1. Introduction
Since COVID-19′s emergence in 2019 and its subsequent global expansion, over 760 million people have been infected, resulting in more than 6.87 million deaths. The pandemic remains a public Health Emergency of International Concern (WHO), and the long–COVID syndrome is a matter of serious concern. There are currently over 200 COVID-19 vaccine candidates in the research and development pipeline to address the worldwide disease burden [
1], and an increasing number of safe and efficacious vaccines have been approved for emergency use [
2].
An ideal SARS-CoV-2 vaccine should elicit broad, potent, and long–lasting immunity to prevent virus replication in the nasopharynx, shedding, and transmission. Various vaccine approaches have been successfully used to combat the pandemic, and new innovative strategies against SARS-CoV-2 and emerging variants are under active development [
3]. Among them, recombinant adenoviruses (AdVs) are a promising platform for SARS-CoV-2 vaccine development due to their mature manufacturing process, high gene–transfer potential, and immunogenicity [
4]. Their safety has been an asset in the development of several prophylactic vaccines and immunotherapeutics [
5]. However, the high seroprevalence of human adenovirus vectors and pre–existing immunity, in particular against adenovirus 5 in the population, are potential limitations to their clinical application [
6]. Therefore, the selection of adenoviral vectors from non–human primates, such as chimpanzees and gorillas, should circumvent interference from pre–existing anti–human adenovirus immunity.
Currently, SARS-CoV-2 vaccines are primarily administered intramuscularly, which triggers strong protective humoral and cell–mediated immune responses [
7]. However, COVID-19 is a mucosally transmitted disease. SARS-CoV-2 infects and persists in the upper respiratory tract, where the nasal epithelium contains the highest concentration of the angiotensin–converting enzyme 2 (ACE2) receptor that promotes virus replication [
8]. Conventional intramuscular immunization reduces the severity of disease and symptomatic cases, but it does not elicit secretory IgA (sIgA) responses at mucosal sites and, thus, does not provide significant protection against SARS-CoV-2 infection, shedding, and transmission [
9].
This is why current efforts focus on the development of COVID-19 vaccines that can be directly administered to the mucosa of both the upper and lower respiratory tracts to induce sIgA, long–term lung resident memory T cells (T
RM), and sterilizing immunity [
10]. Pre–clinical and clinical studies have shown that intranasal (IN) vaccination produces strong local mucosal immunity as well as systemic protection comparable to intramuscular immunization [
10]. Various IN vaccines against SARS-CoV-2 are under intensive investigation, with 12 candidates reaching clinical trials at different stages [
10]. Furthermore, individuals with high levels of wild–type spike–specific mucosal IgA were shown to be at a lesser risk of subsequent Omicron breakthrough infection [
11], and broadly neutralizing IgA antibodies elicited by mucosal tissues from Wuhan COVID-19 convalescent patients were found to potently neutralize Omicron BA.1 and BA.2 infections [
12].
Although intranasal administration of influenza vaccines has been recommended, it is worth noting that the vast majority of vaccine droplets land in the anterior nose, with only a trace of them landing in the middle turbinate region [
13]. Viral shedding and aerosols in the upper airways are the key contributors to viral spread [
14]. An intranasal vaccination strategy, however, is not optimal to prevent SARS-CoV-2′s airborne transmission. Indeed, Altimmune announced that the development of its intranasal COVID-19 vaccine (AdCOVID) was terminated due to poor results from its phase I clinical trial [
15]. Phase I trial data published by Oxford–AstraZeneca revealed that intranasal ChAdOx1 administration induced neither a consistent mucosal immune response nor a robust systemic response [
16]. Therefore, direct pulmonary immunization using aerosol–generating technologies is potentially a more effective strategy to elicit immunity in both the upper and lower respiratory tracts. Aerosolization for pulmonary immunization not only eliminates the need for needles [
17], but it is also superior to intramuscular injection [
11] and intranasal administration [
12] to protect against a variety of respiratory pathogen infections at their sites of entry [
18]. An aerosolized human adenovirus 5–based COVID-19 vaccine elicited robust systemic and mucosal immunity against SARS-CoV-2 variants in rhesus macaques [
19]. Preliminary results from recent phase I trials showed that two doses of the aerosolized adenovirus type–5 vector–based COVID-19 vaccine elicited neutralizing antibody responses similar to those obtained with one dose of intramuscular injection [
15]. Another aerosol–inhaled COVID-19 vaccine developed by McMaster University that provides robust protection against both the ancestral strain and variants of SARS-CoV-2 has also moved to clinical trials (NCT05094609) [
20].
There is compelling evidence that the protective immunity induced by two doses of current COVID-19 vaccines rapidly wanes over time, in particular in the elderly and vulnerable individuals with underlying medical conditions [
21]. This situation is compounded by the emergence of the highly transmissible SARS-CoV-2 Omicron sublineages that escape vaccine–induced neutralizing immunity [
22]. Booster vaccination with a third or fourth dose of a homologous or heterologous vaccine has been recommended to recall protective immunity in previously vaccinated individuals and broaden the spectrum of cross–neutralizing antibodies against variants of concern (VOCs) [
23]. In particular, heterologous boosting with an inhaled adenovirus–based COVID-19 vaccine of subjects previously immunized with an inactivated vaccine produced high cross–neutralizing antibody titers against Omicron subvariants [
24].
To broaden the efficacy of our SARS-CoV-2 vaccines, we engineered two chimpanzee adenovirus vectors (ChAd) expressing a stabilized SARS-CoV-2 prefusion spike glycoprotein either from the Wuhan–Hu–1 (BV-AdCoV-1) or the Omicron BA.1 (C68–COA04) strain. We first compared the humoral and mucosal responses to BV-AdCoV-1 in cynomolgus monkeys using different vaccination modalities, then in rhesus macaques using different vaccine doses. We next investigated whether homologous boosting with a monovalent BV-AdCoV-1 or heterologous boosting with the Omicron BA.1 vector (C68–COA04) could optimize cross–protective immunity against WT, Delta, and Omicron BA.1. BA.2 and BA.4/5 strains in BV-AdCoV-1–vaccinated rhesus macaques compared with a bivalent (BV-AdCoV-1/C68–COA04) booster vaccination. Our findings show that the administration of an aerosolized boosting with either BV-AdCoV-1 or C68–COA04 has the potential to optimize and broaden cross–neutralizing antibody responses against SARS-CoV-2 variants of concern, including Omicron sublineages.
2. Materials and Methods
2.1. Cells
African green monkey kidney cells (Vero) and human embryonic kidney cells (HEK293A cells) were purchased from American Type Culture Collection (ATCC, Rocville, MD, USA). Both cell lines were maintained in Dulbecco’s modified Eagle’s medium (DMEM, Gibco, Carlsbad, CA, USA) supplemented with 5% fetal bovine serum (FBS) (Gibco, Carlsbad, CA, USA) and 1% penicillin/streptomycin (Gibco, Carlsbad, CA, USA). Cells were cultured at 37 °C in a humid atmosphere incubator with 5% CO2.
2.2. Animals
Eight male cynomolgus monkeys (weighing 2.7–3.8 kg) were purchased from Guangxi XiongSen Experimental Animal Breeding Primate Development Co., Ltd., Guigang, China, and the immunogenicity study was conducted in JOINN Laboratories (Suzhou) Co., Ltd., Suzhou, China. A total of 30 rhesus macaques (females/males ratio of 1:1, weighing 3.0–6.0 kg) were purchased from Suzhou Xishan Zhongke Laboratory Animal Co., Ltd., Suzhou, China, and the immunogenicity studies were conducted in Suzhou Guochen BioTek Co., Ltd., Suzhou, China. All animals were housed individually in a 12 h light and dark cycle at 18–26 °C. Water and food were provided daily in sufficient quantities.
2.3. Ethic Statement
The vaccination protocol for the eight cynomolgus monkeys was approved by the Animal Ethics Committee (No. ACU21–1193) of JOINN Laboratories (Suzhou) Co., Ltd., Suzhou, China.
The vaccination protocol for the 30 rhesus macaques was approved by the Animal Ethics Committee (No. IACUC–21061501) of Suzhou Guochen BioTek Co., Ltd., Suzhou, China.
The vaccination protocol for 18 rhesus macaques was approved by the Animal Ethics Committee (No. IACUC–22042001) of Suzhou Guochen BioTek Co., Ltd., Suzhou, China.
The challenge protocol for 12 rhesus macaques was approved by the Institutional Animal Care and Use Committee of the Institute of Medical Biology, Chinese Academy of Medicine Sciences & Peking Union Medical College, Kunming, China (Approval Number: DWSP202107020).
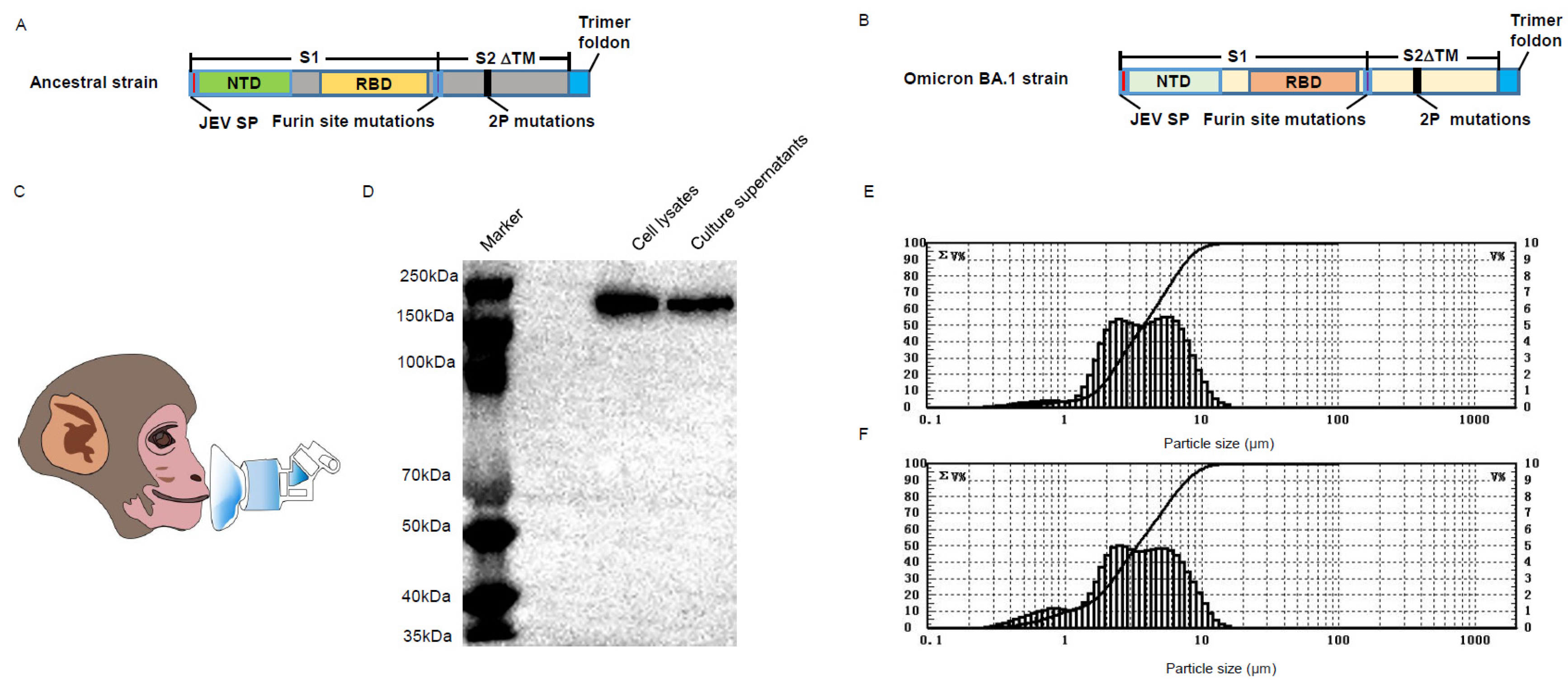
2.4. Construction of Chimpanzee Adenovirus Vectors
2.4.1. Generation of BV-AdCoV-1
The BV-AdCoV-1 vector expressing the ancestral Wuhan Hu–1 prefusion spike gene was constructed as previously described [
25] and shown to protect golden Syrian hamsters against SARS-CoV-2 infection without disease enhancement post–challenge. The particle size distribution of aerosols generated by nebulizing BV-AdCoV-1 was determined using a Winner 311XP laser light scattering particle analyzer (Jinan, China).
2.4.2. Generation of C68–COA04 (Omicron BA.1)
The COA04 gene cassette contains a 2P–based prefusion spike gene of the Omicron BA.1 variant (GenBank ID: UFO69279.1). As for the construction of BV-AdCoV-1, the spike gene was modified to (1) encode mutations of amino acids 982/983 to prolines; (2) substitute the furin cleavage site at residues 679–68 aa with GlySerAlaSer; (3) replace the original signal peptide (1–12 amino acids) with the Japanese encephalitis virus envelope signal peptide (Js SP); (4) delete the gene segments coding for the transmembrane and intracellular domains (1205–1270 aa) of the spike protein; (5) add the trimerization sequence from T4 phage fibritin to the C–terminus of the spike extracellular domain to stabilize the trimer.
The codon–optimized prefusion spike gene cassette was synthesized by Genecreate (Wuhan, China) and inserted into the chimpanzee AdC68 vector (Suzhou Xiangyi Biotechnology Co., Ltd., Suzhou, China) to yield the pAdC68–COA04 plasmid. After linearization, pAdC68–COA04 was transfected into 85% confluent HEK293A cells to rescue the recombinant adenovirus. After several amplifications of the rescued adenovirus in HEK293A cells, the virus was purified by anion–exchange and gel–filtration chromatography. The purified virus was named C68–COA04. Virus particle numbers and virus titers were determined by UV260 and ICC50 methods, respectively.
After virus propagation, the purified Omicron pre–S protein was analyzed by SDS–PAGE under reducing conditions followed by Western blotting using a monoclonal anti–Omicron virus spike antibody (Sino Biological Inc., Beijing, China, Cat 40592–MM117: 1:500). Protein bands were detected using Tanon 5200 Chemiluminescent Imaging System (Tanon, Shanghai, China).
The presence of replication–competent adenoviruses (RCAs) was assessed after three passages of the recombinant chimpanzee adenovirus vectors in A549 cells that do not express E1. The particle size distribution of aerosols generated by nebulizing C68–COA04 was determined using a Winner 311XP laser light scattering particle analyzer (Jinan, China).
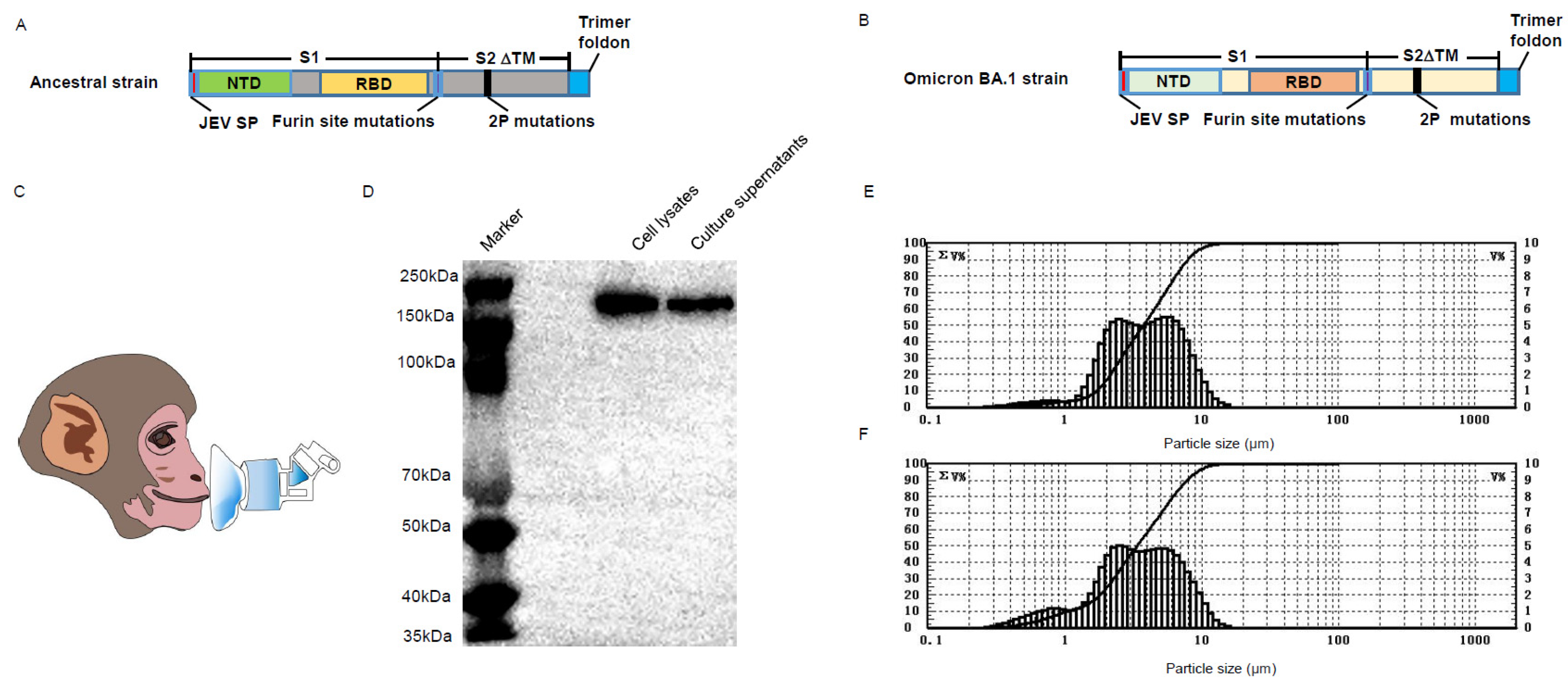
2.5. Immunogenicity Study in Cynomolgus Monkeys
To compare the immune responses induced by aerosol or intranasal vaccine administration, eight male cynomolgus monkeys were divided into 2 groups (n = 4). Animals in group 1 received an intranasal dose of BV-AdCoV-1 (5 × 1010 VP/dose), and animals in group 2 were immunized by aerosol inhalation (IH) of BV-AdCoV-1 (5 × 1010 VP/dose). All monkeys were vaccinated using a two–dose regimen on day 0 (D0) and day 28 (D28). Aerosol inhalation devices were provided by Aerogen Ltd., Galway, Ireland, and intranasal devices by Wuxi NEST Biotechnology Co., Ltd., Wuxi, China.
The sera were collected to measure serum binding and neutralizing antibody titers on days –1 (D–1), 14 (D14), 26 (D26), 42 (D42), and 56 (D56). Bronchoalveolar lavage fluids (BALFs) and nasal lavage fluids (NLFs) were also collected to determine binding anti–wild type spike IgA antibody titers. Briefly, a catheter was introduced into the trachea after the animal was sacrificed, and the catheter and trachea were then stitched together. With a syringe, sodium chloride was progressively delivered to the lungs at a volume of 10–15 mL/kg. The alveoli were lavaged 3 times for approximately 10 s. Then, BALFs were collected and stored at −80 °C for subsequent assays.
To collect NLFs, 1 mL of 0.9% sterile sodium chloride was injected with a syringe into one nostril while pinching the other one. The fluid was aspirated back into the syringe. The procedure was repeated 3–4 times. The collected fluid was then used to lavage the other nostril. Finally, the NLFs were collected into a sterile container and stored at −80 °C for subsequent assays.
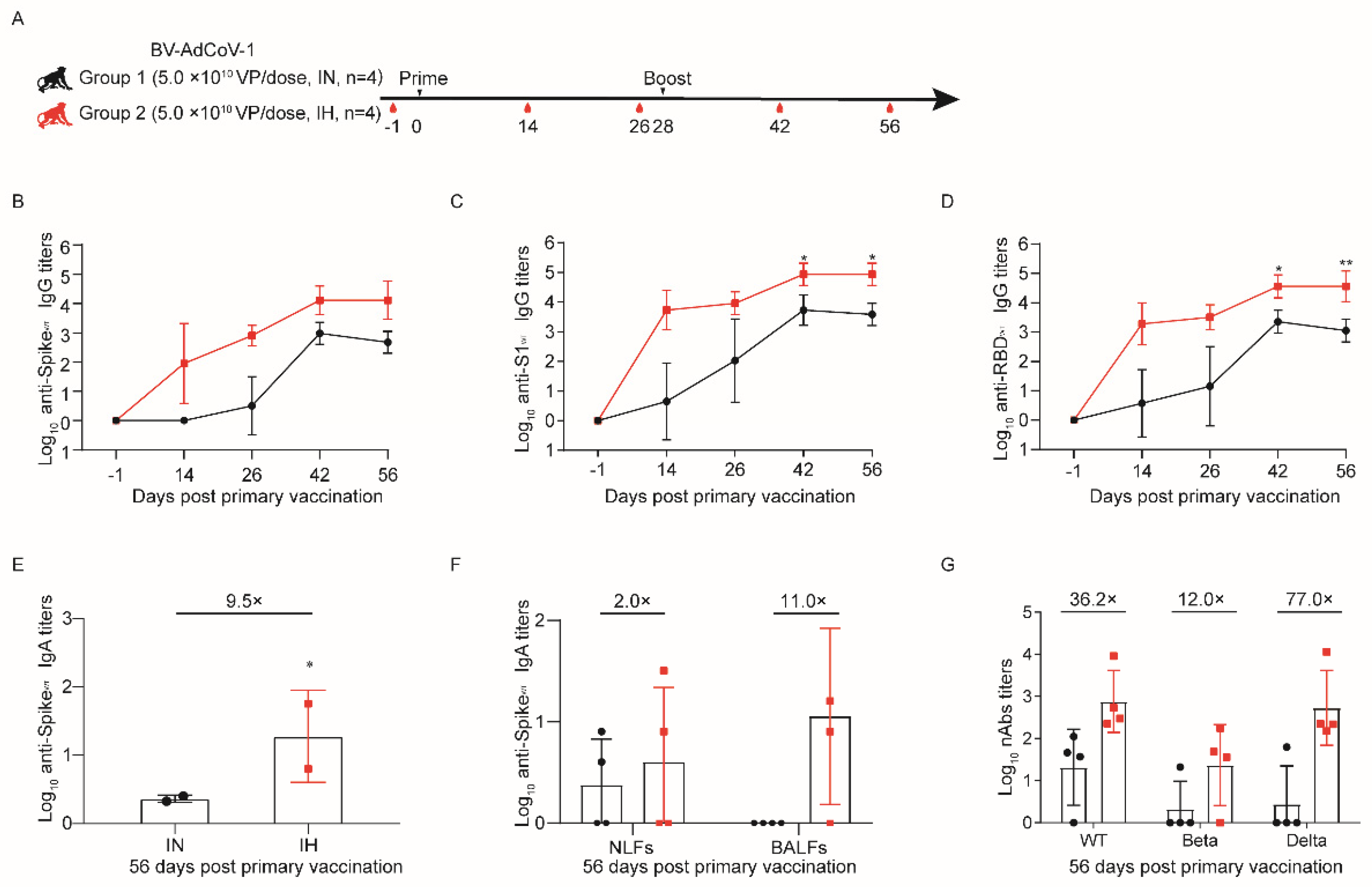
2.6. Immunogenicity Study Design in Rhesus Macaques
2.6.1. Vaccination and Challenge
Thirty rhesus macaques were randomly assigned to six different vaccination regimens: a negative control group (NC) (0.9% sterile sodium chloride (Saline), aerosol inhalation,
n = 6), group 1 (0.6 × 10
10 VP/dose, aerosol inhalation,
n = 4), group 2 (2 × 10
10 VP/dose, aerosol inhalation,
n = 4), group 3 (5 × 10
10 VP/dose, aerosol inhalation,
n = 6), group 4 (10 × 10
10 VP/dose, aerosol inhalation,
n = 6), and group 5 (5 × 10
10 VP/dose, intramuscular administration,
n = 4). Animals in each group were half males and half females. The details about the grouping and vaccination schedule are presented in
Supplementary Table S1. Animals were all vaccinated with BV-AdCoV-1 using a two–dose regimen on day 0 (D0) and day 28 (D28). Blood samples were collected on D–1, D14, and D26 (two days before boost 1) and D42 (14 days after boost 1). Serum binding and neutralizing antibody titers were measured at each time point. Aerosol inhalation devices were provided by Aerogen Ltd. Galway, Ireland.
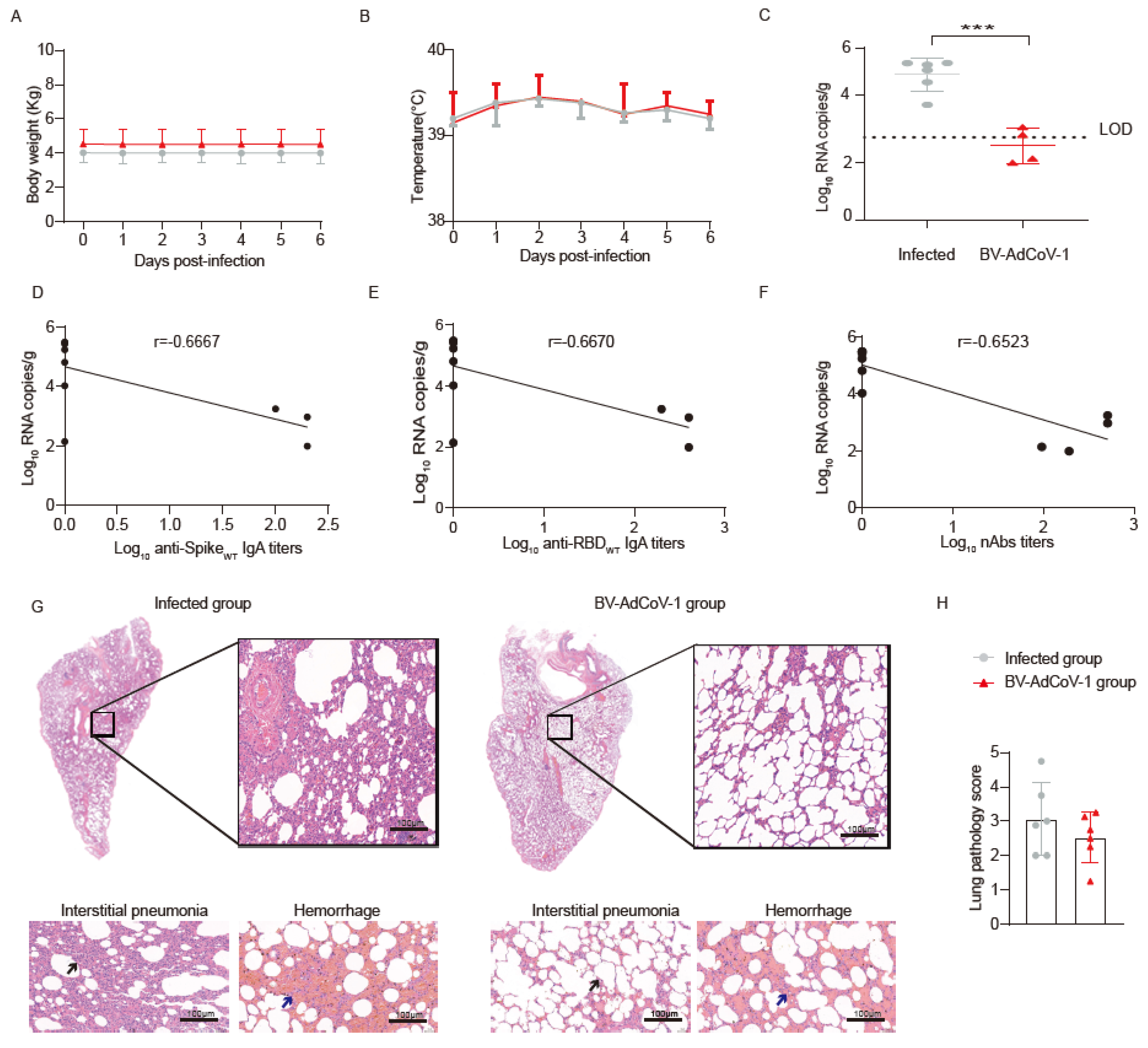
The animals in the NC group and group 3 (twelve in total) were transferred to the Institute of Medical Biology, Chinese Academy of Medicine Sciences & Peking Union Medical College. All experiments with live SARS-CoV-2 in NHPs were performed in an ABSL–3 facility. On D53, the 12 animals in the NC group and group 3 were challenged and referred to as the infected and vaccinated groups, respectively. Briefly, the animals were administered 1 × 106 PFUs in 1 mL (500 μL intranasally and 500 μL intratracheally) of the SARS-CoV-2 GD108# strain, which is similar to the Wuhan Hu–1 strain. Weight and body temperature were recorded daily. The nasal, throat, and anal swabs were collected every two days for RT–qPCR analysis to determine the viral load. All animals were sacrificed 7 days post–challenge. The lungs were collected for Hematoxylin–Eosin (HE) staining and histopathology analysis. Additionally, selected tissues were analyzed for viral loads, and levels of cytokines were measured in sera and lung homogenates.
2.6.2. Durability of Antibody Response and Sequential Immunization
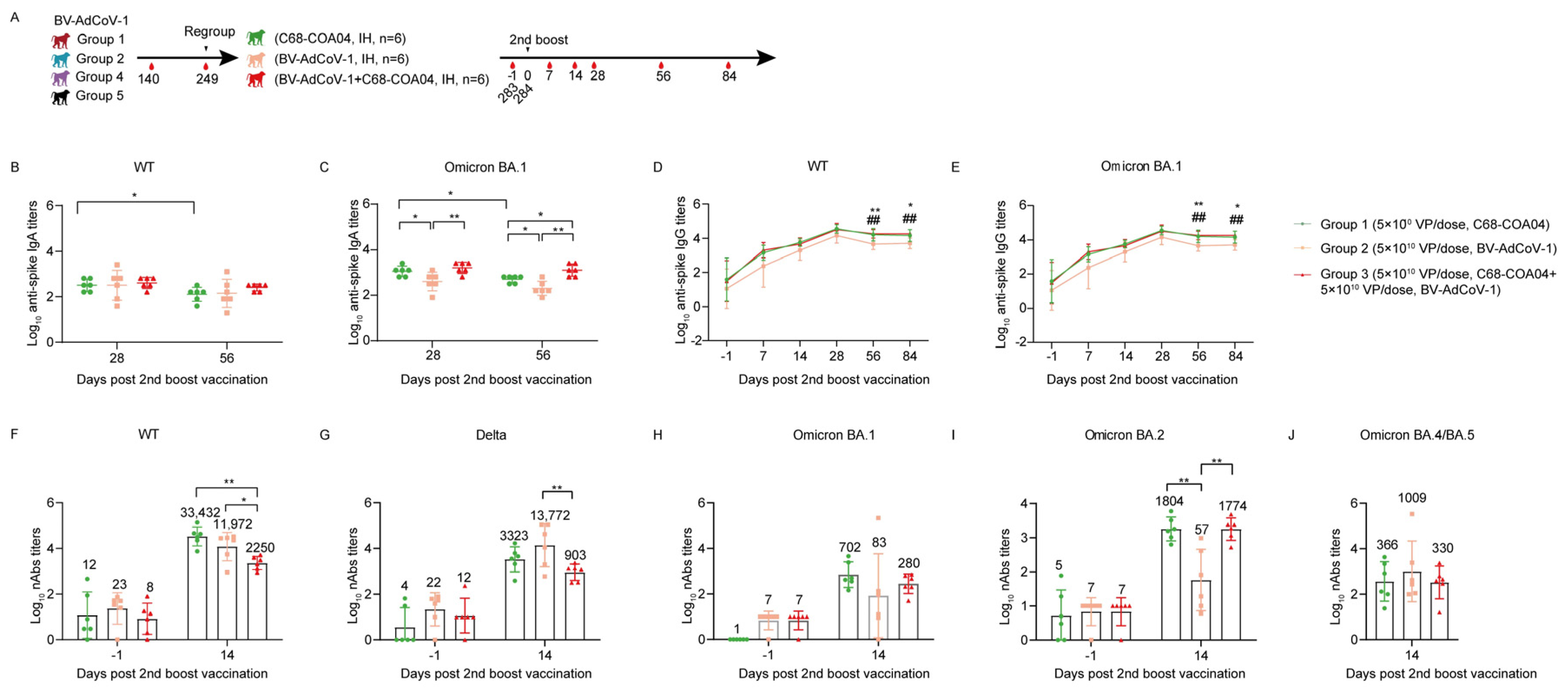
The remaining 18 animals were periodically monitored to explore the durability of the antibody response until D249. After 8 months, the 18 rhesus macaques were regrouped into three groups, among which binding antibody titers did not differ significantly: Group 1: six animals received a second booster of C68–COA04 vaccine (5 × 1010 VPs) on D284; Group 2: six animals received BV-AdCoV-1 (5 × 1010 VPs); and Group 3: six animals received a combination of C68–COA04 (5 × 1010 VPs)/BV-AdCoV-1 (5 × 1010 VPs) vaccine. Inhalation devices were provided by Dongguan Aidisy Machinery and Electronic Equipment Co., Ltd., Dongguan, China. Sera were collected on D–1, D7, D14, D28, D56, and D84 to measure binding and neutralizing antibody titers against SARS-CoV-2 variants of concern (VOCs).
2.7. Enzyme–Linked Immunosorbent Assay (ELISA)
Immune sera, BALFs, or NLFs were analyzed by indirect ELISA for binding IgG and IgA titers as previously described [
26]. Briefly, 96–well plates (Cat#3590; Corning, New York, NY, USA) were coated over–night at 4 °C with 2 µg/mL of either the SARS-CoV-2 Spike, S1, or RBD protein (Genscript Biotech Corporation, Nanjing, China) from the ancestral virus or the Omicron BA.1 strain (Sino Biological Inc., Beijing, China) in 0.05 M carbonate–bicarbonate buffer (pH 9.6), before blockade with 200 µL of blocking buffer (PBST containing 0.05% Tween–20 and 10% skimmed milk) at 37 °C for 2 h. The sera at a starting dilution fold of 1:100 and BALFs and NLFs at a starting dilution fold of 1:4 were 2–fold serially diluted with blocking buffer. Then, 100 μL of serially diluted samples were added to the wells, and plates were further incubated for an hour at 37 °C. After washing the plates with PBST three times, horseradish peroxidase (HRP)–conjugated goat anti–human IgG antibody (Invitrogen, cat# A18805; 1:10000) or (HRP)–conjugated goat anti–human IgA antibody (Invitrogen, cat# A18781, 1:1000) was added to the wells, and the plates were further incubated for an additional 1 h at 37 °C. After three washes with PBST, 100 μL/well of a 3, 3′, 5, 5′–Tetramethylbenzidine (TMB) solution was added. The plates were incubated in the dark for 15 min at room temperature, and reactions were terminated by adding 100 μL/well of 1 M H
2SO
4 before the absorbance values were read at 450 nm. Pre–immune serum (100–fold diluted) was used as the negative control, and an anti–SARS-CoV-2 serum prepared by BravoVax Biotech Co. Ltd., Wuhan (China) was used as a positive control. The cutoff value was calculated as being 2.1 times the mean OD
450 values obtained for samples from the negative control. The endpoint titer was calculated as the highest sample dilution at which the OD
450 value was equal to or greater than the cutoff value.
2.8. Enzyme–Linked Immunospot (ELISpot) Assay
To evaluate the T–cell responses after the second boost with either C68–COA04, BV-AdCoV-1, or the C68–COA04/BV-AdCoV-1 combination vaccine, blood samples from the 18 rhesus macaques were collected on D283. Peripheral blood mononuclear cells (PBMCs) were separated by Ficoll–Hypaque density gradient centrifugation. The numbers of PBMCs secreting IFN–γ and IL–4 were determined using IFN–γ and IL–4 ELISpot kits (3421M–4AST–2, 3410–2APW–2, Mabtech, Sweden) according to the manufacturer’s instructions. Briefly, 50 μL of PBMCs (5 × 106 cells/mL) were added in duplicate to ELISpot plates pre–coated with anti–IFN–γ or anti–IL–4 antibodies. A Wuhan–Hu–1 or Omicron spike peptide pool (15–mers overlapping by 11–amino acid residues at a final concentration of 2 μg/mL per peptide) (GenScript Inc., Nanjing, China) was used to re–stimulate immune cells. An anti–CD3 monoclonal antibody (1:1000, Mabtech, Nacka Strand, Sweden) served as a positive control, and an FBS–free medium as a negative control. Cells were incubated at 37 °C in 5% CO2 for 48 h, then the cell suspension was discarded, and 100 μL of either anti–IFN–γ antibody conjugated to Alkaline phosphatase (ALP) (7–B6–1–ALP, 1:1000, Mabtech, Nacka Strand, Sweden) or anti–IL4 antibody conjugated to ALP (IL4–Ⅱ–ALP, 1:300, Mabtech, Sweden) was added to the plates. The plates were further incubated at room temperature for 2 h. The spots were visualized by adding BCIP/NBT–plus substrate (Cat#3650–10, Mabtech, Nacka Strand, Sweden) and counted using an automated ELISpot reader (Mabtech, Nacka Strand, Sweden).
2.9. Luciferase–Based Pseudovirus Neutralization Assay
Sera were inactivated for 30 min at 56 °C, then 4–fold serially diluted from a starting dilution fold of 1:20 with cell culture medium. 100 μL of diluted sera were incubated with 50 μL of luciferase–expressing SARS-CoV-2 pseudoviruses (1.3 × 104 CCID50/mL) for one hour at 37 °C, and then the mixtures were distributed to the plates (150 μL/well). Vero cells (2 × 104 in 100 μL) were then added and incubated at 37 °C for another 20–28 h. The cells were lysed, and the luciferase activity was measured by the Britelite Plus Reporter Gene Assay System (PerkinElmer, Waltham, MA, USA). The relative light unit (RLU) values were read using a chemiluminescence detector (EnSight, PerkinElmer, Waltham, MA, USA). The IC50 neutralizing antibody titer was calculated by the Reed–Muench method. Each serum sample was assayed at six dilutions in duplicates. Ancestral SARS-CoV-2 (WT), Delta, and Beta pseudoviruses were purchased from Beijing YunLing Biotechnology Co., Ltd., Beijing, China. Ancestral SARS-CoV-2 (WT), Delta, and Omicron subvariants (BA.1, BA.2, and BA.4/BA.5) pseudoviruses used in the Omicron vector immunogenicity study were donated by Wuhan University, Wuhan, China. The first WHO International Standard for anti–SARS-CoV-2 immunoglobulin (NIBSC, 20/268, high 20/150) was used to determine neutralizing antibody titers.
2.10. Cytopathic Effect (CPE)–Based Micro–Neutralization Assay
Sera were inactivated for 30 min at 56 °C. Based on virus–induced cytopathic effects, a microneutralization (MN) assay was used to assess the ability of macaques’ immune sera to neutralize authentic SARS-CoV-2. The inactivated sera at a starting dilution of 1:4 were 3–fold serially diluted and added to the 96–well plate pre–filled with 100 CCID50 in 50 μL per well of SARS-CoV-2 GD108# strain. After incubating the plates at 37 °C for 2 h, 100 μL of 1 × 105 /mL Vero cell suspension (1 × 104 in 100 μL) was added to the wells. The plates were then incubated for 5 days at 37 °C in 5% CO2. The cytopathic effects were then observed and recorded. The assays were performed in duplicates. The neutralizing antibody titers were defined as the reciprocal of the highest serum dilution at which a 50% reduction in CPE was observed.
2.11. Tissue Homogenization
Monkey tissues from the lungs were collected and cut into small pieces (~1–2 g). After being washed twice with cold PBS, the tissues were homogenized in 20 volumes of cold PBS using a glass homogenizer (Bead Ruptor 24 Elite, OMNI, Kennesaw, GA, USA). The homogenates were centrifuged at 7000× g for 10 min, and the supernatants were collected and stored at −80 °C for cytokine assays.
2.12. RT–qPCR
The viral load was measured by real–time quantitative PCR (RT–qPCR) after reverse transcription by targeting a region on the E gene of SARS-CoV-2 (E–Sarbeco, Sangon Biotech, Shanghai, China). Primers and probe sequences are listed in
Supplementary Table S2. Briefly, total RNA was extracted from 0.1 g of the homogenized lung, nasal mucosa, trachea, bronchus, lung lymph nodes, neck lymph nodes, spleen, brain, heart, liver, kidney, intestine lymph nodes, and ovaries/testes tissues, as well as from nasal, throat and anal swabs of rhesus monkeys using the TaKaRa MiniBEST Viral RNA/DNA Extraction Kit Ver.5.0 (TaKaRa, Shiga, Japan). Gene expression was quantitatively assessed using the CFX96 Touch Real–Time P PCR Detection System (Bio–Rad, Hercules, CA, USA). The virus abundance was determined using the 2
−ΔΔCt method [
27]. The data were normalized against the γ–actin house–keeping gene.
2.13. Cytokine Analyses
The cytokine levels in immune sera and lung homogenates were determined using the Non–Human Primate Cytokine and Chemokine–30 Plex Procartaplex Panel (Cat# EPX010–40420–901, Thermo Fisher Scientific, Carlsbad, CA, USA) according to the manufacturer’s protocol. In short, lung tissues were mechanically homogenized and centrifuged at 16,000× g for 10 min at 4 °C. The supernatants of the tissue homogenates were collected and diluted to 10 mg protein/mL with PBS. Then, 25 µL of diluted lung homogenates or sera and 25 µL of Universal Assay buffer were added to wells of a 96–well plate containing magnetic beads coated with different anti–cytokine capture antibodies. The plates were sealed after incubation on a shaker at 500 rpm for 60 min at room temperature in the dark. The beads were washed by putting the 96–well plate on a flat magnet for 30 s, after which the fluid was discarded by using an automated plate washer. The magnet was removed, and the beads were resuspended in 25 µL of a mixture of biotinylated detection antibodies. After washing the plates twice, 50 μL of streptavidin–R–phycoerythrin solution was added to each well for another 30 min incubation at room temperature. Beads were washed twice, and 20 μL of Reading Buffer was added to each well. After a 5 min incubation, the plates were read using the Luminex 100/200 Magpix system (Luminex Corporation, Austin, TX, USA).
2.14. Hematoxylin–Eosin (HE) Staining and Histopathology Scoring
Lung tissues from the left upper lobe, left lower lobe, right upper lobe, and right lower lobe were collected for HE histopathology analysis. Sections of 5 μm were cut from paraffin–embedded tissues, deparaffinized in xylene, passed through 100% ethanol, and rehydrated in tap water. Samples were stained with hematoxylin, decolorized with 0.125% HCl/70% ethanol, blued in blue returning liquid (Servicebio Technology Co., Ltd., Wuhan, China), counterstained with eosin, dehydrated, and mounted in Micromount (Servicebio Technology Co., Ltd., Wuhan, China) cover slipping medium at room temperature.
The lung lobes were used for pathological analyses and scored semi–quantitatively as Grade 0 (Within normal limits); Grade 1 (Minimal); Grade 2 (Slight), Grade 3 (Moderate), Grade 4 (Significant), and Grade 5 (Severe) [
28]. Scoring was performed based on the number of lung lobes affected and the degrees of inflammatory and hemorrhagic lesions. An average lung lobe score was calculated by combining scores from each criterion. The digital images of HE–stained slides were examined under a microscope (Nikon Eclipse E100, Tokyo, Japan).
2.15. Chimpanzee Adenovirus Vector Neutralization Assay
Serum samples were serially diluted and incubated with an AdC68 vector expressing the green fluorescence protein (rAdC68–GFP, 1.2 × 10
5 TCID
50/well) for 1 h at 37 °C. The mixtures were inoculated to HEK293A cells (2 × 10
4 cells/well). After incubation at 37 °C in 5% CO
2 for 48 h, the fluorescent spots were counted under a fluorescence microscope (Olympus BX–60) with a 450 nm excitation. The half–maximal inhibitory concentration (IC
50) values were calculated according to the Reed and Muench method [
29].
2.16. Statistical Analysis
The data expressed as means ± standard deviation (SD) were processed using GraphPad software 9.0 (GraphPad Software, San Diego, CA, USA). Differences were identified using two or three–way ANOVA and considered significant when p < 0.05. Correlations were assessed by a non–parametric Spearman correlation test.
4. Discussion
The objective of the study was to evaluate the efficacy of mucosal administration of optimized chimpanzee adenovirus vectors expressing prefusion–stabilized spike proteins at inducing immunoprotection against SARS-CoV-2 and broad cross–neutralization of variants of concern.
Simian adenovirus vectors are a promising vaccine platform [
32] to induce sterilizing immunity against SARS-CoV-2, not only because they have a good Th1 immunogenicity profile and are easy to administer mucosally but also because they have a low seroprevalence in humans [
33] and thus circumvent interference by pre–existing anti–human Ad5 immunity [
34]. In addition, chimpanzee ChAd68–based vectors induce sustained immunity [
35]. We selected the replication–deficient AdCh68 adenovirus vector, which is serologically and phylogenetically distinct from ChAd–Y25 used to engineer ChAdOx1 to express SARS-CoV-2 pre–fusion spike proteins that are more immunogenic than their native counterparts [
36]. The two purified AdCh68–based vectors expressing either the Wuhan–Hu–1 or the Omicron BA.1 prefusion spikes used in this study were found to be free of RCA after three passages in A549 cells and thus are safe for clinical use. Both pre–S proteins secreted in the supernatants of vector–infected HEK293 cells formed non–covalent stabilized trimers as judged by Western blotting, size exclusion chromatography, and transmission electron microscopy.
Four adenovirus–based COVID-19 vaccines, Ad26.CoV2.S (Johnson & Johnson, New Brunswick, NJ, USA), Vaxzevria (ChAdOx1 nCoV–19, Oxford/AstraZeneca, Oxford, UK), Convidecia (Ad5–nCoV, CanSinoBIO, Tianjin, China), Sputnik V (Ad5/Ad26, Gamaleya Institute, Moscow, Russia) approved for human use are administered intramuscularly. Intranasal SARS-CoV-2 vaccines have been shown to reduce viral load and subsequent viral transmission and are currently under intensive development [
10].
ChAd–based vaccines administered intranasally efficiently protected the respiratory tract of mice, rats, hamsters, and rabbits against SARS-CoV-2 infection [
37]. Similar protective effects have also been observed in non–human primates. A single intranasal administration of a simian ChAd–36 vector expressing a pre–S protein resulted in the production of robust humoral and mucosal responses that prevented or limited SARS-CoV-2 infection in rhesus macaques [
38]. The mucosal administration of a live–attenuated respiratory syncytial virus expressing a chimeric SARS-CoV-2 spike in African green monkeys elicited mucosal IgA in the nose, reduced virus shedding by more than 200–fold and protected animals against viral challenge [
39]. However, the approved ChAdOx1 vaccine, which expresses a native spike and induces protective humoral and cellular responses when administered intramuscularly, failed to elicit substantial mucosal or systemic responses in human vaccinees when administered intranasally.
Several studies have established the superiority of intranasal immunization and nebulization inhalation over intramuscular injection in inducing protective immunity against SARS-CoV-2 in animal models [
26,
40]. However, intranasal vaccination can only deliver vaccines to the upper respiratory tract. In contrast, aerosol inhalation [
10] efficiently delivers vaccines to both the upper and lower respiratory tracts and thus elicits stronger mucosal and systemic immune responses than intranasal immunization in animals and humans [
21]. We first confirmed in cynomolgus macaques, which are a good surrogate for humans in inhalation studies [
41], that aerosol inhalation outperformed vaccination by intranasal administration. No adverse events were observed upon vaccination. Immunization with the BV-AdCoV-1 vector (5 × 10
10 VPs) expressing a stabilized trimeric pre–fusion spike from the ancestral Wuhan Hu–1 strain consistently induced 10– to 20–fold higher anti–spike, anti–S1, anti–RBD, and neutralizing antibody responses than intranasal administration. Nebulization inhalation also produced higher S– and RBD–specific serum monomeric mIgA responses and secretory sIgA in nasal and bronchoalveolar fluids than intranasal immunization, which failed to elicit detectable sIgA in nasal fluids under the test conditions used in this study. Kim et al. [
42] reported that only intranasal or sublingual but not intramuscular immunization with an adenovirus 5 vector expressing S1–induced neutralizing IgA antibodies in BALFs. However, it was reported that serum anti–spike IgA levels in subjects immunized IM with the BNT162b2 mRNA vaccine correlated with anti–RBD and neutralization antibodies. Muller et al. [
43] proposed that high serum IgA responses might be associated with more effective protection against breakthrough infections. Early SARS-CoV-2–specific humoral response in humans is dominated by IgA antibodies that greatly contribute to virus neutralization [
17]. Dimeric and polymeric sIgA linked by the J piece predominate in the airways and play a major role in virus neutralization at the nasopharyngeal entry site [
17]. Zhang et al. [
44] reported that RBD–specific IgA in convalescent sera correlated with IgG responses and that if IgA monomers were less neutralizing than IgG antibodies, dimeric IgA in secretions were 15 times more potent neutralizing antibodies than their IgG counterparts. Human IgA dimers were also shown to efficiently neutralize Omicron BA.1 and BA.2 variants [
45]. In the present study, we found that anti–RBD IgA moderately correlated (r = 0.5755) with virus neutralization and noticed a moderate inverse correlation (r = −0.6667, r = −0.6670) between lung viral load and serum anti–S/RBD IgA antibodies. We were not able to assess the relative percentages of monomeric versus dimeric sIgA in BALFs due to the lack of commercial anti–chimpanzee J pieces and anti–secretory component antibodies.
Pseudovirus cross–neutralizing antibody titers against the Delta strain were similar to those obtained for the ancestral strain and 77 times higher than those elicited by IN immunization. Meanwhile, the cross–neutralizing antibody GMT against the Beta strain was 12 times higher than that elicited by IN immunization. These results indicate that aerosol inhalation elicits stronger cross–neutralizing antibody responses and mucosal immunity than intranasal administration.
Aerosol inhalation was, therefore, used in rhesus monkeys to perform comparative immunogenicity studies with intramuscular vaccination. A dose–escalation study with BV-AdCoV-1 showed that two administrations of 5 × 10
10 VPs were necessary and optimal to induce significantly higher spike– and RBD–specific IgG antibody levels than those elicited by intramuscular immunization. This two–dose regimen also produced serum–binding anti–S and anti–RBD IgA antibodies. Strong positive correlations were established between S– (r = 0.9615) or RBD–specific (r = 0.9576) IgG and neutralizing antibody titers, which are the best correlates of protection. The level of neutralizing antibodies was found to inversely correlate with the viral load (r = −0.6523). High positive correlations of neutralizing antibody titers with anti–spike and anti–RBD IgG antibodies and with ACE–2 binding inhibition were also observed in mRNA–vaccinees and infected patients. These immunological markers are thus good predictors of the protective potency of SARS-CoV-2 vaccines [
46]. The 5 × 10
10 dose of BV-AdCoV-1 elicited neutralization titers (GMT 266) comparable to those obtained for the WHO reference and in the range of those correlating with high protective efficacy, namely GMTs of 50 to 500 in rhesus macaques [
47] and above GMT of 100 in humans [
48]. Moreover, two 0.6 × 10
10 VP doses only of inhaled vector were sufficient to generate pseudovirus neutralization antibody titers equivalent to those obtained with a 10–fold higher dose (5 × 10
10 VPs) of the same vector injected intramuscularly, allowing for vaccine–sparing for mass immunization. Two 5 × 10
10 VP doses of aerosolized BV-AdCoV-1 also elicited higher levels of cross–neutralizing antibodies against SARS-CoV-2 Delta and, to a lesser extent, against the Beta variant than IM administration. Furthermore, aerosolized immunization with the ChAd vector expressing prefusion spike protein markedly reduced the viral load in the lungs and nasal swabs and protected animals against pathology. Interestingly, although COVID-19 vaccines consistently induce low neutralizing antibody titers against the Beta strain, they can still protect rhesus macaques in a Beta variant challenge model [
49].
T–cell adaptive immunity plays a critical role in virus clearance, reducing disease severity and long–term antiviral immunity. The induction of strong T–cell responses is a distinct feature of the ChAd vectored vaccines [
50]. Vaccination with ChAdOx1 nCoV–19 promotes robust cellular immunity and the expansion of Th1 cells in macaques [
51]. Similar responses were observed in humans vaccinated with the ChAdOx1 vaccine [
43], and a polyfunctional Th1 response potentially resilient to spike point mutations was observed in AZD1222/ChAdOx1–vaccinated adults, whereas CD4
+ Th2 responses were not detected [
52]. In our study, BV-AdCoV-1 expressing prefusion spikes predominantly induced Th1 responses as judged by the high numbers of IFN–γ and very low numbers of IL–4–producing PBMCs after spike peptide re–stimulation. Such a response might contribute to protection in case of reduced neutralizing antibody titers or low antibody avidity. Although not observed so far with spike–based COVID-19 vaccines, immunopotentiation occurring in animals immunized with SARS–CoV–1 has been linked to the production of insufficient amounts of antibodies and a skewed immune response toward T–helper cell type 2 (Th2). The BV-AdCoV-1 vector, which elicits robust neutralizing antibodies and weak Th2 responses, did not cause vaccine–associated enhanced disease. Most pro–inflammatory cytokines were hardly detectable in the sera and lungs of vaccinated animals after viral challenge, and there was no increased secretion of TNF–α and IFN–γ in sera nor of IL–5 in the lungs compared to non–vaccinated monkeys.
The prime–boost schedules of COVID-19 vaccines have unambiguously demonstrated their safety, clinical effectiveness, and efficacy [
25]. However, recent reports have consistently highlighted that immunity begins to decline 6–8 months after two vaccine doses [
1], exposing vaccinees to re–infection or breakthrough infections, in particular by emerging SARS-CoV-2 variants. Adenovirus vectors are known to induce durable antibody responses and long–lasting T–cell immunity [
53]. We found that six months after the second BV-AdCoV-1 dose, anti–RBD and anti–S antibody GMTs in 18 rhesus macaques from different immunization groups plateaued before waning down to lower levels. Since the emergence of the highly transmissible Omicron BA.1 in 2021, Omicron sub–lineages, such as BA.2 and BA.4/5, have successively rapidly spread worldwide and escaped persistent immunity in convalescent patients [
54] as well as neutralization in fully vaccinated individuals [
55]. Three exposures to SARS-CoV-2 spike by either infection or vaccination were found to elicit superior neutralizing activity to all variants of concern [
56], and a third dose of the COVID-19 vaccine is now required to boost pre–established immune responses [
23]. We indeed observed that a second homologous booster of BV-AdCoV-1 on D284 dramatically enhanced pre–existing mucosal and humoral immunity against SARS-CoV-2 WT and the Delta strain and that the efficacy of this second booster was not affected by anti–adenovirus vector immunity.
Several studies have revealed that boosting fully vaccinated animals or human subjects with a heterologous vaccine type was more efficient than administering a third dose of homologous immunogen [
57]. Priming of macaques with a chimpanzee adenovirus vector ChAd–S expressing a full–length ancestral protein followed by an inactivated virus vaccine [
49] significantly improved the immune responses and neutralized Omicron variants. Heterologous prime–boost immunization with ChAdOx1–nCoV and the mRNA BNT162b2 vaccine achieved very high neutralizing antibody titers and Th1 responses in humans [
58]. Aerosol inhalation of a hAd5–vectored vaccine expressing the Wuha–Hu–1 spike generated stronger neutralizing responses against the D614, BA.2, and BA.5 strains in subjects having received an inactivated virus vaccine than homologous vaccination [
37]. In the present study, we assessed the merits of a heterologous prime–boost schedule using the same ChAd68 vector backbone to express two divergent SARS-CoV-2 spikes. We tested whether the C68–COA04 vector expressing the Omicron BA.1 pre–S protein could enhance the pre–existing neutralizing antibody responses provided by BV-AdCoV-1 against the ancestral strain and SARS-CoV-2 VOCs in particular against the epidemic Omicron sublineages which have recently spread worldwide. A second boost with C68–COA04 induced IgA responses against WT and BA.1 spikes and significantly increased pre–existing binding IgG titers against both strains and neutralizing antibodies against the ancestral strain, more than the monovalent BV-AdCoV-1 alone, perhaps due to part to antigenic imprinting. The booster dose also dramatically enhanced neutralizing immunity against WT, Delta, and Omicron variants BA.1 and BA.2. The bivalent vaccine combining both vectors did not improve the immune responses obtained with its components. But interestingly, regardless of the vaccine used as a boost, the prime–boot regimens used in the study elicited stronger neutralizing antibody responses against BA.2 than against BA.1, which is in line with the results of a previous report that three doses of homologous mRNA vaccination or a heterologous adenovirus/mRNA schedule weakly neutralize the BA.1 sub–lineage compared to BA.2 [
59]. The two monovalent and the bivalent vaccines elicited substantial cross–neutralizing antibody responses against Omicron BA.4/5 variants. Indeed, homologous booster with vector–based vaccines expressing the same antigen is less effective than heterologous booster with the same vector–based vaccines expressing different immunogens [
60]. The heterologous immunization approach is thus a potentially efficacious approach to controlling infection from the Delta strain, the Omicron sublineages, and the hybrid “Deltacron” variant [
60].



 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}