
Development and Optimal Immune Strategy of an Alum-Stabilized Pickering emulsion for Cancer Vaccines

 , ,
, ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. Mice
2.4. Preparation of APE
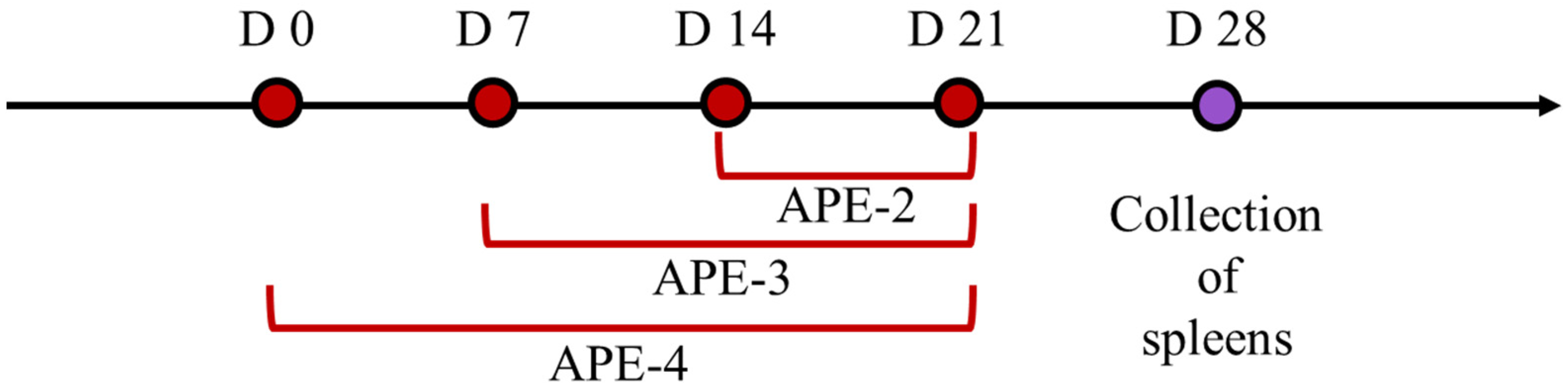
2.5. Vaccination Study
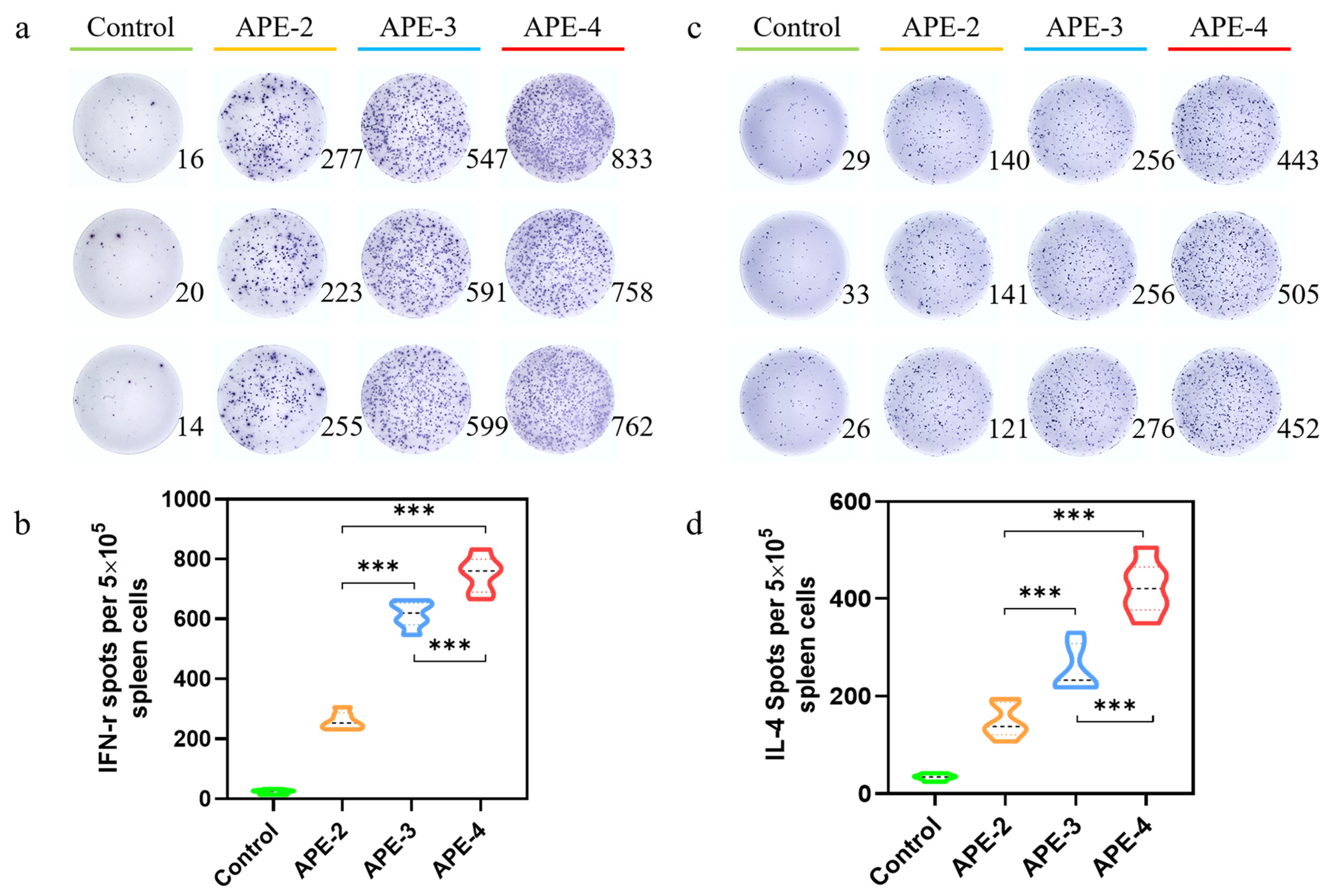
2.6. ELISpot Evaluations
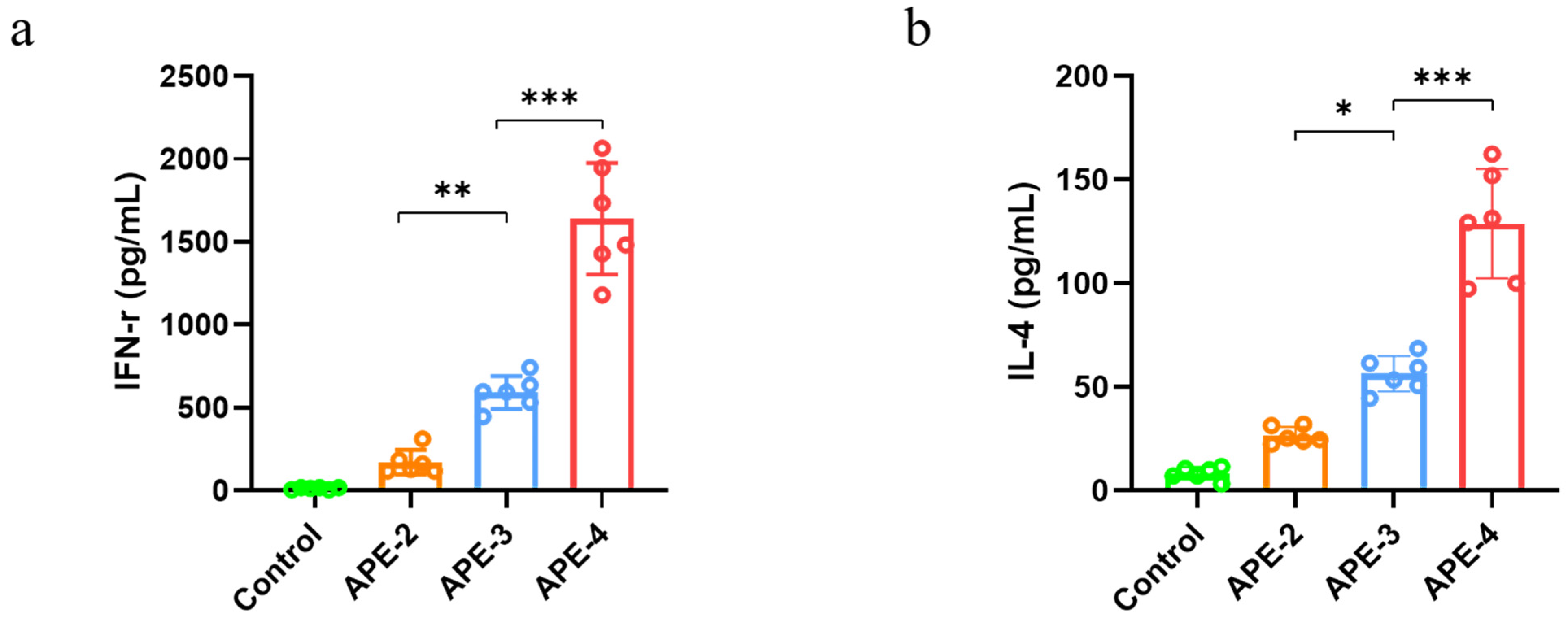
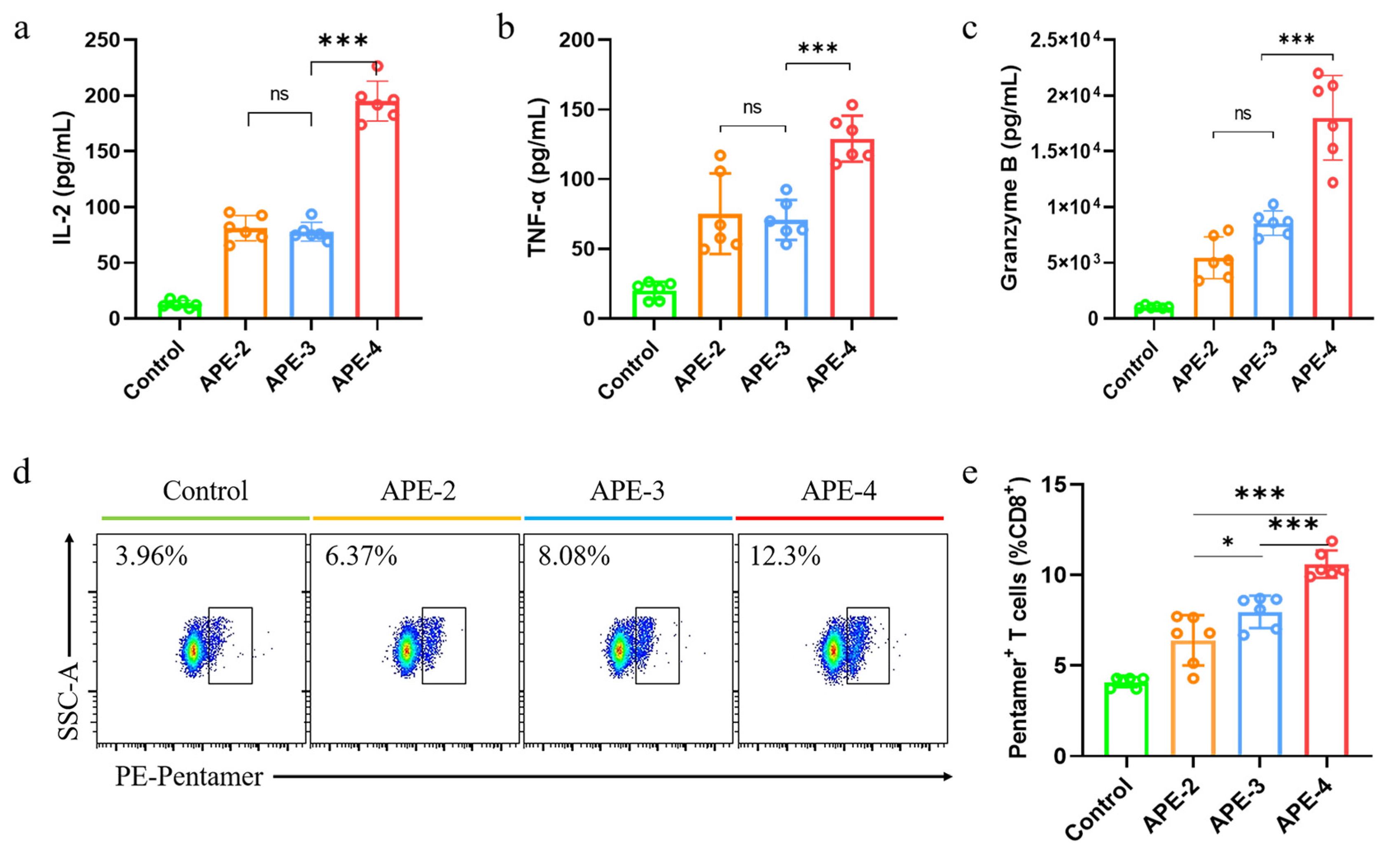
2.7. Cytokine Secretion and Immune Cells in the Spleen
2.8. Cytokine Secretion and Immune Cells in Tumors and TDLNs
2.9. Evaluation of Health
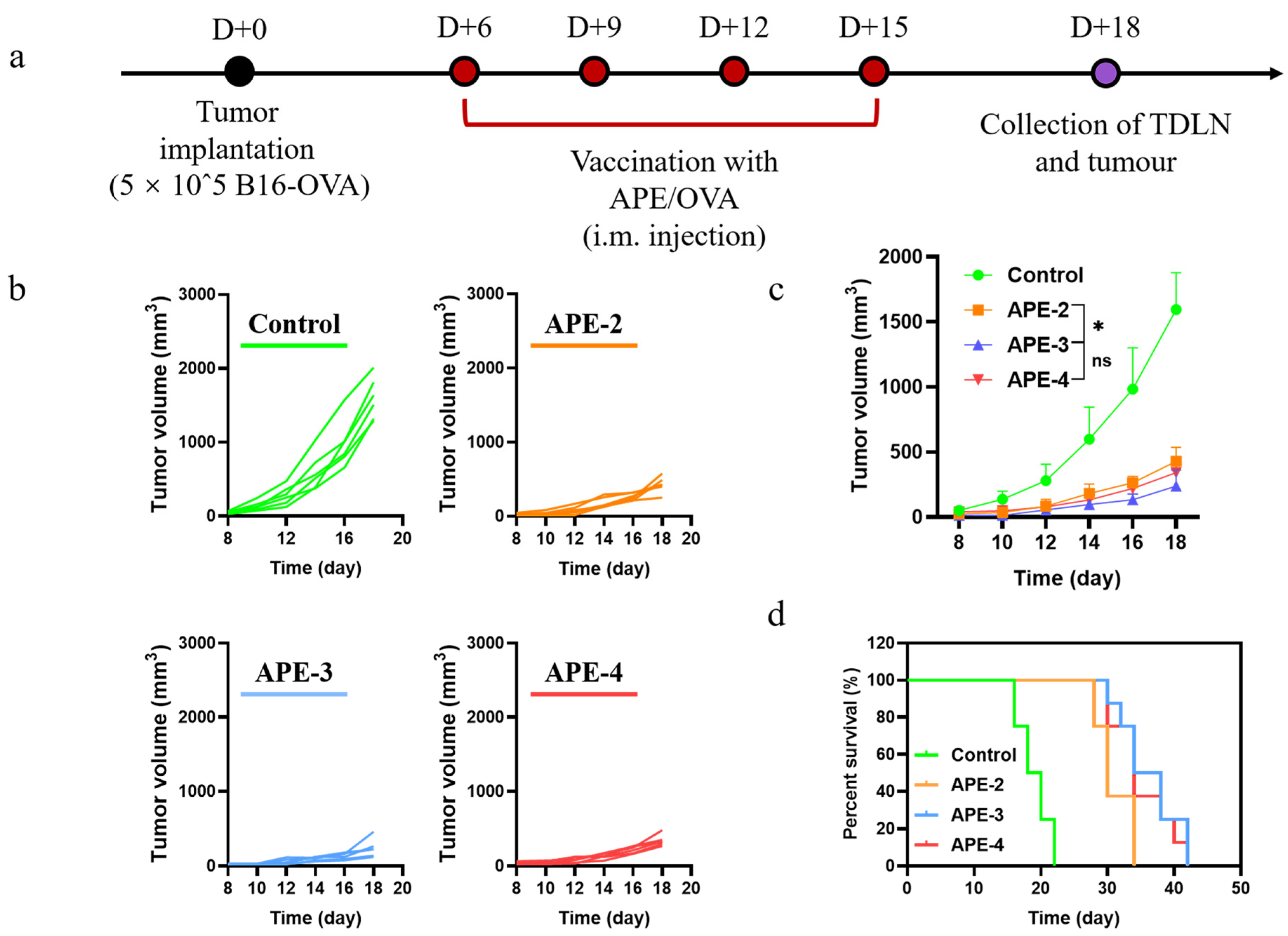
2.10. Evaluation of Tumor Growth Inhibition Using a B16-OVA-Bearing Mouse Model
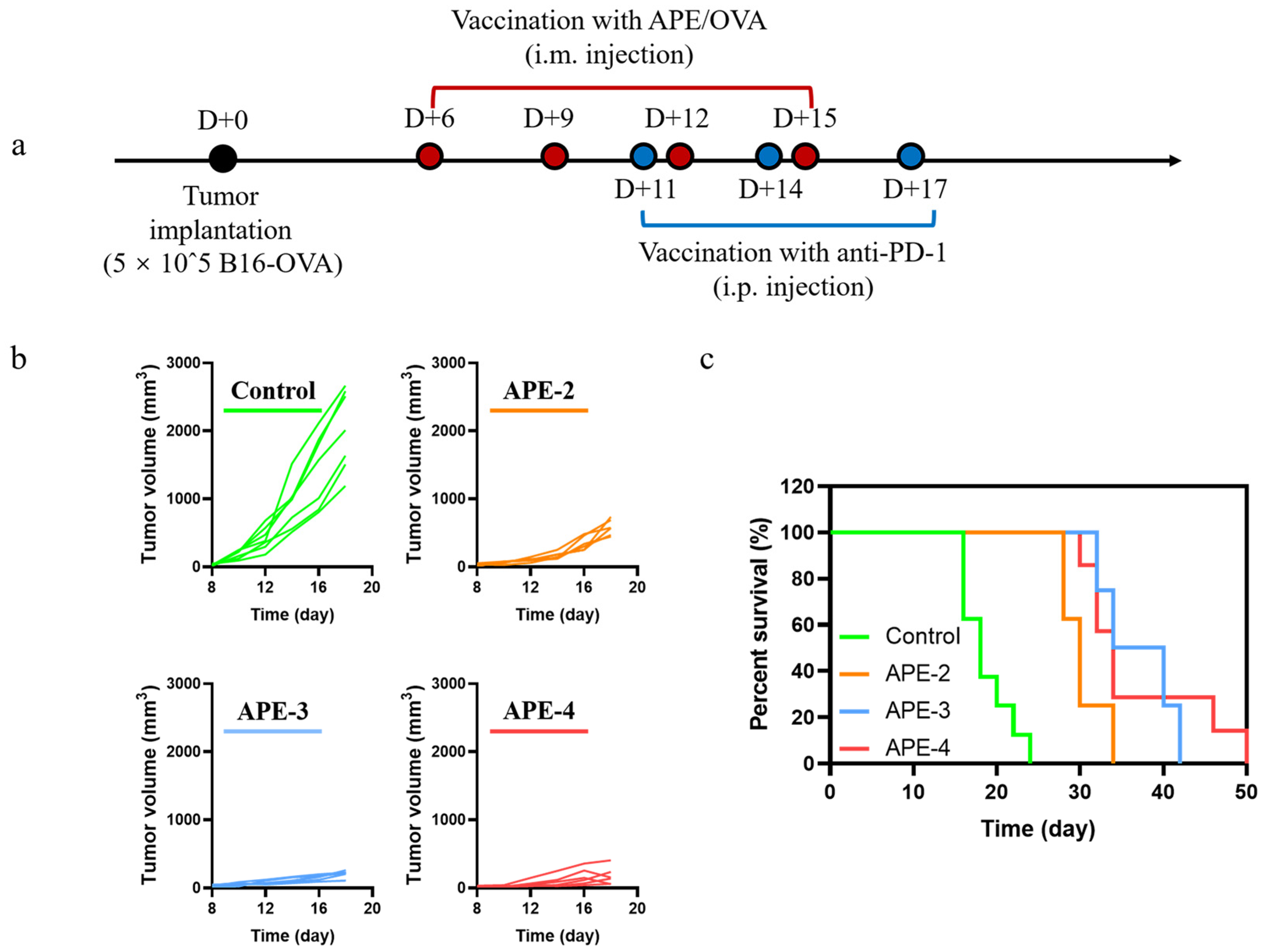
2.11. Combination with Anti-PD-1 Antibody Using a B16-OVA-Bearing Mouse Model
2.12. Statistical Analysis
3. Results
3.1. Alum-Stabilized Pickering Emulsion Preparation and Characterization
3.2. Adjuvant Effects of Alum-Stabilized Pickering Emulsion as a Tumor Vaccination
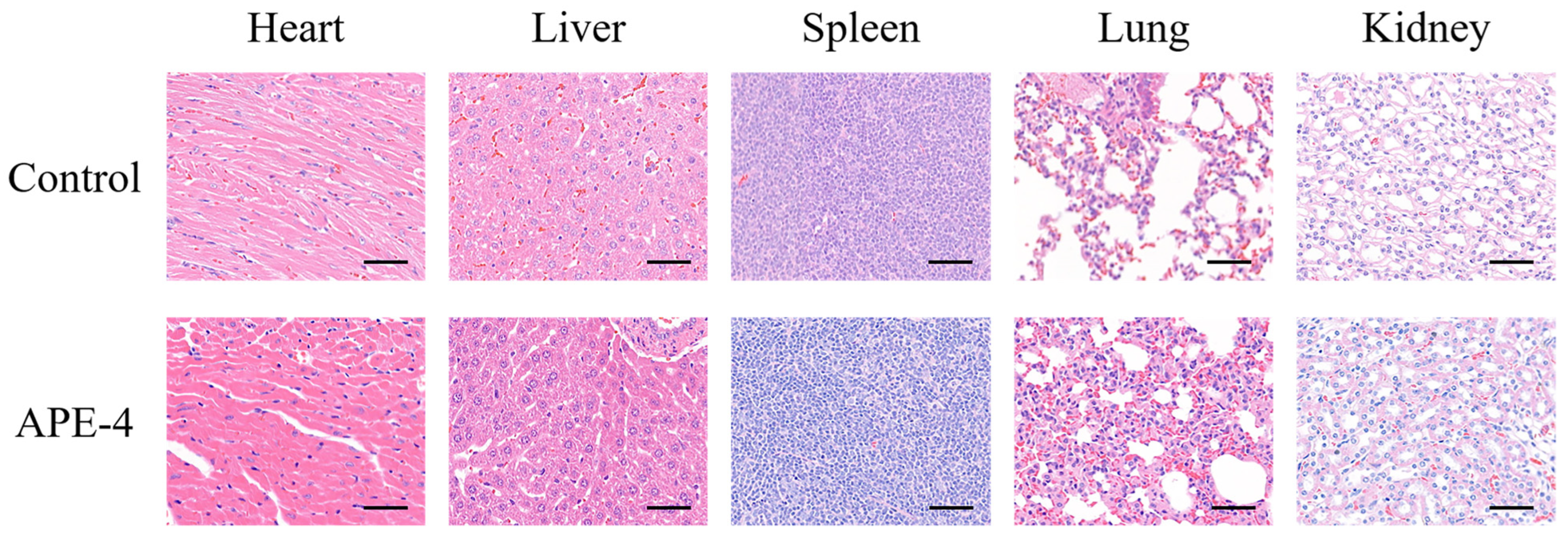
3.3. The Safety Profile of APE after Multiple Immunizations
3.4. Anti-Tumor Therapeutic Efficacy
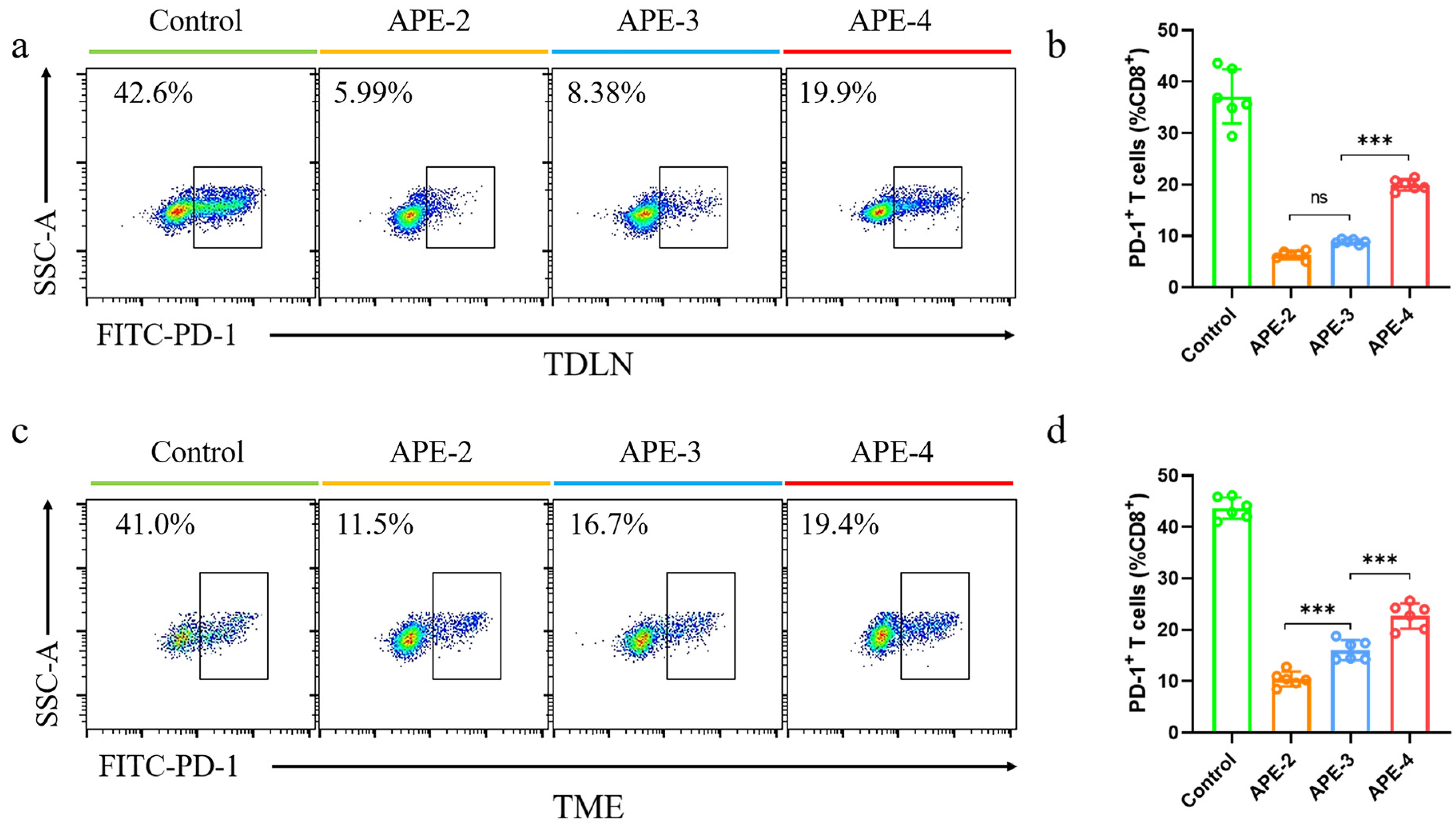
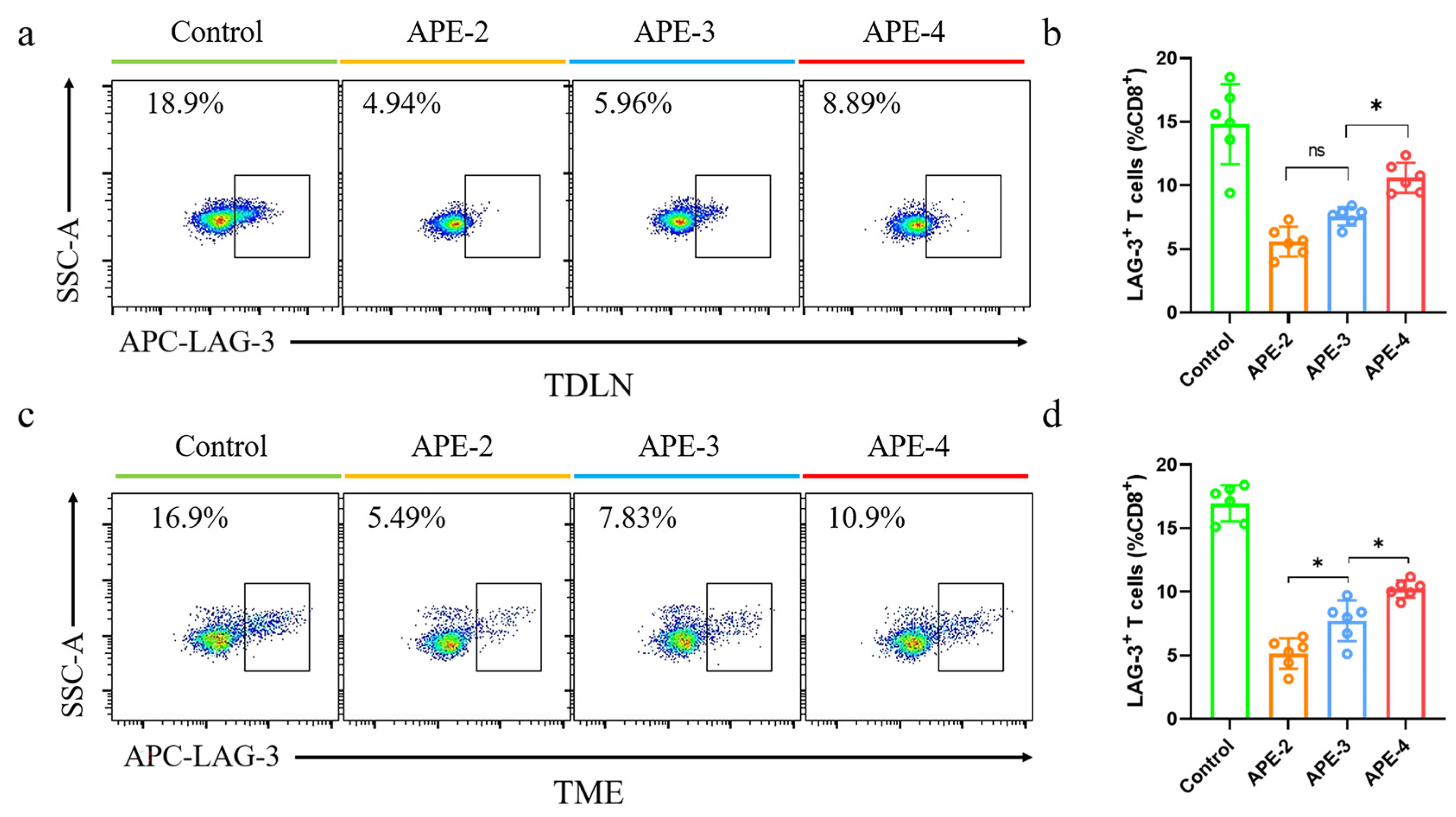
3.5. Immune Responses in TDLNs and TME in the B16-OVA Tumor Model
3.6. Anti-Tumor Therapeutic Efficacy of APE Combined with Anti-PD-1 Therapy
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stäger, S.; Alexander, J.; Kirby, A.C.; Botto, M.; Rooijen, N.V.; Smith, D.F.; Brombacher, F.; Kaye, P.M. Natural Antibodies and Complement Are Endogenous Adjuvants for Vaccine-Induced CD8+ T-Cell Responses. Nat. Med. 2003, 9, 1287–1292. [Google Scholar] [CrossRef] [PubMed]
- Xi, X.; Ye, T.; Wang, S.; Na, X.; Wang, J.; Qing, S.; Gao, X.; Wang, C.; Li, F.; Wei, W.; et al. Self-Healing Microcapsules Synergetically Modulate Immunization Microenvironments for Potent Cancer Vaccination. Sci. Adv. 2020, 6, eaay7735. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Moon, J. Nanoparticle Drug Delivery Systems Designed to Improve Cancer Vaccines and Immunotherapy. Vaccines 2015, 3, 662–685. [Google Scholar] [CrossRef] [PubMed]
- Mizukoshi, E.; Nakagawa, H.; Tamai, T.; Kitahara, M.; Fushimi, K.; Nio, K.; Terashima, T.; Iida, N.; Arai, K.; Yamashita, T.; et al. Peptide Vaccine-Treated, Long-Term Surviving Cancer Patients Harbor Self-Renewing Tumor-Specific CD8+ T Cells. Nat. Commun. 2022, 13, 3123. [Google Scholar] [CrossRef] [PubMed]
- Rastogi, I.; Muralidhar, A.; McNeel, D.G. Vaccines as Treatments for Prostate Cancer. Nat. Rev. Urol, 2023; Epub ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Jiang, Q.; Zhao, X.; Zhao, R.; Wang, Y.; Wang, Y.; Liu, J.; Shang, Y.; Zhao, S.; Wu, T.; et al. A DNA Nanodevice-Based Vaccine for Cancer Immunotherapy. Nat. Mater. 2021, 20, 421–430. [Google Scholar] [CrossRef]
- Macagno, A.; Napolitani, G.; Lanzavecchia, A.; Sallusto, F. Duration, Combination and Timing: The Signal Integration Model of Dendritic Cell Activation. Trends Immunol. 2007, 28, 227–233. [Google Scholar] [CrossRef]
- Irvine, D.J.; Swartz, M.A.; Szeto, G.L. Engineering Synthetic Vaccines Using Cues from Natural Immunity. Nat. Mater. 2013, 12, 978–990. [Google Scholar] [CrossRef]
- Von Andrian, U.H.; Mempel, T.R. Homing and Cellular Traffic in Lymph Nodes. Nat. Rev. Immunol. 2003, 3, 867–878. [Google Scholar] [CrossRef]
- Xia, Y.; Fan, Q.; Hao, D.; Wu, J.; Ma, G.; Su, Z. Chitosan-Based Mucosal Adjuvants: Sunrise on the Ocean. Vaccine 2015, 33, 5997–6010. [Google Scholar] [CrossRef]
- Pulendran, B. The Varieties of Immunological Experience: Of Pathogens, Stress, and Dendritic Cells. Annu. Rev. Immunol. 2015, 33, 563–606. [Google Scholar] [CrossRef]
- Rosalia, R.A.; Cruz, L.J.; Van Duikeren, S.; Tromp, A.T.; Silva, A.L.; Jiskoot, W.; De Gruijl, T.; Löwik, C.; Oostendorp, J.; Van Der Burg, S.H.; et al. CD40-Targeted Dendritic Cell Delivery of PLGA-Nanoparticle Vaccines Induce Potent Anti-Tumor Responses. Biomaterials 2015, 40, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Green, J.J.; Elisseeff, J.H. Mimicking Biological Functionality with Polymers for Biomedical Applications. Nature 2016, 540, 386–394. [Google Scholar] [CrossRef]
- Xia, Y.; Wu, J.; Wei, W.; Du, Y.; Wan, T.; Ma, X.; An, W.; Guo, A.; Miao, C.; Yue, H.; et al. Exploiting the Pliability and Lateral Mobility of Pickering Emulsion for Enhanced Vaccination. Nat. Mater. 2018, 17, 187–194. [Google Scholar] [CrossRef]
- Du, Y.; Song, T.; Wu, J.; Gao, X.-D.; Ma, G.; Liu, Y.; Xia, Y. Engineering Mannosylated Pickering Emulsions for the Targeted Delivery of Multicomponent Vaccines. Biomaterials 2022, 280, 121313. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Wu, N.; Gao, Y.; Wang, X.; Wu, J.; Ma, G. Alum Pickering Emulsion as Effective Adjuvant to Improve Malaria Vaccine Efficacy. Vaccines 2021, 9, 1244. [Google Scholar] [CrossRef]
- Cao, F.; Peng, S.; An, Y.; Xu, K.; Zheng, T.; Dai, L.; Ogino, K.; Ngai, T.; Xia, Y.; Ma, G. Inside-out Assembly of Viral Antigens for the Enhanced Vaccination. Signal Transduct. Target. Ther. 2023, 8, 189. [Google Scholar] [CrossRef]
- Van Der Burg, S.H.; Arens, R.; Ossendorp, F.; Van Hall, T.; Melief, C.J.M. Vaccines for Established Cancer: Overcoming the Challenges Posed by Immune Evasion. Nat. Rev. Cancer 2016, 16, 219–233. [Google Scholar] [CrossRef] [PubMed]
- Watkins, E.A.; Antane, J.T.; Roberts, J.L.; Lorentz, K.M.; Zuerndorfer, S.; Dunaif, A.C.; Bailey, L.J.; Tremain, A.C.; Nguyen, M.; De Loera, R.C.; et al. Persistent Antigen Exposure via the Eryptotic Pathway Drives Terminal T Cell Dysfunction. Sci. Immunol. 2021, 6, eabe1801. [Google Scholar] [CrossRef]
- Wherry, E.J.; Kurachi, M. Molecular and Cellular Insights into T Cell Exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef]
- Wherry, E.J. T Cell Exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef]
- Fuc, E.; Złotkowska, D.; Wasilewska, E.; Wróblewska, B. OVA-Experienced CD4+ T Cell Transfer and Chicken Protein Challenge Affect the Immune Response to OVA in a Murine Model. Int. J. Mol. Sci. 2021, 22, 6573. [Google Scholar] [CrossRef]
- Peng, S.; Cao, F.; Xia, Y.; Gao, X.; Dai, L.; Yan, J.; Ma, G. Particulate Alum via Pickering Emulsion for an Enhanced COVID-19 Vaccine Adjuvant. Adv. Mater. 2020, 32, 2004210. [Google Scholar] [CrossRef]
- Pang, G.; Liu, Y.; Wang, Y.; Wang, Y.; Wang, F.; Zhao, J.; Zhang, L.W. Endotoxin contamination in ovalbumin as viewed from a nano-immunotherapy perspective. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2022, 14, e1747. [Google Scholar] [CrossRef] [PubMed]
- Orr, M.T.; Khandhar, A.P.; Seydoux, E.; Liang, H.; Gage, E.; Mikasa, T.; Beebe, E.L.; Rintala, N.D.; Persson, K.H.; Ahniyaz, A.; et al. Reprogramming the Adjuvant Properties of Aluminum Oxyhydroxide with Nanoparticle Technology. NPJ Vaccines 2019, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- Moyer, T.J.; Kato, Y.; Abraham, W.; Chang, J.Y.H.; Kulp, D.W.; Watson, N.; Turner, H.L.; Menis, S.; Abbott, R.K.; Bhiman, J.N.; et al. Engineered Immunogen Binding to Alum Adjuvant Enhances Humoral Immunity. Nat. Med. 2020, 26, 430–440. [Google Scholar] [CrossRef]
- Liu, J.; Guo, S.; Jin, Z.; Zhao, K. Adjuvanted Quaternized Chitosan Composite Aluminum Nanoparticles-Based Vaccine Formulation Promotes Immune Responses in Chickens. Vaccine 2023, 41, 2982–2989. [Google Scholar] [CrossRef] [PubMed]
- HogenEsch, H.; O’Hagan, D.T.; Fox, C.B. Optimizing the Utilization of Aluminum Adjuvants in Vaccines: You Might Just Get What You Want. NPJ Vaccines 2018, 3, 51. [Google Scholar] [CrossRef]
- Teplensky, M.H.; Evangelopoulos, M.; Dittmar, J.W.; Forsyth, C.M.; Sinegra, A.J.; Wang, S.; Mirkin, C.A. Multi-Antigen Spherical Nucleic Acid Cancer Vaccines. Nat. Biomed. Eng. 2023; Epub ahead of print. [Google Scholar] [CrossRef]
- Schaefer, J.T.; Patterson, J.W.; Deacon, D.H.; Smolkin, M.E.; Petroni, G.R.; Jackson, E.M.; Slingluff, C.L. Dynamic Changes in Cellular Infiltrates with Repeated Cutaneous Vaccination: A Histologic and Immunophenotypic Analysis. J. Transl. Med. 2010, 8, 79. [Google Scholar] [CrossRef]
- Beatty, G.L.; Gladney, W.L. Immune Escape Mechanisms as a Guide for Cancer Immunotherapy. Clin. Cancer Res. 2015, 21, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Mamuti, M.; Wang, H. Therapeutic Vaccines for Cancer Immunotherapy. ACS Biomater. Sci. Eng. 2020, 6, 6036–6052. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Size (nm) | Zeta Potential (mV) | OVA Loading Efficiency 1 (%) | |
|---|---|---|---|
| Alum | 847.0 ± 18.9 | +17.1 ± 0.3 | 97.9 ± 0.4 |
| APE | 2761.0 ± 179.4 | +30.6 ± 2.4 | 98.1 ± 0.6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peng, S.; Yan, Y.; Ngai, T.; Li, J.; Ogino, K.; Xia, Y. Development and Optimal Immune Strategy of an Alum-Stabilized Pickering emulsion for Cancer Vaccines. Vaccines 2023, 11, 1169. https://doi.org/10.3390/vaccines11071169
Peng S, Yan Y, Ngai T, Li J, Ogino K, Xia Y. Development and Optimal Immune Strategy of an Alum-Stabilized Pickering emulsion for Cancer Vaccines. Vaccines. 2023; 11(7):1169. https://doi.org/10.3390/vaccines11071169
Chicago/Turabian StylePeng, Sha, Yumeng Yan, To Ngai, Jianjun Li, Kenji Ogino, and Yufei Xia. 2023. "Development and Optimal Immune Strategy of an Alum-Stabilized Pickering emulsion for Cancer Vaccines" Vaccines 11, no. 7: 1169. https://doi.org/10.3390/vaccines11071169
APA StylePeng, S., Yan, Y., Ngai, T., Li, J., Ogino, K., & Xia, Y. (2023). Development and Optimal Immune Strategy of an Alum-Stabilized Pickering emulsion for Cancer Vaccines. Vaccines, 11(7), 1169. https://doi.org/10.3390/vaccines11071169