
Nanocapsules Comprised of Purified Protein: Construction and Applications in Vaccine Research

 , ,
, ,  and
and
Abstract
1. Introduction
2. Materials and Methods
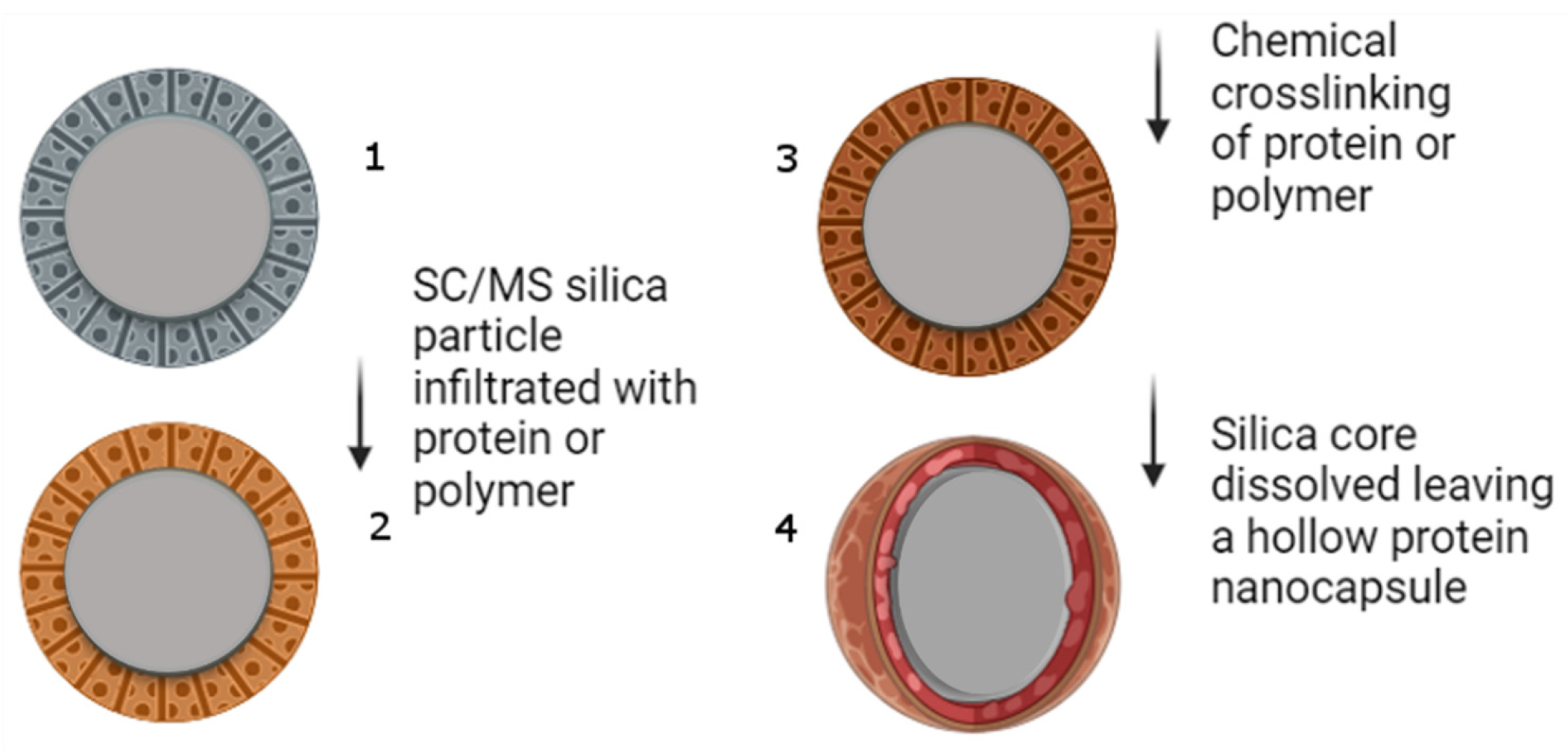
2.1. Preparation of Silica Templates
2.2. Preparation of Proteins
- H1N1 influenza haemagglutinin fragment c27: pET45b: c27. A gene segment encoding amino acids 301–404 of the Haemagglutinin gene from A/Puerto Rico/8-SV8/1934(H1N1) (Accession # AEX92912.1) was inserted in frame with the leader sequence encoded by the pET-45b vector, using standard cloning procedures. The encoded protein had the following sequence, with the vector encoded sequence underlined:MAHHHHHHVGTGSNDDDDKSPDPGAINSSLPYQNIHPVTIGECPKYVRSAKLRMVTGLRNIPSIQSRGLFGAIAGFIEGGWTGMIDGWYGYHHQNEQGSGYAADQKSTQNAINGITNKVNTVIEKMN
- H. pylori urease A subunit: pRSETa-Ure A. The construction and expression of this protein has been previously described [13].
- 3.
- The HIV gp120 protein sequence was taken from Uniprot (ID: P04578) and was reverse translated and codon optimised for mammalian expression using the IDT codon optimisation tool (Integrated DNA Technologies, Coralville, IA, USA). For protein detection and purification purposes, a 6xHis-tag was added to the N-terminus of the gp120 sequence, and the native signal peptide was added to the N-terminus of the 6xHis-tag. The gp120 protein was 1554 aa in length (inclusive of the 6xHis-tag). The 6539 bp codon optimised construct was purchased in mammalian expression vector pcDNA3.1(-)zeo (Genscript, Piscataway, NJ, USA) and delivered as lyophilised plasmid. The mammalian cell line HEK293 was used for recombinant protein expression using the Freestyle293 Expression System (ThermoFisher, Waltham, MA, USA), as recommended by the manufacturer.
2.3. Nanocapsule Synthesis and Characterisation Protocols
2.4. Vaccine Trial: H. pylori UreA Nanocapsules
2.5. Tissue Analyses and Bacterial Burden Post-Challenge
2.6. Evaluation of Dose-Dependent Cytotoxicity of gp120 Nanocapsules
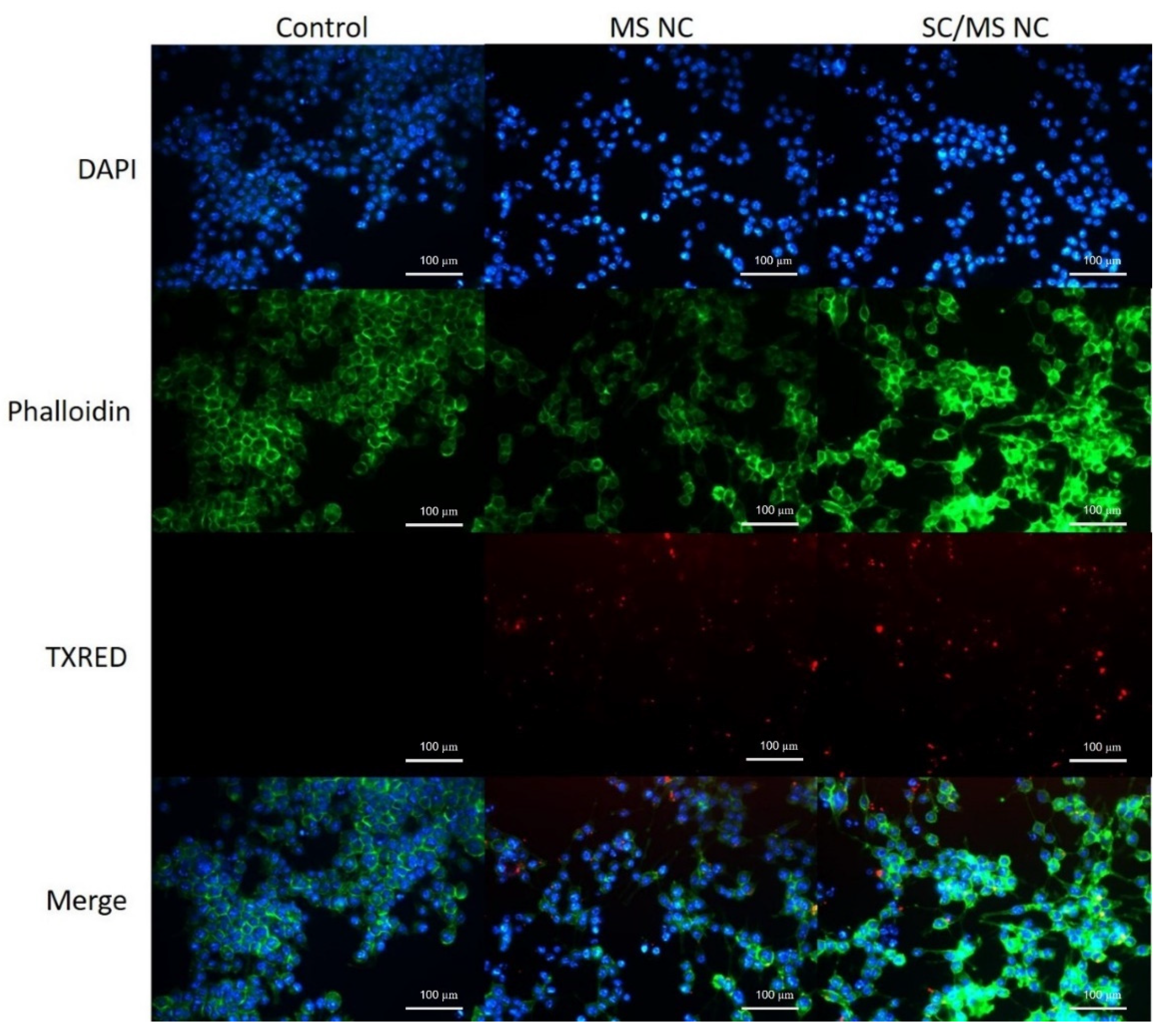
2.7. Characterisation of gp120 Nanocapsule Uptake by Fluorescence Microscopy
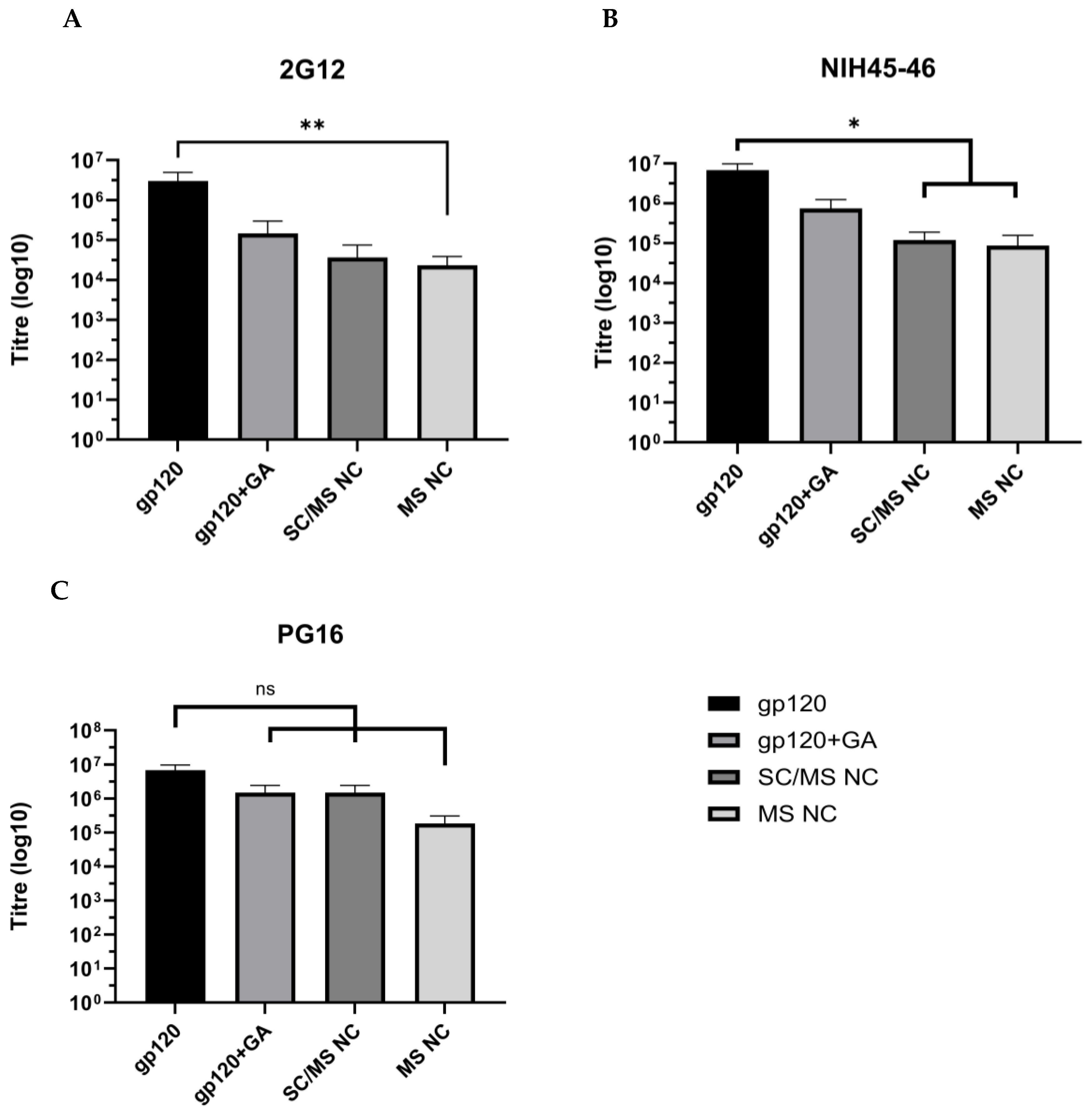
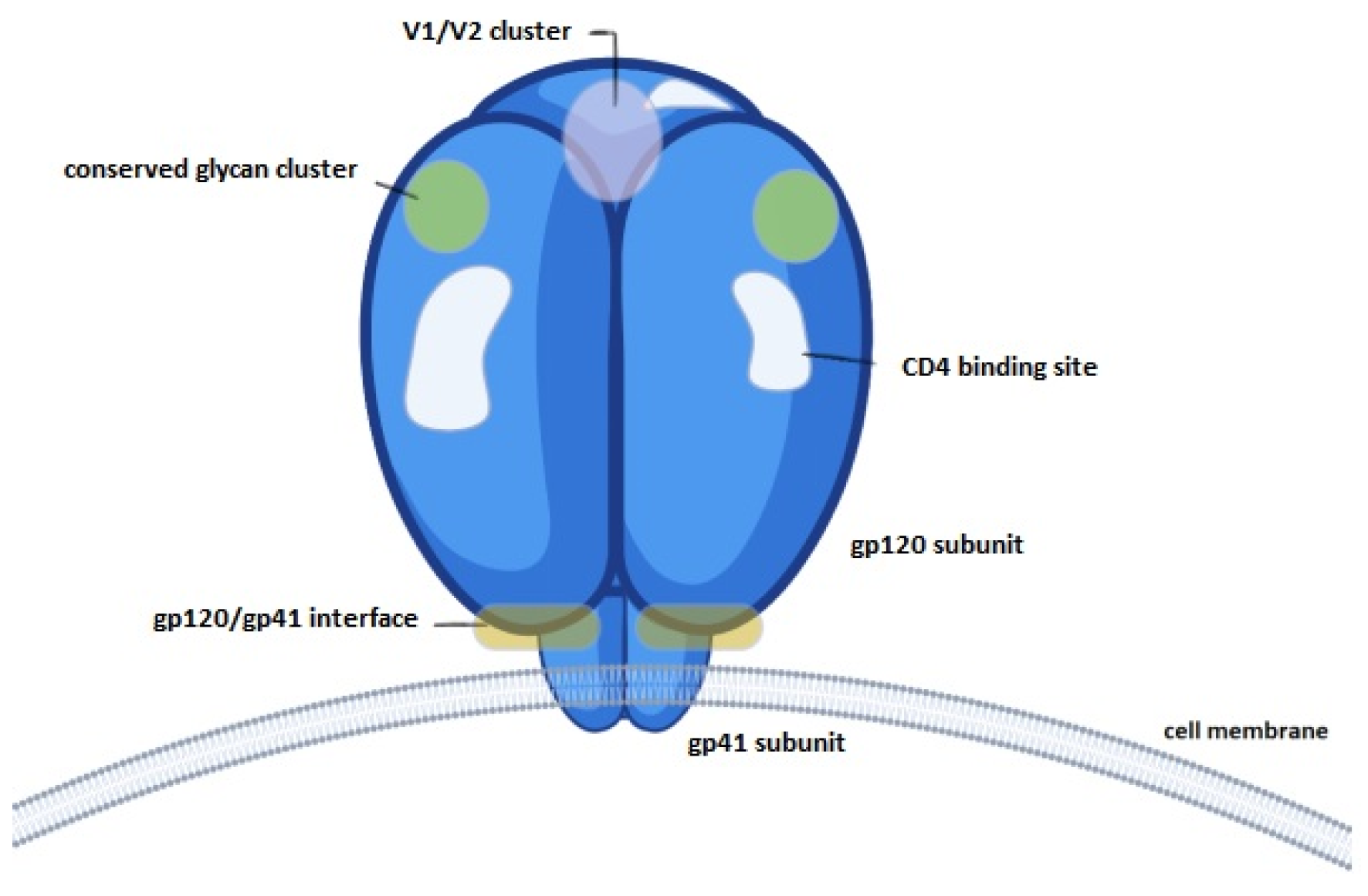
2.8. Evaluation of Epitope Availability of gp120 Nanocapsules via Indirect Enzyme-Linked Immunosorbent Assay (ELISA)
3. Results
3.1. Construction and Characterization of the Nanocapsules
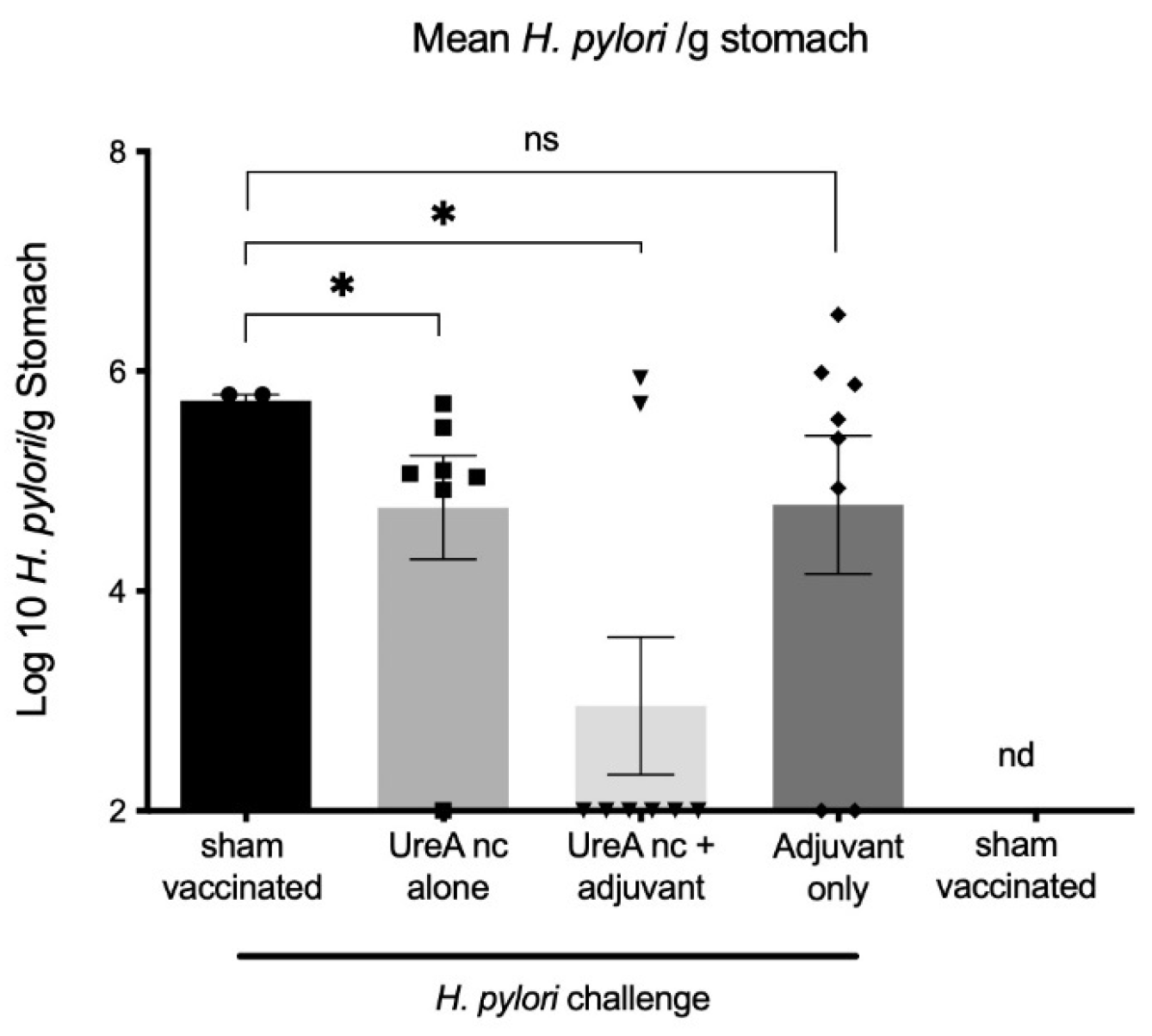
3.2. Vaccine Trial
3.2.1. H. pylori Colonisation
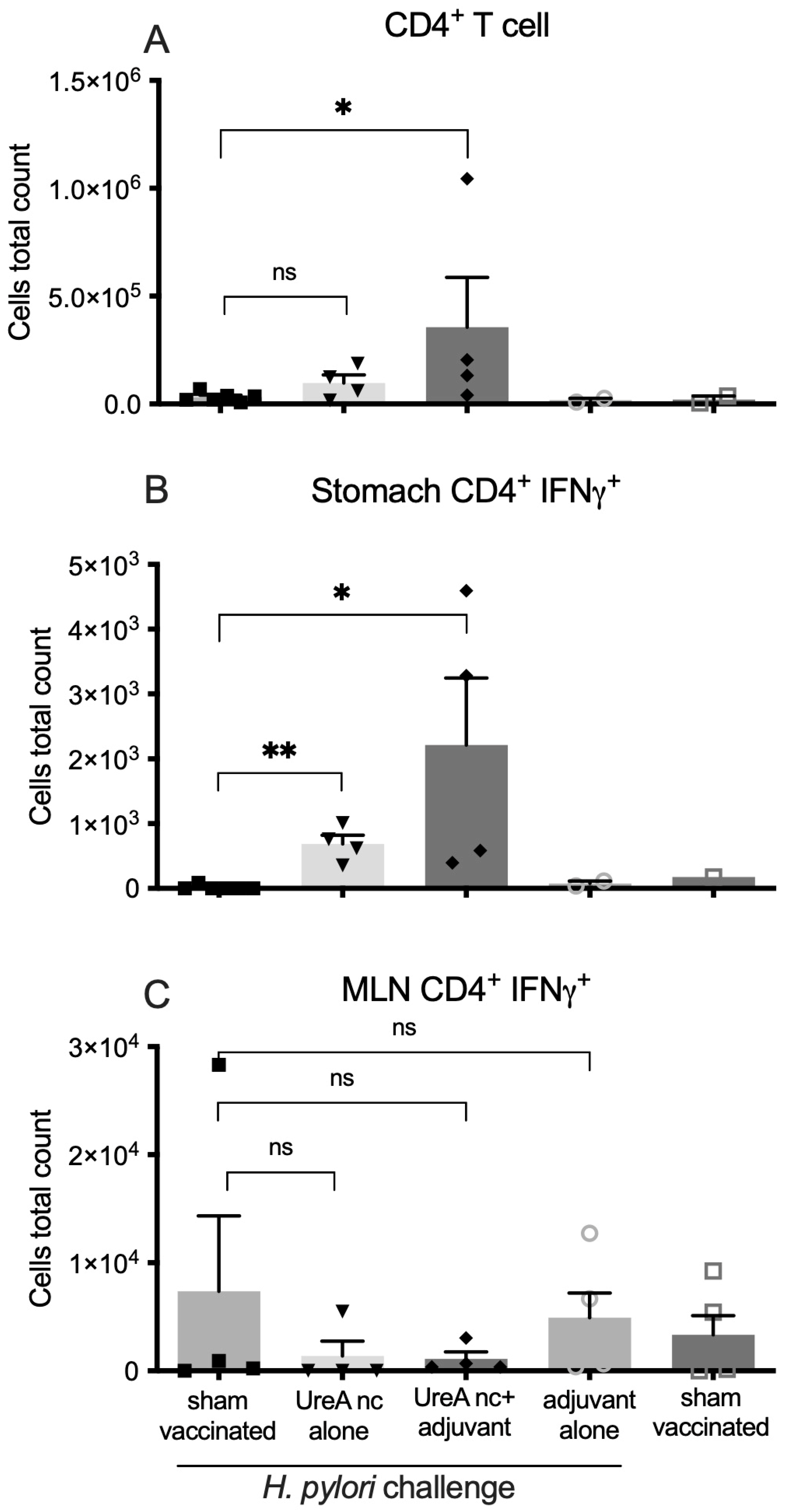
3.2.2. Characterisation of Immune Cell Populations
3.3. Evaluation of gp120 Nanocapsules
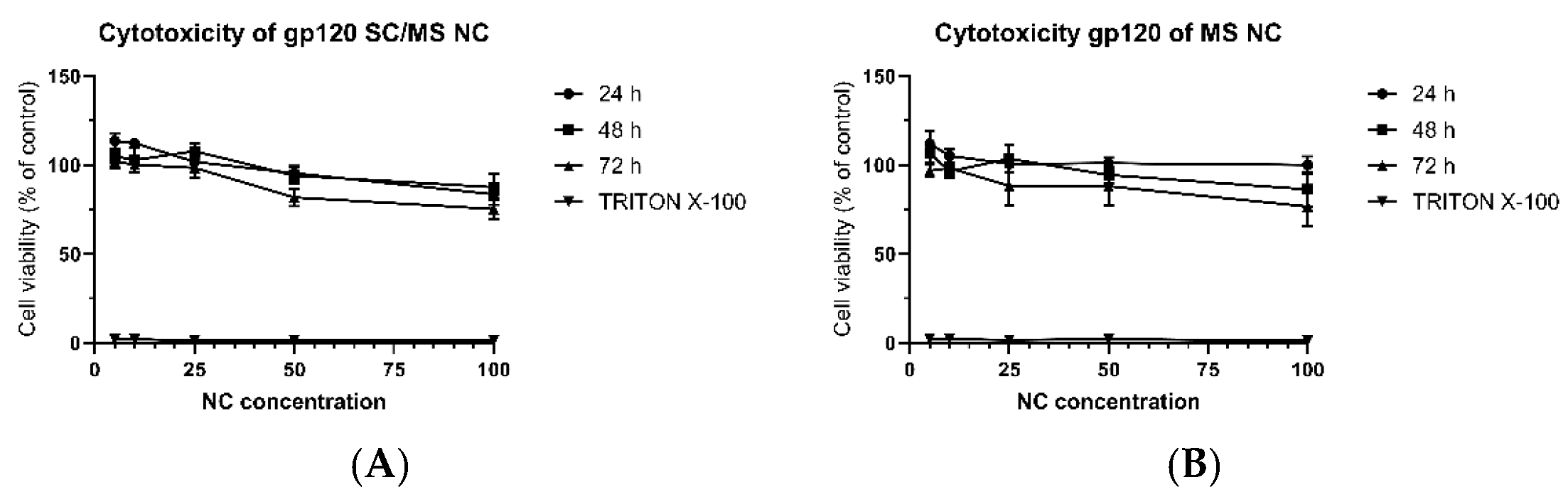
3.3.1. Dose-Dependent Cytotoxicity
3.3.2. Characterisation of gp120 Nanocapsule Uptake by Fluorescence Microscopy
3.3.3. Evaluation of Epitope Availability of gp120 Nanocapsules via Indirect ELISA
4. Discussion
5. Conclusions
- This study demonstrates the formation of nanocapsules compromised of cross-linked protein only;
- Nanocapsules can be formed from a variety of proteins of differing masses and isoelectric points;
- Different-sized nanocapsules can be created.
- UreA nanocapsules were able to induce protective responses in a mouse model of H. pylori infection;
- gp120 nanocapsules retained the availability of B-cell epitopes;
- This technique may be a universally applicable platform for the formation or protein-only nanoparticles.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, E.Y.; Sarmadi, M.; Ying, B.; Jaklenec, A.; Langer, R. Recent advances in nano- and micro-scale carrier systems for controlled delivery of vaccines. Biomaterials 2023, 303, 122345. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Luo, Y.; Lovell, J.F. Vaccine approaches for antigen capture by liposomes. Expert Rev. Vaccines 2023, 22, 1022–1040. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Li, Z.; Jaklenec, A.; Hu, Q. Vaccine delivery systems toward lymph nodes. Adv. Drug Deliv. Rev. 2021, 179, 113914. [Google Scholar] [CrossRef] [PubMed]
- Manolova, V.; Flace, A.; Bauer, M.; Schwarz, K.; Saudan, P.; Bachmann, M.F. Nanoparticles target distinct dendritic cell populations according to their size. Eur. J. Immunol. 2008, 38, 1404–1413. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, Y.; Zhou, Z. Lipid Nanoparticle-Based Delivery System—A Competing Place for mRNA Vaccines. ACS Omega 2024, 9, 6219–6234. [Google Scholar] [CrossRef] [PubMed]
- Kudsiova, L.; Lansley, A.; Scutt, G.; Allen, M.; Bowler, L.; Williams, S.; Lippett, S.; Stafford, S.; Tarzi, M.; Cross, M.; et al. Stability testing of the Pfizer-BioNTech BNT162b2 COVID-19 vaccine: A translational study in UK vaccination centres. BMJ Open Sci. 2021, 5, e100203. [Google Scholar] [CrossRef]
- Dey, A.; Chozhavel Rajanathan, T.M.; Chandra, H.; Pericherla, H.P.R.; Kumar, S.; Choonia, H.S.; Bajpai, M.; Singh, A.K.; Sinha, A.; Saini, G.; et al. Immunogenic potential of DNA vaccine candidate, ZyCoV-D against SARS-CoV-2 in animal models. Vaccine 2021, 39, 4108–4116. [Google Scholar] [CrossRef]
- Francis, J.E.; Skakic, I.; Majumdar, D.; Taki, A.C.; Shukla, R.; Walduck, A.; Smooker, P.M. Solid Lipid Nanoparticles Delivering a DNA Vaccine Encoding Helicobacter pylori Urease A Subunit: Immune Analyses before and after a Mouse Model of Infection. Int. J. Mol. Sci. 2024, 25, 1076. [Google Scholar] [CrossRef]
- Khobragade, A.; Bhate, S.; Ramaiah, V.; Deshpande, S.; Giri, K.; Phophle, H.; Supe, P.; Godara, I.; Revanna, R.; Nagarkar, R.; et al. Efficacy, safety, and immunogenicity of the DNA SARS-CoV-2 vaccine (ZyCoV-D): The interim efficacy results of a phase 3, randomised, double-blind, placebo-controlled study in India. Lancet 2022, 399, 1313–1321. [Google Scholar] [CrossRef]
- Taki, A.; Smooker, P. Small Wonders-The Use of Nanoparticles for Delivering Antigen. Vaccines 2015, 3, 638–661. [Google Scholar] [CrossRef]
- Wang, Y.; Bansal, V.; Zelikin, A.N.; Caruso, F. Templated synthesis of single-component polymer capsules and their application in drug delivery. Nano Lett. 2008, 8, 1741–1745. [Google Scholar] [CrossRef] [PubMed]
- Taki, A.C.; Francis, J.E.; Skakic, I.; Dekiwadia, C.; McLean, T.R.; Bansal, V.; Smooker, P.M. Protein-only nanocapsules induce cross-presentation in dendritic cells, demonstrating potential as an antigen delivery system. Nanomedicine 2020, 28, 102234. [Google Scholar] [CrossRef] [PubMed]
- Skakic, I.; Francis, J.E.; Dekiwadia, C.; Aibinu, I.; Huq, M.; Taki, A.C.; Walduck, A.; Smooker, P.M. An Evaluation of Urease A Subunit Nanocapsules as a Vaccine in a Mouse Model of Helicobacter pylori Infection. Vaccines 2023, 11, 1652. [Google Scholar] [CrossRef]
- Skakic, I.; Francis, J.E.; Smooker, P.M. Design and Synthesis of Protein-Based Nanocapsule Vaccines. Methods Mol. Biol. 2022, 2412, 339–354. [Google Scholar] [PubMed]
- Becher, D.; Deutscher, M.E.; Simpfendorfer, K.R.; Wijburg, O.L.; Pederson, J.S.; Lew, A.M.; Strugnell, R.A.; Walduck, A.K. Local recall responses in the stomach involving reduced regulation and expanded help mediate vaccine-induced protection against Helicobacter pylori in mice. Eur. J. Immunol. 2010, 40, 2778–2790. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.; O’Rourke, J.; De Ungria, M.C.; Robertson, B.; Daskalopoulos, G.; Dixon, M.F. A standardized mouse model of Helicobacter pylori infection: Introducing the Sydney strain. Gastroenterology 1997, 112, 1386–1397. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.P.; Pedersen, J.; Zhan, Y.; Lew, A.M.; Pearse, M.J.; Wijburg, O.L.; Strugnell, R.A. CD8+ T cells are associated with severe gastritis in Helicobacter pylori-infected mice in the absence of CD4+ T cells. Infect. Immun. 2008, 76, 1289–1297. [Google Scholar] [CrossRef]
- Tan, M.P.; Kaparakis, M.; Galic, M.; Pedersen, J.; Pearse, M.; Wijburg, O.L.C.; Janssen, P.H.; Strugnell, R.A. Chronic Helicobacter pylori infection does not significantly alter the microbiota of the murine stomach. Appl. Environ. Microbiol. 2007, 73, 1010–1013. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Walker, L.M.; Phogat, S.K.; Chan-Hui, P.Y.; Wagner, D.; Phung, P.; Goss, J.L.; Wrin, T.; Simek, M.D.; Fling, S.; Mitcham, J.L.; et al. Broad and potent neutralizing antibodies from an African donor reveal a new HIV-1 vaccine target. Science 2009, 326, 285–289. [Google Scholar] [CrossRef]
- Diskin, R.; Scheid, J.F.; Marcovecchio, P.M.; West, A.P., Jr.; Klein, F.; Gao, H.; Gnanapragasam, P.N.; Abadir, A.; Seaman, M.S.; Nussenzweig, M.C.; et al. Increasing the potency and breadth of an HIV antibody by using structure-based rational design. Science 2011, 334, 1289–1293. [Google Scholar] [CrossRef]
- Trkola, A.; Purtscher, M.; Muster, T.; Ballaun, C.; Buchacher, A.; Sullivan, N.; Srinivasan, K.; Sodroski, J.; Moore, J.P.; Katinger, H. Human monoclonal antibody 2G12 defines a distinctive neutralization epitope on the gp120 glycoprotein of human immunodeficiency virus type 1. J. Virol. 1996, 70, 1100–1108. [Google Scholar] [CrossRef] [PubMed]
- Bansal, A.; D’Souza, B.; Kapoor, D.N.; Singh, P.; Starr, G.; Muppireddy, K.K. Crafting Immunological Response Using Particulate Vaccines. Crit. Rev. Ther. Drug Carrier Syst. 2022, 39, 49–82. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Ali, N. Nanovaccine: An emerging strategy. Expert Rev. Vaccines 2021, 20, 1273–1290. [Google Scholar] [CrossRef] [PubMed]
- Goethals, E.C.; Elbaz, A.; Lopata, A.L.; Bhargava, S.K.; Bansal, V. Decoupling the effects of the size, wall thickness, and porosity of curcumin-loaded chitosan nanocapsules on their anticancer efficacy: Size is the winner. Langmuir 2013, 29, 658–666. [Google Scholar] [CrossRef] [PubMed]
- Varma, S.; Dey, S.; Palanisamy, D. Cellular Uptake Pathways of Nanoparticles: Process of Endocytosis and Factors Affecting their Fate. Curr. Pharm. Biotechnol. 2022, 23, 679–706. [Google Scholar] [CrossRef] [PubMed]
- Pappo, J.; Torrey, D.; Castriotta, L.; Savinainen, A.; Kabok, Z.; Ibraghimov, A. Helicobacter pylori infection in immunized mice lacking major histocompatibility complex class I and class II functions. Infect. Immun. 1999, 67, 337–341. [Google Scholar] [CrossRef]
- Zolla-Pazner, S. Identifying epitopes of HIV-1 that induce protective antibodies. Nat. Rev. Immunol. 2004, 4, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Sahay, B.; Nguyen, C.Q.; Yamamoto, J.K. Conserved HIV Epitopes for an Effective HIV Vaccine. J. Clin. Cell Immunol. 2017, 8, 518. [Google Scholar] [CrossRef] [PubMed]
- Scanlan, C.N.; Pantophlet, R.; Wormald, M.R.; Ollmann Saphire, E.; Stanfield, R.; Wilson, I.A.; Katinger, H.; Dwek, R.A.; Rudd, P.M.; Burton, D.R. The broadly neutralizing anti-human immunodeficiency virus type 1 antibody 2G12 recognizes a cluster of alpha1-->2 mannose residues on the outer face of gp120. J. Virol. 2002, 76, 7306–7321. [Google Scholar] [CrossRef]
- Kwong, P.D.; Wyatt, R.; Robinson, J.; Sweet, R.W.; Sodroski, J.; Hendrickson, W.A. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 1998, 393, 648–659. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Vaccine Day 0 and Boost Day 14 | Dose per Vaccination | Challenged (Day 35) |
|---|---|---|---|
| Sham-vaccinated | PBS | 50 µL PBS | Yes |
| UreA nanocapsules | Nanocapsules without adjuvant | 10 µg of nanocapsule in 50 µL PBS | Yes |
| UreA nanocapsules + adjuvant | Nanocapsules with TiterMax® Gold adjuvant | 10 µg of nanocapsule in 50 µL TiterMax/PBS | Yes |
| Adjuvant only | TiterMax® Gold adjuvant only | 50 µL TiterMax/PBS | Yes |
| Sham-vaccinated, no challenge | PBS | 50 µL PBS | No |
| Forward | 5′-CTTAACCATAGAACTGCATTTGAAACTAC-3′ |
| Reverse | 5′-GGTCGCCTTCGCAATGAGTA-3′ |
| Protein | MW (Including Tag) | pI (Including Tag) | TEM Diameter | Hydrodynamic Size | Zeta Potential |
|---|---|---|---|---|---|
| Ovalbumin | 44.5 (native) | 4.5 | 516 ± 20 nm (SC/MS) 41 ± 2.5 nm (MS) | 410 nm (SC/MS) 48 nm (MS) | Not determined |
| C27 | 13.6 kDa | 6.81 | Approx. 500 nm (SC/MS) | Not determined | Not determined |
| Urease A | 28 kDa | 7.4 | 523.3 ± 13.62 nm (SC/MS) 40 ± 9.21 nm (MS) | 543 nm (SC/MS) 51nm (MS) | −17 mV (SC/MS) −14 mV (MS) |
| gp120 | 120 kDa | 4.97 | 520.2 ± 15.32 nm (SC/MS) 56.6 ± 6.17 nm (MS) | 487 nm (SC/MS) 56 nm (MS) | −15 mV (SC/MS) −14 mV (MS) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skakic, I.; Taki, A.C.; Francis, J.E.; Dekiwadia, C.; Van, T.T.H.; Joe, C.C.D.; Phan, T.; Lovrecz, G.; Gorry, P.R.; Ramsland, P.A.; et al. Nanocapsules Comprised of Purified Protein: Construction and Applications in Vaccine Research. Vaccines 2024, 12, 410. https://doi.org/10.3390/vaccines12040410
Skakic I, Taki AC, Francis JE, Dekiwadia C, Van TTH, Joe CCD, Phan T, Lovrecz G, Gorry PR, Ramsland PA, et al. Nanocapsules Comprised of Purified Protein: Construction and Applications in Vaccine Research. Vaccines. 2024; 12(4):410. https://doi.org/10.3390/vaccines12040410
Chicago/Turabian StyleSkakic, Ivana, Aya C. Taki, Jasmine E. Francis, Chaitali Dekiwadia, Thi Thu Hao Van, Carina C. D. Joe, Tram Phan, George Lovrecz, Paul R. Gorry, Paul A. Ramsland, and et al. 2024. "Nanocapsules Comprised of Purified Protein: Construction and Applications in Vaccine Research" Vaccines 12, no. 4: 410. https://doi.org/10.3390/vaccines12040410
APA StyleSkakic, I., Taki, A. C., Francis, J. E., Dekiwadia, C., Van, T. T. H., Joe, C. C. D., Phan, T., Lovrecz, G., Gorry, P. R., Ramsland, P. A., Walduck, A. K., & Smooker, P. M. (2024). Nanocapsules Comprised of Purified Protein: Construction and Applications in Vaccine Research. Vaccines, 12(4), 410. https://doi.org/10.3390/vaccines12040410