
Regulatory T Cells in the Tumor Microenvironment and Cancer Progression: Role and Therapeutic Targeting


Abstract
:1. Introduction
- Suppressive cytokines (TGF-β, IL-10, IL-35)
- Immune checkpoints and inhibitory receptors (CTLA-4, PD-1, LAG-3, TIM-3, ICOS, TIGIT, IDO)
- Direct cytotoxicity (perforin/granzyme-mediated)
- Metabolic disruption of T effector cell activity (IL-2 consumption)
- Induction of tolerogenic DCs, which promote T cell exhaustion and expansion
2. Tregs in Cancer
- Recruitment: Tregs are recruited into tumors in response to chemokines secreted by tumor cells and innate immune cells; key chemokine-chemokine receptor combinations include CCL17/22-CCR4, CCL5-CCR5, CCL28-CCR10 and CXCL9/10/11-CXCR3.
- Expansion: Tregs can be expanded in situ, and proliferate efficiently in response to tumor-derived factors (TGF-β, IL-10) within the TME.
- Conversion: Generation of suppressive Tregs from non-suppressive CD25− conventional T cells (Tconv) driven by tumor-derived transforming growth factor-beta (TGF-β) and adenosine; this has mainly been studied in murine models and the contribution of Treg induction to Treg accumulation within the TME in human cancer remains to be confirmed.
2.1. Immunosuppressive Roles of Tumor-Infiltrating Tregs in Cancer
Enhanced Suppression in the TME
- CD45RA−FoxP3hi activated “effector” Tregs
- CD45RA+FoxP3lo resting Tregs
- cytokine-secreting CD45RA−FoxP3lo non-suppressive T cells, or “non-Tregs”
2.2. TME-Treg Crosstalk
2.3. Protective Role of Tregs in Inflammation/Cancer
2.4. Treg Subsets and Heterogeneity
2.5. Antigen Specificity of Tregs
2.6. Tregs by Cancer Stage
2.6.1. Cancer Initiation and Establishment
2.6.2. Tumor Metastasis
2.6.3. Remission/Recurrence
3. Clinical Evidence for Treg Therapeutic Targeting
3.1. Immune Checkpoint Inhibition and Tregs
3.1.1. Anti-CTLA-4
3.1.2. Anti-PD-1
3.2. Effect of Radiotherapy and Chemotherapy on Tregs
4. Challenges of Targeting Tregs in Humans
4.1. Misclassification of Tregs
4.2. Systemic versus Specific Subset Depletion
4.3. Tregs in the Immune Context
5. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| Ab | antibody |
| ADCC | antibody-dependent cell-mediated cytotoxicity |
| ADCP | antibody-dependent cellular phagocytosis |
| Ag | antigen |
| APC | antigen presenting cell |
| CCR | C-C motif chemokine receptor |
| CD25 | IL-2 receptor alpha chain |
| COX-2 | cyclooxygenase-2 |
| CRC | colorectal cancer |
| CRT | chemo-radiotherapy |
| CTL | CD8+ cytotoxic t cell |
| CTLA-4 | cytotoxic T-lymphocyte-associated protein 4 |
| CTX | cyclophosphamide |
| CXCR | CXC chemokine receptor |
| eTreg | effector Treg |
| FcγRIII | Fc gamma receptor III |
| FoxP3 | forkhead box P3 |
| GARP | glycoprotein A repetitions predominant |
| GITR | glucocorticoid-induced TNFR-related protein |
| HAVCR2 | Hepatitis A virus cellular receptor 2 |
| HBV/HCV | hepatitis B/C virus |
| HCC | hepatocellular carcinoma |
| HNC | head and neck cancer |
| HNSCC | head and neck squamous cell carcinoma |
| HPV | human papilloma virus |
| ICI | immune checkpoint inhibition |
| ICOS | Inducible T-cell costimulator |
| IDO | indoleamine 2,3-dioxygenase |
| IFN-γ | interferon gamma |
| IL-10/35 | Interleukin 10/35 |
| LAG-3 | lymphocyte activation gene-3 |
| LAP | latency-associated peptide |
| mAb | monoclonal antibody |
| MDSC | myeloid-derived suppressor cell |
| NK cell | natural killer cell |
| NRP1 | neuropilin 1 |
| PD-1 | programmed death 1 |
| PD-L1 | programmed death-ligand 1 |
| PGE-2 | prostaglandin E-2 |
| pTreg | peripheral Treg |
| RANK | receptor activator of nuclear factor kappa-B |
| RANKL | receptor activator of nuclear factor kappa-B ligand |
| RCC | renal cell carcinoma |
| RT | radiotherapy |
| S1P | sphingosine-1-phosphate |
| S1PR1 | sphingosine-1-phosphate receptor 1 |
| TAA | tumor-associated antigen |
| Tconv | conventional T cell |
| TDLN | tumor-draining lymph node |
| Teff | CD4+ T effector cell |
| TEX | tumor-derived exosome |
| TGF-β | Transforming growth factor beta |
| Th cell | T helper cell |
| TI | tumor-infiltrating |
| TIGIT | T cell immunoreceptor with Ig and ITIM domains |
| TILs | tumor-infiltrating lymphocytes |
| TIM-3 | T-cell immunoglobulin and mucin-domain containing-3 |
| TME | tumor microenvironment |
| Treg | regulatory T cell |
| tTreg | thymic-derived Treg |
| VEGF-A | vascular endothelial growth factor-A |
| γδ T cell | gamma delta T cell |
References
- Sakaguchi, S.; Yamaguchi, T.; Nomura, T.; Ono, M. Regulatory T cells and immune tolerance. Cell 2008, 133, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.M.; Bautista, J.L.; Hsieh, C.S. Thymic and peripheral differentiation of regulatory T cells. Adv. Immunol. 2011, 112, 25–71. [Google Scholar] [PubMed]
- Yadav, M.; Stephan, S.; Bluestone, J.A. Peripherally induced tregs—Role in immune homeostasis and autoimmunity. Front. Immunol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Ono, M.; Setoguchi, R.; Yagi, H.; Hori, S.; Fehervari, Z.; Shimizu, J.; Takahashi, T.; Nomura, T. Foxp3+CD25+CD4+ natural regulatory T cells in dominant self-tolerance and autoimmune disease. Immunol. Rev. 2006, 212, 8–27. [Google Scholar] [CrossRef] [PubMed]
- Weiner, H.L.; da Cunha, A.P.; Quintana, F.; Wu, H. Oral tolerance. Immunol. Rev. 2011, 241, 241–259. [Google Scholar] [CrossRef] [PubMed]
- Shevach, E.M. Mechanisms of foxp3+ T regulatory cell-mediated suppression. Immunity 2009, 30, 636–645. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Oberle, N.; Krammer, P.H. Molecular mechanisms of treg-mediated T cell suppression. Front. Immunol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Miyara, M.; Yoshioka, Y.; Kitoh, A.; Shima, T.; Wing, K.; Niwa, A.; Parizot, C.; Taflin, C.; Heike, T.; Valeyre, D.; et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity 2009, 30, 899–911. [Google Scholar] [CrossRef] [PubMed]
- Duhen, T.; Duhen, R.; Lanzavecchia, A.; Sallusto, F.; Campbell, D.J. Functionally distinct subsets of human FOXP3+ Treg cells that phenotypically mirror effector Th cells. Blood 2012, 119, 4430–4440. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Maiella, S.; Xhaard, A.; Pang, Y.; Wenandy, L.; Larghero, J.; Becavin, C.; Benecke, A.; Bianchi, E.; Socie, G.; et al. Multiparameter single-cell profiling of human CD4+FOXP3+ regulatory T-cell populations in homeostatic conditions and during graft-versus-host disease. Blood 2013, 122, 1802–1812. [Google Scholar] [CrossRef] [PubMed]
- Mason, G.M.; Lowe, K.; Melchiotti, R.; Ellis, R.; de Rinaldis, E.; Peakman, M.; Heck, S.; Lombardi, G.; Tree, T.I. Phenotypic Complexity of the Human Regulatory T Cell Compartment Revealed by Mass Cytometry. J. Immunol. 2015, 195, 2030–2037. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Wing, J.B.; Sakaguchi, S. Two modes of immune suppression by Foxp3(+) regulatory T cells under inflammatory or non-inflammatory conditions. Semin. Immunol. 2011, 23, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Vignali, D.A. Mechanisms of T(reg) Suppression: Still a Long Way to Go. Front. Immunol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Elkord, E.; Alcantar-Orozco, E.M.; Dovedi, S.J.; Tran, D.Q.; Hawkins, R.E.; Gilham, D.E. T regulatory cells in cancer: Recent advances and therapeutic potential. Expert Opin. Biol. Ther. 2010, 10, 1573–1586. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, H.; Sakaguchi, S. Regulatory T cells in cancer immunotherapy. Curr. Opin. Immunol. 2014, 27, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, B.; Abd Al Samid, M.; al-Ramadi, B.K.; Elkord, E. Phenotypic alterations, clinical impact and therapeutic potential of regulatory T cells in cancer. Expert Opin. Biol. Ther. 2014, 14, 931–945. [Google Scholar] [CrossRef] [PubMed]
- Curiel, T.J.; Coukos, G.; Zou, L.; Alvarez, X.; Cheng, P.; Mottram, P.; Evdemon-Hogan, M.; Conejo-Garcia, J.R.; Zhang, L.; Burow, M.; et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 2004, 10, 942–949. [Google Scholar] [CrossRef] [PubMed]
- Leffers, N.; Gooden, M.J.; de Jong, R.A.; Hoogeboom, B.N.; ten Hoor, K.A.; Hollema, H.; Boezen, H.M.; van der Zee, A.G.; Daemen, T.; Nijman, H.W. Prognostic significance of tumor-infiltrating T-lymphocytes in primary and metastatic lesions of advanced stage ovarian cancer. Cancer Immunol. Immunother. 2009, 58, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Du, Z.; Yang, F.; Di, Y.; Li, J.; Zhou, Z.; Pillarisetty, V.G.; Fu, D. FOXP3+ lymphocyte density in pancreatic cancer correlates with lymph node metastasis. PLoS ONE 2014, 9, e106741. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Xu, X.; Guo, S.; Zhang, C.; Tang, Y.; Tian, Y.; Ni, B.; Lu, B.; Wang, H. An increased abundance of tumor-infiltrating regulatory T cells is correlated with the progression and prognosis of pancreatic ductal adenocarcinoma. PLoS ONE 2014, 9, e91551. [Google Scholar] [CrossRef] [PubMed]
- Tao, H.; Mimura, Y.; Aoe, K.; Kobayashi, S.; Yamamoto, H.; Matsuda, E.; Okabe, K.; Matsumoto, T.; Sugi, K.; Ueoka, H. Prognostic potential of FOXP3 expression in non-small cell lung cancer cells combined with tumor-infiltrating regulatory T cells. Lung Cancer 2012, 75, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Sayour, E.J.; McLendon, P.; McLendon, R.; De Leon, G.; Reynolds, R.; Kresak, J.; Sampson, J.H.; Mitchell, D.A. Increased proportion of FoxP3+ regulatory T cells in tumor infiltrating lymphocytes is associated with tumor recurrence and reduced survival in patients with glioblastoma. Cancer Immunol. Immunother. 2015, 64, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.Z.; Novak, A.J.; Stenson, M.J.; Witzig, T.E.; Ansell, S.M. Intratumoral CD4+CD25+ regulatory T-cell-mediated suppression of infiltrating CD4+ T cells in B-cell non-Hodgkin lymphoma. Blood 2006, 107, 3639–3646. [Google Scholar] [CrossRef] [PubMed]
- DeLeeuw, R.J.; Kost, S.E.; Kakal, J.A.; Nelson, B.H. The prognostic value of FoxP3+ tumor-infiltrating lymphocytes in cancer: A critical review of the literature. Clin. Cancer Res. 2012, 18, 3022–3029. [Google Scholar] [CrossRef] [PubMed]
- Shang, B.; Liu, Y.; Jiang, S.J.; Liu, Y. Prognostic value of tumor-infiltrating FoxP3+ regulatory T cells in cancers: A systematic review and meta-analysis. Sci. Rep. 2015. [Google Scholar] [CrossRef] [PubMed]
- Rasku, M.A.; Clem, A.L.; Telang, S.; Taft, B.; Gettings, K.; Gragg, H.; Cramer, D.; Lear, S.C.; McMasters, K.M.; Miller, D.M.; et al. Transient T cell depletion causes regression of melanoma metastases. J. Transl. Med. 2008. [Google Scholar] [CrossRef] [PubMed]
- Ladoire, S.; Arnould, L.; Apetoh, L.; Coudert, B.; Martin, F.; Chauffert, B.; Fumoleau, P.; Ghiringhelli, F. Pathologic complete response to neoadjuvant chemotherapy of breast carcinoma is associated with the disappearance of tumor-infiltrating foxp3+ regulatory T cells. Clin. Cancer Res. 2008, 14, 2413–2420. [Google Scholar] [CrossRef] [PubMed]
- Rech, A.J.; Mick, R.; Martin, S.; Recio, A.; Aqui, N.A.; Powell, D.J., Jr.; Colligon, T.A.; Trosko, J.A.; Leinbach, L.I.; Pletcher, C.H.; et al. CD25 blockade depletes and selectively reprograms regulatory T cells in concert with immunotherapy in cancer patients. Sci. Transl. Med. 2012. [Google Scholar] [CrossRef] [PubMed]
- Haas, M.; Dimmler, A.; Hohenberger, W.; Grabenbauer, G.G.; Niedobitek, G.; Distel, L.V. Stromal regulatory T-cells are associated with a favourable prognosis in gastric cancer of the cardia. BMC Gastroenterol. 2009. [Google Scholar] [CrossRef] [PubMed]
- Salama, P.; Phillips, M.; Grieu, F.; Morris, M.; Zeps, N.; Joseph, D.; Platell, C.; Iacopetta, B. Tumor-infiltrating FOXP3+ T regulatory cells show strong prognostic significance in colorectal cancer. J. Clin. Oncol. 2009, 27, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Ladoire, S.; Martin, F.; Ghiringhelli, F. Prognostic role of FOXP3+ regulatory T cells infiltrating human carcinomas: The paradox of colorectal cancer. Cancer Immunol. Immunother. 2011, 60, 909–918. [Google Scholar] [CrossRef] [PubMed]
- Ondondo, B.; Jones, E.; Godkin, A.; Gallimore, A. Home sweet home: The tumor microenvironment as a haven for regulatory T cells. Front. Immunol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.H. Reining in FoxP3(+) regulatory T cells by the sphingosine 1-phosphate-S1P1 axis. Immunol. Cell Biol. 2009, 87, 502–504. [Google Scholar] [CrossRef] [PubMed]
- Priceman, S.J.; Shen, S.; Wang, L.; Deng, J.; Yue, C.; Kujawski, M.; Yu, H. S1PR1 is crucial for accumulation of regulatory T cells in tumors via STAT3. Cell. Rep. 2014, 6, 992–999. [Google Scholar] [CrossRef] [PubMed]
- Fridman, W.H.; Pages, F.; Sautes-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Giraldo, N.A.; Becht, E.; Remark, R.; Damotte, D.; Sautes-Fridman, C.; Fridman, W.H. The immune contexture of primary and metastatic human tumours. Curr. Opin. Immunol. 2014, 27, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Kakita, N.; Kanto, T.; Itose, I.; Kuroda, S.; Inoue, M.; Matsubara, T.; Higashitani, K.; Miyazaki, M.; Sakakibara, M.; Hiramatsu, N.; et al. Comparative analyses of regulatory T cell subsets in patients with hepatocellular carcinoma: A crucial role of CD25(−)FOXP3(−) T cells. Int. J. Cancer 2012, 131, 2573–2583. [Google Scholar] [CrossRef] [PubMed]
- Schuler, P.J.; Schilling, B.; Harasymczuk, M.; Hoffmann, T.K.; Johnson, J.; Lang, S.; Whiteside, T.L. Phenotypic and functional characteristics of CD4+CD39+FOXP3+ and CD4+CD39+FOXP3neg T-cell subsets in cancer patients. Eur. J. Immunol. 2012, 42, 1876–1885. [Google Scholar] [CrossRef] [PubMed]
- Jie, H.B.; Gildener-Leapman, N.; Li, J.; Srivastava, R.M.; Gibson, S.P.; Whiteside, T.L.; Ferris, R.L. Intratumoral regulatory T cells upregulate immunosuppressive molecules in head and neck cancer patients. Br. J. Cancer 2013, 109, 2629–2635. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.C.; Mahalingam, J.; Chiang, J.M.; Su, P.J.; Chu, Y.Y.; Lai, H.Y.; Fang, J.H.; Huang, C.T.; Chiu, C.T.; Lin, C.Y. Activated but not resting regulatory T cells accumulated in tumor microenvironment and correlated with tumor progression in patients with colorectal cancer. Int. J. Cancer 2013, 132, 1341–1350. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, D.; Nishikawa, H.; Maeda, Y.; Nishioka, M.; Tanemura, A.; Katayama, I.; Ezoe, S.; Kanakura, Y.; Sato, E.; Fukumori, Y.; et al. Anti-CCR4 mAb selectively depletes effector-type FoxP3+CD4+ regulatory T cells, evoking antitumor immune responses in humans. Proc. Natl. Acad. Sci. USA 2013, 110, 17945–17950. [Google Scholar] [CrossRef] [PubMed]
- Pedroza-Gonzalez, A.; Verhoef, C.; Ijzermans, J.N.; Peppelenbosch, M.P.; Kwekkeboom, J.; Verheij, J.; Janssen, H.L.; Sprengers, D. Activated tumor-infiltrating CD4+ regulatory T cells restrain antitumor immunity in patients with primary or metastatic liver cancer. Hepatology 2013, 57, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Yang, Y.; Chen, Z.; Jiang, Z.; Gu, Y.; Liu, Y.; Xu, S.; Lin, C.; Pan, Z.; Zhou, W.; et al. Human hepatocellular carcinoma-infiltrating CD4(+)CD69(+)Foxp3(-) regulatory T cell suppresses T cell response via membrane-bound TGF-beta1. J. Mol. Med. (Berl.) 2014, 92, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Scurr, M.; Ladell, K.; Besneux, M.; Christian, A.; Hockey, T.; Smart, K.; Bridgeman, H.; Hargest, R.; Phillips, S.; Davies, M.; et al. Highly prevalent colorectal cancer-infiltrating LAP(+) Foxp3(−) T cells exhibit more potent immunosuppressive activity than Foxp3(+) regulatory T cells. Mucosal Immunol. 2014, 7, 428–439. [Google Scholar] [CrossRef] [PubMed]
- Kurose, K.; Ohue, Y.; Sato, E.; Yamauchi, A.; Eikawa, S.; Isobe, M.; Nishio, Y.; Uenaka, A.; Oka, M.; Nakayama, E. Increase in activated Treg in TIL in lung cancer and in vitro depletion of Treg by ADCC using an antihuman CCR4 mAb (KM2760). J. Thorac. Oncol. 2015, 10, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Amedei, A.; Niccolai, E.; Benagiano, M.; Della Bella, C.; Cianchi, F.; Bechi, P.; Taddei, A.; Bencini, L.; Farsi, M.; Cappello, P.; et al. Ex vivo analysis of pancreatic cancer-infiltrating T lymphocytes reveals that ENO-specific Tregs accumulate in tumor tissue and inhibit Th1/Th17 effector cell functions. Cancer Immunol. Immunother. 2013, 62, 1249–1260. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.; He, H.W.; Wang, J.X.; Cai, X.Y.; Li, Y.W.; Zhou, J.; Cheng, Y.F.; Jin, J.J.; Fan, J.; Qiu, S.J. The functional impairment of HCC-infiltrating gammadelta T cells, partially mediated by regulatory T cells in a TGFbeta- and IL-10-dependent manner. J. Hepatol. 2013, 58, 977–983. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.L.; Chen, L.; Li, M.X.; Dong, P.; Xue, J.; Wang, J.; Zhang, T.T.; Wang, X.A.; Zhang, F.M.; Ge, H.L.; et al. Elevated expression of Foxp3 in tumor-infiltrating Treg cells suppresses T-cell proliferation and contributes to gastric cancer progression in a COX-2-dependent manner. Clin. Immunol. 2010, 134, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Zhang, Y.; Zhang, J.P.; Liang, J.; Li, L.; Zheng, L. Tim-3 expression defines regulatory T cells in human tumors. PLoS ONE 2013, 8, e58006. [Google Scholar] [CrossRef] [PubMed]
- Bu, M.; Shen, Y.; Seeger, W.L.; An, S.; Qi, R.; Sanderson, J.A.; Cai, Y. Ovarian carcinoma-infiltrating regulatory T cells were more potent suppressors of CD8(+) T cell inflammation than their peripheral counterparts, a function dependent on TIM3 expression. Tumour Biol. 2016, 37, 3949–3956. [Google Scholar] [CrossRef] [PubMed]
- Nirschl, C.J.; Drake, C.G. Molecular pathways: Coexpression of immune checkpoint molecules: Signaling pathways and implications for cancer immunotherapy. Clin. Cancer Res. 2013, 19, 4917–4924. [Google Scholar] [CrossRef] [PubMed]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Rifkin, D.B. Latent transforming growth factor-beta (TGF-beta) binding proteins: Orchestrators of TGF-beta availability. J. Biol. Chem. 2005, 280, 7409–7412. [Google Scholar] [CrossRef] [PubMed]
- Tran, D.Q.; Andersson, J.; Wang, R.; Ramsey, H.; Unutmaz, D.; Shevach, E.M. GARP (LRRC32) is essential for the surface expression of latent TGF-beta on platelets and activated FOXP3+ regulatory T cells. Proc. Natl. Acad. Sci. USA 2009, 106, 13445–13450. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Tang, D.N.; Fu, T.; Sharma, P. Identification of human regulatory T cells in the setting of T-cell activation and anti-CTLA-4 immunotherapy on the basis of expression of latency-associated peptide. Cancer Discov. 2012, 2, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, J.; Lin, C.Y.; Chiang, J.M.; Su, P.J.; Chu, Y.Y.; Lai, H.Y.; Fang, J.H.; Huang, C.T.; Lin, Y.C. CD4(+) T cells expressing latency-associated peptide and Foxp3 are an activated subgroup of regulatory T cells enriched in patients with colorectal cancer. PLoS ONE 2014, 9, e108554. [Google Scholar] [CrossRef] [PubMed]
- Abd Al Samid, M.; Chaudhary, B.; Khaled, Y.S.; Ammori, B.J.; Elkord, E. Combining FoxP3 and Helios with GARP/LAP markers can identify expanded Treg subsets in cancer patients. Oncotarget 2016, 7, 14083–14094. [Google Scholar] [PubMed]
- Whiteside, T.L. What are regulatory T cells (Treg) regulating in cancer and why? Semin. Cancer Biol. 2012, 22, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Sundstrom, P.; Stenstad, H.; Langenes, V.; Ahlmanner, F.; Theander, L.; Ndah, T.G.; Fredin, K.; Borjesson, L.; Gustavsson, B.; Bastid, J.; et al. Regulatory T Cells from Colon Cancer Patients Inhibit Effector T-cell Migration through an Adenosine-Dependent Mechanism. Cancer Immunol. Res. 2016, 4, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Schuler, P.J.; Saze, Z.; Hong, C.S.; Muller, L.; Gillespie, D.G.; Cheng, D.; Harasymczuk, M.; Mandapathil, M.; Lang, S.; Jackson, E.K.; et al. Human CD4+ CD39+ regulatory T cells produce adenosine upon co-expression of surface CD73 or contact with CD73+ exosomes or CD73+ cells. Clin. Exp. Immunol. 2014, 177, 531–543. [Google Scholar] [CrossRef] [PubMed]
- Dunne, M.R.; Ryan, C.; Nolan, B.; Tosetto, M.; Geraghty, R.; Winter, D.C.; O’Connell, P.R.; Hyland, J.M.; Doherty, G.A.; Sheahan, K.; et al. Enrichment of Inflammatory IL-17 and TNF-alpha Secreting CD4(+) T Cells within Colorectal Tumors despite the Presence of Elevated CD39(+) T Regulatory Cells and Increased Expression of the Immune Checkpoint Molecule, PD-1. Front. Oncol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Deaglio, S.; Dwyer, K.M.; Gao, W.; Friedman, D.; Usheva, A.; Erat, A.; Chen, J.F.; Enjyoji, K.; Linden, J.; Oukka, M.; et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J. Exp. Med. 2007, 204, 1257–1265. [Google Scholar] [CrossRef] [PubMed]
- Strauss, L.; Bergmann, C.; Szczepanski, M.; Gooding, W.; Johnson, J.T.; Whiteside, T.L. A unique subset of CD4+CD25highFoxp3+ T cells secreting interleukin-10 and transforming growth factor-beta1 mediates suppression in the tumor microenvironment. Clin. Cancer Res. 2007, 13, 4345–4354. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Nishikawa, H.; Wada, H.; Nagano, Y.; Sugiyama, D.; Atarashi, K.; Maeda, Y.; Hamaguchi, M.; Ohkura, N.; Sato, E.; et al. Two FOXP3CD4 T cell subpopulations distinctly control the prognosis of colorectal cancers. Nat. Med. 2016, 22, 679–684. [Google Scholar] [CrossRef] [PubMed]
- Mailloux, A.W.; Young, M.R. Regulatory T-cell trafficking: From thymic development to tumor-induced immune suppression. Crit. Rev. Immunol. 2010, 30, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Kavanagh, B.; O’Brien, S.; Lee, D.; Hou, Y.; Weinberg, V.; Rini, B.; Allison, J.P.; Small, E.J.; Fong, L. CTLA4 blockade expands FoxP3+ regulatory and activated effector CD4+ T cells in a dose-dependent fashion. Blood 2008, 112, 1175–1183. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Joh, T.; Uike, N.; Yamamoto, K.; Utsunomiya, A.; Yoshida, S.; Saburi, Y.; Miyamoto, T.; Takemoto, S.; Suzushima, H.; et al. Defucosylated anti-CCR4 monoclonal antibody (KW-0761) for relapsed adult T-cell leukemia-lymphoma: A multicenter phase II study. J. Clin. Oncol. 2012, 30, 837–842. [Google Scholar] [CrossRef] [PubMed]
- Ogura, M.; Ishida, T.; Hatake, K.; Taniwaki, M.; Ando, K.; Tobinai, K.; Fujimoto, K.; Yamamoto, K.; Miyamoto, T.; Uike, N.; et al. Multicenter phase II study of mogamulizumab (KW-0761), a defucosylated anti-cc chemokine receptor 4 antibody, in patients with relapsed peripheral T-cell lymphoma and cutaneous T-cell lymphoma. J. Clin. Oncol. 2014, 32, 1157–1163. [Google Scholar] [CrossRef] [PubMed]
- Kurose, K.; Ohue, Y.; Wada, H.; Iida, S.; Ishida, T.; Kojima, T.; Doi, T.; Suzuki, S.; Isobe, M.; Funakoshi, T.; et al. Phase Ia Study of FoxP3+ CD4 Treg Depletion by Infusion of a Humanized Anti-CCR4 Antibody, KW-0761, in Cancer Patients. Clin. Cancer Res. 2015, 21, 4327–4336. [Google Scholar] [CrossRef] [PubMed]
- Ni, X.; Jorgensen, J.L.; Goswami, M.; Challagundla, P.; Decker, W.K.; Kim, Y.H.; Duvic, M.A. Reduction of regulatory T cells by Mogamulizumab, a defucosylated anti-CC chemokine receptor 4 antibody, in patients with aggressive/refractory mycosis fungoides and Sezary syndrome. Clin. Cancer Res. 2015, 21, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Li, Y.; Zhu, B. T-cell exhaustion in the tumor microenvironment. Cell. Death Dis. 2015, 6, e1792. [Google Scholar] [CrossRef] [PubMed]
- Lindau, D.; Gielen, P.; Kroesen, M.; Wesseling, P.; Adema, G.J. The immunosuppressive tumour network: Myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology 2013, 138, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Pedroza-Pacheco, I.; Madrigal, A.; Saudemont, A. Interaction between natural killer cells and regulatory T cells: Perspectives for immunotherapy. Cell. Mol. Immunol. 2013, 10, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Ghiringhelli, F.; Menard, C.; Terme, M.; Flament, C.; Taieb, J.; Chaput, N.; Puig, P.E.; Novault, S.; Escudier, B.; Vivier, E.; et al. CD4+CD25+ regulatory T cells inhibit natural killer cell functions in a transforming growth factor-beta-dependent manner. J. Exp. Med. 2005, 202, 1075–1085. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.C.; Li, C.H.; Chu, L.H.; Huang, P.S.; Sheu, B.C.; Huang, S.C. Regulatory T Cells Suppress Natural Killer Cell Immunity in Patients With Human Cervical Carcinoma. Int. J. Gynecol. Cancer 2016, 26, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Khaled, Y.S.; Ammori, B.J.; Elkord, E. Myeloid-derived suppressor cells in cancer: Recent progress and prospects. Immunol. Cell Biol. 2013, 91, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Dannenmann, S.R.; Thielicke, J.; Stockli, M.; Matter, C.; von Boehmer, L.; Cecconi, V.; Hermanns, T.; Hefermehl, L.; Schraml, P.; Moch, H.; et al. Tumor-associated macrophages subvert T-cell function and correlate with reduced survival in clear cell renal cell carcinoma. Oncoimmunology 2013, 2, e23562. [Google Scholar] [CrossRef] [PubMed]
- Tiemessen, M.M.; Jagger, A.L.; Evans, H.G.; van Herwijnen, M.J.; John, S.; Taams, L.S. CD4+CD25+Foxp3+ regulatory T cells induce alternative activation of human monocytes/macrophages. Proc. Natl. Acad. Sci. USA 2007, 104, 19446–19451. [Google Scholar] [CrossRef] [PubMed]
- Fujimura, T.; Kambayashi, Y.; Aiba, S. Crosstalk between regulatory T cells (Tregs) and myeloid derived suppressor cells (MDSCs) during melanoma growth. Oncoimmunology 2012, 1, 1433–1434. [Google Scholar] [CrossRef] [PubMed]
- Pyzer, A.R.; Cole, L.; Rosenblatt, J.; Avigan, D.E. Myeloid-derived suppressor cells as effectors of immune suppression in cancer. Int. J. Cancer 2016. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, R.A.; von Andrian, U.H. How tolerogenic dendritic cells induce regulatory T cells. Adv. Immunol. 2010, 108, 111–165. [Google Scholar] [PubMed]
- Pedroza-Gonzalez, A.; Zhou, G.; Vargas-Mendez, E.; Boor, P.P.; Mancham, S.; Verhoef, C.; Polak, W.G.; Grunhagen, D.; Pan, Q.; Janssen, H.; et al. Tumor-infiltrating plasmacytoid dendritic cells promote immunosuppression by Tr1 cells in human liver tumors. Oncoimmunology 2015, 4, e1008355. [Google Scholar] [PubMed]
- Burr, S.P.; Dazzi, F.; Garden, O.A. Mesenchymal stromal cells and regulatory T cells: The Yin and Yang of peripheral tolerance? Immunol. Cell Biol. 2013, 91, 12–18. [Google Scholar] [PubMed]
- Taflin, C.; Favier, B.; Baudhuin, J.; Savenay, A.; Hemon, P.; Bensussan, A.; Charron, D.; Glotz, D.; Mooney, N. Human endothelial cells generate Th17 and regulatory T cells under inflammatory conditions. Proc. Natl. Acad. Sci. USA 2011, 108, 2891–2896. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Kishore, M.; Gittens, B.; Wang, G.; Coe, D.; Komarowska, I.; Infante, E.; Ridley, A.J.; Cooper, D.; Perretti, M.; et al. Self-recognition of the endothelium enables regulatory T-cell trafficking and defines the kinetics of immune regulation. Nat. Commun. 2014. [Google Scholar] [CrossRef] [PubMed]
- Brinkman, C.C.; Iwami, D.; Hritzo, M.K.; Xiong, Y.; Ahmad, S.; Simon, T.; Hippen, K.L.; Blazar, B.R.; Bromberg, J.S. Treg engage lymphotoxin beta receptor for afferent lymphatic transendothelial migration. Nat. Commun. 2016. [Google Scholar] [CrossRef] [PubMed]
- Maganto-Garcia, E.; Bu, D.X.; Tarrio, M.L.; Alcaide, P.; Newton, G.; Griffin, G.K.; Croce, K.J.; Luscinskas, F.W.; Lichtman, A.H.; Grabie, N. Foxp3+-inducible regulatory T cells suppress endothelial activation and leukocyte recruitment. J. Immunol. 2011, 187, 3521–3529. [Google Scholar] [CrossRef] [PubMed]
- Giatromanolaki, A.; Bates, G.J.; Koukourakis, M.I.; Sivridis, E.; Gatter, K.C.; Harris, A.L.; Banham, A.H. The presence of tumor-infiltrating FOXP3+ lymphocytes correlates with intratumoral angiogenesis in endometrial cancer. Gynecol. Oncol. 2008, 110, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Facciabene, A.; Peng, X.; Hagemann, I.S.; Balint, K.; Barchetti, A.; Wang, L.P.; Gimotty, P.A.; Gilks, C.B.; Lal, P.; Zhang, L.; et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature 2011, 475, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Muller, L.; Mitsuhashi, M.; Simms, P.; Gooding, W.E.; Whiteside, T.L. Tumor-derived exosomes regulate expression of immune function-related genes in human T cell subsets. Sci. Rep. 2016. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. Induced regulatory T cells in inhibitory microenvironments created by cancer. Expert Opin. Biol. Ther. 2014, 14, 1411–1425. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Tang, J.; Cao, H.; Fan, H.; Li, B. Tissue resident regulatory T cells: Novel therapeutic targets for human disease. Cell. Mol. Immunol. 2015, 12, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Trinchieri, G. Cancer and inflammation: An old intuition with rapidly evolving new concepts. Annu. Rev. Immunol. 2012, 30, 677–706. [Google Scholar] [CrossRef] [PubMed]
- Bremnes, R.M.; Al-Shibli, K.; Donnem, T.; Sirera, R.; Al-Saad, S.; Andersen, S.; Stenvold, H.; Camps, C.; Busund, L.T. The role of tumor-infiltrating immune cells and chronic inflammation at the tumor site on cancer development, progression, and prognosis: Emphasis on non-small cell lung cancer. J. Thorac. Oncol. 2011, 6, 824–833. [Google Scholar] [CrossRef] [PubMed]
- Smigiel, K.S.; Srivastava, S.; Stolley, J.M.; Campbell, D.J. Regulatory T-cell homeostasis: Steady-state maintenance and modulation during inflammation. Immunol. Rev. 2014, 259, 40–59. [Google Scholar] [CrossRef] [PubMed]
- Erdman, S.E.; Rao, V.P.; Olipitz, W.; Taylor, C.L.; Jackson, E.A.; Levkovich, T.; Lee, C.W.; Horwitz, B.H.; Fox, J.G.; Ge, Z.; et al. Unifying roles for regulatory T cells and inflammation in cancer. Int. J. Cancer 2010, 126, 1651–1665. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, C.; Strauss, L.; Wang, Y.; Szczepanski, M.J.; Lang, S.; Johnson, J.T.; Whiteside, T.L. T regulatory type 1 cells in squamous cell carcinoma of the head and neck: Mechanisms of suppression and expansion in advanced disease. Clin. Cancer Res. 2008, 14, 3706–3715. [Google Scholar] [CrossRef] [PubMed]
- Betts, G.; Jones, E.; Junaid, S.; El-Shanawany, T.; Scurr, M.; Mizen, P.; Kumar, M.; Jones, S.; Rees, B.; Williams, G.; et al. Suppression of tumour-specific CD4(+) T cells by regulatory T cells is associated with progression of human colorectal cancer. Gut 2012, 61, 1163–1171. [Google Scholar] [CrossRef] [PubMed]
- Gavin, M.A.; Torgerson, T.R.; Houston, E.; DeRoos, P.; Ho, W.Y.; Stray-Pedersen, A.; Ocheltree, E.L.; Greenberg, P.D.; Ochs, H.D.; Rudensky, A.Y. Single-cell analysis of normal and FOXP3-mutant human T cells: FOXP3 expression without regulatory T cell development. Proc. Natl. Acad. Sci. USA 2006, 103, 6659–6664. [Google Scholar] [CrossRef] [PubMed]
- Tran, D.Q.; Ramsey, H.; Shevach, E.M. Induction of FOXP3 expression in naive human CD4+FOXP3 T cells by T-cell receptor stimulation is transforming growth factor-beta dependent but does not confer a regulatory phenotype. Blood 2007, 110, 2983–2990. [Google Scholar] [CrossRef] [PubMed]
- Santegoets, S.J.; Dijkgraaf, E.M.; Battaglia, A.; Beckhove, P.; Britten, C.M.; Gallimore, A.; Godkin, A.; Gouttefangeas, C.; de Gruijl, T.D.; Koenen, H.J.; et al. Monitoring regulatory T cells in clinical samples: consensus on an essential marker set and gating strategy for regulatory T cell analysis by flow cytometry. Cancer Immunol. Immunother. 2015, 64, 1271–1286. [Google Scholar] [CrossRef] [PubMed]
- Adeegbe, D.O.; Nishikawa, H. Natural and induced T regulatory cells in cancer. Front. Immunol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Elkord, E.; Abd Al Samid, M.; Chaudhary, B. Helios, and not FoxP3, is the marker of activated Tregs expressing GARP/LAP. Oncotarget 2015, 6, 20026–20036. [Google Scholar] [CrossRef] [PubMed]
- Marshall, N.A.; Christie, L.E.; Munro, L.R.; Culligan, D.J.; Johnston, P.W.; Barker, R.N.; Vickers, M.A. Immunosuppressive regulatory T cells are abundant in the reactive lymphocytes of Hodgkin lymphoma. Blood 2004, 103, 1755–1762. [Google Scholar] [CrossRef] [PubMed]
- Dobrzanski, M.J.; Rewers-Felkins, K.A.; Samad, K.A.; Quinlin, I.S.; Phillips, C.A.; Robinson, W.; Dobrzanski, D.J.; Wright, S.E. Immunotherapy with IL-10- and IFN-gamma-producing CD4 effector cells modulate “Natural” and “Inducible” CD4 TReg cell subpopulation levels: Observations in four cases of patients with ovarian cancer. Cancer Immunol. Immunother. 2012, 61, 839–854. [Google Scholar] [CrossRef] [PubMed]
- Wing, J.B.; Sakaguchi, S. Multiple treg suppressive modules and their adaptability. Front. Immunol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Redjimi, N.; Raffin, C.; Raimbaud, I.; Pignon, P.; Matsuzaki, J.; Odunsi, K.; Valmori, D.; Ayyoub, M. CXCR3+ T regulatory cells selectively accumulate in human ovarian carcinomas to limit type I immunity. Cancer Res. 2012, 72, 4351–4360. [Google Scholar] [CrossRef] [PubMed]
- Savage, P.A.; Leventhal, D.S.; Malchow, S. Shaping the repertoire of tumor-infiltrating effector and regulatory T cells. Immunol. Rev. 2014, 259, 245–258. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.C.; Robbins, P.F. Cancer immunotherapy targeting neoantigens. Semin. Immunol. 2016, 28, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Francois, V.; Ottaviani, S.; Renkvist, N.; Stockis, J.; Schuler, G.; Thielemans, K.; Colau, D.; Marchand, M.; Boon, T.; Lucas, S.; et al. The CD4(+) T-cell response of melanoma patients to a MAGE-A3 peptide vaccine involves potential regulatory T cells. Cancer Res. 2009, 69, 4335–4345. [Google Scholar] [CrossRef] [PubMed]
- Fourcade, J.; Sun, Z.; Kudela, P.; Janjic, B.; Kirkwood, J.M.; El-Hafnawy, T.; Zarour, H.M. Human tumor antigen-specific helper and regulatory T cells share common epitope specificity but exhibit distinct T cell repertoire. J. Immunol. 2010, 184, 6709–6718. [Google Scholar] [CrossRef] [PubMed]
- Horn, T.; Grab, J.; Schusdziarra, J.; Schmid, S.; Maurer, T.; Nawroth, R.; Wolf, P.; Pritsch, M.; Gschwend, J.E.; Kubler, H.R.; et al. Antitumor T cell responses in bladder cancer are directed against a limited set of antigens and are modulated by regulatory T cells and routine treatment approaches. Int. J. Cancer 2013, 133, 2145–2156. [Google Scholar] [CrossRef] [PubMed]
- Ayyoub, M.; Pignon, P.; Classe, J.M.; Odunsi, K.; Valmori, D. CD4+ T effectors specific for the tumor antigen NY-ESO-1 are highly enriched at ovarian cancer sites and coexist with, but are distinct from, tumor-associated Treg. Cancer Immunol. Res. 2013, 1, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Bindea, G.; Mlecnik, B.; Tosolini, M.; Kirilovsky, A.; Waldner, M.; Obenauf, A.C.; Angell, H.; Fredriksen, T.; Lafontaine, L.; Berger, A.; et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 2013, 39, 782–795. [Google Scholar] [CrossRef] [PubMed]
- Gobert, M.; Treilleux, I.; Bendriss-Vermare, N.; Bachelot, T.; Goddard-Leon, S.; Arfi, V.; Biota, C.; Doffin, A.C.; Durand, I.; Olive, D.; et al. Regulatory T cells recruited through CCL22/CCR4 are selectively activated in lymphoid infiltrates surrounding primary breast tumors and lead to an adverse clinical outcome. Cancer Res. 2009, 69, 2000–2009. [Google Scholar] [CrossRef] [PubMed]
- Faget, J.; Biota, C.; Bachelot, T.; Gobert, M.; Treilleux, I.; Goutagny, N.; Durand, I.; Leon-Goddard, S.; Blay, J.Y.; Caux, C.; et al. Early detection of tumor cells by innate immune cells leads to T(reg) recruitment through CCL22 production by tumor cells. Cancer Res. 2011, 71, 6143–6152. [Google Scholar] [CrossRef] [PubMed]
- Darrasse-Jeze, G.; Podsypanina, K. How numbers, nature, and immune status of foxp3(+) regulatory T-cells shape the early immunological events in tumor development. Front. Immunol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Halvorsen, E.C.; Mahmoud, S.M.; Bennewith, K.L. Emerging roles of regulatory T cells in tumour progression and metastasis. Cancer Metastasis Rev. 2014, 33, 1025–1041. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.; Zhang, W.; Strasner, A.; Grivennikov, S.; Cheng, J.Q.; Hoffman, R.M.; Karin, M. Tumour-infiltrating regulatory T cells stimulate mammary cancer metastasis through RANKL-RANK signalling. Nature 2011, 470, 548–553. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Zhou, Z.; Gu, W.; Peng, F.; Li, J. Treg increases HepG2 cell growth by RANK-RANKL pathway. J. Immunother. Cancer 2014. [Google Scholar] [CrossRef]
- Whiteside, T. The role of regulatory T cells in cancer immunology. Immunotargets Ther. 2015, 4, 159–171. [Google Scholar] [CrossRef] [PubMed]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [PubMed]
- Pere, H.; Tanchot, C.; Bayry, J.; Terme, M.; Taieb, J.; Badoual, C.; Adotevi, O.; Merillon, N.; Marcheteau, E.; Quillien, V.R.; et al. Comprehensive analysis of current approaches to inhibit regulatory T cells in cancer. Oncoimmunology 2012, 1, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Ghiringhelli, F.; Larmonier, N.; Schmitt, E.; Parcellier, A.; Cathelin, D.; Garrido, C.; Chauffert, B.; Solary, E.; Bonnotte, B.; Martin, F. CD4+CD25+ regulatory T cells suppress tumor immunity but are sensitive to cyclophosphamide which allows immunotherapy of established tumors to be curative. Eur. J. Immunol. 2004, 34, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Lutsiak, M.E.; Semnani, R.T.; De Pascalis, R.; Kashmiri, S.V.; Schlom, J.; Sabzevari, H. Inhibition of CD4(+)25+ T regulatory cell function implicated in enhanced immune response by low-dose cyclophosphamide. Blood 2005, 105, 2862–2868. [Google Scholar] [CrossRef] [PubMed]
- Walter, S.; Weinschenk, T.; Stenzl, A.; Zdrojowy, R.; Pluzanska, A.; Szczylik, C.; Staehler, M.; Brugger, W.; Dietrich, P.Y.; Mendrzyk, R.; et al. Multipeptide immune response to cancer vaccine IMA901 after single-dose cyclophosphamide associates with longer patient survival. Nat. Med. 2012, 18, 1254–1261. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Ahmadzadeh, M.; Lu, Y.C.; Liewehr, D.J.; Dudley, M.E.; Liu, F.; Schrump, D.S.; Steinberg, S.M.; Rosenberg, S.A.; Robbins, P.F. Levels of peripheral CD4(+)FoxP3(+) regulatory T cells are negatively associated with clinical response to adoptive immunotherapy of human cancer. Blood 2012, 119, 5688–5696. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Kluger, H.; Callahan, M.K.; Postow, M.A.; Rizvi, N.A.; Lesokhin, A.M.; Segal, N.H.; Ariyan, C.E.; Gordon, R.A.; Reed, K.; et al. Nivolumab plus ipilimumab in advanced melanoma. N. Engl. J. Med. 2013, 369, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Sznol, M.; McDermott, D.F.; Kluger, H.M.; Carvajal, R.D.; Sharfman, W.H.; Brahmer, J.R.; Lawrence, D.P.; Atkins, M.B.; Powderly, J.D.; et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J. Clin. Oncol. 2014, 32, 1020–1030. [Google Scholar] [CrossRef] [PubMed]
- Postow, M.A.; Callahan, M.K.; Wolchok, J.D. Immune Checkpoint Blockade in Cancer Therapy. J. Clin. Oncol. 2015, 33, 1974–1982. [Google Scholar] [CrossRef] [PubMed]
- Ansell, S.M.; Lesokhin, A.M.; Borrello, I.; Halwani, A.; Scott, E.C.; Gutierrez, M.; Schuster, S.J.; Millenson, M.M.; Cattry, D.; Freeman, G.J.; et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N. Engl. J. Med. 2015, 372, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Maker, A.V.; Attia, P.; Rosenberg, S.A. Analysis of the cellular mechanism of antitumor responses and autoimmunity in patients treated with CTLA-4 blockade. J. Immunol. 2005, 175, 7746–7754. [Google Scholar] [CrossRef] [PubMed]
- O’Mahony, D.; Morris, J.C.; Quinn, C.; Gao, W.; Wilson, W.H.; Gause, B.; Pittaluga, S.; Neelapu, S.; Brown, M.; Fleisher, T.A.; et al. A pilot study of CTLA-4 blockade after cancer vaccine failure in patients with advanced malignancy. Clin. Cancer Res. 2007, 13, 958–964. [Google Scholar] [CrossRef] [PubMed]
- Menard, C.; Ghiringhelli, F.; Roux, S.; Chaput, N.; Mateus, C.; Grohmann, U.; Caillat-Zucman, S.; Zitvogel, L.; Robert, C. Ctla-4 blockade confers lymphocyte resistance to regulatory T-cells in advanced melanoma: Surrogate marker of efficacy of tremelimumab? Clin. Cancer Res. 2008, 14, 5242–5249. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Comin-Anduix, B.; Economou, J.S.; Donahue, T.R.; de la Rocha, P.; Morris, L.F.; Jalil, J.; Dissette, V.B.; Shintaku, I.P.; Glaspy, J.A.; et al. Intratumoral immune cell infiltrates, FoxP3, and indoleamine 2,3-dioxygenase in patients with melanoma undergoing CTLA4 blockade. Clin. Cancer Res. 2009, 15, 390–399. [Google Scholar] [CrossRef] [PubMed]
- Ralph, C.; Elkord, E.; Burt, D.J.; O’Dwyer, J.F.; Austin, E.B.; Stern, P.L.; Hawkins, R.E.; Thistlethwaite, F.C. Modulation of lymphocyte regulation for cancer therapy: A phase II trial of tremelimumab in advanced gastric and esophageal adenocarcinoma. Clin. Cancer Res. 2010, 16, 1662–1672. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Burt, D.J.; Ralph, C.; Thistlethwaite, F.C.; Hawkins, R.E.; Elkord, E. Tremelimumab (anti-CTLA4) mediates immune responses mainly by direct activation of T effector cells rather than by affecting T regulatory cells. Clin. Immunol. 2011, 138, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.S.; Hamid, O.; Chasalow, S.D.; Wu, D.Y.; Parker, S.M.; Galbraith, S.; Gnjatic, S.; Berman, D. Ipilimumab increases activated T cells and enhances humoral immunity in patients with advanced melanoma. J. Immunother. 2012, 35, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Tarhini, A.A.; Butterfield, L.H.; Shuai, Y.; Gooding, W.E.; Kalinski, P.; Kirkwood, J.M. Differing patterns of circulating regulatory T cells and myeloid-derived suppressor cells in metastatic melanoma patients receiving anti-CTLA4 antibody and interferon-alpha or TLR-9 agonist and GM-CSF with peptide vaccination. J. Immunother. 2012, 35, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Tarhini, A.A.; Edington, H.; Butterfield, L.H.; Lin, Y.; Shuai, Y.; Tawbi, H.; Sander, C.; Yin, Y.; Holtzman, M.; Johnson, J.; et al. Immune monitoring of the circulation and the tumor microenvironment in patients with regionally advanced melanoma receiving neoadjuvant ipilimumab. PLoS ONE 2014, 9, e87705. [Google Scholar] [CrossRef] [PubMed]
- Smyth, M.J.; Ngiow, S.F.; Teng, M.W. Targeting regulatory T cells in tumor immunotherapy. Immunol. Cell Biol. 2014, 92, 473–474. [Google Scholar] [CrossRef] [PubMed]
- Furness, A.J.; Vargas, F.A.; Peggs, K.S.; Quezada, S.A. Impact of tumour microenvironment and Fc receptors on the activity of immunomodulatory antibodies. Trends Immunol. 2014, 35, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Jie, H.B.; Schuler, P.J.; Lee, S.C.; Srivastava, R.M.; Argiris, A.; Ferrone, S.; Whiteside, T.L.; Ferris, R.L. CTLA-4(+) Regulatory T Cells Increased in Cetuximab-Treated Head and Neck Cancer Patients Suppress NK Cell Cytotoxicity and Correlate with Poor Prognosis. Cancer Res. 2015, 75, 2200–2210. [Google Scholar] [CrossRef] [PubMed]
- Romano, E.; Kusio-Kobialka, M.; Foukas, P.G.; Baumgaertner, P.; Meyer, C.; Ballabeni, P.; Michielin, O.; Weide, B.; Romero, P.; Speiser, D.E. Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patients. Proc. Natl. Acad. Sci. USA 2015, 112, 6140–6145. [Google Scholar] [CrossRef] [PubMed]
- Hou, T.Z.; Qureshi, O.S.; Wang, C.J.; Baker, J.; Young, S.P.; Walker, L.S.; Sansom, D.M. A transendocytosis model of CTLA-4 function predicts its suppressive behavior on regulatory T cells. J. Immunol. 2015, 194, 2148–2159. [Google Scholar] [CrossRef] [PubMed]
- Sarnaik, A.A.; Yu, B.; Yu, D.; Morelli, D.; Hall, M.; Bogle, D.; Yan, L.; Targan, S.; Solomon, J.; Nichol, G.; et al. Extended dose ipilimumab with a peptide vaccine: Immune correlates associated with clinical benefit in patients with resected high-risk stage IIIc/IV melanoma. Clin. Cancer Res. 2011, 17, 896–906. [Google Scholar] [CrossRef] [PubMed]
- Bjoern, J.; Juul Nitschke, N.; Zeeberg Iversen, T.; Schmidt, H.; Fode, K.; Svane, I.M. Immunological correlates of treatment and response in stage IV malignant melanoma patients treated with Ipilimumab. Oncoimmunology 2016, 5, e1100788. [Google Scholar] [CrossRef] [PubMed]
- Liakou, C.I.; Kamat, A.; Tang, D.N.; Chen, H.; Sun, J.; Troncoso, P.; Logothetis, C.; Sharma, P. CTLA-4 blockade increases IFNgamma-producing CD4+ICOShi cells to shift the ratio of effector to regulatory T cells in cancer patients. Proc. Natl. Acad. Sci. USA 2008, 105, 14987–14992. [Google Scholar] [CrossRef] [PubMed]
- Martens, A.; Wistuba-Hamprecht, K.; Yuan, J.; Postow, M.A.; Wong, P.; Capone, M.; Madonna, G.; Khammari, A.; Schilling, B.; Sucker, A.; et al. Increases in absolute lymphocytes and circulating CD4+ and CD8+ T cells are associated with positive clinical outcome of melanoma patients treated with ipilimumab. Clin. Cancer Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Lau, R.; Yu, D.; Zhu, W.; Korman, A.; Weber, J. PD1 blockade reverses the suppression of melanoma antigen-specific CTL by CD4+ CD25(Hi) regulatory T cells. Int. Immunol. 2009, 21, 1065–1077. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Thudium, K.B.; Han, M.; Wang, X.T.; Huang, H.; Feingersh, D.; Garcia, C.; Wu, Y.; Kuhne, M.; Srinivasan, M.; et al. In vitro characterization of the anti-PD-1 antibody nivolumab, BMS-936558, and in vivo toxicology in non-human primates. Cancer Immunol. Res. 2014, 2, 846–856. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.S.; Kudchadkar, R.R.; Yu, B.; Gallenstein, D.; Horak, C.E.; Inzunza, H.D.; Zhao, X.; Martinez, A.J.; Wang, W.; Gibney, G.; et al. Safety, efficacy, and biomarkers of nivolumab with vaccine in ipilimumab-refractory or -naive melanoma. J. Clin. Oncol. 2013, 31, 4311–4318. [Google Scholar] [CrossRef] [PubMed]
- Gibney, G.T.; Kudchadkar, R.R.; DeConti, R.C.; Thebeau, M.S.; Czupryn, M.P.; Tetteh, L.; Eysmans, C.; Richards, A.; Schell, M.J.; Fisher, K.J.; et al. Safety, correlative markers, and clinical results of adjuvant nivolumab in combination with vaccine in resected high-risk metastatic melanoma. Clin. Cancer Res. 2015, 21, 712–720. [Google Scholar] [CrossRef] [PubMed]
- Tsai, K.K.; Daud, A.I. Nivolumab plus ipilimumab in the treatment of advanced melanoma. J. Hematol. Oncol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Bowyer, S.; Prithviraj, P.; Lorigan, P.; Larkin, J.; McArthur, G.; Atkinson, V.; Millward, M.; Khou, M.; Diem, S.; Ramanujam, S.; et al. Efficacy and toxicity of treatment with the anti-CTLA-4 antibody ipilimumab in patients with metastatic melanoma after prior anti-PD-1 therapy. Br. J. Cancer 2016, 114, 1084–1089. [Google Scholar] [CrossRef] [PubMed]
- Lumniczky, K.; Safrany, G. The impact of radiation therapy on the antitumor immunity: Local effects and systemic consequences. Cancer Lett. 2015, 356, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Di Maggio, F.M.; Minafra, L.; Forte, G.I.; Cammarata, F.P.; Lio, D.; Messa, C.; Gilardi, M.C.; Bravata, V. Portrait of inflammatory response to ionizing radiation treatment. J. Inflamm. (Lond.) 2015. [Google Scholar] [CrossRef] [PubMed]
- Persa, E.; Balogh, A.; Safrany, G.; Lumniczky, K. The effect of ionizing radiation on regulatory T cells in health and disease. Cancer Lett. 2015, 368, 252–261. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, A.; Buzzonetti, A.; Martinelli, E.; Fanelli, M.; Petrillo, M.; Ferrandina, G.; Scambia, G.; Fattorossi, A. Selective changes in the immune profile of tumor-draining lymph nodes after different neoadjuvant chemoradiation regimens for locally advanced cervical cancer. Int. J. Radiat. Oncol. Biol. Phys. 2010, 76, 1546–1553. [Google Scholar] [CrossRef] [PubMed]
- Fadul, C.E.; Fisher, J.L.; Gui, J.; Hampton, T.H.; Cote, A.L.; Ernstoff, M.S. Immune modulation effects of concomitant temozolomide and radiation therapy on peripheral blood mononuclear cells in patients with glioblastoma multiforme. Neuro Oncol. 2011, 13, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Schuler, P.J.; Harasymczuk, M.; Schilling, B.; Saze, Z.; Strauss, L.; Lang, S.; Johnson, J.T.; Whiteside, T.L. Effects of adjuvant chemoradiotherapy on the frequency and function of regulatory T cells in patients with head and neck cancer. Clin. Cancer Res. 2013, 19, 6585–6596. [Google Scholar] [CrossRef] [PubMed]
- Sekar, D.; Hahn, C.; Brune, B.; Roberts, E.; Weigert, A. Apoptotic tumor cells induce IL-27 release from human DCs to activate Treg cells that express CD69 and attenuate cytotoxicity. Eur. J. Immunol. 2012, 42, 1585–1598. [Google Scholar] [CrossRef] [PubMed]
- Postow, M.A.; Callahan, M.K.; Barker, C.A.; Yamada, Y.; Yuan, J.; Kitano, S.; Mu, Z.; Rasalan, T.; Adamow, M.; Ritter, E.; et al. Immunologic correlates of the abscopal effect in a patient with melanoma. N. Engl. J. Med. 2012, 366, 925–931. [Google Scholar] [CrossRef] [PubMed]
- Golden, E.B.; Demaria, S.; Schiff, P.B.; Chachoua, A.; Formenti, S.C. An abscopal response to radiation and ipilimumab in a patient with metastatic non-small cell lung cancer. Cancer Immunol. Res. 2013, 1, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Twyman-Saint Victor, C.; Rech, A.J.; Maity, A.; Rengan, R.; Pauken, K.E.; Stelekati, E.; Benci, J.L.; Xu, B.; Dada, H.; Odorizzi, P.M.; et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 2015, 520, 373–377. [Google Scholar] [CrossRef] [PubMed]
- Janik, J.E.; Morris, J.C.; O’Mahony, D.; Pittaluga, S.; Jaffe, E.S.; Redon, C.E.; Bonner, W.M.; Brechbiel, M.W.; Paik, C.H.; Whatley, M.; et al. 90Y-daclizumab, an anti-CD25 monoclonal antibody, provided responses in 50% of patients with relapsed Hodgkin’s lymphoma. Proc. Natl. Acad. Sci. USA 2015, 112, 13045–13050. [Google Scholar] [CrossRef] [PubMed]
- S Ghiringhelli, F.; Menard, C.; Puig, P.E.; Ladoire, S.; Roux, S.; Martin, F.; Solary, E.; Le Cesne, A.; Zitvogel, L.; Chauffert, B. Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol. Immunother. 2007, 56, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Domschke, C.; Stoiber, N.; Schott, S.; Heil, J.; Rom, J.; Blumenstein, M.; Thum, J.; Sohn, C.; Schneeweiss, A.; et al. Metronomic cyclophosphamide treatment in metastasized breast cancer patients: Immunological effects and clinical outcome. Cancer Immunol. Immunother. 2012, 61, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Cha, E.; Klinger, M.; Hou, Y.; Cummings, C.; Ribas, A.; Faham, M.; Fong, L. Improved survival with T cell clonotype stability after anti-CTLA-4 treatment in cancer patients. Sci. Transl. Med. 2014. [Google Scholar] [CrossRef] [PubMed]
- Kvistborg, P.; Philips, D.; Kelderman, S.; Hageman, L.; Ottensmeier, C.; Joseph-Pietras, D.; Welters, M.J.; van der Burg, S.; Kapiteijn, E.; Michielin, O.; et al. Anti-CTLA-4 therapy broadens the melanoma-reactive CD8+ T cell response. Sci. Transl. Med. 2014. [Google Scholar] [CrossRef] [PubMed]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef] [PubMed]
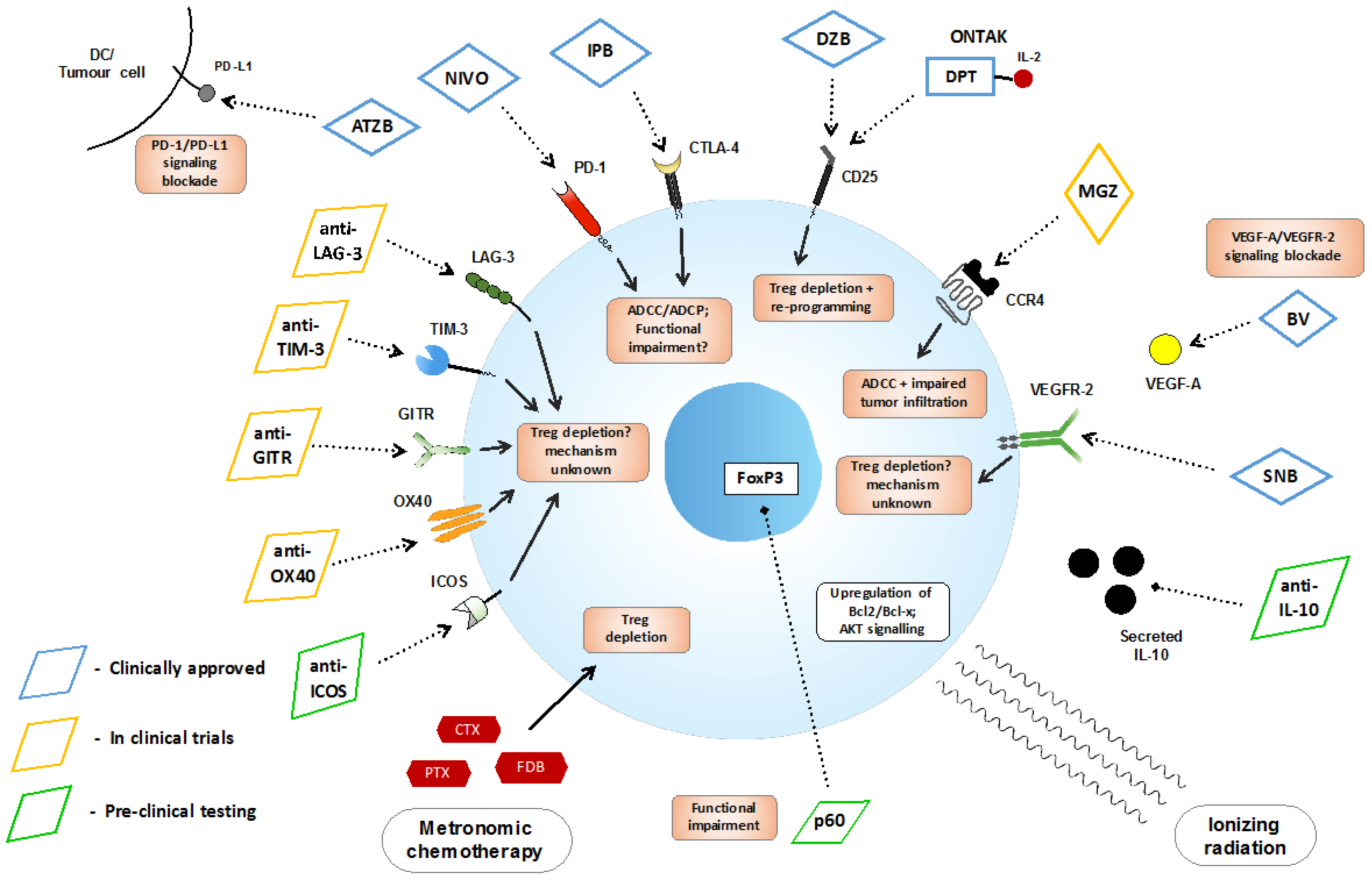

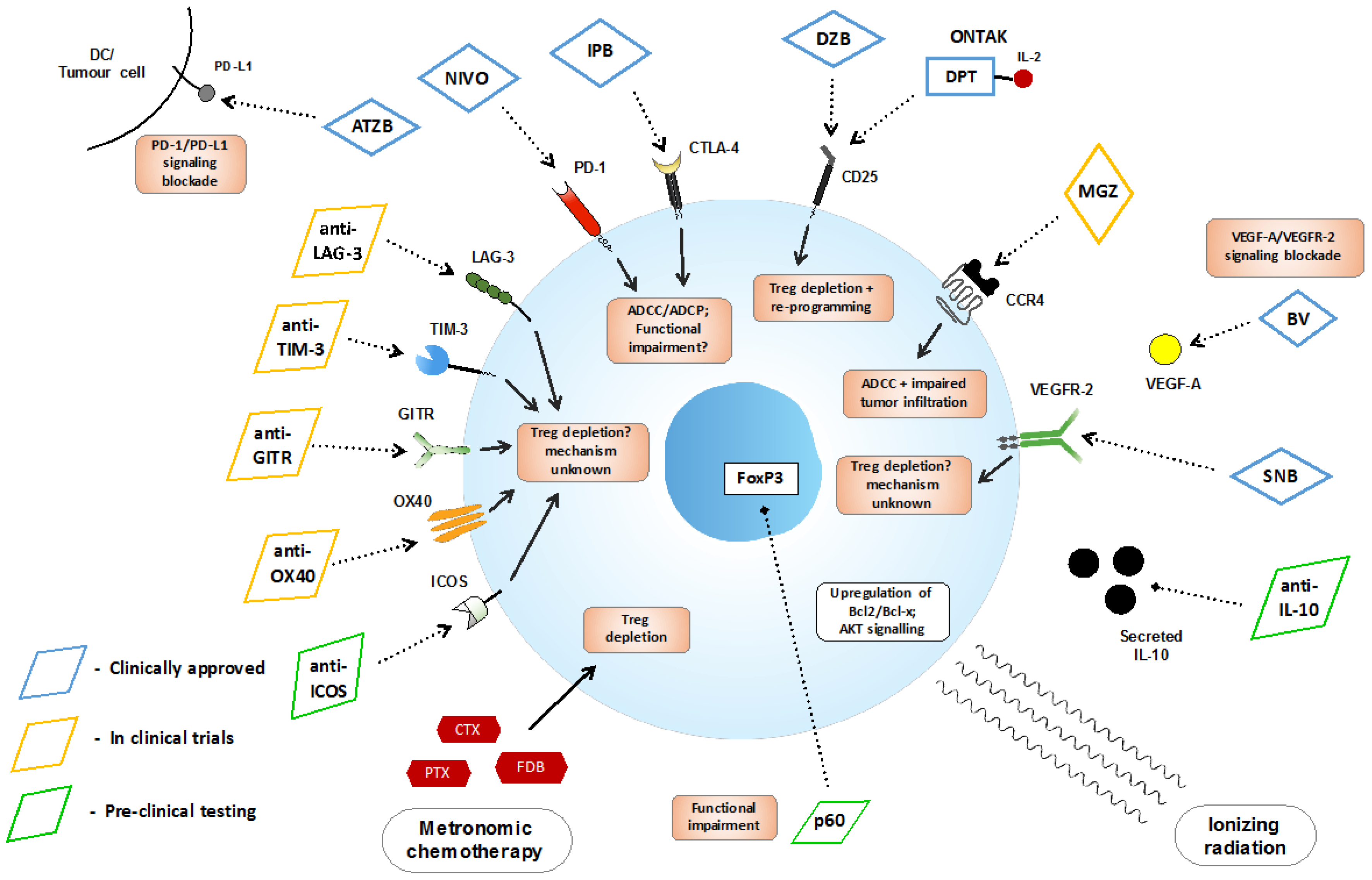{kind=link}
| Therapy | Modality |
|---|---|
| Low-dose chemotherapy | Treg depletion |
| CD25-targetted Abs | Treg depletion |
| Immune checkpoint inhibition (ICI) | Functional targeting + Treg depletion |
| Chemokine receptor blockade | Functional targeting + Treg depletion |
| Blockade of suppressive mechanisms & soluble mediators (IL-10/TGF-β) | Functional targeting |
| Cancer | Treg Markers | PB/TILs | Functional Analysis | Expanded? | Survival | Ref. |
|---|---|---|---|---|---|---|
| Resected stage IIIc/IV melanoma (n = 75) | CD25+ | PB | Suppressive; no effect after treatment | No change | N/A | [147] |
| Unresectable stage III/IV melanoma (n = 80) | CD25hiCD127loFoxP3+ | PB | N/A | No change at weeks 4 & 12 | N/A | [139] |
| Stage IV malignant melanoma | CD25+FoxP3+ | PB | N/A | Decreased | No statistical link | [148] |
| Bladder cancer patients prior to radical cystectomy (n = 6) | CD25+FoxP3+ | PB | Suppressive pre/post- treatment | Overall decrease; variable initial response | N/A | [149] |
| Bladder cancer patients prior to radical cystectomy (n = 6) | CD25+FoxP3+ | TILs | NA | Increase in ICOS+ Teff : FoxP3+ Treg ratio | N/A | [149] |
| Bladder cancer (n = 12) | CD25+LAP+/FoxP3+/CD127lo | PB | CD25+LAP+, but not CD25+CD127lo, suppressive post-treatment | CD25+LAP+ increased in patient subset | N/A | [55] |
| Metastatic RCC or metastatic melanoma (n = 10) | CD25+FoxP3+ | PB | Suppressive pre/post-treatment | No change; increase in activated T cells | N/A | [133] |
| Progressive metastatic hormone-refractory prostate cancer (n = 24) | CD127loCD25hi | PB | Suppressive post-treatment | Increased, and Ki67+ | N/A (study* ongoing) | [66] |
| Stage III/IV melanoma (n = 37) | CD25HIFoxp3⁺ | PB | N/A | Increased at 6 weeks post-treatment | Associated with improved PFS | [141] |
| Stage III/IV melanoma (n = 10) | CD25HIFoxp3⁺ | TILs | N/A | Variable | Inverse trend between Treg & clinical benefit | [141] |
| Stage IV melanoma (n = 82) | CD127loCD25+FoxP3+ | PB | N/A | Increased over 14 weeks of treatment | Higher than median Tregs associated with better survival | [150] |
| Cancer | Treg marker | PB/TILs | Functional Analysis | Expanded? | Survival | Ref. |
|---|---|---|---|---|---|---|
| DTIC-treated stage IV melanoma (n = 10) | CD25+CD127− or FoxP3+ | PB | Suppressive pre-treatment; transient resistance to Treg suppression post-treatment | Increase in absolute Treg count, but not proportion | Treatment-induced transient Treg resistance associated with better survival | [135] |
| Stage III/IV melanoma, combined with IFN-α2b (n = 37) | CD25hiFoxP3+ or CD25hiCD39+ | PB | N/A | Both subsets Increased | N/A | [140] |
| Metastatic melanoma (n = 7) | CD25+FoxP3+ | TILs | N/A | Variable | N/A | [136] |
| Cancer | Treg Markers | PB/TILs | Functional Analysis | Expanded? | Survival | Ref. |
|---|---|---|---|---|---|---|
| Unresectable stage III/IV melanoma (n = 90); IPB-naive (n = 34) or IPB-refractory (n = 56) | CD25+CD127loFoxP3+ | PB | N/A | Decreased in responders & stable patients; increased in non-responders | Increased Tregs associated with progression at 12 weeks | [153] |
| Stage IIIc/IV melanoma (n = 33) | CD127loFoxP3+ | PB | N/A | Expanded in PB at 12 & 24 weeks | Trend towards lower Tregs in non-relapsing patients | [154] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chaudhary, B.; Elkord, E. Regulatory T Cells in the Tumor Microenvironment and Cancer Progression: Role and Therapeutic Targeting. Vaccines 2016, 4, 28. https://doi.org/10.3390/vaccines4030028
Chaudhary B, Elkord E. Regulatory T Cells in the Tumor Microenvironment and Cancer Progression: Role and Therapeutic Targeting. Vaccines. 2016; 4(3):28. https://doi.org/10.3390/vaccines4030028
Chicago/Turabian StyleChaudhary, Belal, and Eyad Elkord. 2016. "Regulatory T Cells in the Tumor Microenvironment and Cancer Progression: Role and Therapeutic Targeting" Vaccines 4, no. 3: 28. https://doi.org/10.3390/vaccines4030028
APA StyleChaudhary, B., & Elkord, E. (2016). Regulatory T Cells in the Tumor Microenvironment and Cancer Progression: Role and Therapeutic Targeting. Vaccines, 4(3), 28. https://doi.org/10.3390/vaccines4030028