

Designing a Multi-Epitope Subunit Vaccine against VP1 Major Coat Protein of JC Polyomavirus


 , ,
, ,
Abstract
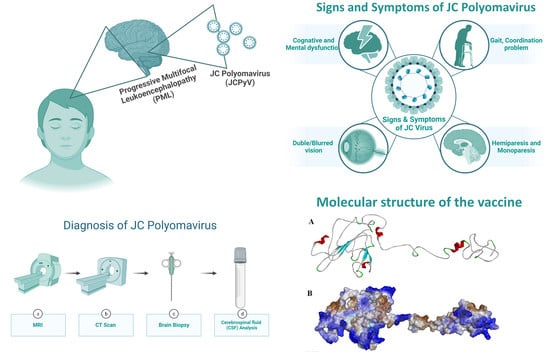
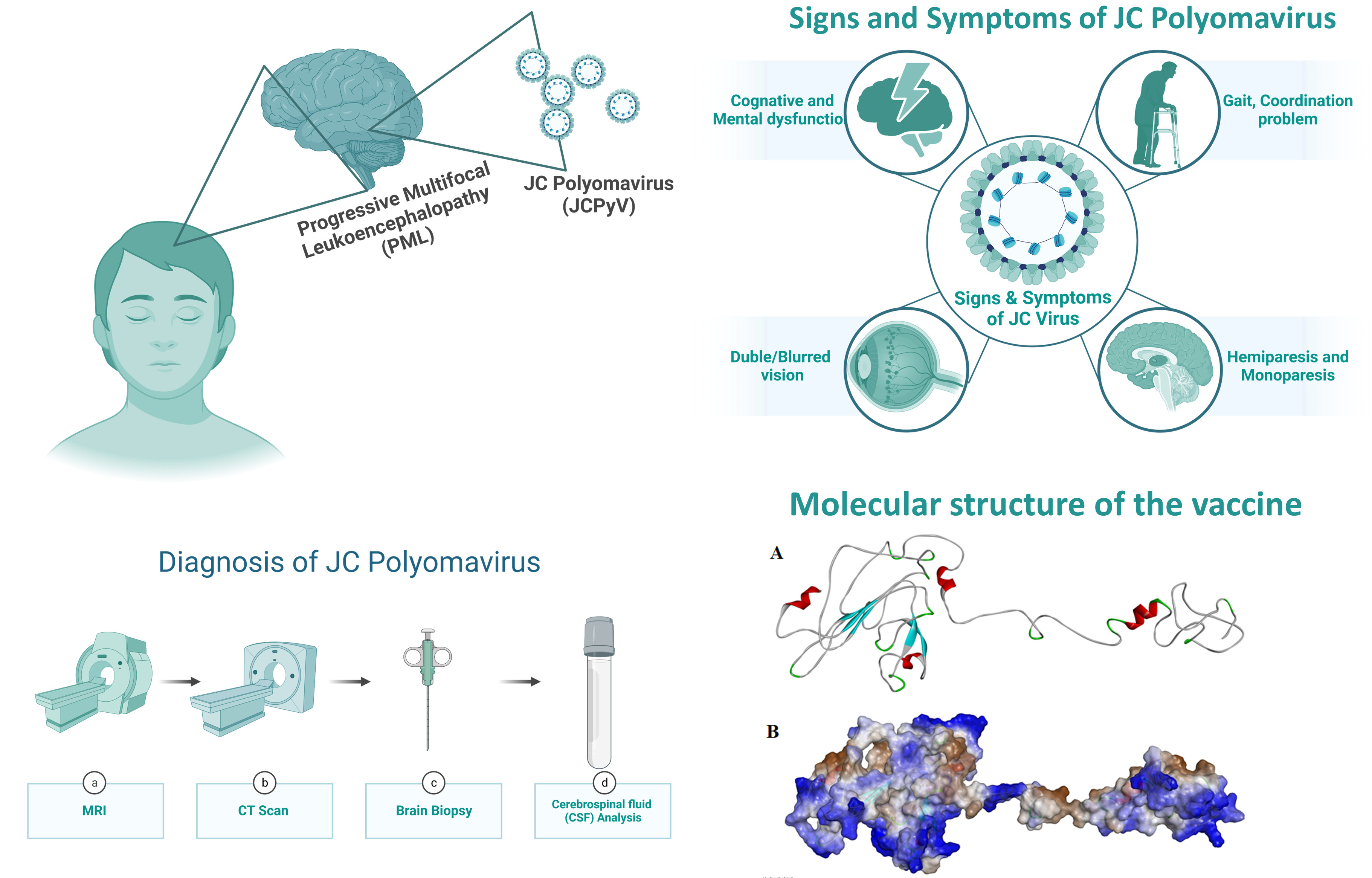
1. Introduction
2. Methodology
2.1. VP1 Protein Sequence Retrieval and Physicochemical Analysis
2.2. B Cell Epitopes Prediction
2.3. Helper T Lymphocyte (HTL) Epitopes Prediction
2.4. Cytotoxic T-Lymphocyte (CTL) Epitopes Prediction
2.5. Computational Construction of Vaccine and Its Physiochemical Analysis
2.6. Vaccine 3D Structure Prediction and Validation
2.7. Interaction of Vaccine with TLR-4 Using Docking
2.8. Molecular Dynamic (MD) Simulation
2.9. Immune Simulation Response of Vaccine Protein
3. Results and Discussion
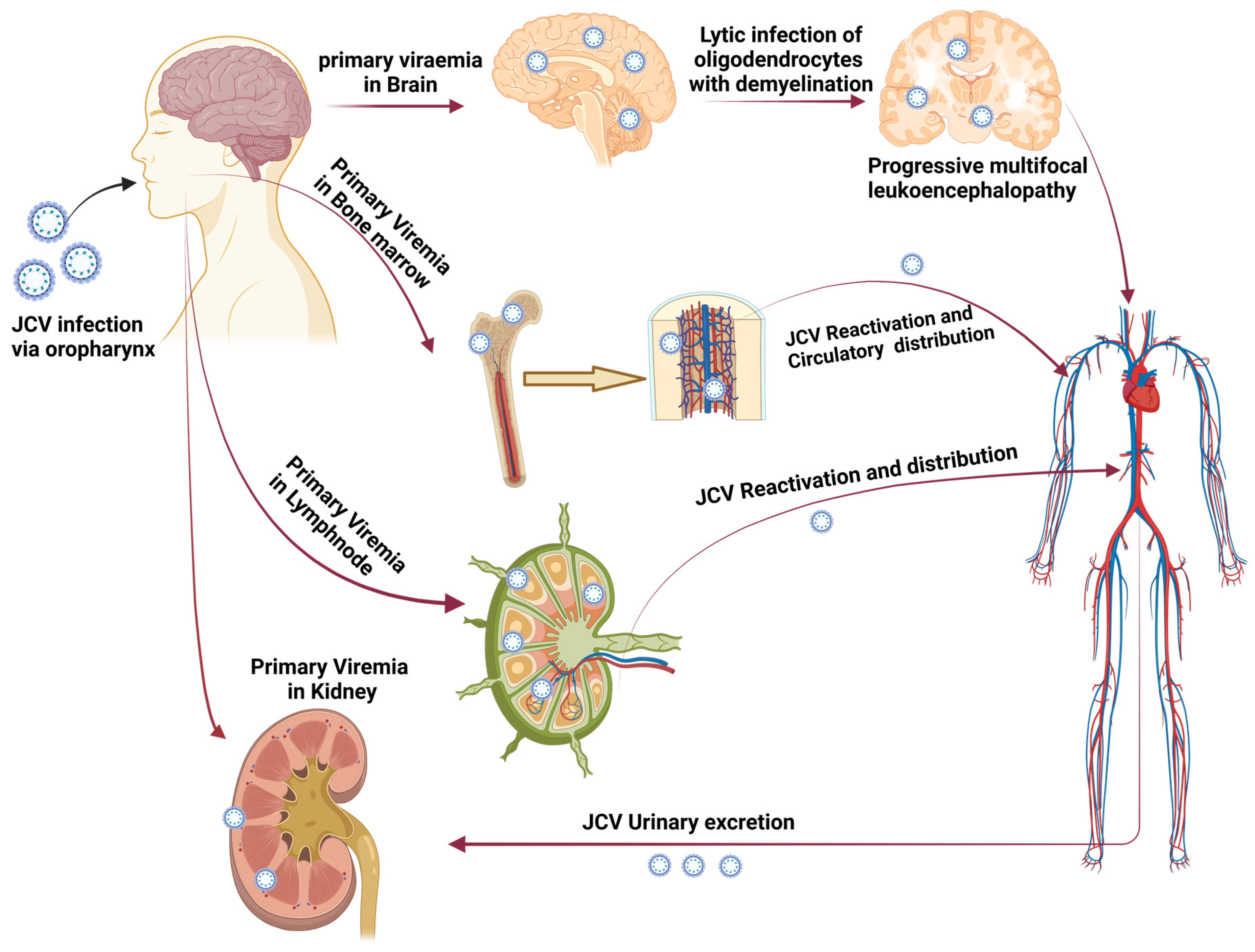
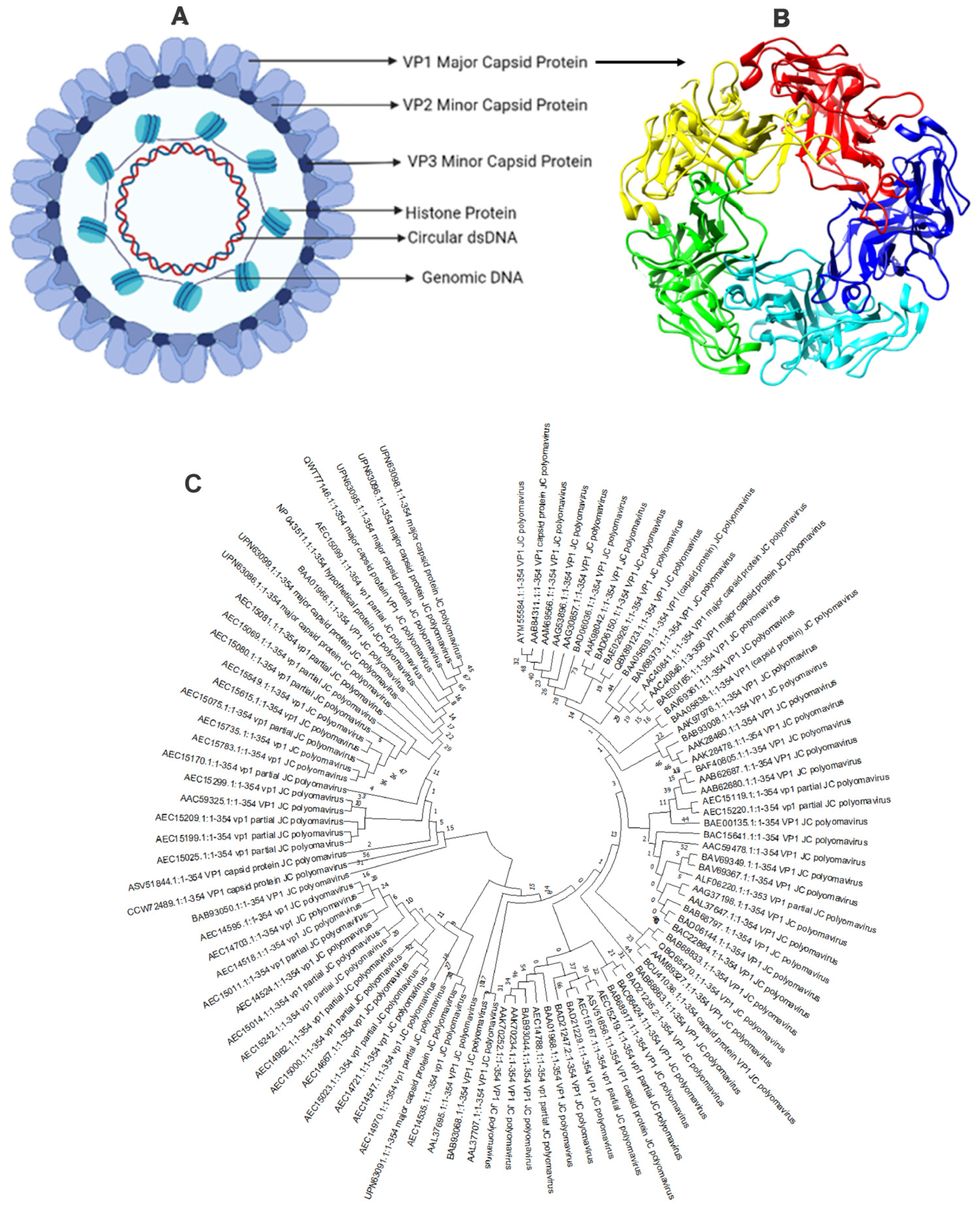
3.1. Sequence Retrieval and Analysis
3.1.1. B Cell Epitope Prediction
3.1.2. HTL Epitope Prediction
3.1.3. CTL Epitope Prediction
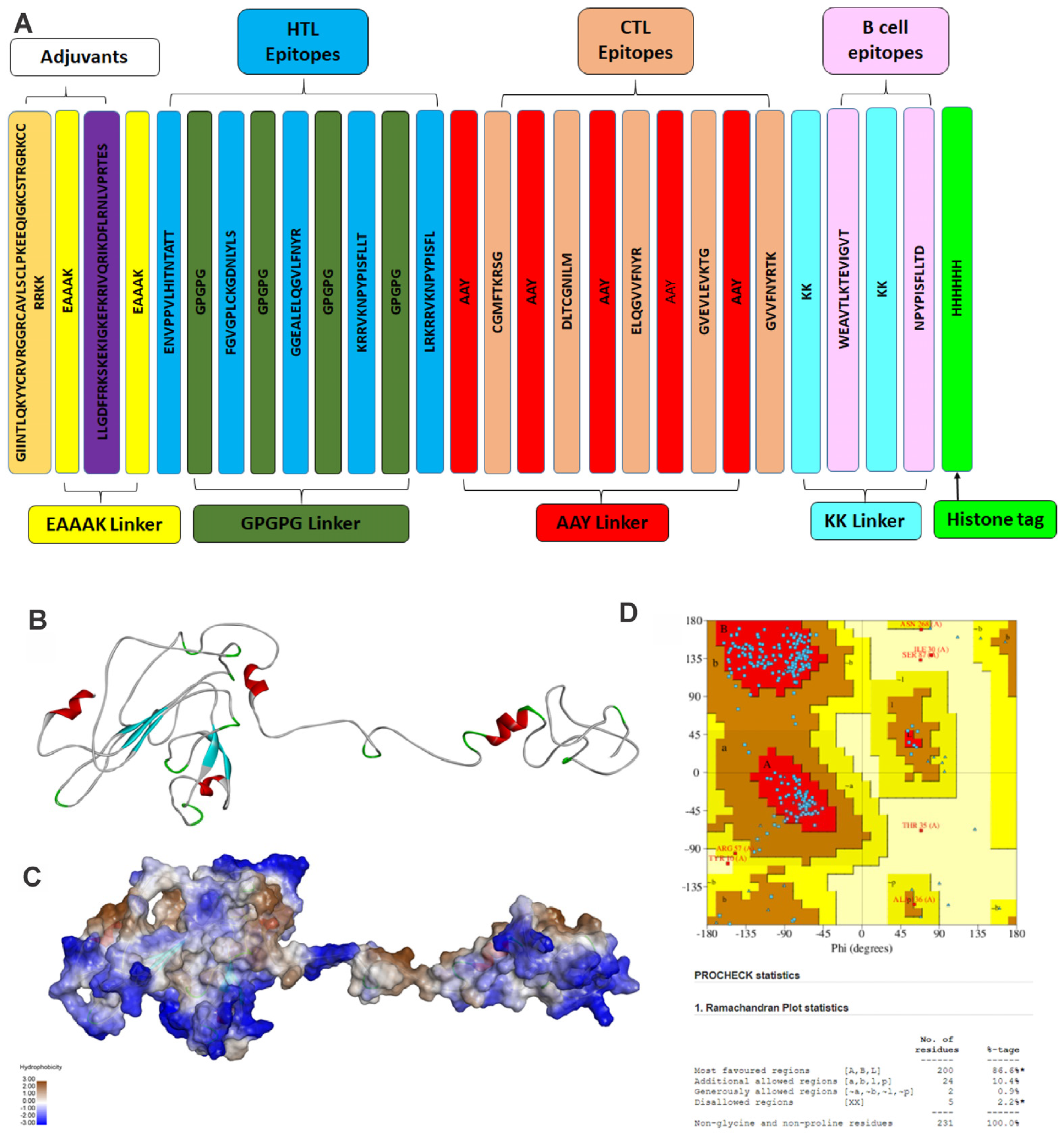
3.1.4. Design of the Linear Vaccine Construct and Physiochemical Analysis
3.1.5. Vaccine Tertiary Structure Prediction and Validation
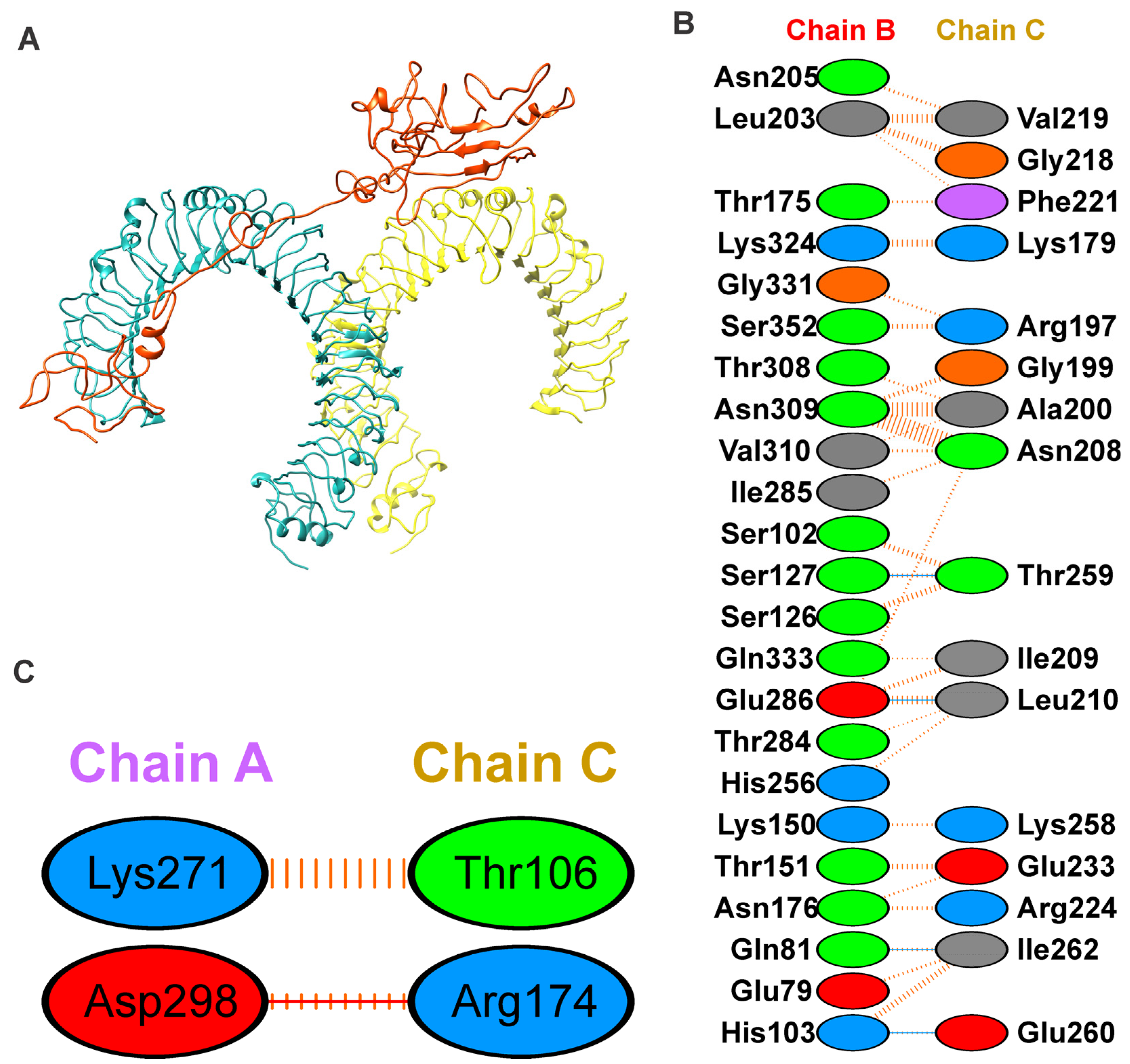
3.1.6. Binding Mode of Vaccine Construct with TLR-4
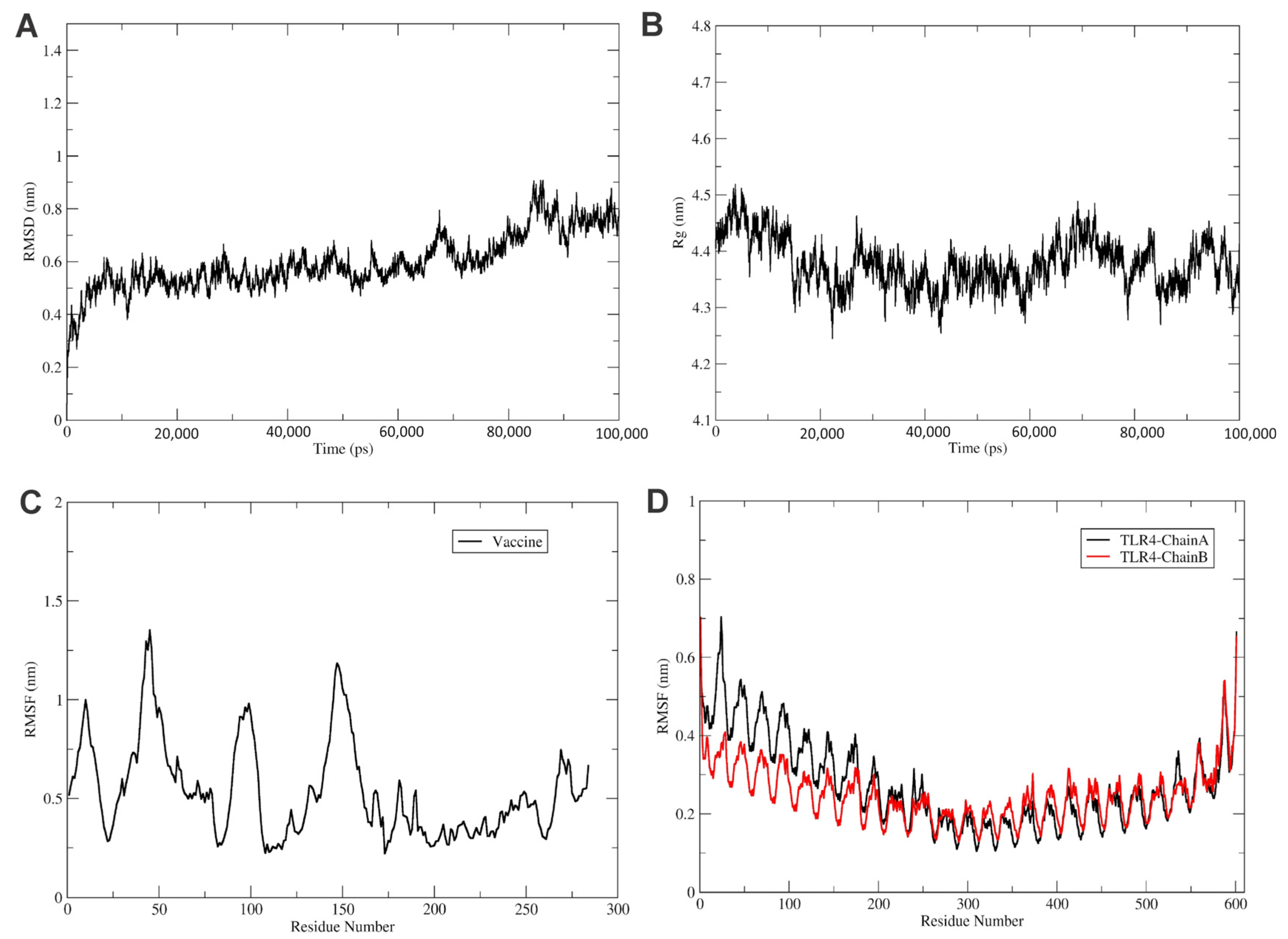
3.2. Molecular Dynamic Simulations
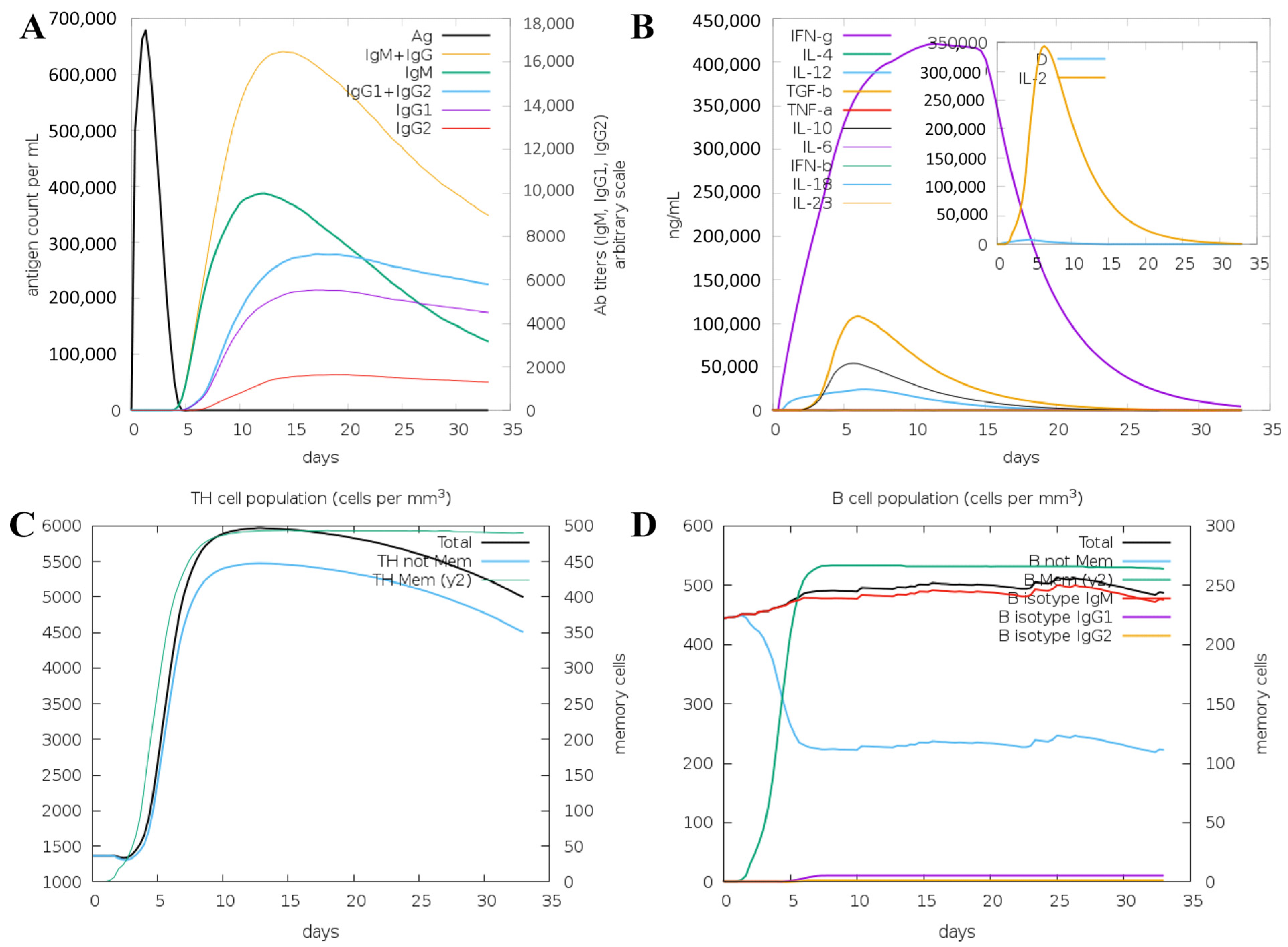
3.3. Immune System Simulation
4. Discussion
5. Conclusions
Limitation
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Imperiale, M.J. JC Polyomavirus: Let’s Please Respect Privacy. J. Virol. 2018, 92, e00561-18. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, A.L.; Atwood, W.J. Fifty Years of JC Polyomavirus: A Brief Overview and Remaining Questions. Viruses 2020, 12, 969. [Google Scholar] [CrossRef] [PubMed]
- Shackelton, L.A.; Rambaut, A.; Pybus, O.G.; Holmes, E.C. JC virus evolution and its association with human populations. J. Virol. 2006, 80, 9928–9933. [Google Scholar] [CrossRef]
- Hildreth, J.E.; Alcendor, D.J. JC polyomavirus and transplantation: Implications for virus reactivation after immunosuppression in transplant patients and the occurrence of PML disease. Transplantology 2021, 2, 37–48. [Google Scholar] [CrossRef]
- Boothpur, R.; Brennan, D.C. Human polyoma viruses and disease with emphasis on clinical BK and JC. J. Clin. Virol. 2010, 47, 306–312. [Google Scholar] [CrossRef]
- Khalili, A.; Craigie, M.; Donadoni, M.; Sariyer, I.K. Host-Immune Interactions in JC Virus Reactivation and Development of Progressive Multifocal Leukoencephalopathy (PML). J. Neuroimmune Pharmacol. 2019, 14, 649–660. [Google Scholar] [CrossRef]
- Harypursat, V.; Zhou, Y.; Tang, S.; Chen, Y. JC Polyomavirus, progressive multifocal leukoencephalopathy and immune reconstitution inflammatory syndrome: A review. AIDS Res. Ther. 2020, 17, 37. [Google Scholar] [CrossRef]
- Nelson, C.D.; Derdowski, A.; Maginnis, M.S.; O’Hara, B.A.; Atwood, W.J. The VP1 subunit of JC polyomavirus recapitulates early events in viral trafficking and is a novel tool to study polyomavirus entry. Virology 2012, 428, 30–40. [Google Scholar] [CrossRef]
- Hornikova, L.; Fraiberk, M.; Man, P.; Janovec, V.; Forstova, J. VP1, the major capsid protein of the mouse polyomavirus, binds microtubules, promotes their acetylation and blocks the host cell cycle. FEBS J. 2017, 284, 301–323. [Google Scholar] [CrossRef]
- Mayberry, C.L.; Nelson, C.D.S.; Maginnis, M.S. JC Polyomavirus Attachment and Entry: Potential Sites for PML Therapeutics. Curr. Clin. Microbiol. Rep. 2017, 4, 132–141. [Google Scholar] [CrossRef]
- Neu, U.; Maginnis, M.S.; Palma, A.S.; Stroh, L.J.; Nelson, C.D.; Feizi, T.; Atwood, W.J.; Stehle, T. Structure-function analysis of the human JC polyomavirus establishes the LSTc pentasaccharide as a functional receptor motif. Cell Host Microbe 2010, 8, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Assetta, B.; Morris-Love, J.; Gee, G.V.; Atkinson, A.L.; O’Hara, B.A.; Maginnis, M.S.; Haley, S.A.; Atwood, W.J. Genetic and Functional Dissection of the Role of Individual 5-HT(2) Receptors as Entry Receptors for JC Polyomavirus. Cell Rep. 2019, 27, 1960–1966.e6. [Google Scholar] [CrossRef] [PubMed]
- Pietropaolo, V.; Prezioso, C.; Bagnato, F.; Antonelli, G. John Cunningham virus: An overview on biology and disease of the etiological agent of the progressive multifocal leukoencephalopathy. New Microbiol. 2018, 41, 179–186. [Google Scholar] [PubMed]
- WHO. Listings of WHO’s Response to COVID-19; World Health Organization: Geneva, Switzerland, 2020. [Google Scholar]
- Pandey, R.K.; Dahiya, S.; Mahita, J.; Sowdhamini, R.; Prajapati, V.K. Vaccination and immunization strategies to design Aedes aegypti salivary protein based subunit vaccine tackling Flavivirus infection. Int. J. Biol. Macromol. 2019, 122, 1203–1211. [Google Scholar] [CrossRef] [PubMed]
- Pandey, R.K.; Ojha, R.; Aathmanathan, V.S.; Krishnan, M.; Prajapati, V.K. Immunoinformatics approaches to design a novel multi-epitope subunit vaccine against HIV infection. Vaccine 2018, 36, 2262–2272. [Google Scholar] [CrossRef] [PubMed]
- Pandey, R.K.; Ojha, R.; Dipti, K.; Kumar, R.; Prajapati, V.K. Immunoselective algorithm to devise multi-epitope subunit vaccine fighting against human cytomegalovirus infection. Infect. Genet. Evol. 2020, 82, 104282. [Google Scholar] [CrossRef]
- Ojha, R.; Pareek, A.; Pandey, R.K.; Prusty, D.; Prajapati, V.K. Strategic Development of a Next-Generation Multi-Epitope Vaccine to Prevent Nipah Virus Zoonotic Infection. ACS Omega 2019, 4, 13069–13079. [Google Scholar] [CrossRef]
- Sun, X.; Zhang, Y.; Zhou, Y.; Lian, X.; Yan, L.; Pan, T.; Jin, T.; Xie, H.; Liang, Z.; Qiu, W.; et al. NPCDR: Natural product-based drug combination and its disease-specific molecular regulation. Nucleic Acids Res. 2022, 50, D1324–D1333. [Google Scholar] [CrossRef]
- UniProt, C. UniProt: A hub for protein information. Nucleic Acids Res. 2015, 43, D204–D212. [Google Scholar] [CrossRef]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.E.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server. Methods Mol. Biol. 1999, 112, 531–552. [Google Scholar] [CrossRef]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Vita, R.; Overton, J.A.; Greenbaum, J.A.; Ponomarenko, J.; Clark, J.D.; Cantrell, J.R.; Wheeler, D.K.; Gabbard, J.L.; Hix, D.; Sette, A.; et al. The immune epitope database (IEDB) 3.0. Nucleic Acids Res. 2015, 43, D405–D412. [Google Scholar] [CrossRef]
- Gupta, S.; Kapoor, P.; Chaudhary, K.; Gautam, A.; Kumar, R.; Open Source Drug Discovery Consortium; Raghava, G.P. In silico approach for predicting toxicity of peptides and proteins. PLoS ONE 2013, 8, e73957. [Google Scholar] [CrossRef]
- Dimitrov, I.; Naneva, L.; Doytchinova, I.; Bangov, I. AllergenFP: Allergenicity prediction by descriptor fingerprints. Bioinformatics 2014, 30, 846–851. [Google Scholar] [CrossRef]
- Lundegaard, C.; Lamberth, K.; Harndahl, M.; Buus, S.; Lund, O.; Nielsen, M. NetMHC-3.0: Accurate web accessible predictions of human, mouse and monkey MHC class I affinities for peptides of length 8-11. Nucleic Acids Res. 2008, 36, W509–W512. [Google Scholar] [CrossRef]
- Dimitrov, I.; Flower, D.R.; Doytchinova, I. AllerTOP—A server for in silico prediction of allergens. BMC Bioinform. 2013, 14 (Suppl. 6), S4. [Google Scholar] [CrossRef]
- Solanki, S.S.; Singh, P.; Kashyap, P.; Sansi, M.S.; Ali, S.A. Promising role of defensins peptides as therapeutics to combat against viral infection. Microb. Pathog. 2021, 155, 104930. [Google Scholar] [CrossRef]
- Durr, U.H.; Sudheendra, U.S.; Ramamoorthy, A. LL-37, the only human member of the cathelicidin family of antimicrobial peptides. Biochim. Biophys. Acta 2006, 1758, 1408–1425. [Google Scholar] [CrossRef]
- Greer, A.; Zenobia, C.; Darveau, R.P. Defensins and LL-37: A review of function in the gingival epithelium. Periodontology 2000 2013, 63, 67–79. [Google Scholar] [CrossRef]
- Ko, J.; Park, H.; Heo, L.; Seok, C. GalaxyWEB server for protein structure prediction and refinement. Nucleic Acids Res. 2012, 40, W294–W297. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.; MacArthur, M.; Moss, D.; Thornton, J. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Kuzmich, N.N.; Sivak, K.V.; Chubarev, V.N.; Porozov, Y.B.; Savateeva-Lyubimova, T.N.; Peri, F. TLR4 signaling pathway modulators as potential therapeutics in inflammation and sepsis. Vaccines 2017, 5, 34. [Google Scholar] [CrossRef] [PubMed]
- Aboudounya, M.M.; Heads, R.J. COVID-19 and toll-like receptor 4 (TLR4): SARS-CoV-2 may bind and activate TLR4 to increase ACE2 expression, facilitating entry and causing hyperinflammation. Mediat. Inflamm. 2021, 2021, 8874339. [Google Scholar] [CrossRef] [PubMed]
- Janovec, V.; Ryabchenko, B.; Skarkova, A.; Pokorna, K.; Rosel, D.; Brabek, J.; Weber, J.; Forstova, J.; Hirsch, I.; Huerfano, S. TLR4-Mediated Recognition of Mouse Polyomavirus Promotes Cancer-Associated Fibroblast-Like Phenotype and Cell Invasiveness. Cancers 2021, 13, 2076. [Google Scholar] [CrossRef]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock and SymmDock: Servers for rigid and symmetric docking. Nucleic Acids Res. 2005, 33, W363–W367. [Google Scholar] [CrossRef]
- Mashiach, E.; Schneidman-Duhovny, D.; Andrusier, N.; Nussinov, R.; Wolfson, H.J. FireDock: A web server for fast interaction refinement in molecular docking. Nucleic Acids Res. 2008, 36, W229–W232. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Kumbhar, B.V.; Bhandare, V.V.; Panda, D.; Kunwar, A. Delineating the interaction of combretastatin A-4 with alphabeta tubulin isotypes present in drug resistant human lung carcinoma using a molecular modeling approach. J. Biomol. Struct. Dyn. 2020, 38, 426–438. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N·log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- DeLano, W.L. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Cryst. 2002, 40, 82–92. [Google Scholar]
- Rapin, N.; Lund, O.; Castiglione, F. Immune system simulation online. Bioinformatics 2011, 27, 2013–2014. [Google Scholar] [CrossRef]
- Kaleeckal Mathew, O.; Sowdhamini, R. PIMA: Protein-Protein interactions in Macromolecular Assembly—A web server for its Analysis and Visualization. Bioinformation 2016, 12, 9–11. [Google Scholar] [CrossRef]
- Wollebo, H.S.; White, M.K.; Gordon, J.; Berger, J.R.; Khalili, K. Persistence and pathogenesis of the neurotropic polyomavirus JC. Ann. Neurol. 2015, 77, 560–570. [Google Scholar] [CrossRef]
- Lauver, M.D.; Lukacher, A.E. JCPyV VP1 Mutations in Progressive MultifocalLeukoencephalopathy: Altering Tropismor Mediating Immune Evasion? Viruses 2020, 12, 1156. [Google Scholar] [CrossRef]
- Hajighahramani, N.; Nezafat, N.; Eslami, M.; Negahdaripour, M.; Rahmatabadi, S.S.; Ghasemi, Y. Immunoinformatics analysis and in silico designing of a novel multi-epitope peptide vaccine against Staphylococcus aureus. Infect. Genet. Evol. 2017, 48, 83–94. [Google Scholar] [CrossRef]
- Pandey, R.K.; Ojha, R.; Mishra, A.; Kumar Prajapati, V. Designing B- and T-cell multi-epitope based subunit vaccine using immunoinformatics approach to control Zika virus infection. J. Cell. Biochem. 2018, 119, 7631–7642. [Google Scholar] [CrossRef]
- Sharma, S.; Kumari, V.; Kumbhar, B.V.; Mukherjee, A.; Pandey, R.; Kondabagil, K. Immunoinformatics approach for a novel multi-epitope subunit vaccine design against various subtypes of Influenza A virus. Immunobiology 2021, 226, 152053. [Google Scholar] [CrossRef]
- Cyster, J.G.; Allen, C.D.C. B Cell Responses: Cell Interaction Dynamics and Decisions. Cell 2019, 177, 524–540. [Google Scholar] [CrossRef]
- Jespersen, M.C.; Mahajan, S.; Peters, B.; Nielsen, M.; Marcatili, P. Antibody Specific B-Cell Epitope Predictions: Leveraging Information From Antibody-Antigen Protein Complexes. Front. Immunol. 2019, 10, 298. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L. Multi-epitope vaccines: A promising strategy against tumors and viral infections. Cell. Mol. Immunol. 2018, 15, 182–184. [Google Scholar] [CrossRef] [PubMed]
- Raza, A.; Asif Rasheed, M.; Raza, S.; Tariq Navid, M.; Afzal, A.; Jamil, F. Prediction and analysis of multi epitope based vaccine against Newcastle disease virus based on haemagglutinin neuraminidase protein. Saudi J. Biol. Sci. 2022, 29, 3006–3014. [Google Scholar] [CrossRef]
- Chauhan, V.; Rungta, T.; Goyal, K.; Singh, M.P. Designing a multi-epitope based vaccine to combat Kaposi Sarcoma utilizing immunoinformatics approach. Sci. Rep. 2019, 9, 2517. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Length | Antigenicity | Allergenicity | Toxicity |
|---|---|---|---|---|
| WEAVTLKTEVIGVT | 14 | Antigenic | Non-Allergen | Non-Toxin |
| NPYPISFLLTD | 11 | Antigenic | Non-Allergen | Non-Toxic |
| Peptide | Length | Antigenicity | Allergenicity | Toxicity |
|---|---|---|---|---|
| ENVPPVLHINTATT | 15 | Antigenic | Non-Allergen | Non-Toxin |
| KRRVKNPYISFLLT | 15 | Antigenic | Non-Allergen | Non-Toxin |
| LRKRRVKNPYISFL | 15 | Antigenic | Non-Allergen | Non-Toxin |
| FGVGPLCKGDNLYLS | 15 | Antigenic | Non-Allergen | Non-Toxin |
| GGEALELQGVLFNYR | 15 | Antigenic | Non-Allergen | Non-Toxin |
| Peptide | Length | Antigenicity | Allergenicity | Toxicity |
|---|---|---|---|---|
| CGMFTKRSG | 9 | Antigenic | Non-Allergen | Non-Toxin |
| DLTCGNILM | 9 | Antigenic | Non-Allergen | Non-Toxin |
| ELQGVVFNYR | 10 | Antigenic | Non-Allergen | Non-Toxin |
| GVEVLEVKTG | 10 | Antigenic | Non-Allergen | Non-Toxin |
| GVVFNYRTK | 9 | Antigenic | Non-Allergen | Non-Toxin |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kanse, S.; Khandelwal, M.; Pandey, R.K.; Khokhar, M.; Desai, N.; Kumbhar, B.V. Designing a Multi-Epitope Subunit Vaccine against VP1 Major Coat Protein of JC Polyomavirus. Vaccines 2023, 11, 1182. https://doi.org/10.3390/vaccines11071182
Kanse S, Khandelwal M, Pandey RK, Khokhar M, Desai N, Kumbhar BV. Designing a Multi-Epitope Subunit Vaccine against VP1 Major Coat Protein of JC Polyomavirus. Vaccines. 2023; 11(7):1182. https://doi.org/10.3390/vaccines11071182
Chicago/Turabian StyleKanse, Sukhada, Mehak Khandelwal, Rajan Kumar Pandey, Manoj Khokhar, Neetin Desai, and Bajarang Vasant Kumbhar. 2023. "Designing a Multi-Epitope Subunit Vaccine against VP1 Major Coat Protein of JC Polyomavirus" Vaccines 11, no. 7: 1182. https://doi.org/10.3390/vaccines11071182
APA StyleKanse, S., Khandelwal, M., Pandey, R. K., Khokhar, M., Desai, N., & Kumbhar, B. V. (2023). Designing a Multi-Epitope Subunit Vaccine against VP1 Major Coat Protein of JC Polyomavirus. Vaccines, 11(7), 1182. https://doi.org/10.3390/vaccines11071182