
Evaluation of Peptide-Based Vaccines Against Group A Streptococcus in Staphylococcus aureus-Infected Mice

 ,
,  ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Equipment
2.3. Peptide Synthesis
2.4. Preparation of Vaccines
2.5. Characterization of Vaccines: Size, Shape, and Structural Conformation
2.6. In Vivo Assessment in Mice
2.7. Evaluation of Serological Immune Responses: Immunogenicity (Against Peptides), Avidity (Against GAS), and the Cross-Reactivity Between the Two Bacteria
2.8. Opsonization Assay
2.9. Data Analysis
3. Results
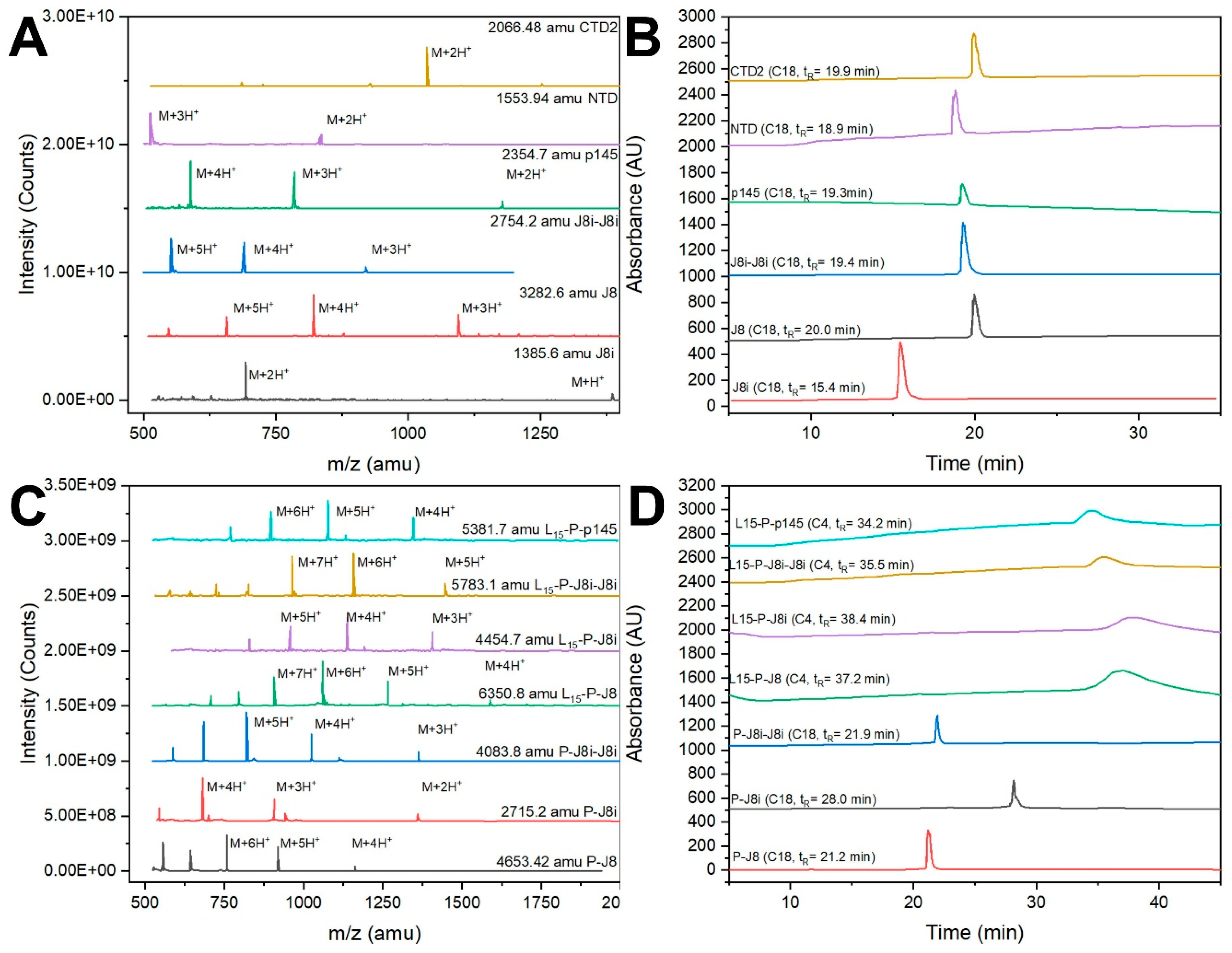
3.1. Peptide Synthesis and Characterization
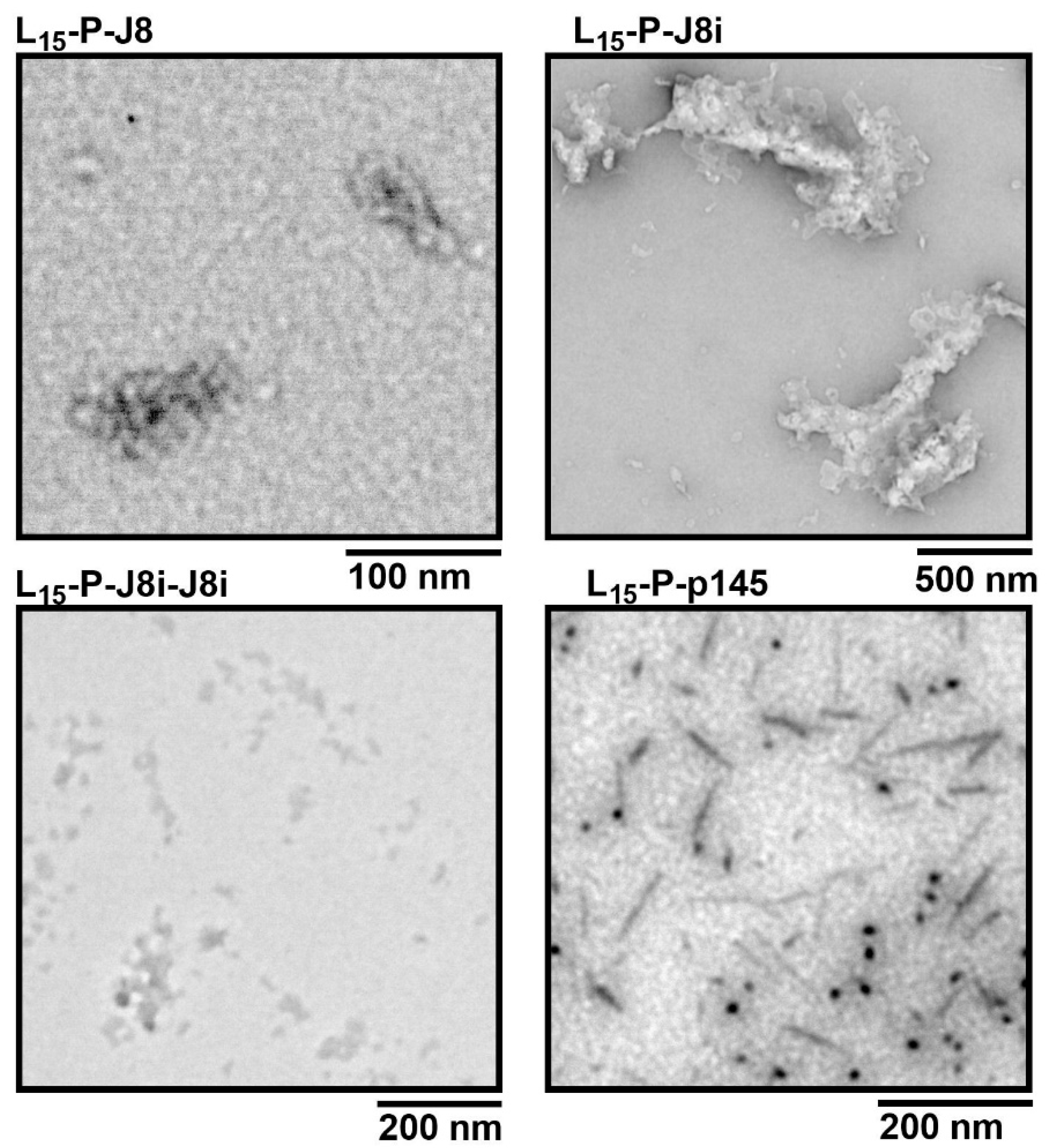
3.2. Size and Shape of Self-Assembled Nanovaccines
3.3. Secondary Structural Evaluation of Peptide Antigens and Vaccines
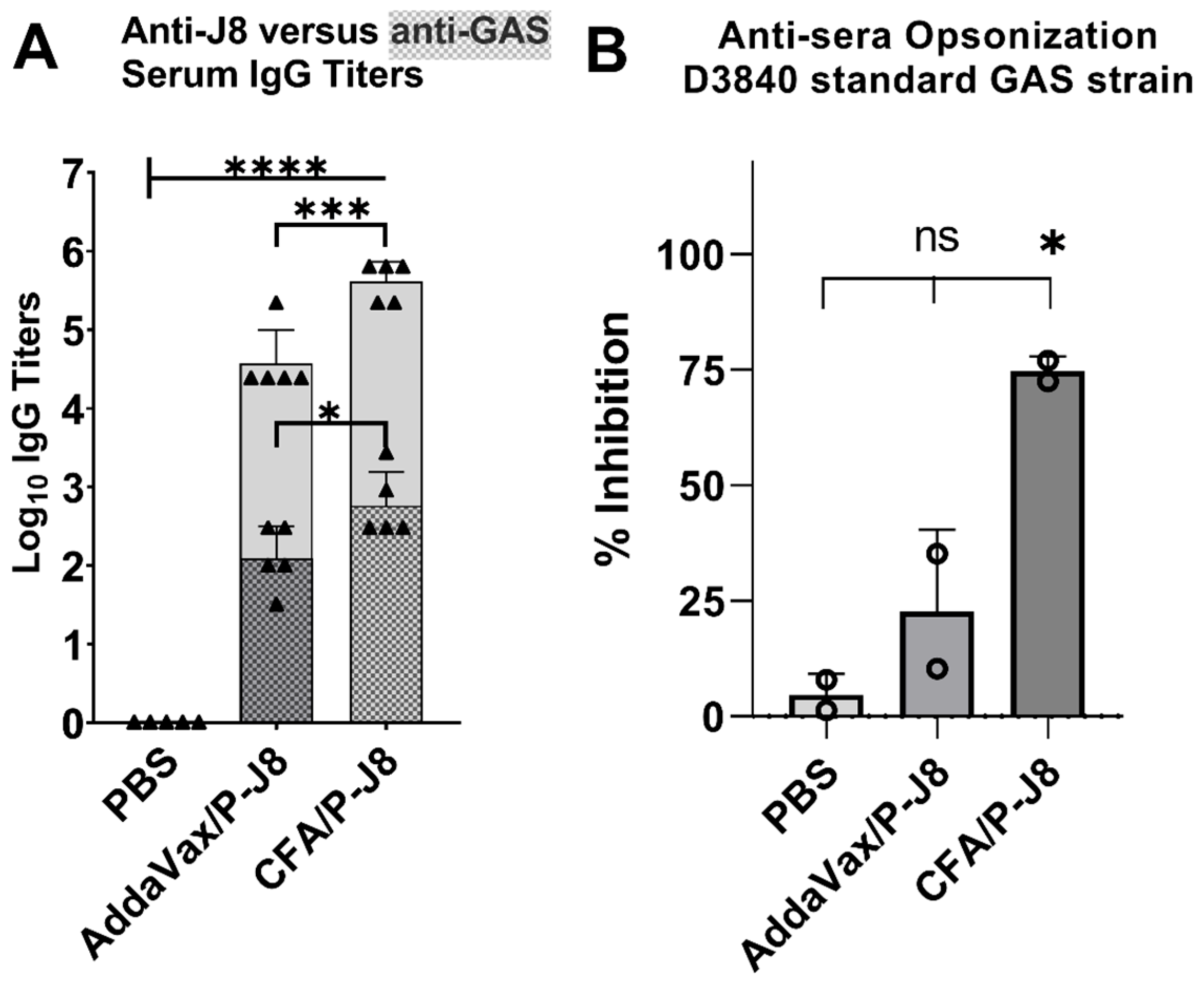
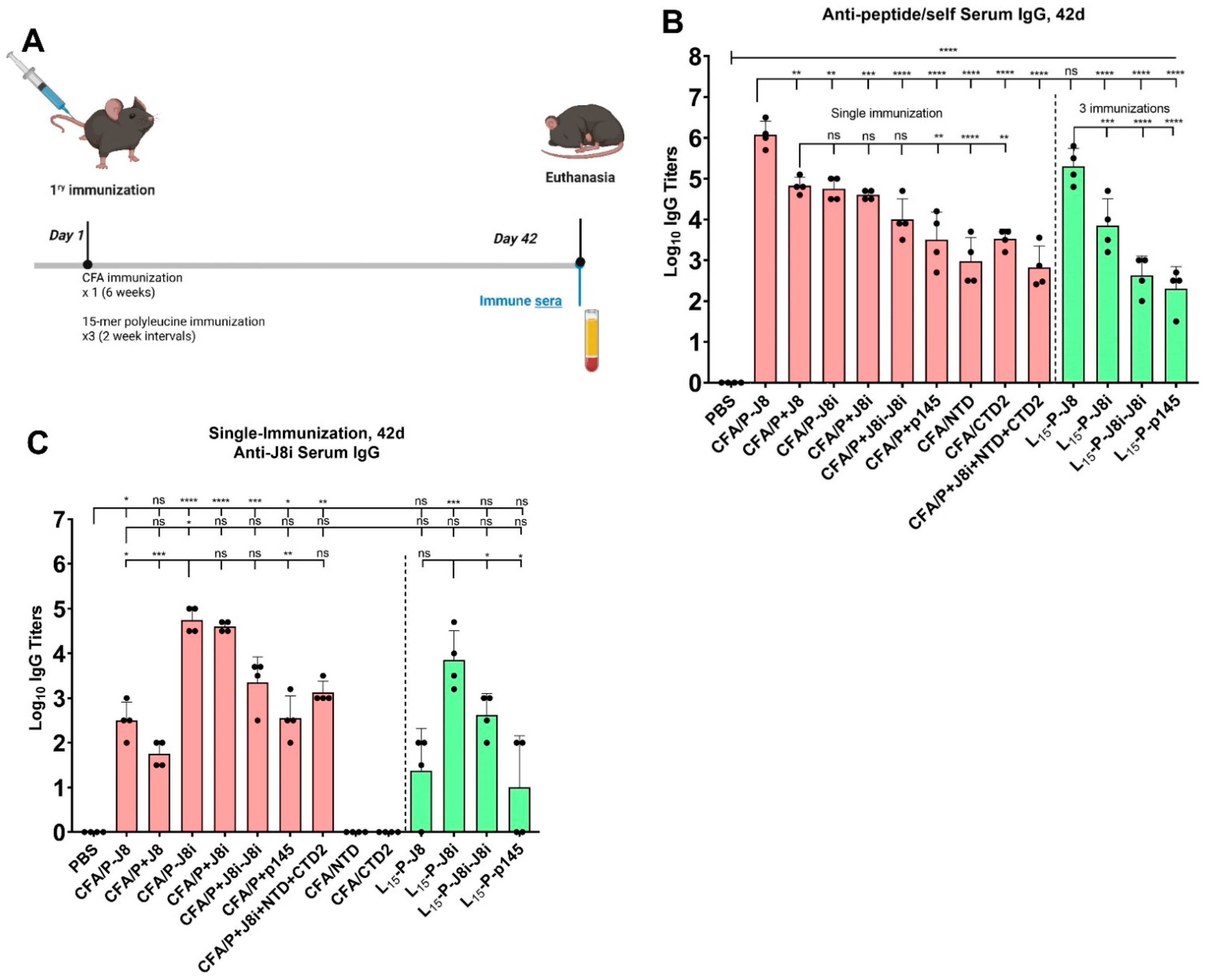
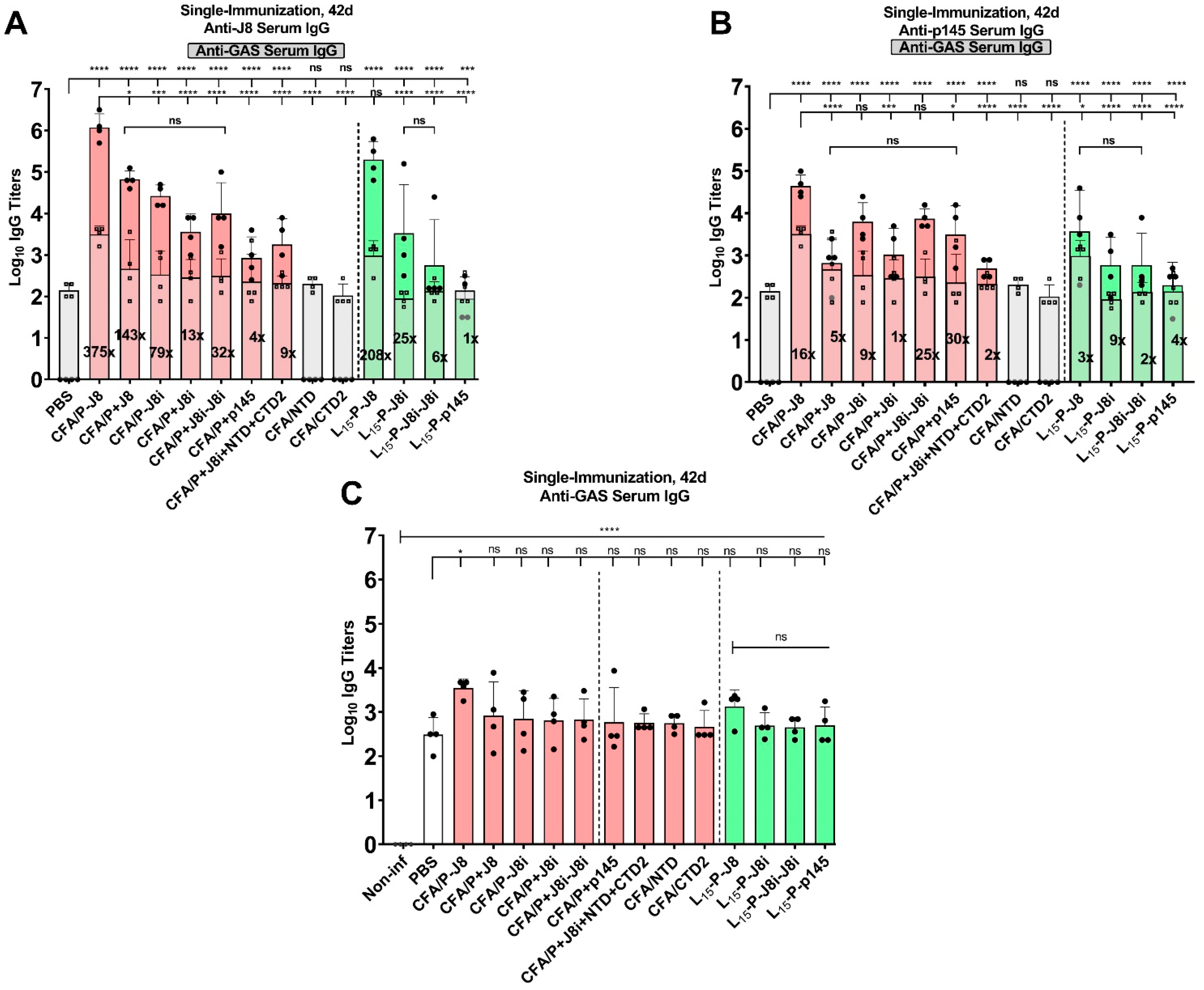
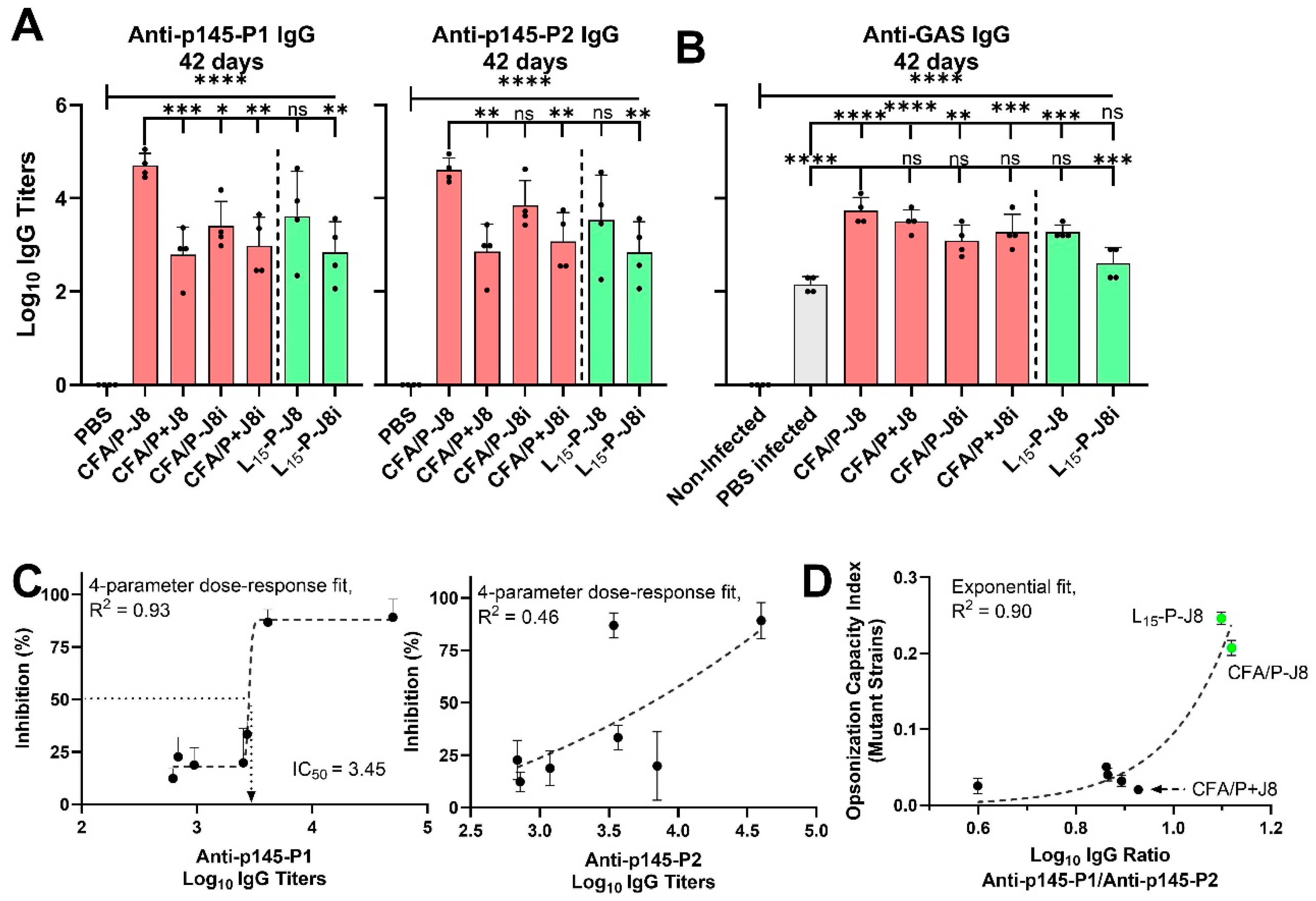
3.4. Immunological Evaluation of Mouse Sera
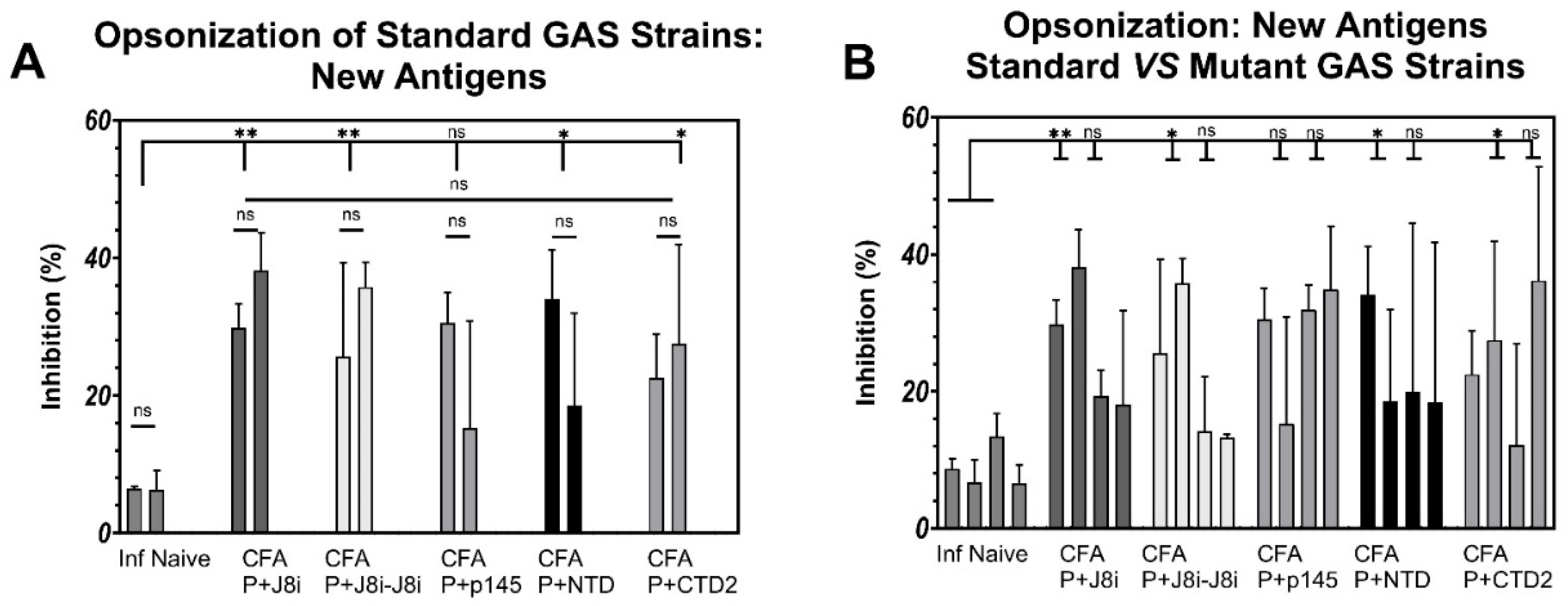
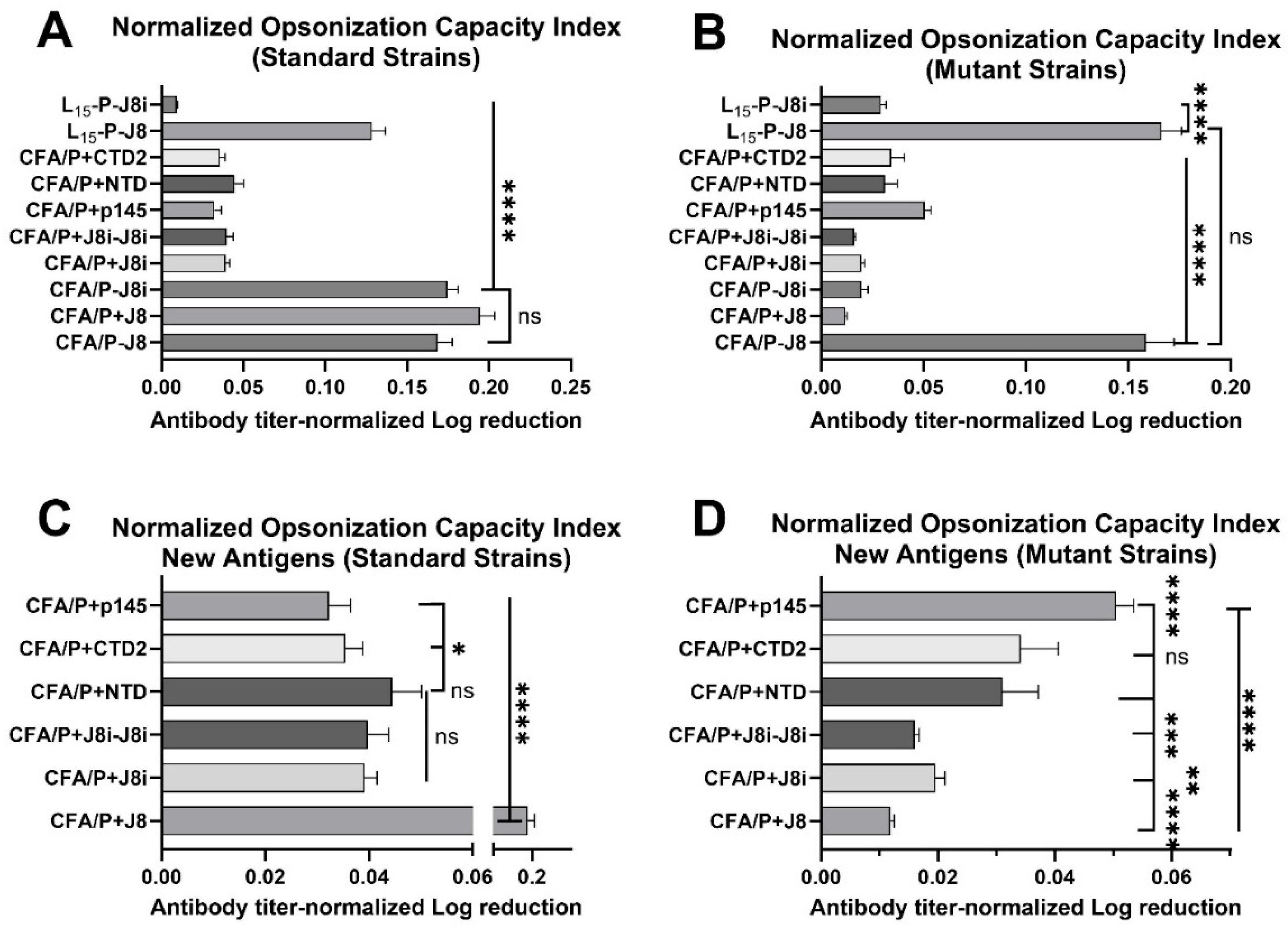
3.5. Opsonization Efficacy Evaluation of Mouse Sera
4. Discussion
4.1. Size and Secondary Structural Characterization of Synthesized Peptide Constructs
4.2. The Immunological Evaluation of Mouse Sera: Immunogenicity and Immune Response Quality
4.3. In Vitro Opsonization Efficacy of Mouse Sera Against Standard and Mutant GAS Strains
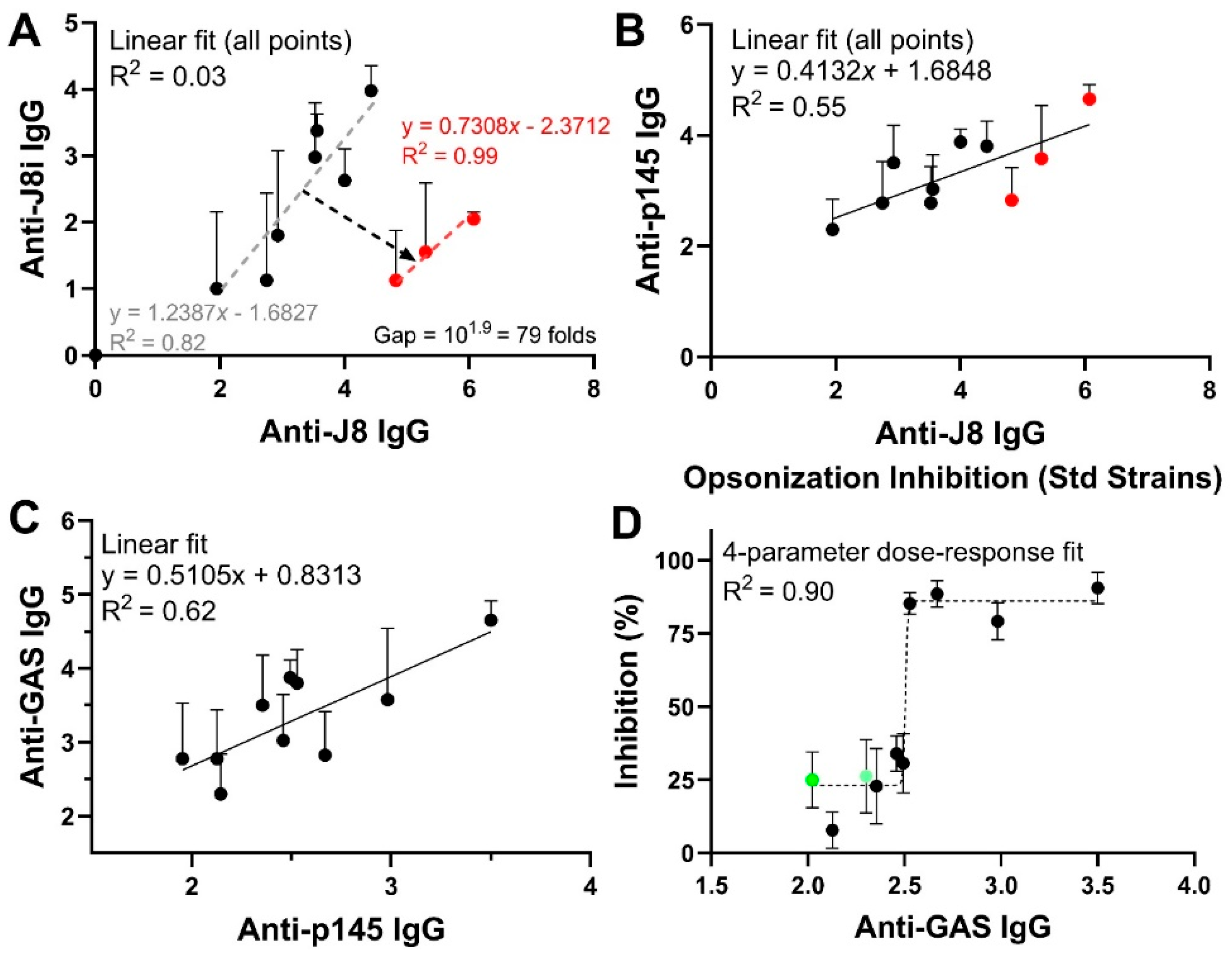
4.4. Relationships Among Immunological Responses and Opsonic Bactericidal Activity
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sanyahumbi, S. Streptococcus pyogenes: Basic Biology to Clinical Manifestations. In Streptococcus pyogenes: Basic Biology to Clinical Manifestations; Ferretti, J.J., Stevens, D.L., Fischetti, V.A., Eds.; University of Oklahoma Health Sciences Center: Oklahoma City, OK, USA, 2016. [Google Scholar]
- Vekemans, J.; Gouvea-Reis, F.; Kim, J.H.; Excler, J.L.; Smeesters, P.R.; O’Brien, K.L.; Van Beneden, C.A.; Steer, A.C.; Carapetis, J.R.; Kaslow, D.C. The Path to Group A Streptococcus Vaccines: World Health Organization Research and Development Technology Roadmap and Preferred Product Characteristics. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2019, 69, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.R.; McIntyre, L.; Mutreja, A.; Lacey, J.A.; Lees, J.A.; Towers, R.J.; Duchêne, S.; Smeesters, P.R.; Frost, H.R.; Price, D.J.; et al. Atlas of group A streptococcal vaccine candidates compiled using large-scale comparative genomics. Nat. Genet. 2019, 51, 1035–1043. [Google Scholar] [CrossRef] [PubMed]
- Musser, J.M.; Beres, S.B.; Zhu, L.; Olsen, R.J.; Vuopio, J.; Hyyryläinen, H.L.; Gröndahl-Yli-Hannuksela, K.; Kristinsson, K.G.; Darenberg, J.; Henriques-Normark, B.; et al. Reduced In Vitro Susceptibility of Streptococcus pyogenes to β-Lactam Antibiotics Associated with Mutations in the pbp2x Gene Is Geographically Widespread. J. Clin. Microbiol. 2020, 58, 10-1128. [Google Scholar] [CrossRef] [PubMed]
- Tedijanto, C.; Olesen, S.W.; Grad, Y.H.; Lipsitch, M. Estimating the proportion of bystander selection for antibiotic resistance among potentially pathogenic bacterial flora. Proc. Natl. Acad. Sci. USA 2018, 115, E11988–E11995. [Google Scholar] [CrossRef]
- Azuar, A.; Li, Z.; Shibu, M.A.; Zhao, L.; Luo, Y.; Shalash, A.O.; Khalil, Z.G.; Capon, R.J.; Hussein, W.M.; Toth, I.; et al. Poly(hydrophobic amino acid)-Based Self-Adjuvanting Nanoparticles for Group A Streptococcus Vaccine Delivery. J. Med. Chem. 2021, 64, 2648–2658. [Google Scholar] [CrossRef]
- Skwarczynski, M.; Zhao, G.; Boer, J.C.; Ozberk, V.; Azuar, A.; Cruz, J.G.; Giddam, A.K.; Khalil, Z.G.; Pandey, M.; Shibu, M.A.; et al. Poly(amino acids) as a potent self-adjuvanting delivery system for peptide-based nanovaccines. Sci. Adv. 2020, 6, eaax2285. [Google Scholar] [CrossRef]
- Walkinshaw, D.R.; Wright, M.E.E.; Mullin, A.E.; Excler, J.-L.; Kim, J.H.; Steer, A.C. The Streptococcus pyogenes vaccine landscape. npj Vaccines 2023, 8, 16. [Google Scholar] [CrossRef]
- Russo, B.T.; Ayinuola, Y.A.; Singh, D.; Carothers, K.; Fischetti, V.A.; Flores-Mireles, A.L.; Lee, S.W.; Ploplis, V.A.; Liang, Z.; Castellino, F.J. The M Protein of Streptococcus pyogenes Strain AP53 Retains Cell Surface Functional Plasminogen Binding after Inactivation of the Sortase A Gene. J. Bacteriol. 2020, 202, 10-1128. [Google Scholar] [CrossRef]
- Brouwer, S.; Rivera-Hernandez, T.; Curren, B.F.; Harbison-Price, N.; De Oliveira, D.M.P.; Jespersen, M.G.; Davies, M.R.; Walker, M.J. Pathogenesis, epidemiology and control of Group A Streptococcus infection. Nat. Rev. Microbiol. 2023, 21, 431–447. [Google Scholar] [CrossRef]
- Kehoe, M.A.; Miller, L.; Poirier, T.P.; Whitnack, E.; Beachey, E.H.; Robinson, J.H.; Pinkney, M. Molecular Biology of Group A Streptococcal M Proteins. In Pathogenesis of Wound and Biomaterial-Associated Infections; Wadström, T., Eliasson, I., Holder, I., Ljungh, Å., Eds.; Springer: London, UK, 1990; pp. 47–54. [Google Scholar]
- Caparon, M.G.; Stephens, D.S.; Olsén, A.; Scott, J.R. Role of M protein in adherence of group A streptococci. Infect. Immun. 1991, 59, 1811–1817. [Google Scholar] [CrossRef]
- Ryan, P.A.; Pancholi, V.; Fischetti, V.A. Group A streptococci bind to mucin and human pharyngeal cells through sialic acid-containing receptors. Infect. Immun. 2001, 69, 7402–7412. [Google Scholar] [CrossRef] [PubMed]
- Walker, M.J.; Barnett, T.C.; McArthur, J.D.; Cole, J.N.; Gillen, C.M.; Henningham, A.; Sriprakash, K.S.; Sanderson-Smith, M.L.; Nizet, V. Disease manifestations and pathogenic mechanisms of Group A Streptococcus. Clin. Microbiol. Rev. 2014, 27, 264–301. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Sakota, V.; Jackson, D.; Franklin, A.R.; Beall, B. Array of M protein gene subtypes in 1064 recent invasive group A streptococcus isolates recovered from the active bacterial core surveillance. J. Infect. Dis. 2003, 188, 1587–1592. [Google Scholar] [CrossRef] [PubMed]
- Massell, B.F.; Honikman, L.H.; Amezcua, J. Rheumatic fever following streptococcal vaccination. Report of three cases. JAMA 1969, 207, 1115–1119. [Google Scholar] [CrossRef]
- Sekuloski, S.; Batzloff, M.R.; Griffin, P.; Parsonage, W.; Elliott, S.; Hartas, J.; O’Rourke, P.; Marquart, L.; Pandey, M.; Rubin, F.A.; et al. Evaluation of safety and immunogenicity of a group A streptococcus vaccine candidate (MJ8VAX) in a randomized clinical trial. PLoS ONE 2018, 13, e0198658. [Google Scholar] [CrossRef]
- Hayman, W.A.; Brandt, E.R.; Relf, W.A.; Cooper, J.; Saul, A.; Good, M.F. Mapping the minimal murine T cell and B cell epitopes within a peptide vaccine candidate from the conserved region of the M protein of group A streptococcus. Int. Immunol. 1997, 9, 1723–1733. [Google Scholar] [CrossRef]
- Pruksakorn, S.; Currie, B.; Brandt, E.; Phornphutkul, C.; Hunsakunachai, S.; Manmontri, A.; Robinson, J.H.; Kehoe, M.A.; Galbraith, A.; Good, M.F. Identification of T cell autoepitopes that cross-react with the C-terminal segment of the M protein of group A streptococci. Int. Immunol. 1994, 6, 1235–1244. [Google Scholar] [CrossRef]
- Pastural, É.; McNeil, S.A.; MacKinnon-Cameron, D.; Ye, L.; Langley, J.M.; Stewart, R.; Martin, L.H.; Hurley, G.J.; Salehi, S.; Penfound, T.A.; et al. Safety and immunogenicity of a 30-valent M protein-based group a streptococcal vaccine in healthy adult volunteers: A randomized, controlled phase I study. Vaccine 2020, 38, 1384–1392. [Google Scholar] [CrossRef]
- Dale, J.B.; Penfound, T.A.; Chiang, E.Y.; Walton, W.J. New 30-valent M protein-based vaccine evokes cross-opsonic antibodies against non-vaccine serotypes of group A streptococci. Vaccine 2011, 29, 8175–8178. [Google Scholar] [CrossRef]
- Brandt, E.R.; Hayman, W.A.; Currie, B.; Carapetis, J.; Wood, Y.; Jackson, D.C.; Cooper, J.; Melrose, W.D.; Saul, A.J.; Good, M.F. Opsonic human antibodies from an endemic population specific for a conserved epitope on the M protein of group A streptococci. Immunology 1996, 89, 331–337. [Google Scholar] [CrossRef]
- Pandey, M.; Wykes, M.N.; Hartas, J.; Good, M.F.; Batzloff, M.R. Long-Term Antibody Memory Induced by Synthetic Peptide Vaccination Is Protective against Streptococcus pyogenes Infection and Is Independent of Memory T Cell Help. J. Immunol. 2013, 190, 2692–2701. [Google Scholar] [CrossRef] [PubMed]
- Batzloff, M.R.; Hayman, W.A.; Davies, M.R.; Zeng, M.; Pruksakorn, S.; Brandt, E.R.; Good, M.F. Protection against group A streptococcus by immunization with J8-diphtheria toxoid: Contribution of J8- and diphtheria toxoid-specific antibodies to protection. J. Infect. Dis. 2003, 187, 1598–1608. [Google Scholar] [CrossRef] [PubMed]
- Shalash, A.O.; Hussein, W.M.; Nahar, U.J.; Wang, J.; Lu, L.; Azuar, A.; Kiong, J.J.E.; Koirala, P.; Khalil, Z.G.; Toth, I. Rational Development of a Novel Emulgel Adjuvant for Single-Shot Effective Vaccination: A Multivariate Analysis Approach. Adv. Mater. 2025, 2506496. [Google Scholar] [CrossRef] [PubMed]
- Pandey, M.; Powell, J.; Calcutt, A.; Zaman, M.; Phillips, Z.N.; Ho, M.F.; Batzloff, M.R.; Good, M.F. Physicochemical characterisation, immunogenicity and protective efficacy of a lead streptococcal vaccine: Progress towards Phase I trial. Sci. Rep. 2017, 7, 13786. [Google Scholar] [CrossRef]
- Schulz, D.; Grumann, D.; Trübe, P.; Pritchett-Corning, K.; Johnson, S.; Reppschläger, K.; Gumz, J.; Sundaramoorthy, N.; Michalik, S.; Berg, S.; et al. Laboratory Mice Are Frequently Colonized with Staphylococcus aureus and Mount a Systemic Immune Response-Note of Caution for In vivo Infection Experiments. Front. Cell. Infect. Microbiol. 2017, 7, 152. [Google Scholar] [CrossRef]
- Tong, S.Y.; Davis, J.S.; Eichenberger, E.; Holland, T.L.; Fowler, V.G., Jr. Staphylococcus aureus infections: Epidemiology, pathophysiology, clinical manifestations, and management. Clin. Microbiol. Rev. 2015, 28, 603–661. [Google Scholar] [CrossRef]
- Shalash, A.O.; Becker, L.; Yang, J.; Giacomin, P.; Pearson, M.; Hussein, W.M.; Loukas, A.; Toth, I.; Skwarczynski, M. Development of a peptide vaccine against hookworm infection: Immunogenicity, efficacy, and immune correlates of protection. J. Allergy Clin. Immunol. 2022, 150, 157–169.e110. [Google Scholar] [CrossRef]
- Greenfield, N.J. Using circular dichroism spectra to estimate protein secondary structure. Nat. Protoc. 2007, 1, 2876–2890. [Google Scholar] [CrossRef]
- Wijesundara, N.M.; Lee, S.F.; Cheng, Z.; Davidson, R.; Rupasinghe, H.P.V. Carvacrol exhibits rapid bactericidal activity against Streptococcus pyogenes through cell membrane damage. Sci. Rep. 2021, 11, 1487. [Google Scholar] [CrossRef]
- Toniolo, C.; Bonora, G.M.; Lüscher, I.F.; Schneider, C.H. Chain-length dependence for secondary structure formation of homo-oligopeptides from epsilon-tert.-butyloxycarbonyl-L-lysine with a lipophilic C-terminal group. Int. J. Pept. Protein Res. 1984, 23, 47–54. [Google Scholar] [CrossRef]
- Gazzara, J.A.; Phillips, M.C.; Lund-Katz, S.; Palgunachari, M.N.; Segrest, J.P.; Anantharamaiah, G.M.; Snow, J.W. Interaction of class A amphipathic helical peptides with phospholipid unilamellar vesicles. J. Lipid Res. 1997, 38, 2134–2146. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Peptide Sequence | Analytical Data | Purity | Yield |
|---|---|---|---|---|
| J8 | QAEDKVKQSREAKKQVEKALKQLEDKVQ Molecular weight: 3282.6 g/mol. | ESI-MS: [M + 3H]+3 m/z 1095.8 (cal. 1095.3), [M + 4H]+4 m/z 821.0 (cal. 821.5); HPLC, tR = 20.0 min (C18 column, 0–100% solvent B, 53 min) | 98% | 53% |
| J8i | SREAKKQVEKAL Molecular weight: 1385.6 g/mol. | ESI-MS: [M + H]+ m/z 1387.9 (cal. 1387.6), [M + 2H]+2 m/z 693.5 (cal. 694.5); HPLC, tR = 15.4 min (C18 column, 0–100% solvent B, 53 min) | 97% | 55% |
| J8i-J8i | SREAKKQVEKALSREAKKQVEKAL Molecular weight: 2754.2 g/mol. | ESI-MS: [M + 3H]+3 m/z 920.0 (cal. 919.1), [M + 4H]+4 m/z 690.6 (cal. 689.6), [M + 5H]+5 m/z 552.0 (cal. 551.8); HPLC, tR = 19.4 min (C18 column, 0–100% solvent B, 53 min) | 93% | 57% |
| P145 | LRRDLDASREAKKQVEKALE Molecular weight: 2354.7 g/mol. | ESI-MS: [M + 2H]+2 m/z 1177.9 (cal. 1178.4), [M + 3H]+3 m/z 785.1 (cal. 785.9), [M + 4H]+4 m/z 588.7 (cal. 589.7); HPLC, tR = 19.3 min (C4 column, 0–100% solvent B, 53 min) | 95% | 52% |
| NTD | LRKLKKGTASVAVAL Molecular weight: 1553.9 g/mol. | ESI-MS: [M + 2H]+2 m/z 777.7 (cal. 778.0), [M + 3H]+3 m/z 518.5 (cal. 519.0); HPLC, tR = 18.9 min (C18 column, 0–100% solvent B, 53 min) | 96% | 55% |
| CTD2 | KALKEQLAKQAEELAKLR Molecular weight: 2066.5 g/mol. | ESI-MS: [M + 2H]+2 m/z 1034.8 (cal. 1034.3), [M + 3H]+3 m/z 685.6 (cal. 689.8); HPLC, tR = 19.9 min (C18 column, 0–100% solvent B, 53 min) | 97% | 53% |
| P-J8 | AKFVAAWTLKAAA-QAEDKVKQSREAKKQVEKALKQLEDKVQ Molecular weight: 4653.4 g/mol. | ESI-MS: [M + 4H]+4 m/z 1164.8 (cal. 1164.4), [M + 5H]+5 m/z 932.2 (cal. 931.7), [M + 6H]+6 m/z:776.9 (cal. 776.6); HPLC, tR = 21.2 min (C18 column, 0–100% solvent B, 53 min) | 96% | 43% |
| P-J8i | AKFVAAWTLKAAA-SREAKKQVEKAL Molecular weight: 2715.2 g/mol. | ESI-MS: [M + 2H]+2 m/z 1357.9 (cal. 1358.6), [M + 3H]+3 m/z:907.0 (cal. 906.1), [M + 4H]+4 m/z:681.4 (cal. 679.8); HPLC, tR = 28.0 min (C18 column, 0–100% solvent B, 53 min) | 98% | 45% |
| P-J8i-J8i | AKFVAAWTLKAAA-SREAKKQVEKALSREAKKQVEKAL Molecular weight: 4083.8 g/mol. | ESI-MS: [M + 3H]+3 m/z 1363.1 (cal. 1362.3), [M + 4H]+4 m/z: 1023.1 (cal. 1022.0), [M + 5H]+5 m/z: 818.5 (cal. 817.8); HPLC, tR = 21.9 min (C18 column, 0–100% solvent B, 53 min) | 97% | 46% |
| L15-P-J8 | LLLLLLLLLLLLLLL-AKFVAAWTLKAAA-QAEDKVKQSREAKKQVEKALKQLEDKVQ Molecular weight: 6350.8 g/mol. | ESI-MS: [M + 5H]+5 m/z 1269.8 (cal. 1271.2), [M + 6H]+6 m/z: 1059.7 (cal. 1059.5), [M + 7H]+7 m/z: 908.5 (cal. 908.3); HPLC, tR = 37.2 min (C4 column, 0–100% solvent B, 53 min) | 96% | 23% |
| L15-P-J8i | LLLLLLLLLLLLLLL-AKFVAAWTLKAAA-SREAKKQVEKAL Molecular weight: 4454.7 g/mol. | ESI-MS: [M + 4H]+4 m/z: 1115.2 (cal. 1114.7), [M + 5H]+5 m/z: 892.5 (cal. 891.9); HPLC, tR = 38.4 min (C4 column, 0–100% solvent B, 53 min) | 95% | 18% |
| L15-P-J8i-J8i | LLLLLLLLLLLLLLL-AKFVAAWTLKAAA-SREAKKQVEKALSREAKKQVEKAL Molecular weight: 5783.1 g/mol. | ESI-MS: [M + 5H]+5 m/z 1157.2 (cal. 1157.6), [M + 6H]+6 m/z: 964.8 (cal. 964.9), [M + 7H]+7 m/z: 826.9 (cal. 827.2); HPLC, tR = 35.5 min (C4 column, 0–100% solvent B, 53 min) | 95% | 21% |
| L15-P-p145 | LLLLLLLLLLLLLLL-AKFVAAWTLKAAA-LRRDLDASREAKKQVEKALE Molecular weight: 5381.7 g/mol. | ESI-MS: [M + 4H]+4 m/z 1346.8 (cal. 1346.4), [M + 5H]+5 m/z: 1076.5 (cal. 1077.3), [M + 6H]+6 m/z: 897.5 (cal. 898.0); HPLC, tR = 34.2 min (C4 column, 0–100% solvent B, 53 min) | 96% | 19% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shalash, A.O.; Sun, H.; Cui, Y.; Wang, J.; Arnts, B.; Bauer, J.; Hussein, W.M.; Khalil, Z.G.; Skwarczynski, M.; Toth, I. Evaluation of Peptide-Based Vaccines Against Group A Streptococcus in Staphylococcus aureus-Infected Mice. Vaccines 2025, 13, 632. https://doi.org/10.3390/vaccines13060632
Shalash AO, Sun H, Cui Y, Wang J, Arnts B, Bauer J, Hussein WM, Khalil ZG, Skwarczynski M, Toth I. Evaluation of Peptide-Based Vaccines Against Group A Streptococcus in Staphylococcus aureus-Infected Mice. Vaccines. 2025; 13(6):632. https://doi.org/10.3390/vaccines13060632
Chicago/Turabian StyleShalash, Ahmed O., Haolan Sun, Yiru Cui, Jingwen Wang, Barb Arnts, Jannah Bauer, Waleed M. Hussein, Zeinab G. Khalil, Mariusz Skwarczynski, and Istvan Toth. 2025. "Evaluation of Peptide-Based Vaccines Against Group A Streptococcus in Staphylococcus aureus-Infected Mice" Vaccines 13, no. 6: 632. https://doi.org/10.3390/vaccines13060632
APA StyleShalash, A. O., Sun, H., Cui, Y., Wang, J., Arnts, B., Bauer, J., Hussein, W. M., Khalil, Z. G., Skwarczynski, M., & Toth, I. (2025). Evaluation of Peptide-Based Vaccines Against Group A Streptococcus in Staphylococcus aureus-Infected Mice. Vaccines, 13(6), 632. https://doi.org/10.3390/vaccines13060632