
β-Catenin in Dendritic Cells Negatively Regulates CD8 T Cell Immune Responses through the Immune Checkpoint Molecule Tim-3

 ,
,  , ,
, ,  , and
, and
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Significance
2. Introduction
3. Methods
3.1. Mice and Treatment
3.2. Antibodies and Reagents
3.3. In Vivo Cross-Priming Assays
3.4. Single-Cell RNA Sequencing (scRNAseq) of Primed Pmel-1 CD8 T Cells
3.5. Data Analysis for scRNAseq
3.6. Tumor Cell Lines and Treatment of Tumor-Bearing Mice
3.7. Statistical Analysis
4. Results
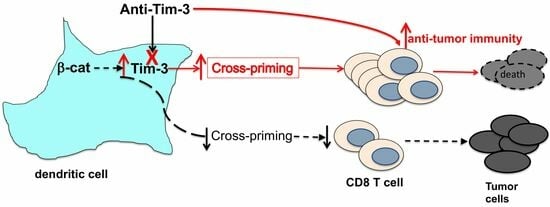
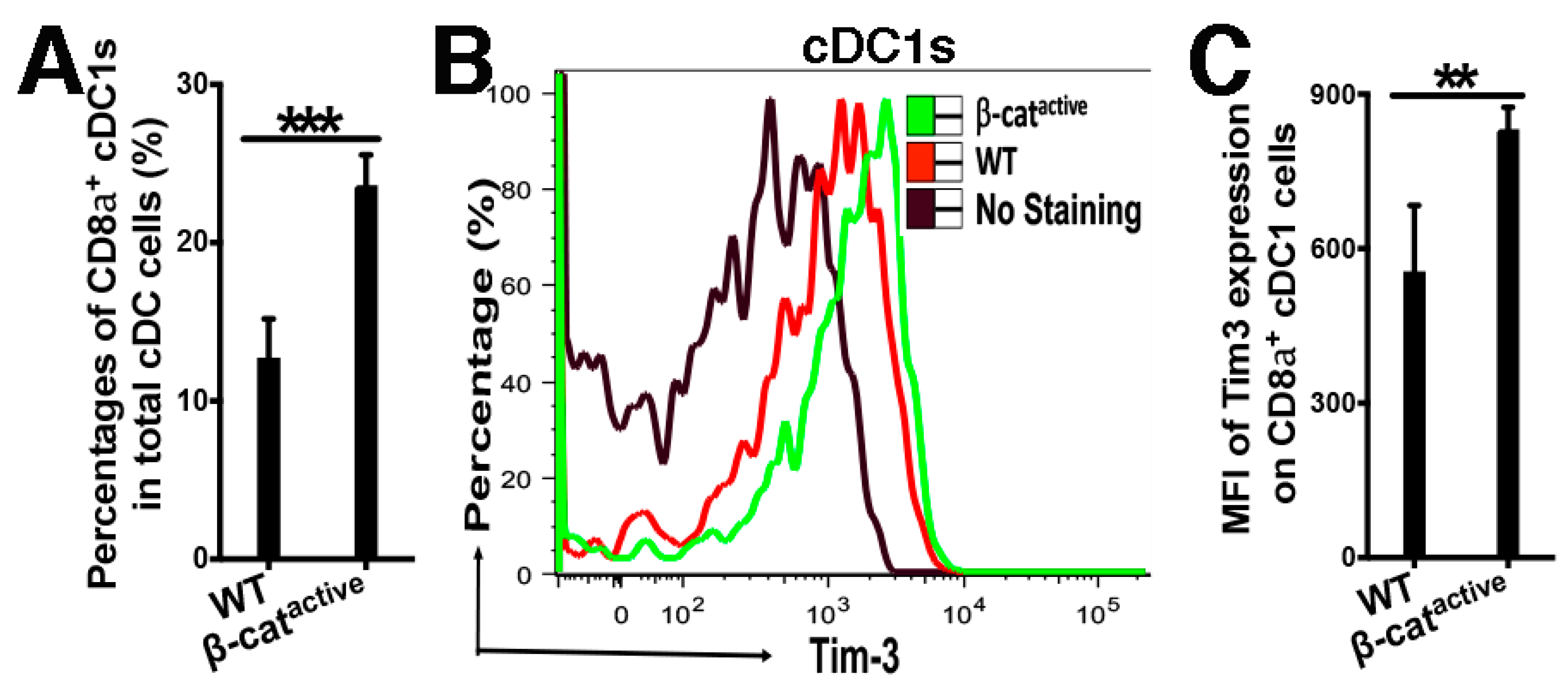
4.1. β-Catenin Upregulates the Expression of the Inhibitory Molecule Tim-3 in DCs
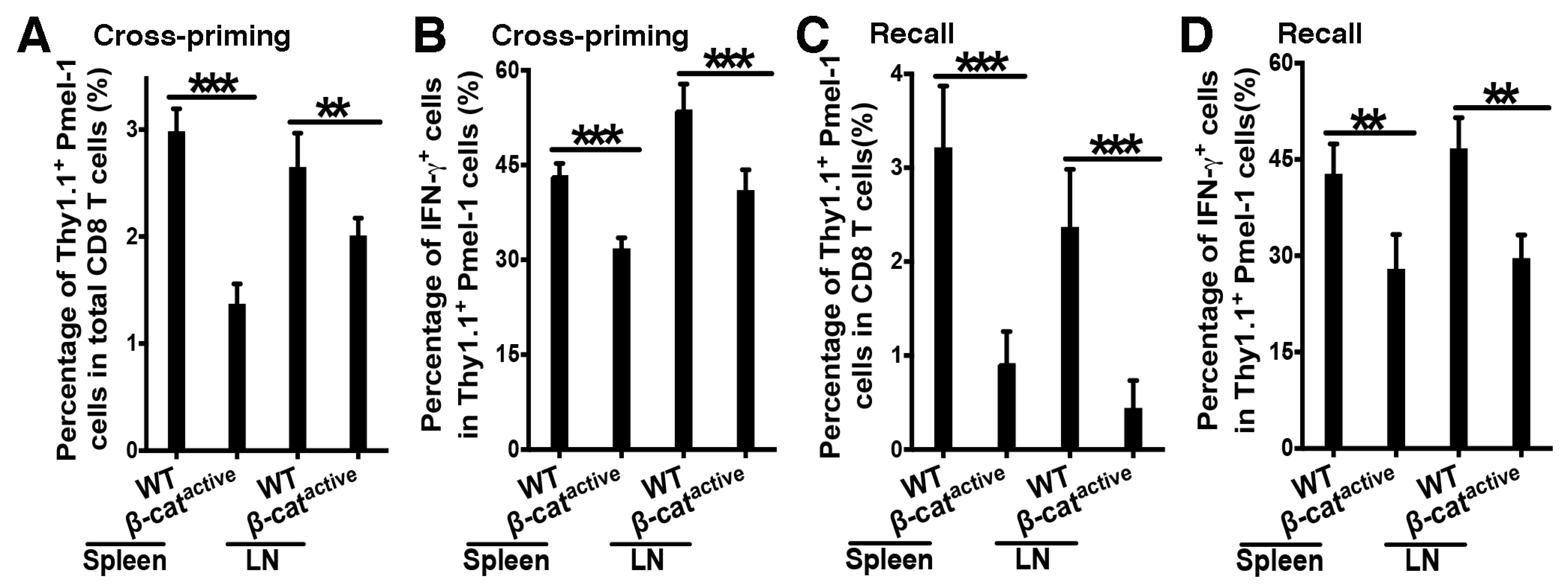
4.2. β-Catenin in DCs Suppresses Tumor Antigen-Specific CD8 T Cell Responses
4.3. Single-Cell RNA Sequencing of Pmel-1 CD8 T Cells Primed in WT and CD11c-β-Cateninactive Mice
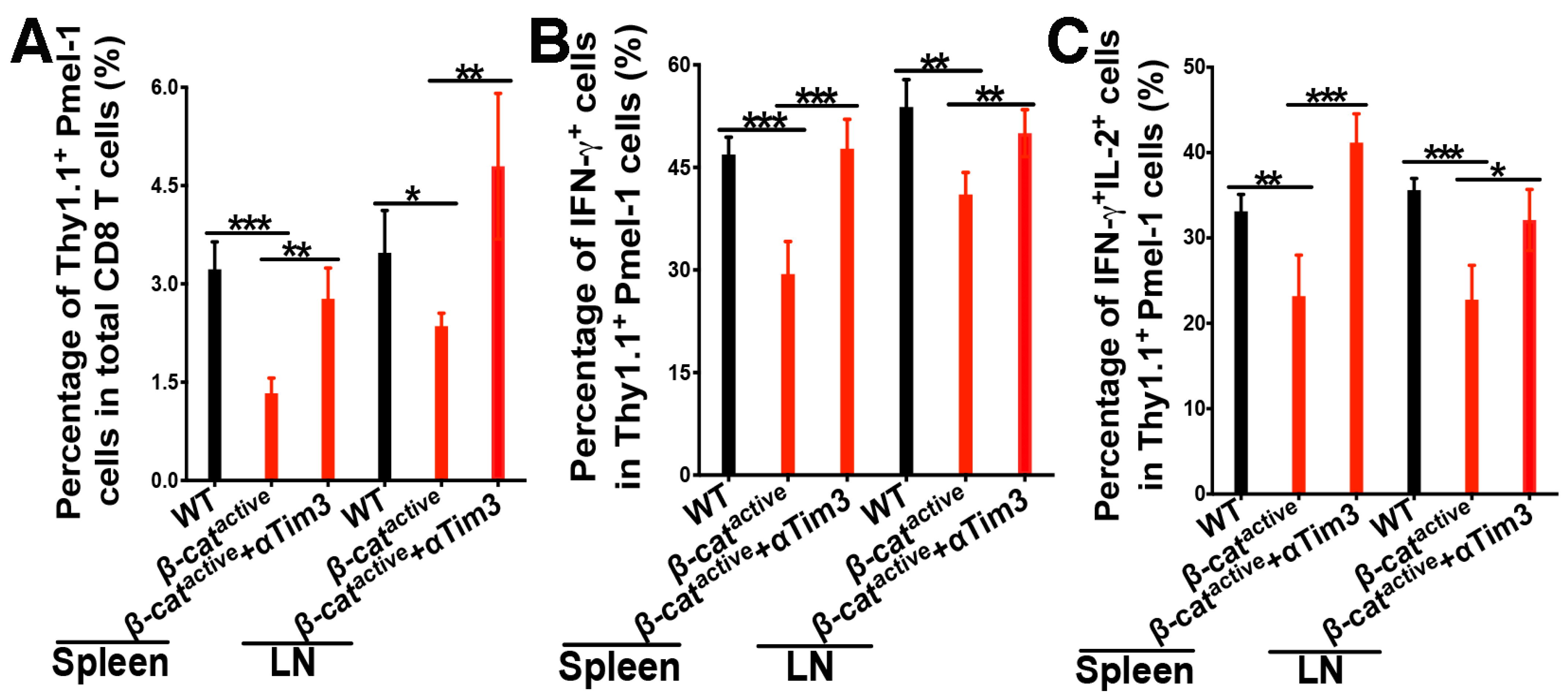
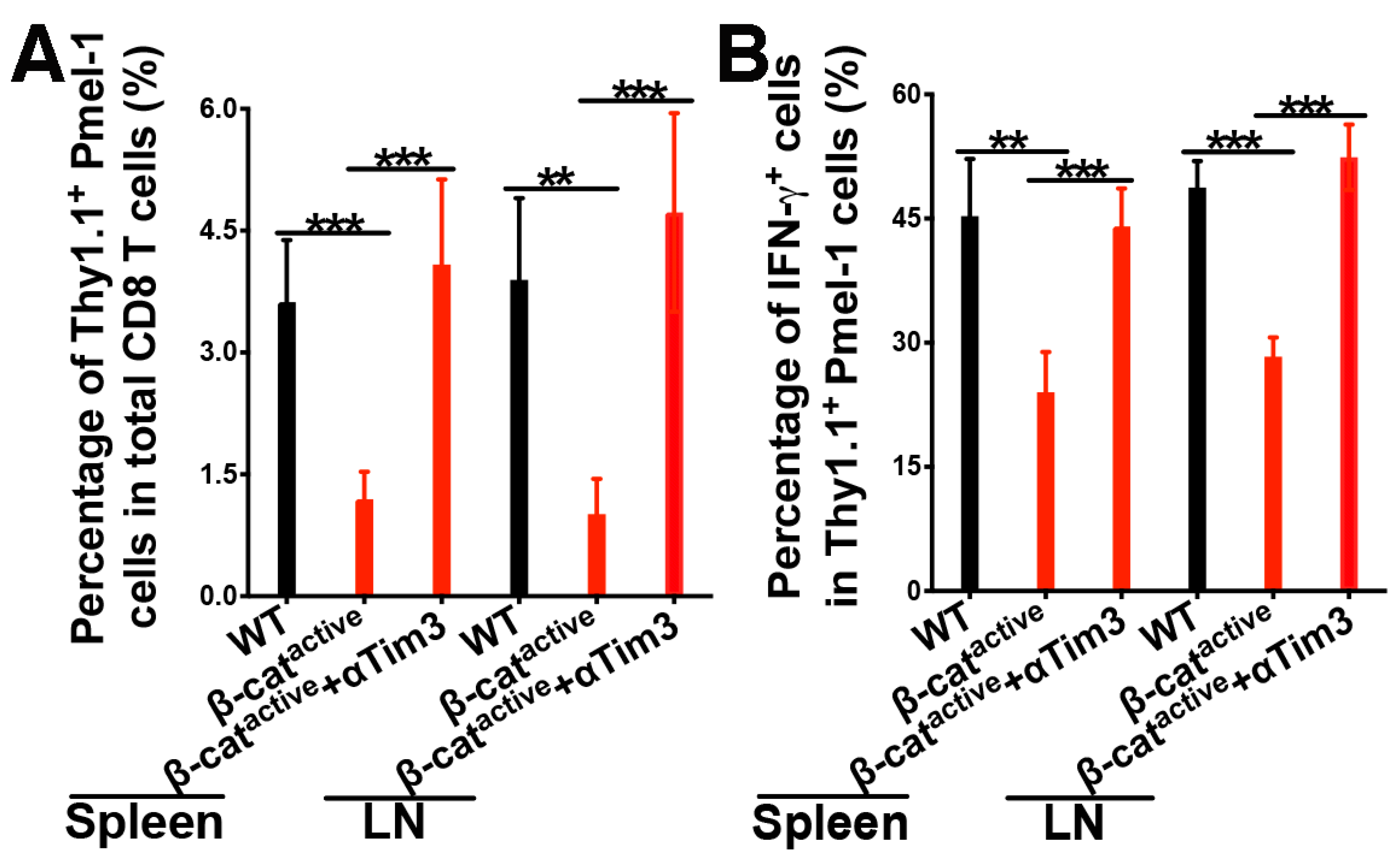
4.4. Anti-Tim-3 Treatment Reverses β-Catenin-Mediated Suppression of CD8 T Cell Responses
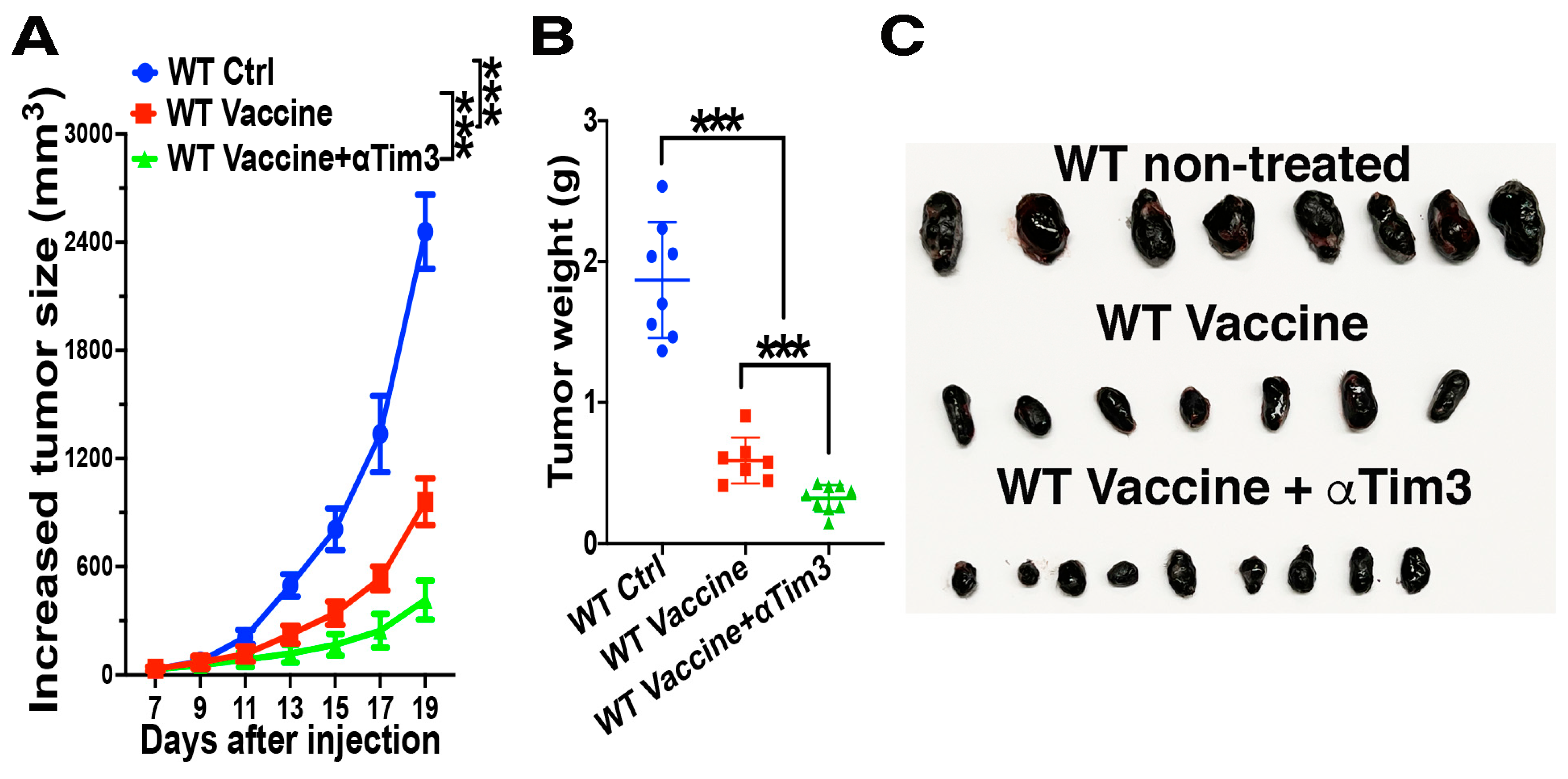
4.5. Combination of Tim-3 Blockade and DC Vaccination Led to Improved DC Vaccine Efficacy
5. Discussion
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jhunjhunwala, S.; Hammer, C.; Delamarre, L. Antigen presentation in cancer: Insights into tumour immunogenicity and immune evasion. Nat. Rev. Cancer 2021, 21, 298–312. [Google Scholar] [CrossRef] [PubMed]
- Murphy, T.L.; Murphy, K.M. Dendritic cells in cancer immunology. Cell Mol. Immunol. 2022, 19, 3–13. [Google Scholar] [CrossRef]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef]
- Fu, C.; Ma, T.; Zhou, L.; Mi, Q.S.; Jiang, A. Dendritic Cell-Based Vaccines Against Cancer: Challenges, Advances and Future Opportunities. Immunol. Investig. 2022, 51, 2133–2158. [Google Scholar] [CrossRef]
- Kleindienst, P.; Brocker, T. Endogenous dendritic cells are required for amplification of T cell responses induced by dendritic cell vaccines in vivo. J. Immunol. 2003, 170, 2817–2823. [Google Scholar] [CrossRef]
- Yewdall, A.W.; Drutman, S.B.; Jinwala, F.; Bahjat, K.S.; Bhardwaj, N. CD8+ T cell priming by dendritic cell vaccines requires antigen transfer to endogenous antigen presenting cells. PLoS ONE 2010, 5, e11144. [Google Scholar] [CrossRef]
- Ferris, S.T.; Ohara, R.A.; Ou, F.; Wu, R.; Huang, X.; Kim, S.; Chen, J.; Liu, T.T.; Schreiber, R.D.; Murphy, T.L.; et al. cDC1 Vaccines Drive Tumor Rejection by Direct Presentation Independently of Host cDC1. Cancer Immunol. Res. 2022, 10, 920–931. [Google Scholar] [CrossRef]
- Veglia, F.; Gabrilovich, D.I. Dendritic cells in cancer: The role revisited. Curr. Opin. Immunol. 2017, 45, 43–51. [Google Scholar] [CrossRef]
- Chrisikos, T.T.; Zhou, Y.; Slone, N.; Babcock, R.; Watowich, S.S.; Li, H.S. Molecular regulation of dendritic cell development and function in homeostasis, inflammation, and cancer. Mol. Immunol. 2019, 110, 24–39. [Google Scholar] [CrossRef]
- Sanchez-Paulete, A.R.; Teijeira, A.; Cueto, F.J.; Garasa, S.; Perez-Gracia, J.L.; Sanchez-Arraez, A.; Sancho, D.; Melero, I. Antigen cross-presentation and T-cell cross-priming in cancer immunology and immunotherapy. Ann. Oncol. 2017, 28, xii44–xii55. [Google Scholar] [CrossRef]
- Bandola-Simon, J.; Roche, P.A. Dysfunction of antigen processing and presentation by dendritic cells in cancer. Mol. Immunol. 2018, 113, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.; Jiang, A. Dendritic Cells and CD8 T Cell Immunity in Tumor Microenvironment. Front. Immunol. 2018, 9, 3059. [Google Scholar] [CrossRef] [PubMed]
- Broz, M.L.; Binnewies, M.; Boldajipour, B.; Nelson, A.E.; Pollack, J.L.; Erle, D.J.; Barczak, A.; Rosenblum, M.D.; Daud, A.; Barber, D.L.; et al. Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell 2014, 26, 638–652. [Google Scholar] [CrossRef] [PubMed]
- Salmon, H.; Idoyaga, J.; Rahman, A.; Leboeuf, M.; Remark, R.; Jordan, S.; Casanova-Acebes, M.; Khudoynazarova, M.; Agudo, J.; Tung, N.; et al. Expansion and Activation of CD103+ Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity 2016, 44, 924–938. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Dai, D.; Horton, B.; Gajewski, T.F. Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell 2017, 31, 711–723.e714. [Google Scholar] [CrossRef] [PubMed]
- Carreno, B.M.; Magrini, V.; Becker-Hapak, M.; Kaabinejadian, S.; Hundal, J.; Petti, A.A.; Ly, A.; Lie, W.R.; Hildebrand, W.H.; Mardis, E.R.; et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 2015, 348, 803–808. [Google Scholar] [CrossRef]
- Jiang, A.; Bloom, O.; Ono, S.; Cui, W.; Unternaehrer, J.; Jiang, S.; Whitney, J.A.; Connolly, J.; Banchereau, J.; Mellman, I. Disruption of E-cadherin-mediated adhesion induces a functionally distinct pathway of dendritic cell maturation. Immunity 2007, 27, 610–624. [Google Scholar] [CrossRef]
- Manicassamy, S.; Reizis, B.; Ravindran, R.; Nakaya, H.; Salazar-Gonzalez, R.M.; Wang, Y.C.; Pulendran, B. Activation of beta-catenin in dendritic cells regulates immunity versus tolerance in the intestine. Science 2010, 329, 849–853. [Google Scholar] [CrossRef] [PubMed]
- Van den Bossche, J.; Malissen, B.; Mantovani, A.; De Baetselier, P.; Van Ginderachter, J.A. Regulation and function of the E-cadherin/catenin complex in cells of the monocyte-macrophage lineage and DCs. Blood 2012, 119, 1623–1633. [Google Scholar] [CrossRef]
- Liang, X.; Fu, C.; Cui, W.; Ober-Blobaum, J.L.; Zahner, S.P.; Shrikant, P.A.; Clausen, B.E.; Flavell, R.A.; Mellman, I.; Jiang, A. beta-catenin mediates tumor-induced immunosuppression by inhibiting cross-priming of CD8+ T cells. J. Leukoc. Biol. 2014, 95, 179–190. [Google Scholar] [CrossRef]
- Hong, Y.; Manoharan, I.; Suryawanshi, A.; Majumdar, T.; Angus-Hill, M.L.; Koni, P.A.; Manicassamy, B.; Mellor, A.L.; Munn, D.H.; Manicassamy, S. beta-catenin promotes regulatory T-cell responses in tumors by inducing vitamin A metabolism in dendritic cells. Cancer Res. 2015, 75, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.; Liang, X.; Cui, W.; Ober-Blobaum, J.L.; Vazzana, J.; Shrikant, P.A.; Lee, K.P.; Clausen, B.E.; Mellman, I.; Jiang, A. beta-Catenin in dendritic cells exerts opposite functions in cross-priming and maintenance of CD8+ T cells through regulation of IL-10. Proc. Natl. Acad. Sci. USA 2015, 112, 2823–2828. [Google Scholar] [CrossRef] [PubMed]
- Holtzhausen, A.; Zhao, F.; Evans, K.S.; Tsutsui, M.; Orabona, C.; Tyler, D.S.; Hanks, B.A. Melanoma-Derived Wnt5a Promotes Local Dendritic-Cell Expression of IDO and Immunotolerance: Opportunities for Pharmacologic Enhancement of Immunotherapy. Cancer Immunol. Res. 2015, 3, 1082–1095. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Xiao, C.; Evans, K.S.; Theivanthiran, T.; DeVito, N.; Holtzhausen, A.; Liu, J.; Liu, X.; Boczkowski, D.; Nair, S.; et al. Paracrine Wnt5a-beta-Catenin Signaling Triggers a Metabolic Program that Drives Dendritic Cell Tolerization. Immunity 2018, 48, 147–160. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.A.; Wu, D.C.; Cheung, J.; Navarro, A.; Xiong, H.; Cubas, R.; Totpal, K.; Chiu, H.; Wu, Y.; Comps-Agrar, L.; et al. PD-L1 expression by dendritic cells is a key regulator of T-cell immunity in cancer. Nat. Cancer 2020, 1, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Mayoux, M.; Roller, A.; Pulko, V.; Sammicheli, S.; Chen, S.; Sum, E.; Jost, C.; Fransen, M.F.; Buser, R.B.; Kowanetz, M.; et al. Dendritic cells dictate responses to PD-L1 blockade cancer immunotherapy. Sci. Transl. Med. 2020, 12, 546–556. [Google Scholar] [CrossRef] [PubMed]
- Dixon, K.O.; Tabaka, M.; Schramm, M.A.; Xiao, S.; Tang, R.; Dionne, D.; Anderson, A.C.; Rozenblatt-Rosen, O.; Regev, A.; Kuchroo, V.K. TIM-3 restrains anti-tumour immunity by regulating inflammasome activation. Nature 2021, 595, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Shah, K.V.; Chien, A.J.; Yee, C.; Moon, R.T. CTLA-4 is a direct target of Wnt/beta-catenin signaling and is expressed in human melanoma tumors. J. Investig. Dermatol. 2008, 128, 2870–2879. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Lee, J.H.; Jiang, H.; Wang, C.; Wang, S.; Zheng, Z.; Shao, F.; Xu, D.; Xia, Y.; Li, J.; et al. Beta-Catenin induces transcriptional expression of PD-L1 to promote glioblastoma immune evasion. J. Exp. Med. 2020, 217, e20191115. [Google Scholar] [CrossRef] [PubMed]
- Karnam, A.; Bonam, S.R.; Rambabu, N.; Wong, S.S.W.; Aimanianda, V.; Bayry, J. Wnt-beta-Catenin Signaling in Human Dendritic Cells Mediates Regulatory T-Cell Responses to Fungi via the PD-L1 Pathway. mBio 2021, 12, e0282421. [Google Scholar] [CrossRef]
- Zhou, L.; Adrianto, I.; Wang, J.; Wu, X.; Datta, I.; Mi, Q.S. Single-Cell RNA-Seq Analysis Uncovers Distinct Functional Human NKT Cell Sub-Populations in Peripheral Blood. Front. Cell Dev. Biol. 2020, 8, 384. [Google Scholar] [CrossRef]
- Wang, J.; Adrianto, I.; Subedi, K.; Liu, T.; Wu, X.; Yi, Q.; Loveless, I.; Yin, C.; Datta, I.; Sant’Angelo, D.B.; et al. Integrative scATAC-seq and scRNA-seq analyses map thymic iNKT cell development and identify Cbfbeta for its commitment. Cell Discov. 2023, 9, 61. [Google Scholar] [CrossRef]
- Zheng, G.X.; Terry, J.M.; Belgrader, P.; Ryvkin, P.; Bent, Z.W.; Wilson, R.; Ziraldo, S.B.; Wheeler, T.D.; McDermott, G.P.; Zhu, J.; et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017, 8, 14049. [Google Scholar] [CrossRef]
- Macosko, E.Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas, A.R.; Kamitaki, N.; Martersteck, E.M.; et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 2015, 161, 1202–1214. [Google Scholar] [CrossRef]
- Satija, R.; Farrell, J.A.; Gennert, D.; Schier, A.F.; Regev, A. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 2015, 33, 495–502. [Google Scholar] [CrossRef]
- Nakao, A.; Yoshihama, M.; Kenmochi, N. RPG: The Ribosomal Protein Gene database. Nucleic Acids Res. 2004, 32, D168–D170. [Google Scholar] [CrossRef]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H., 2nd; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef]
- Ma, H.; Gao, G.; Weber, G.M. Use of DAVID algorithms for clustering custom annotated gene lists in a non-model organism, rainbow trout. BMC Res. Notes 2018, 11, 63. [Google Scholar] [CrossRef]
- Cohen, S.B.; Smith, N.L.; McDougal, C.; Pepper, M.; Shah, S.; Yap, G.S.; Acha-Orbea, H.; Jiang, A.; Clausen, B.E.; Rudd, B.D.; et al. beta-Catenin Signaling Drives Differentiation and Proinflammatory Function of IRF8-Dependent Dendritic Cells. J. Immunol. 2015, 194, 210–222. [Google Scholar] [CrossRef]
- Boyman, O.; Cho, J.H.; Sprent, J. The role of interleukin-2 in memory CD8 cell differentiation. Adv. Exp. Med. Biol. 2010, 684, 28–41. [Google Scholar] [CrossRef]
- Feau, S.; Arens, R.; Togher, S.; Schoenberger, S.P. Autocrine IL-2 is required for secondary population expansion of CD8(+) memory T cells. Nat. Immunol. 2011, 12, 908–913. [Google Scholar] [CrossRef] [PubMed]
- Becht, E.; McInnes, L.; Healy, J.; Dutertre, C.A.; Kwok, I.W.H.; Ng, L.G.; Ginhoux, F.; Newell, E.W. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. 2018, 37, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, K.; Stetler-Stevenson, M.; Wilson, W.; Yuan, C.M. Novel CD19 expression in a peripheral T cell lymphoma: A flow cytometry case report with morphologic correlation. Cytom. B Clin. Cytom. 2009, 76, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Vlaming, M.; Bilemjian, V.; Freile, J.A.; Lourens, H.J.; van Rooij, N.; Huls, G.; van Meerten, T.; de Bruyn, M.; Bremer, E. CD20 positive CD8 T cells are a unique and transcriptionally-distinct subset of T cells with distinct transmigration properties. Sci. Rep. 2021, 11, 20499. [Google Scholar] [CrossRef] [PubMed]
- Yi, Q.; Wang, J.; Liu, T.; Yao, Y.; Loveless, I.; Subedi, K.; Toor, J.; Adrianto, I.; Xiao, H.; Chen, B.; et al. scRNA-Seq and imaging mass cytometry analyses unveil iNKT cells-mediated anti-tumor immunity in pancreatic cancer liver metastasis. Cancer Lett. 2023, 561, 216149. [Google Scholar] [CrossRef]
- Fensterl, V.; Sen, G.C. Interferon-induced Ifit proteins: Their role in viral pathogenesis. J. Virol. 2015, 89, 2462–2468. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, D.L.; Thompson, S.B.; Klarquist, J.; Harbell, M.G.; Kilgore, A.M.; Lasda, E.L.; Hesselberth, J.R.; Hunter, C.A.; Kedl, R.M. Vaccine adjuvant-elicited CD8(+) T cell immunity is co-dependent on T-bet and FOXO1. Cell Rep. 2023, 42, 112911. [Google Scholar] [CrossRef] [PubMed]
- Preiss, N.K.; Kamal, Y.; Wilkins, O.M.; Li, C.; Kolling, F.W.t.; Trask, H.W.; Usherwood, Y.K.; Cheng, C.; Frost, H.R.; Usherwood, E.J. Characterizing control of memory CD8 T cell differentiation by BTB-ZF transcription factor Zbtb20. Life Sci. Alliance 2023, 6, e202201683. [Google Scholar] [CrossRef]
- Overwijk, W.W.; Tsung, A.; Irvine, K.R.; Parkhurst, M.R.; Goletz, T.J.; Tsung, K.; Carroll, M.W.; Liu, C.; Moss, B.; Rosenberg, S.A.; et al. gp100/pmel 17 is a murine tumor rejection antigen: Induction of “self”-reactive, tumoricidal T cells using high-affinity, altered peptide ligand. J. Exp. Med. 1998, 188, 277–286. [Google Scholar] [CrossRef]
- Hawkins, W.G.; Gold, J.S.; Dyall, R.; Wolchok, J.D.; Hoos, A.; Bowne, W.B.; Srinivasan, R.; Houghton, A.N.; Lewis, J.J. Immunization with DNA coding for gp100 results in CD4 T-cell independent antitumor immunity. Surgery 2000, 128, 273–280. [Google Scholar] [CrossRef]
- Gold, J.S.; Ferrone, C.R.; Guevara-Patino, J.A.; Hawkins, W.G.; Dyall, R.; Engelhorn, M.E.; Wolchok, J.D.; Lewis, J.J.; Houghton, A.N. A single heteroclitic epitope determines cancer immunity after xenogeneic DNA immunization against a tumor differentiation antigen. J. Immunol. 2003, 170, 5188–5194. [Google Scholar] [CrossRef] [PubMed]
- Danilo, M.; Chennupati, V.; Silva, J.G.; Siegert, S.; Held, W. Suppression of Tcf1 by Inflammatory Cytokines Facilitates Effector CD8 T Cell Differentiation. Cell Rep. 2018, 22, 2107–2117. [Google Scholar] [CrossRef]
- Vander Lugt, B.; Beck, Z.T.; Fuhlbrigge, R.C.; Hacohen, N.; Campbell, J.J.; Boes, M. TGF-beta suppresses beta-catenin-dependent tolerogenic activation program in dendritic cells. PLoS ONE 2011, 6, e20099. [Google Scholar] [CrossRef] [PubMed]
- Shan, M.; Gentile, M.; Yeiser, J.R.; Walland, A.C.; Bornstein, V.U.; Chen, K.; He, B.; Cassis, L.; Bigas, A.; Cols, M.; et al. Mucus enhances gut homeostasis and oral tolerance by delivering immunoregulatory signals. Science 2013, 342, 447–453. [Google Scholar] [CrossRef]
- Feng, M.; Jin, J.Q.; Xia, L.; Xiao, T.; Mei, S.; Wang, X.; Huang, X.; Chen, J.; Liu, M.; Chen, C.; et al. Pharmacological inhibition of beta-catenin/BCL9 interaction overcomes resistance to immune checkpoint blockades by modulating T(reg) cells. Sci. Adv. 2019, 5, eaau5240. [Google Scholar] [CrossRef] [PubMed]
- DeVito, N.C.; Sturdivant, M.; Thievanthiran, B.; Xiao, C.; Plebanek, M.P.; Salama, A.K.S.; Beasley, G.M.; Holtzhausen, A.; Novotny-Diermayr, V.; Strickler, J.H.; et al. Pharmacological Wnt ligand inhibition overcomes key tumor-mediated resistance pathways to anti-PD-1 immunotherapy. Cell Rep. 2021, 35, 109071. [Google Scholar] [CrossRef] [PubMed]
- Kikushige, Y.; Shima, T.; Takayanagi, S.; Urata, S.; Miyamoto, T.; Iwasaki, H.; Takenaka, K.; Teshima, T.; Tanaka, T.; Inagaki, Y.; et al. TIM-3 is a promising target to selectively kill acute myeloid leukemia stem cells. Cell Stem Cell 2010, 7, 708–717. [Google Scholar] [CrossRef] [PubMed]
- Curigliano, G.; Gelderblom, H.; Mach, N.; Doi, T.; Tai, D.; Forde, P.M.; Sarantopoulos, J.; Bedard, P.L.; Lin, C.C.; Hodi, F.S.; et al. Phase I/Ib Clinical Trial of Sabatolimab, an Anti-TIM-3 Antibody, Alone and in Combination with Spartalizumab, an Anti-PD-1 Antibody, in Advanced Solid Tumors. Clin. Cancer Res. 2021, 27, 3620–3629. [Google Scholar] [CrossRef] [PubMed]
- Zeidan, A.M.; Giagounidis, A.; Sekeres, M.A.; Xiao, Z.; Sanz, G.F.; Hoef, M.V.; Ma, F.; Hertle, S.; Santini, V. STIMULUS-MDS2 design and rationale: A phase III trial with the anti-TIM-3 sabatolimab (MBG453) + azacitidine in higher risk MDS and CMML-2. Future Oncol. 2023, 19, 631–642. [Google Scholar] [CrossRef]
- Dixon, K.O.; Lahore, G.F.; Kuchroo, V.K. Beyond T cell exhaustion: TIM-3 regulation of myeloid cells. Sci. Immunol. 2024, 9, eadf2223. [Google Scholar] [CrossRef]
- Monney, L.; Sabatos, C.A.; Gaglia, J.L.; Ryu, A.; Waldner, H.; Chernova, T.; Manning, S.; Greenfield, E.A.; Coyle, A.J.; Sobel, R.A.; et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature 2002, 415, 536–541. [Google Scholar] [CrossRef] [PubMed]
- de Mingo Pulido, A.; Gardner, A.; Hiebler, S.; Soliman, H.; Rugo, H.S.; Krummel, M.F.; Coussens, L.M.; Ruffell, B. TIM-3 Regulates CD103(+) Dendritic Cell Function and Response to Chemotherapy in Breast Cancer. Cancer Cell 2018, 33, 60–74. [Google Scholar] [CrossRef]
- Yang, M.; Du, W.; Yi, L.; Wu, S.; He, C.; Zhai, W.; Yue, C.; Sun, R.; Menk, A.V.; Delgoffe, G.M.; et al. Checkpoint molecules coordinately restrain hyperactivated effector T cells in the tumor microenvironment. Oncoimmunology 2020, 9, 1708064. [Google Scholar] [CrossRef] [PubMed]
- Carenza, C.; Franzese, S.; Calcaterra, F.; Mavilio, D.; Della Bella, S. Comprehensive Phenotyping of Dendritic Cells in Cancer Patients by Flow Cytometry. Cytometry A 2021, 99, 218–230. [Google Scholar] [CrossRef]
- Chiba, S.; Baghdadi, M.; Akiba, H.; Yoshiyama, H.; Kinoshita, I.; Dosaka-Akita, H.; Fujioka, Y.; Ohba, Y.; Gorman, J.V.; Colgan, J.D.; et al. Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat. Immunol. 2012, 13, 832–842. [Google Scholar] [CrossRef]
- Sakuishi, K.; Apetoh, L.; Sullivan, J.M.; Blazar, B.R.; Kuchroo, V.K.; Anderson, A.C. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J. Exp. Med. 2010, 207, 2187–2194. [Google Scholar] [CrossRef] [PubMed]
- Baghdadi, M.; Nagao, H.; Yoshiyama, H.; Akiba, H.; Yagita, H.; Dosaka-Akita, H.; Jinushi, M. Combined blockade of TIM-3 and TIM-4 augments cancer vaccine efficacy against established melanomas. Cancer Immunol. Immunother. 2013, 62, 629–637. [Google Scholar] [CrossRef] [PubMed]
- Wenthe, J.; Naseri, S.; Hellstrom, A.C.; Moreno, R.; Ullenhag, G.; Alemany, R.; Lovgren, T.; Eriksson, E.; Loskog, A. Immune priming using DC- and T cell-targeting gene therapy sensitizes both treated and distant B16 tumors to checkpoint inhibition. Mol. Ther. Oncolytics 2022, 24, 429–442. [Google Scholar] [CrossRef]
- Luo, J.; Pang, S.; Hui, Z.; Zhao, H.; Xu, S.; Yu, W.; Yang, L.; Sun, Q.; Hao, X.; Wei, F.; et al. Blocking Tim-3 enhances the anti-tumor immunity of STING agonist ADU-S100 by unleashing CD4+ T cells through regulating type 2 conventional dendritic cells. Theranostics 2023, 13, 4836–4857. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fu, C.; Wang, J.; Ma, T.; Yin, C.; Zhou, L.; Clausen, B.E.; Mi, Q.-S.; Jiang, A. β-Catenin in Dendritic Cells Negatively Regulates CD8 T Cell Immune Responses through the Immune Checkpoint Molecule Tim-3. Vaccines 2024, 12, 460. https://doi.org/10.3390/vaccines12050460
Fu C, Wang J, Ma T, Yin C, Zhou L, Clausen BE, Mi Q-S, Jiang A. β-Catenin in Dendritic Cells Negatively Regulates CD8 T Cell Immune Responses through the Immune Checkpoint Molecule Tim-3. Vaccines. 2024; 12(5):460. https://doi.org/10.3390/vaccines12050460
Chicago/Turabian StyleFu, Chunmei, Jie Wang, Tianle Ma, Congcong Yin, Li Zhou, Björn E. Clausen, Qing-Sheng Mi, and Aimin Jiang. 2024. "β-Catenin in Dendritic Cells Negatively Regulates CD8 T Cell Immune Responses through the Immune Checkpoint Molecule Tim-3" Vaccines 12, no. 5: 460. https://doi.org/10.3390/vaccines12050460
APA StyleFu, C., Wang, J., Ma, T., Yin, C., Zhou, L., Clausen, B. E., Mi, Q.-S., & Jiang, A. (2024). β-Catenin in Dendritic Cells Negatively Regulates CD8 T Cell Immune Responses through the Immune Checkpoint Molecule Tim-3. Vaccines, 12(5), 460. https://doi.org/10.3390/vaccines12050460