


Epigenetic Drift Is Involved in the Efficacy of HBV Vaccination
 , ,
, , 
 ,
,  ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Participants and Phenotype
2.2. Sample Processing
2.3. Epivariant Calling and Outlier Detection
- Epivariants: These are characterized by methylation changes that significantly deviate from the population reference IQR, indicating substantial epigenetic divergence from normal levels.
- Hypo-epivariants: These show a reduction in methylation levels that fall below the lower quartile (or minimum IQR threshold), reflecting a loss of methylation compared to the baseline.
- Hyper-epivariants: These show an increase in methylation levels that fall below the upper quartile (or maximum IQR threshold), reflecting a gain of methylation compared to the baseline.
- Non-epivariants: These regions maintain methylation levels within the population reference IQR, indicating epigenetic stability without significant variation.
2.4. Analysis
3. Results
3.1. Characteristics of Cohort
3.2. Epivariant Distribution
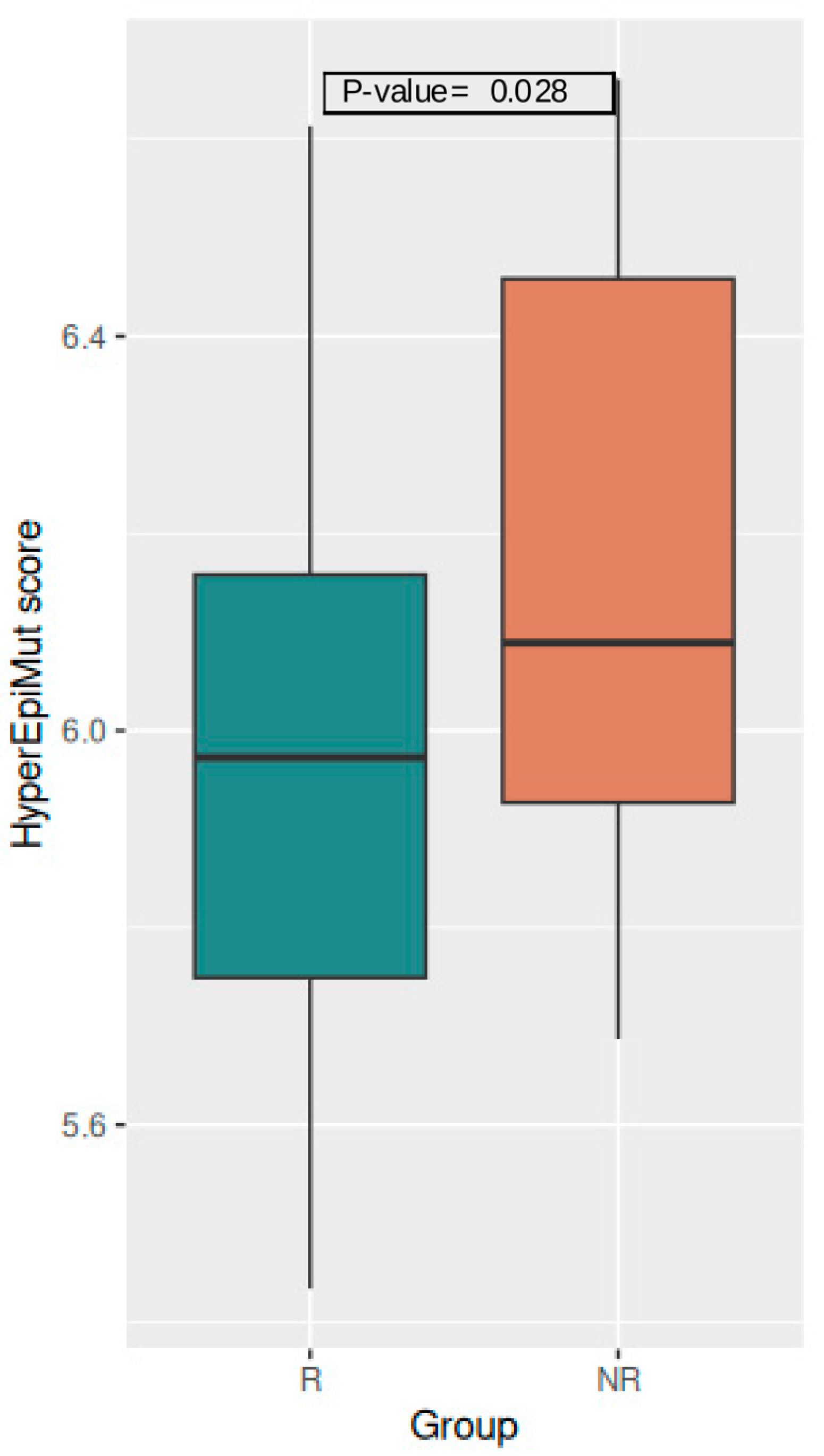
3.3. R and NR Differences in Epivariant Scores
3.3.1. Global Level
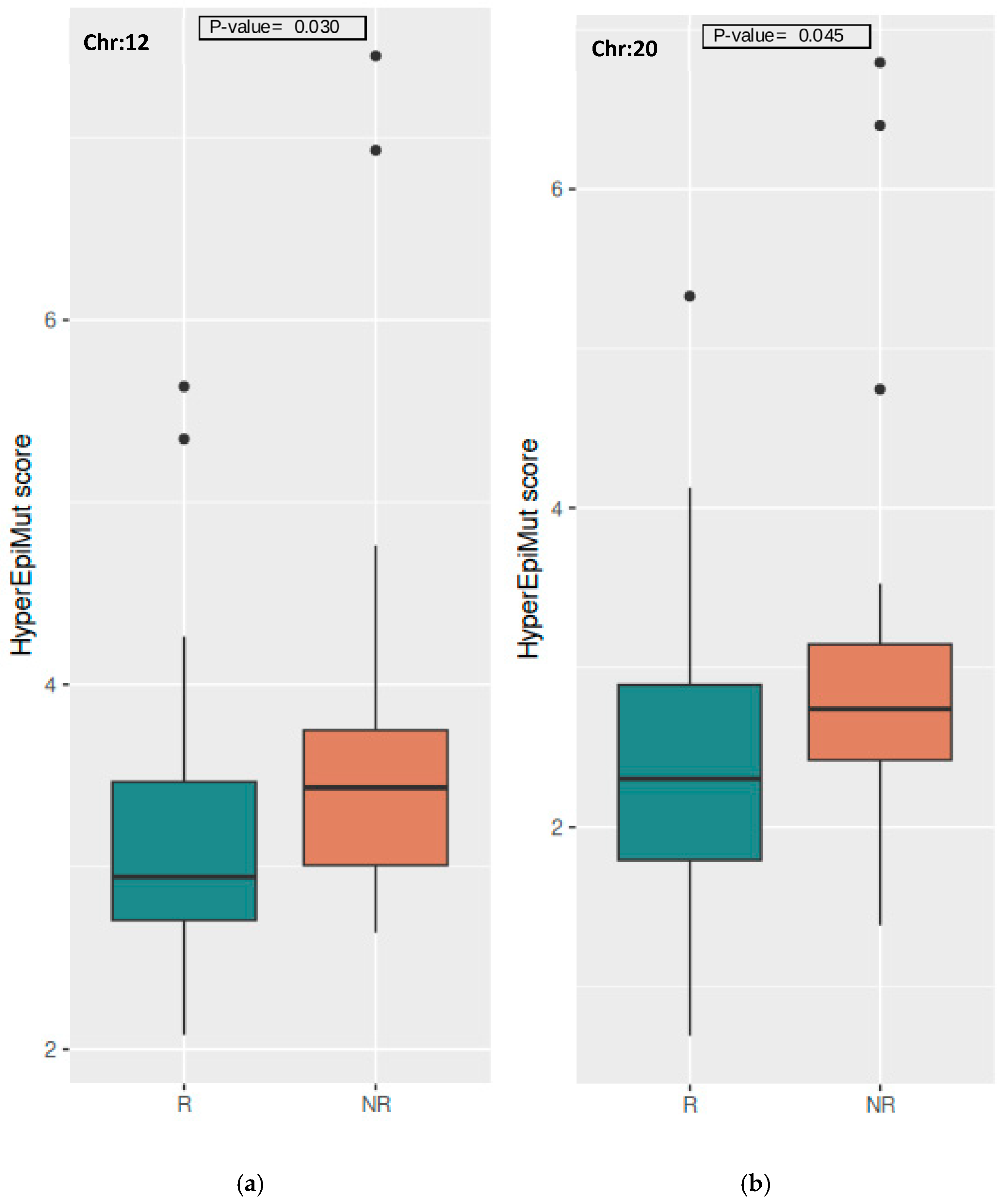
3.3.2. Chromosome Level
3.3.3. Gene Level
3.4. Pathways Associated to HBV Vaccine Response
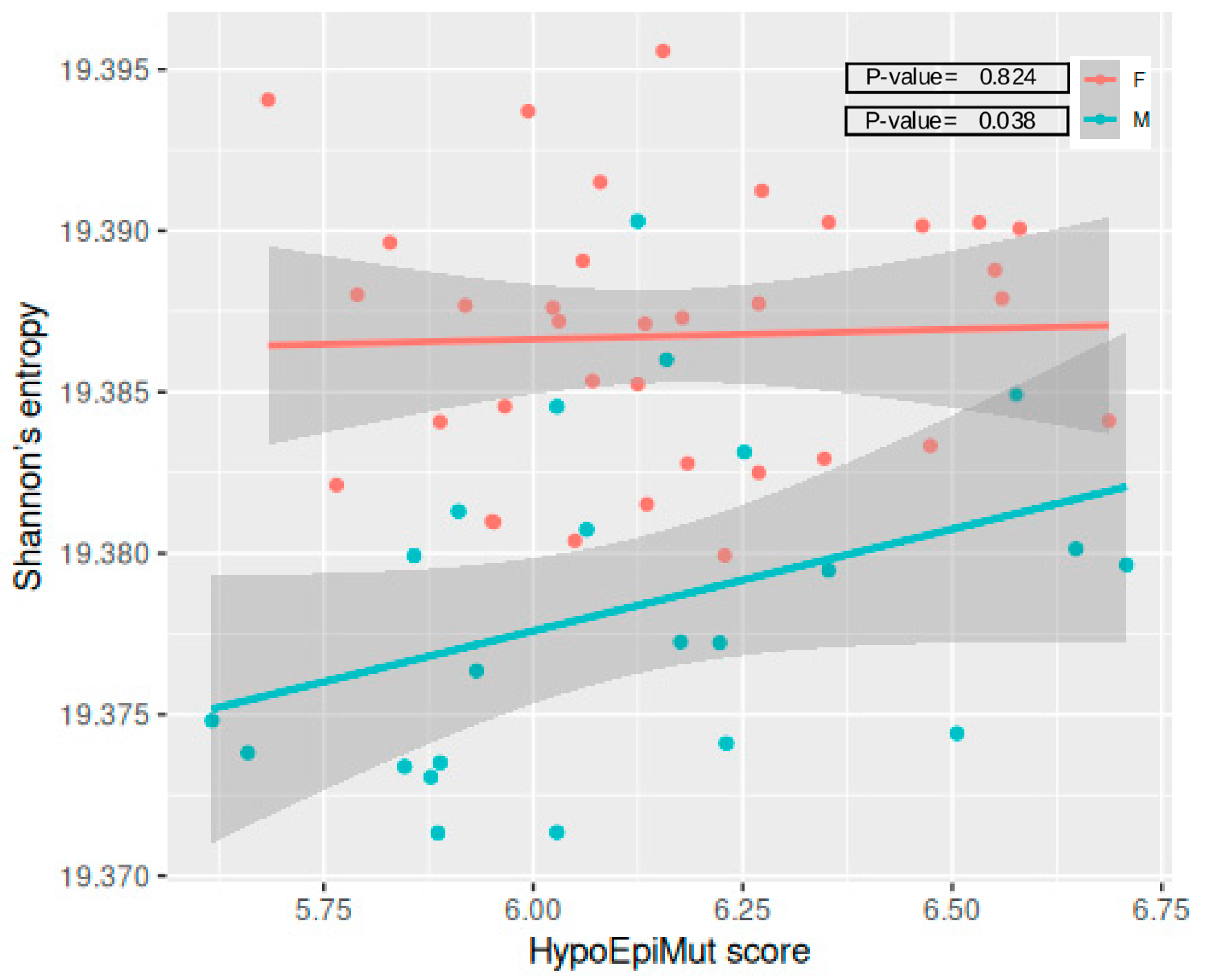
3.5. Epivariants and Epigenetic Aging
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization: WHO. World Health Organization: WHO. Hepatitis B [Internet]. Available online: https://www.who.int/news-room/fact-sheets/detail/hepatitis-b (accessed on 3 January 2023).
- Zimmermann, P.; Curtis, N. Factors That Influence the Immune Response to Vaccination. Clin. Microbiol. Rev. 2019, 32, e00084-18. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowska, K.M.; Anticoli, S.; Salvioli, S.; Calzari, L.; Gentilini, D.; Albano, C.; Di Prinzio, R.R.; Zaffina, S.; Carsetti, R.; Ruggieri, A.; et al. B Cells Isolated from Individuals Who Do Not Respond to the HBV Vaccine Are Characterized by Higher DNA Methylation-Estimated Aging Compared to Responders. Vaccines 2024, 12, 880. [Google Scholar] [CrossRef] [PubMed]
- Pang, A.P.S.; Higgins-Chen, A.T.; Comite, F.; Raica, I.; Arboleda, C.; Went, H.; Mendez, T.; Schotsaert, M.; Dwaraka, V.; Smith, R.; et al. Longitudinal Study of DNA Methylation and Epigenetic Clocks Prior to and Following Test-Confirmed COVID-19 and mRNA Vaccination. Front. Genet. 2022, 13, 819749. [Google Scholar] [CrossRef] [PubMed]
- Gentilini, D.; Garagnani, P.; Pisoni, S.; Bacalini, M.G.; Calzari, L.; Mari, D.; Vitale, G.; Franceschi, C.; Di Blasio, A.M. Stochastic epigenetic mutations (DNA methylation) increase exponentially in human aging and correlate with X chromosome inactivation skewing in females. Aging 2015, 7, 568–578. [Google Scholar] [CrossRef] [PubMed]
- Gentilini, D.; Scala, S.; Gaudenzi, G.; Garagnani, P.; Capri, M.; Cescon, M.; Grazi, G.L.; Bacalini, M.G.; Pisoni, S.; Dicitore, A.; et al. Epigenome-wide association study in hepatocellular carcinoma: Identification of stochastic epigenetic mutations through an innovative statistical approach. Oncotarget 2017, 8, 41890–41902. [Google Scholar] [CrossRef]
- Legge 27 Maggio 1991, n. Legge 27 Maggio 1991, n. 165: Obbligatorietà Della Vaccinazione Contro L’epatite Virale B (Law May 27, 1991, N. 165. Mandatory Vaccination against Viral Hepatitis B). Gazzetta Ufficiale N. 127, 1 June 1991. Available online: https://www.gazzettaufficiale.it/eli/id/1991/06/01/091G0201/sg (accessed on 30 January 2024).
- Chen, Y.-A.; Lemire, M.; Choufani, S.; Butcher, D.T.; Grafodatskaya, D.; Zanke, B.W.; Gallinger, S.; Hudson, T.J.; Weksberg, R. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics 2013, 8, 203–209. [Google Scholar] [CrossRef]
- PiPidsley, R.; Zotenko, E.; Peters, T.J.; Lawrence, M.G.; Risbridger, G.P.; Molloy, P.; Van Djik, S.; Muhlhausler, B.; Stirzaker, C.; Clark, S.J. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol. 2016, 17, 208. [Google Scholar]
- Zhou, W.; Laird, P.W.; Shen, H. Comprehensive characterization, annotation and innovative use of Infinium DNA methylation BeadChip probes. Nucleic Acids Res. 2016, 45, gkw967. [Google Scholar] [CrossRef]
- Gentilini, D.; Somigliana, E.; Pagliardini, L.; Rabellotti, E.; Garagnani, P.; Bernardinelli, L.; Papaleo, E.; Candiani, M.; Di Blasio, A.M.; Viganò, P. Multifactorial analysis of the stochastic epigenetic variability in cord blood confirmed an impact of common behavioral and environmental factors but not of in vitro conception. Clin. Epigenet. 2018, 10, 77. [Google Scholar] [CrossRef]
- Spada, E.; Calzari, L.; Corsaro, L.; Fazia, T.; Mencarelli, M.; Di Blasio, A.M.; Bernardinelli, L.; Zangheri, G.; Vignali, M.; Gentilini, D. Epigenome Wide Association and Stochastic Epigenetic Mutation Analysis on Cord Blood of Preterm Birth. IJMS 2020, 21, 5044. [Google Scholar] [CrossRef]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’Ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [PubMed]
- Klein, S.L.; Jedlicka, A.; Pekosz, A. The Xs and Y of immune responses to viral vaccines. Lancet Infect. Dis. 2010, 10, 338–349. [Google Scholar] [CrossRef] [PubMed]
- Pereira, M.; Gazzinelli, R.T. Regulation of innate immune signaling by IRAK proteins. Front. Immunol. 2023, 14, 1133354. [Google Scholar] [CrossRef] [PubMed]
- Jiang, R.; Zhang, Z.; Zhong, Z.; Zhang, C. Long-non-coding RNA RUSC1-AS1 accelerates osteosarcoma development by miR-101-3p-mediated Notch1 signalling pathway. J. Bone Oncol. 2021, 30, 100382. [Google Scholar] [CrossRef]
- Juste-Dolz, A.; Nascimento, N.M.D.; Monzó, I.; Grau-García, E.; Román-Ivorra, J.A.; Lopez-Paz, J.L.; Escorihuela, J.; Puchades, R.; Morais, S.; Gimenez-Romero, D.; et al. New structural insights into the role of TROVE2 complexes in the on-set and pathogenesis of systemic lupus erythematosus determined by a combination of QCM-D and DPI. Anal Bioanal. Chem. 2019, 411, 4709–4720. [Google Scholar] [CrossRef]
- Cao, Z.; Xia, Z.; Zhou, Y.; Yang, X.; Hao, H.; Peng, N.; Liu, S.; Zhu, Y. Methylcrotonoyl-CoA carboxylase 1 potentiates RLR-induced NF-κB signaling by targeting MAVS complex. Sci. Rep. 2016, 6, 33557. [Google Scholar] [CrossRef]
- Wang, T.; Yu, Q.; Zhang, W.; Gao, L. Comprehensive Analysis of the PROSER2-AS1-Related ceRNA Network and Immune Cell Infiltration in Papillary Thyroid Carcinoma. IJGM 2022, 15, 1647–1663. [Google Scholar] [CrossRef]
- Meijuan, C.; Fang, M.; Qian, W. Dachsous cadherin related 1 (DCHS1) is a novel biomarker for immune infiltration and epithelial-mesenchymal transition in endometrial cancer via pan-cancer analysis. J. Ovarian Res. 2024, 17, 162. [Google Scholar] [CrossRef]
- Anderluh, M.; Berti, F.; Bzducha-Wróbel, A.; Chiodo, F.; Colombo, C.; Compostella, F.; Durlik, K.; Ferhati, X.; Holmdahl, R.; Jovanovic, D.; et al. Emerging glyco-based strategies to steer immune responses. FEBS J. 2021, 288, 4746–4772. [Google Scholar] [CrossRef]
- Anderson, A.C.; Acharya, N. Steroid hormone regulation of immune responses in cancer. Immunometabolism 2022, 4, e00012. [Google Scholar] [CrossRef] [PubMed]
- Di Cara, F.; Andreoletti, P.; Trompier, D.; Vejux, A.; Bülow, M.H.; Sellin, J.; Lizard, G.; Cherkaoui-Malki, M.; Savary, S. Peroxisomes in Immune Response and Inflammation. IJMS 2019, 20, 3877. [Google Scholar] [CrossRef] [PubMed]
- Cabrera-Ortega, A.A.; Feinberg, D.; Liang, Y.; Rossa, C.; Graves, D.T. The Role of Forkhead Box 1 (FOXO1) in the Immune System: Dendritic Cells, T Cells, B Cells, and Hematopoietic Stem Cells. Crit. Rev. Immunol. 2017, 37, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Holbrook, J.; Lara-Reyna, S.; Jarosz-Griffiths, H.; McDermott, M.F. Tumour necrosis factor signalling in health and disease. F1000Research 2019, 8, 111. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | NRs (n = 30) | Rs (n = 41) | p-Value |
|---|---|---|---|
| Sex, F | 15 (50%) | 27 (66%) | 0.272 3 |
| Age 1 | 35.93 (11.70) | 33.45 (7.86) | 0.319 4 |
| B cell count 2 | 2.75 × 106 (2.12 × 106) | 2.10 × 106 (2.10 × 106) | 0.149 5 |
| Gene | Chr | Epivariants | Mean R | Mean NR | p-Value 1 | Adj. p-Value 2 |
|---|---|---|---|---|---|---|
| WDR66 | 12 | hypo + hyper | 0.200 | 0.458 | 0.022 | 0.995 |
| ING4 | 12 | hypo + hyper | 0.086 | 0.333 | 0.033 | 0.995 |
| PUS7L | 12 | hypo + hyper | 0.343 | 1.042 | 0.043 | 0.995 |
| IRAK4 | 12 | hypo + hyper | 0.343 | 1.042 | 0.043 | 0.995 |
| THTPA | 14 | hypo + hyper | 0.086 | 0.250 | 0.010 | 0.997 |
| ZFHX2 | 14 | hypo + hyper | 0.086 | 0.208 | 0.018 | 0.997 |
| SIPA1L1 | 14 | hypo + hyper | 0.086 | 0.250 | 0.035 | 0.997 |
| RUSC1-AS1 | 1 | hypo + hyper | 0.086 | 0.292 | 0.009 | 0.995 |
| UCHL5 | 1 | hypo + hyper | 0.029 | 0.208 | 0.020 | 0.995 |
| EMC1 | 1 | hypo + hyper | 0.029 | 0.167 | 0.027 | 0.995 |
| MRTO4 | 1 | hypo + hyper | 0.029 | 0.167 | 0.027 | 0.995 |
| TROVE2 | 1 | hypo + hyper | 0.029 | 0.167 | 0.030 | 0.995 |
| CYP2J2 | 1 | hypo + hyper | 0.029 | 0.417 | 0.035 | 0.995 |
| EPHB2 | 1 | hypo + hyper | 0.029 | 0.208 | 0.041 | 0.995 |
| DENND2C | 1 | hypo + hyper | 0.057 | 0.167 | 0.048 | 0.995 |
| BDNF-AS | 11 | hypo + hyper | 0.029 | 0.208 | 0.014 | 0.996 |
| LIN7C | 11 | hypo + hyper | 0.029 | 0.208 | 0.014 | 0.996 |
| TBX10 | 11 | hypo + hyper | 0.086 | 0.292 | 0.044 | 0.996 |
| CORO1B | 11 | hypo + hyper | 0.029 | 0.083 | 0.050 | 0.996 |
| SOX2-OT | 3 | hypo + hyper | 0.086 | 0.375 | 0.007 | 0.998 |
| MME | 3 | hypo + hyper | 0.029 | 0.167 | 0.018 | 0.998 |
| MCCC1 | 3 | hypo + hyper | 0.771 | 3.000 | 0.025 | 0.998 |
| AHSG | 3 | hypo + hyper | 0.029 | 0.167 | 0.038 | 0.998 |
| CEP63 | 3 | hypo + hyper | 0.029 | 0.208 | 0.038 | 0.998 |
| PLSCR1 | 3 | hypo + hyper | 0.257 | 0.667 | 0.041 | 0.998 |
| KCNAB1 | 3 | hypo + hyper | 0.029 | 0.250 | 0.050 | 0.998 |
| CKAP4 | 12 | hyper | 0.056 | 0.200 | 0.012 | 0.994 |
| WDR66 | 12 | hyper | 0.139 | 0.400 | 0.042 | 0.994 |
| B4GALT5 | 20 | hyper | 0.028 | 0.200 | 0.027 | 0.996 |
| Gene | Chr | Epivariants | Sex | Median R | Median NR | p-Value 1 | Adj. p-Value 2 |
|---|---|---|---|---|---|---|---|
| PROSER2-AS1 | 10 | hypo + hyper | F | 0.074 | 0.400 | 0.012 | 0.997 |
| DCHS1 | 11 | hypo + hyper | F | 0.037 | 0.133 | 0.020 | 0.999 |
| ZNF232 | 17 | hyper | M | 0.500 | 0.867 | 0.047 | 1.000 |
| SLC37A3 | 7 | hyper | M | 0.214 | 0.600 | 0.045 | 1.000 |
| PPP1R13B | 14 | hyper | M | 0.071 | 0.467 | 0.041 | 1.000 |
| LINC00637 | 14 | hyper | M | 0.071 | 0.467 | 0.041 | 1.000 |
| RTN1 | 14 | hyper | M | 0.071 | 0.600 | 0.045 | 1.000 |
| Pathway | Overlap 1 | p-Value 2 | Combined Score 3 |
|---|---|---|---|
| Other glycan degradation | 7/18 | 0.009 | 17.656 |
| Pentose and glucuronate interconversions | 10/34 | 0.018 | 9.916 |
| Ascorbate and aldarate metabolism | 9/30 | 0.022 | 9.767 |
| Steroid hormone biosynthesis | 14/61 | 0.049 | 5.355 |
| Pathway | Overlap 1 | p-Value 2 | Combined Score 3 |
|---|---|---|---|
| Peroxisome | 30/82 | 0.006 | 9.398 |
| Small cell lung cancer | 31/92 | 0.020 | 6.385 |
| Pancreatic cancer | 26/76 | 0.026 | 6.103 |
| TNF signaling pathway | 36/112 | 0.027 | 5.486 |
| FoxO signaling pathway | 41/131 | 0.030 | 5.132 |
| Cell cycle | 39/124 | 0.031 | 5.110 |
| Mitophagy | 23/68 | 0.039 | 5.309 |
| Colorectal cancer | 28/86 | 0.040 | 4.976 |
| Chronic myeloid leukemia | 25/76 | 0.045 | 4.875 |
| Salmonella infection | 71/249 | 0.048 | 3.896 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferraresi, F.; Anticoli, S.; Salvioli, S.; Pirazzini, C.; Calzari, L.; Gentilini, D.; Albano, C.; Di Prinzio, R.R.; Zaffina, S.; Carsetti, R.; et al. Epigenetic Drift Is Involved in the Efficacy of HBV Vaccination. Vaccines 2024, 12, 1330. https://doi.org/10.3390/vaccines12121330
Ferraresi F, Anticoli S, Salvioli S, Pirazzini C, Calzari L, Gentilini D, Albano C, Di Prinzio RR, Zaffina S, Carsetti R, et al. Epigenetic Drift Is Involved in the Efficacy of HBV Vaccination. Vaccines. 2024; 12(12):1330. https://doi.org/10.3390/vaccines12121330
Chicago/Turabian StyleFerraresi, Francesca, Simona Anticoli, Stefano Salvioli, Chiara Pirazzini, Luciano Calzari, Davide Gentilini, Christian Albano, Reparata Rosa Di Prinzio, Salvatore Zaffina, Rita Carsetti, and et al. 2024. "Epigenetic Drift Is Involved in the Efficacy of HBV Vaccination" Vaccines 12, no. 12: 1330. https://doi.org/10.3390/vaccines12121330
APA StyleFerraresi, F., Anticoli, S., Salvioli, S., Pirazzini, C., Calzari, L., Gentilini, D., Albano, C., Di Prinzio, R. R., Zaffina, S., Carsetti, R., Garagnani, P., Ruggieri, A., & Kwiatkowska, K. M. (2024). Epigenetic Drift Is Involved in the Efficacy of HBV Vaccination. Vaccines, 12(12), 1330. https://doi.org/10.3390/vaccines12121330