca Raton, FL, USA, 2015; pp. 467–486. [Google Scholar]
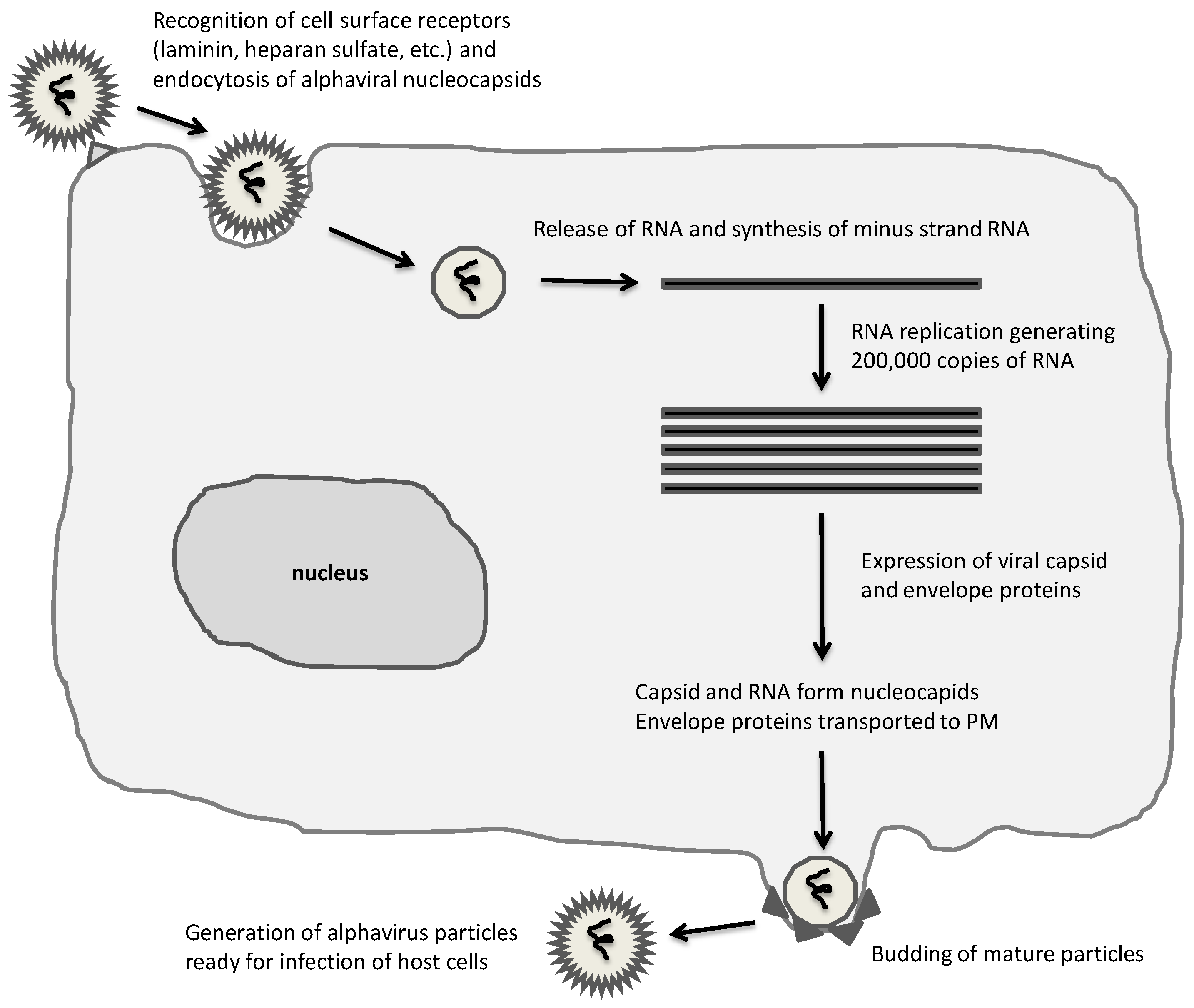
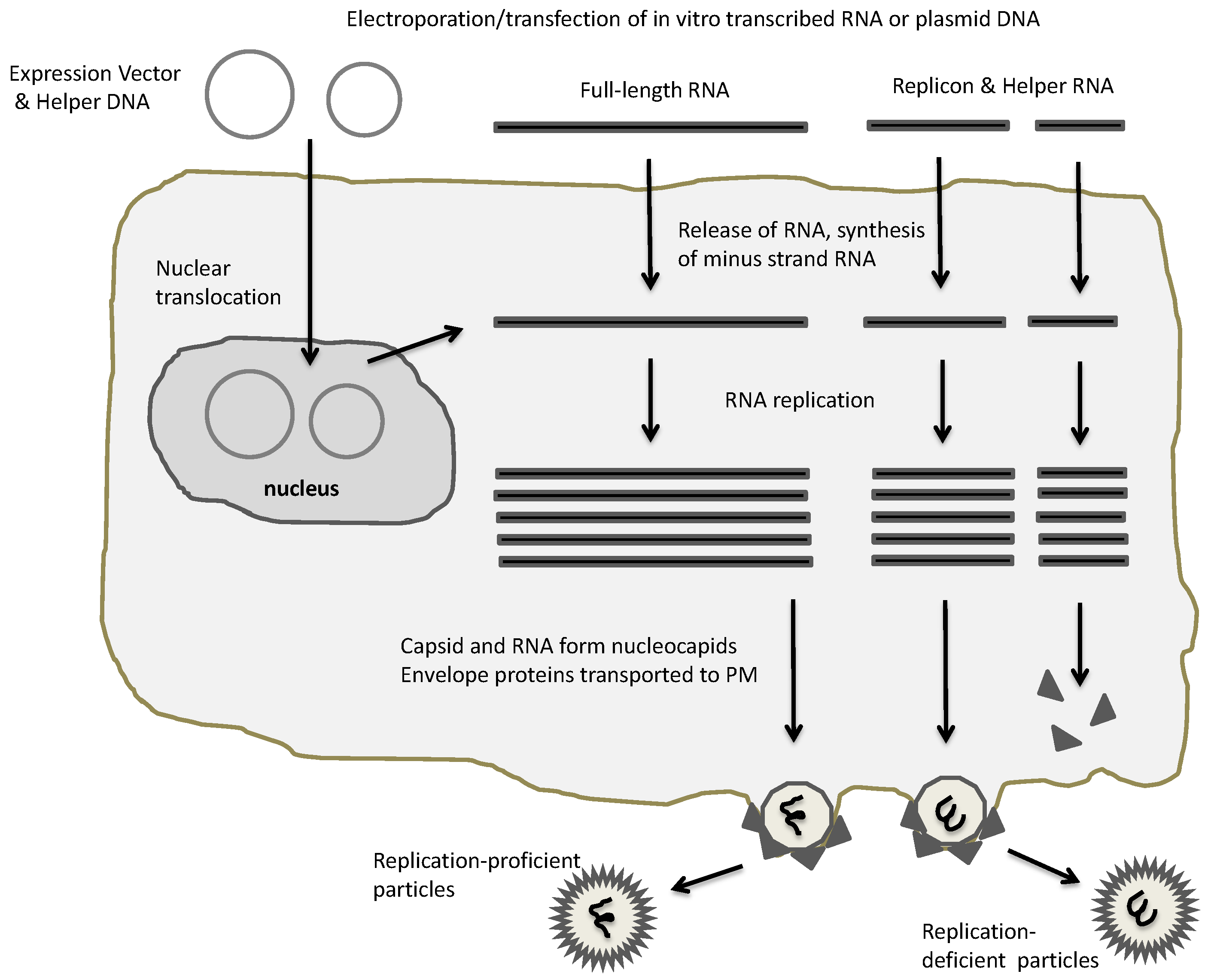
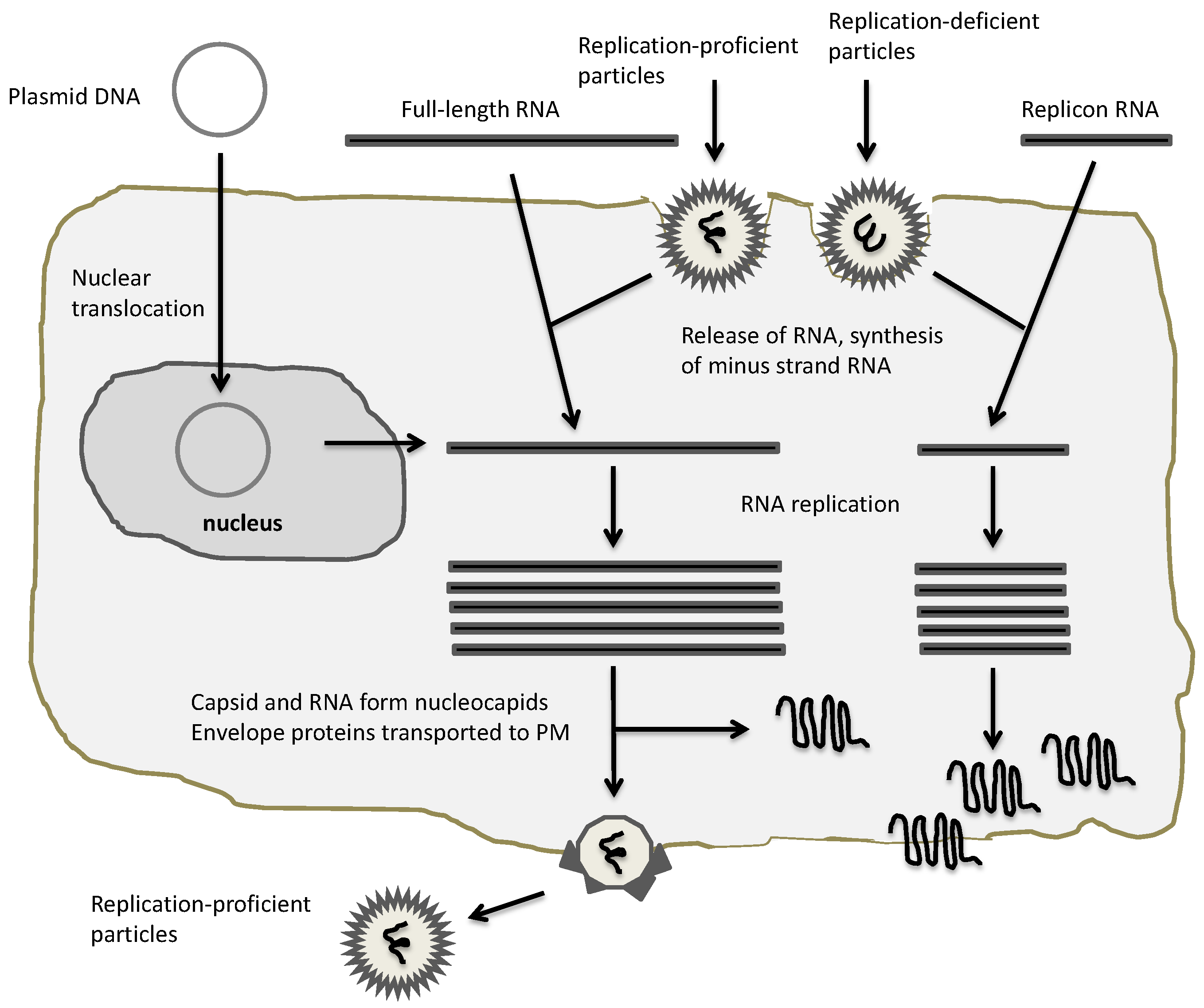

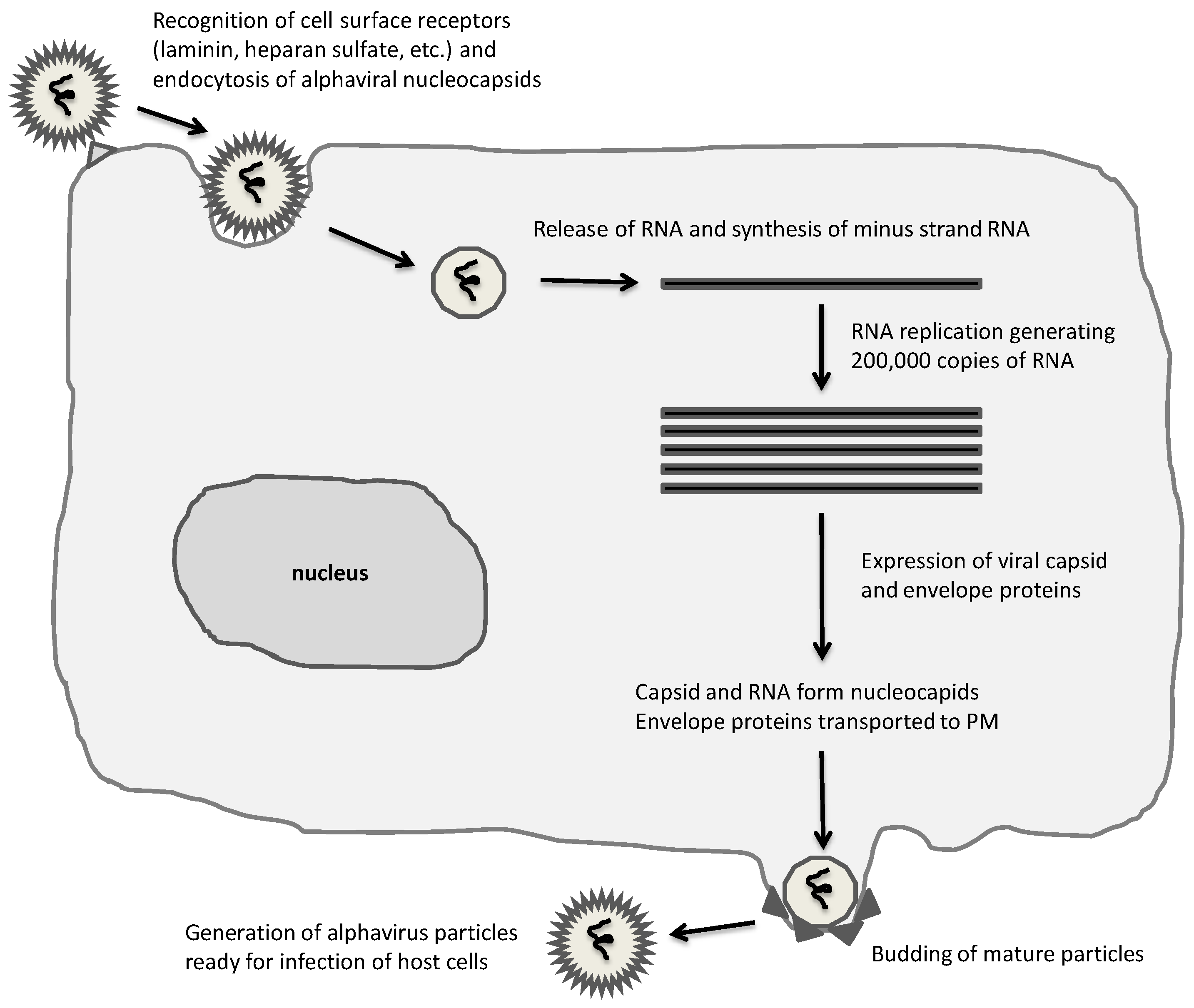{kind=link}
{kind=link}
{kind=link}
| Cancer | Vector/Gene | Effect | Reference |
|---|---|---|---|
| Blood | SIN | tumor targeting, prolonged survival | [80] |
| Bone | SFV/EGFP | tumor regression, improved survival | [75] |
| SFV/EGFP | tumor cell killing | [79] | |
| Brain and neuronal | SFV/IL-12 | tumor regression, improved survival | [77] |
| SFV/EGFP | tumor eradication | [71] | |
| SFV4-miRT124 | tumor regression, improved survival | [96] | |
| SIN/GFP | tumor killing in vivo | [72] | |
| CHIK | apoptosis in U87MG cells | [73] | |
| SIN AR339 | tumor regression in vivo | [68] | |
| Cervix | SIN AR339 | suppression of ascites formation | [65] |
| Colon | SFV/EGFP | tumor regression | [79] |
| SFV/IL12 + PD1 | tumor regression, prolonged survival | [82] | |
| Kidney | encSFV/IL12 | enhanced IL-12 secretion in patients | [105] |
| Liver | M1 | tumor growth inhibition | [83] |
| Lung | SFV/EGFP | tumor regression | [75] |
| Melanoma | SFV/EGFP | tumor regression in mouse, rat | [71] |
| SFV/EGFP | tumor regression in mice | [70] | |
| SFV/IL12 + PD1 | tumor regression, prolonged survival | [82] | |
| encSFV/IL12 | enhanced IL-12 secretion in patients | [105] | |
| Oral | SIN AR339 | apoptosis in OSCC cell lines | [67] |
| Ovarian | SIN AR339 | suppression of ascites formation | [65] |
| SFV + VV/OVA | tumor killing | [81] |
© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lundstrom, K. Oncolytic Alphaviruses in Cancer Immunotherapy. Vaccines 2017, 5, 9. https://doi.org/10.3390/vaccines5020009
Lundstrom K. Oncolytic Alphaviruses in Cancer Immunotherapy. Vaccines. 2017; 5(2):9. https://doi.org/10.3390/vaccines5020009
Chicago/Turabian StyleLundstrom, Kenneth. 2017. "Oncolytic Alphaviruses in Cancer Immunotherapy" Vaccines 5, no. 2: 9. https://doi.org/10.3390/vaccines5020009
APA StyleLundstrom, K. (2017). Oncolytic Alphaviruses in Cancer Immunotherapy. Vaccines, 5(2), 9. https://doi.org/10.3390/vaccines5020009