
Mafosfamide Boosts GMI-HBVac against HBV via Treg Depletion in HBV-Infected Mice


Abstract
1. Introduction
2. Materials and Methods
2.1. Animals, Viruses, and Reagents
2.2. CHB Animal Model
2.3. Immunization
2.4. Delayed-Type Hypersensitivity (DTH) Assay
2.5. Isolation of Splenocytes
2.6. Isolation of T Cells (CD8+ T Cells, CD4+ T Cells)
2.7. Cell Culture for Intracellular Cytokine Analysis
2.8. In Vitro T Cell Proliferation Assay
2.9. Serological and Biochemical Analysis
2.10. HBV DNA Quantitation
2.11. In Vivo Cytotoxic Lysis Assay
2.12. Histology and Immunohistochemistry (IHC) Analysis
2.13. Flow Cytometry (FCM)
2.14. Statistics
3. Results
3.1. Production of rAAV8-1.3HBV-Infected Mice and Immune Tolerance Facilitated by Increased CD4+CD25+Foxp3+ Tregs
3.2. Low Concentrations of Mafosfamide (a Cyclophosphamide) Contribute to Tregs Decrease, with Minimal Detrimental Effects In Vivo
3.3. Immunization of Mafosfamide Plus GMI-HBVac Breaks Immune Tolerance and Enhances the Function of DCs (CD11b+CD11c+) in rAAV8-1.3HBV-Infected Mice
3.4. Mafosfamide Plus GMI-HBVac Induces Robust T-Cell Responses and Facilitates Cytokine Secretion in the Peripheral Immune System
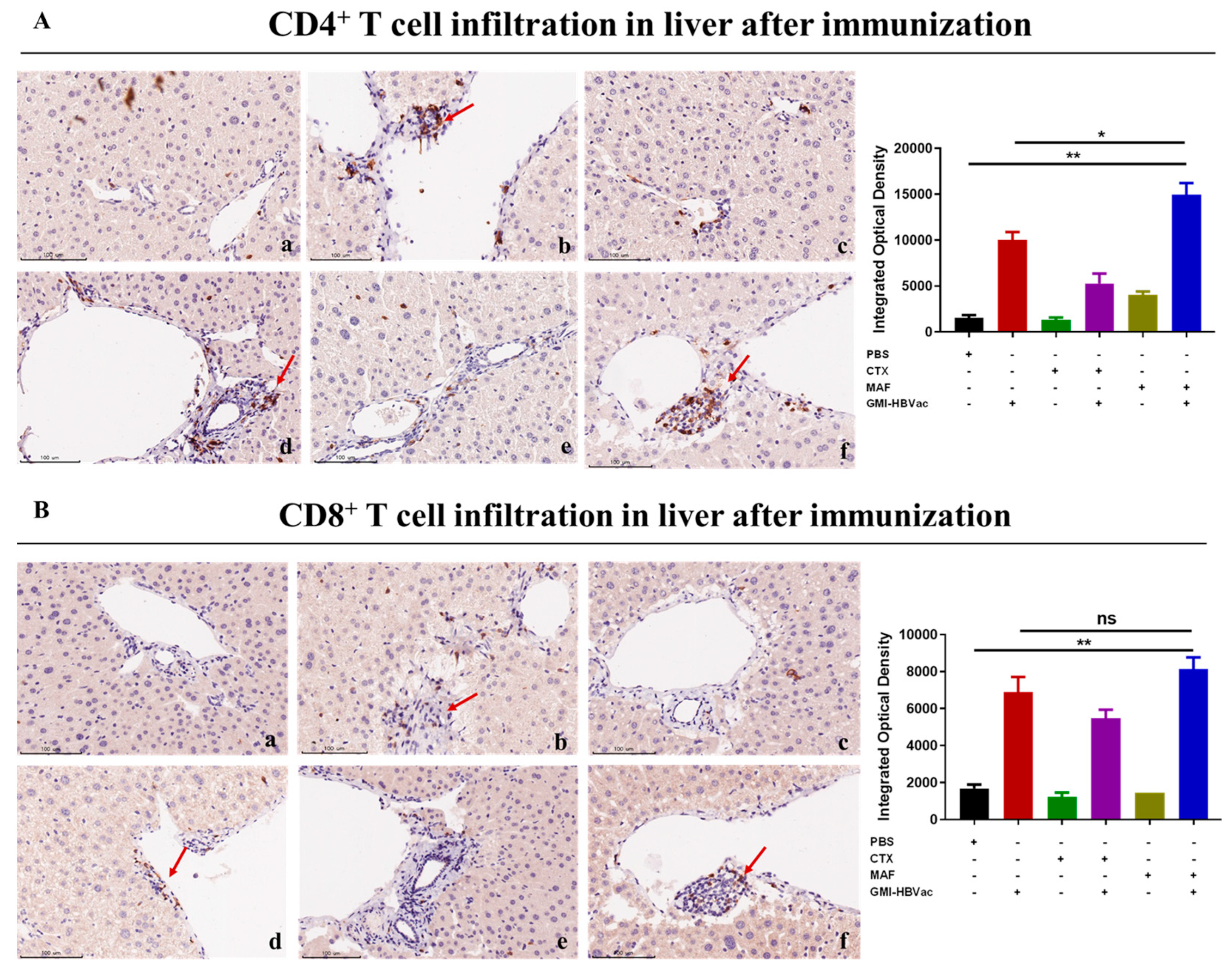
3.5. Mafosfamide Plus GMI-HBVac Promotes the Infiltration of T Cells and Enhances IFN-γ Secretion in the Liver Microenvironment of rAAV8-1.3HBV-Infected Mice
3.6. Mafosfamide Plus GMI-HBVac Vaccination Contributes to HBsAg Seroclearance and HBV-Infected Hepatocyte Clearance in Chronic HBV Infection
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lok, A.S.; McMahon, B.J. Chronic hepatitis B. Hepatology 2001, 34, 1225–1241. [Google Scholar] [CrossRef] [PubMed]
- Sheena, B.S.; Hiebert, L.; Han, H.; Ippolito, H.; Abbasi-Kangevari, M.; Abbasi-Kangevari, Z.; Abbastabar, H.; Abdoli, A.; Ali, H.A.; Adane, M.M.; et al. (GBD 2019 Hepatitis B Collaborators) Global, regional, and national burden of hepatitis B, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet Gastroenterol. Hepatol. 2022, 7, 796–829. [Google Scholar] [CrossRef] [PubMed]
- Razavi-Shearer, D.; Gamkrelidze, I.; Nguyen, M.H.; Chen, D.S.; Van Damme, P.; Abbas, Z.; Abdulla, M.; Abou Rached, A.; Adda, D.; Aho, I.; et al. Global prevalence, treatment, and prevention of hepatitis B virus infection in 2016: A modelling study. Lancet Gastroenterol. Hepatol. 2018, 3, 383–403. [Google Scholar] [CrossRef] [PubMed]
- WHO. Hepatitis B vaccines: WHO position paper recommendations. Vaccine 2010, 28, 589–590. [Google Scholar] [CrossRef] [PubMed]
- WHO. Global Health Sector Strategies Viral Hepatitis 2016–2021. Available online: https://www.who.int/publications/i/item/WHO-HIV-2016.06 (accessed on 17 May 2016).
- WHO. Global Health Sector Strategies on, Respectively, HIV, Viral Hepatitis and Sexually Transmitted Infections for the Period 2022–2030. Available online: https://www.who.int/publications/i/item/9789240053779 (accessed on 18 July 2022).
- Waheed, Y.; Siddiq, M.; Jamil, Z.; Najmi, M.H. Hepatitis elimination by 2030: Progress and challenges. World J. Gastroenterol. 2018, 24, 4959–4961. [Google Scholar] [CrossRef] [PubMed]
- Nayagam, S.; Thursz, M.; Sicuri, E.; Conteh, L.; Wiktor, S.; Low-Beer, D.; Hallett, T.B. Requirements for global elimination of hepatitis B: A modelling study. Lancet Infect. Dis. 2016, 16, 1399–1408. [Google Scholar] [CrossRef]
- Marangoni, F.; Zhakyp, A.; Corsini, M.; Geels, S.N.; Carrizosa, E.; Thelen, M.; Mani, V.; Prüßmann, J.N.; Warner, R.D.; Ozga, A.J.; et al. Expansion of tumor-associated Treg cells upon disruption of a CTLA-4-dependent feedback loop. Cell 2021, 184, 3998–4015.e19. [Google Scholar] [CrossRef]
- Yang, J.; Li, L. CD4+CD25+ regulatory T-cells: A potential target for treating patients with chronic HBV infection. Cell. Mol. Immunol. 2017, 14, 800–801. [Google Scholar] [CrossRef]
- Stoop, J.N.; van der Molen, R.G.; Baan, C.C.; van der Laan, L.J.W.; Kuipers, E.J.; Kusters, J.G.; Janssen, H.L.A. Regulatory T Cells Contribute to the Impaired Immune Response in Patients with Chronic Hepatitis B Virus Infection. Hepatology 2005, 41, 771–778. [Google Scholar] [CrossRef]
- Peng, G.; Li, S.; Wu, W.; Sun, Z.; Chen, Y.; Chen, Z. Circulating CD4+ CD25+ regulatory T cells correlate with chronic hepatitis B infection. Immunology 2008, 123, 57–65. [Google Scholar] [CrossRef]
- Vargas, F.A.; Furness, A.J.; Solomon, I.; Joshi, K.; Mekkaoui, L.; Lesko, M.H.; Rota, E.M.; Dahan, R.; Georgiou, A.; Sledzinska, A.; et al. Fc-Optimized Anti-CD25 Depletes Tumor-Infiltrating Regulatory T Cells and Synergizes with PD-1 Blockade to Eradicate Established Tumors. Immunity 2017, 46, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Buchan, S.L.; Dou, L.; Remer, M.; Booth, S.G.; Dunn, S.N.; Lai, C.; Semmrich, M.; Teige, I.; Mårtensson, L.; Penfold, C.A.; et al. Antibodies to Costimulatory Receptor 4-1BB Enhance Anti-tumor Immunity via T Regulatory Cell Depletion and Promotion of CD8 T Cell Effector Function. Immunity 2018, 49, 958–970. [Google Scholar] [CrossRef] [PubMed]
- Mills, K.H. Regulatory T cells: Friend or foe in immunity to infection? Nat. Rev. Immunol. 2004, 4, 841–855. [Google Scholar] [CrossRef] [PubMed]
- Joller, N.; Lozano, E.; Burkett, P.R.; Patel, B.; Xiao, S.; Zhu, C.; Xia, J.; Tan, T.G.; Sefik, E.; Yajnik, V.; et al. Treg Cells Expressing the Coinhibitory Molecule TIGIT Selectively Inhibit Proinflammatory Th1 and Th17 Cell Responses. Immunity 2014, 40, 569–581. [Google Scholar] [CrossRef]
- Motoyoshi, Y.; Kaminoda, K.; Saitoh, O.; Hamasaki, K.; Nakao, K.; Ishii, N.; Nagayama, Y.; Eguchi, K. Different mechanisms for antitumor effects of low- and high-dose cyclophosphamide. Oncol. Rep. 2006, 16, 141–146. [Google Scholar]
- Emadi, A.; Jones, R.J.; Brodsky, R.A. Cyclophosphamide and cancer: Golden anniversary. Nat. Rev. Clin. Oncol. 2009, 6, 638–647. [Google Scholar] [CrossRef]
- Sistigu, A.; Viaud, S.; Chaput, N.; Bracci, L.; Proietti, E.; Zitvogel, L. Immunomodulatory effects of cyclophosphamide and implementations for vaccine design. Semin. Immunopathol. 2011, 33, 369–383. [Google Scholar] [CrossRef]
- Joy, M.S.; La, M.; Wang, J.; Bridges, A.S.; Hu, Y.; Hogan, S.L.; Frye, R.F.; Blaisdell, J.; Goldstein, J.A.; Dooley, M.A.; et al. Cyclophosphamide and 4-hydroxycyclophosphamide pharmacokinetics in patients with glomerulonephritis secondary to lupus and small vessel vasculitis. Br. J. Clin. Pharmacol. 2012, 74, 445–455. [Google Scholar] [CrossRef]
- Goldstein, M.; Roos, W.P.; Kaina, B. Apoptotic death induced by the cyclophosphamide analogue mafosfamide in human lymphoblastoid cells: Contribution of DNA replication, transcription inhibition and Chk/p53 signaling. Toxicol. Appl. Pharmacol. 2008, 229, 20–32. [Google Scholar] [CrossRef]
- Helft, J.; Böttcher, J.; Chakravarty, P.; Zelenay, S.; Huotari, J.; Schraml, B.U.; Goubau, D.; e Sousa, C.R. GM-CSF Mouse Bone Marrow Cultures Comprise a Heterogeneous Population of CD11c+MHC II+ Macrophages and Dendritic Cells. Immunity 2015, 42, 1197–1211. [Google Scholar] [CrossRef]
- Becher, B.; Tugues, S.; Greter, M. GM-CSF: From Growth Factor to Central Mediator of Tissue Inflammation. Immunity 2016, 45, 963–973. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Y.; Xu, Y.; Lew, A.M. The regulation of the development and function of dendritic cell subsets by GM-CSF: More than a hematopoietic growth factor. Mol. Immunol. 2012, 52, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Lucifora, J.; Xia, Y.; Reisinger, F.; Zhang, K.; Stadler, D.; Cheng, X.; Sprinzl, M.F.; Koppensteiner, H.; Makowska, Z.; Volz, T.; et al. Specific and Nonhepatotoxic Degradation of Nuclear Hepatitis B Virus cccDNA. Science 2014, 343, 1221–1228. [Google Scholar] [CrossRef] [PubMed]
- Gibbert, K.; Schlaak, J.; Yang, D.; Dittmer, U. IFN-a subtypes: Distinct biological activities in antiviral therapy. Br. J. Pharmacol. 2013, 168, 1048–1058. [Google Scholar] [CrossRef]
- Zhao, W.; Zhao, G.; Zhang, S.; Wang, X.; Yu, X.; Wang, B. Clearance of HBeAg and HBsAg of HBV in mice model by a recombinant HBV vaccine combined with GM-CSF and IFN-α as an effective therapeutic vaccine adjuvant. Oncotarget 2018, 9, 34213–34228. [Google Scholar] [CrossRef]
- Wang, X.; Dong, A.; Xiao, J.; Zhou, X.; Mi, H.; Xu, H.; Zhang, J.; Wang, B. Overcoming HBV immune tolerance to eliminate HBsAg-positive hepatocytes via pre-administration of GM-CSF as a novel adjuvant for a hepatitis B vaccine in HBV transgenic mice. Cell. Mol. Immunol. 2016, 13, 850–861. [Google Scholar] [CrossRef]
- Coccia, M.; Collignon, C.; Hervé, C.; Chalon, A.; Welsby, I.; Detienne, S.; Van Helden, M.J.; Dutta, S.; Genito, C.J.; Waters, N.C.; et al. Cellular and molecular synergy in AS01-adjuvanted vaccines results in an early IFN-γ response promoting vaccine immunogenicity. Nat. Partn. J. Vaccines 2017, 2, 25. [Google Scholar]
- Li, H.J.; Zhai, N.C.; Song, H.X.; Yang, Y.; Cui, A.; Li, T.Y.; Tu, Z.K. The Role of Immune Cells in Chronic HBV Infection. J. Clin. Transl. Hepatol. 2015, 3, 277–283. [Google Scholar]
- Guidotti, L.G.; Inverso, D.; Sironi, L.; Di Lucia, P.; Fioravanti, J.; Ganzer, L.; Fiocchi, A.; Vacca, M.; Aiolfi, R.; Sammicheli, S.; et al. Immunosurveillance of the liver by intravascular effector CD8(+) T cells. Cell 2015, 161, 486–500. [Google Scholar] [CrossRef]
- Cheng, Y.; Gunasegaran, B.; Singh, H.D.; Dutertre, C.-A.; Loh, C.Y.; Lim, J.Q.; Crawford, J.C.; Lee, H.K.; Zhang, X.; Lee, B.; et al. Non-terminally exhausted tumor-resident memory HBV-specific T cell responses correlate with relapse-free survival in hepatocellular carcinoma. Immunity 2021, 54, 1825–1840. [Google Scholar] [CrossRef]
- Biswas, S.; Rust, L.N.; Wettengel, J.M.; Yusova, S.; Fischer, M.; Carson, J.N.; Johnson, J.; Wei, L.; Thode, T.; Kaadige, M.R.; et al. Long-term hepatitis B virus infection of rhesus macaques requires suppression of host immunity. Nat. Commun. 2022, 13, 2995. [Google Scholar] [CrossRef] [PubMed]
- Gocher-Demske, A.M.; Cui, J.; Szymczak-Workman, A.L.; Vignali, K.M.; Latini, J.N.; Pieklo, G.P.; Kimball, J.C.; Avery, L.; Cipolla, E.M.; Huckestein, B.R.; et al. IFNγ-induction of TH1-like regulatory T cells controls antiviral responses. Nat. Immunol. 2023, 24, 841–854. [Google Scholar] [CrossRef]
- Gao, J.; Shi, L.Z.; Zhao, H.; Chen, J.; Xiong, L.; He, Q.; Chen, T.; Roszik, J.; Bernatchez, C.; Woodman, S.E.; et al. Loss of IFN-gama Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell 2016, 167, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Crouse, J.; Kalinke, U.; Oxenius, A. Regulation of antiviral T cell responses by type I interferons. Nat. Rev. Immunol. 2015, 15, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Watashi, K.; Liang, G.; Iwamoto, M.; Marusawa, H.; Uchida, N.; Daito, T.; Kitamura, K.; Muramatsu, M.; Ohashi, H.; Kiyohara, T.; et al. Interleukin-1 and tumor necrosis factor-α trigger restriction of hepatitis B virus infection via a cytidine deaminase activation-induced cytidine deaminase (AID). J. Biol. Chem. 2013, 288, 31715–31727. [Google Scholar] [CrossRef] [PubMed]
- Jergović, M.; Coplen, C.P.; Uhrlaub, J.L.; Besselsen, D.G.; Cheng, S.; Smithey, M.J.; Nikolich-Žugich, J. Infection-induced type I interferons critically modulate the homeostasis and function of CD8+ naïve T cells. Nat. Commun. 2021, 12, 5303. [Google Scholar] [CrossRef] [PubMed]
- Solans, L.; Debrie, A.S.; Borkner, L.; Aguiló, N.; Thiriard, A.; Coutte, L.; Uranga, S.; Trottein, F.; Martín, C.; Mills, K.H.; et al. IL-17-dependent SIgA-mediated protection against nasal Bordetella pertussis infection by live attenuated BPZE1 vaccine. Mucosal Immunol. 2018, 11, 1753–1762. [Google Scholar] [CrossRef]
- Fang, Z.; Li, J.; Yu, X.; Zhang, D.; Ren, G.; Shi, B.; Wang, C.; Kosinska, A.D.; Wang, S.; Zhou, X.; et al. Polarization of Monocytic Myeloid-Derived Suppressor Cells by Hepatitis B Surface Antigen Is Mediated via ERK/IL-6/STAT3 Signaling Feedback and Restrains the Activation of T Cells in Chronic Hepatitis B Virus Infection. J. Immunol. 2015, 195, 4873–4883. [Google Scholar] [CrossRef]
- Pallett, L.J.; Gill, U.S.; Quaglia, A.; Sinclair, L.V.; Jover-Cobos, M.; Schurich, A.; Singh, K.P.; Thomas, N.; Das, A.; Chen, A.; et al. Metabolic regulation of hepatitis B immunopathology by myeloid-derived suppressor cells. Nat. Med. 2015, 21, 591–600. [Google Scholar] [CrossRef]
- Zoulim, F.; Fournier, C.; Habersetzer, F.; Sprinzl, M.; Pol, S.; Coffin, C.S.; Leroy, V.; Ma, M.; Wedemeyer, H.; Lohse, A.W.; et al. Safety and immunogenicity of single and multiple injections of the therapeutic vaccine TG1050 in NUC- suppressed chronic hepatitis B (CHB) patients: Unblinded analysis of a double- blind, placebo- controlled phase 1b study [abstract]. Hepatology 2018, 68, 252A. [Google Scholar]
- Hu, Y.; Wang, X.M.; Wang, Z.M.; Gao, D.; Zhang, L.; Wu, M.; Zhang, H.; Hu, Z.; Qiu, S.; Chen, H.; et al. A phase 1 clinical trial of therapeutic vaccine t101 in chronic hepatitis b patients: A randomized, double- blind, placebo controlled, single and multiple injections, dose escalation study [abstract]. J. Hepatol. 2019, 70, e153. [Google Scholar] [CrossRef]
- Boni, C.; Janssen, H.L.; Rossi, M.; Yoon, S.K.; Vecchi, A.; Barili, V.; Yoshida, E.M.; Trinh, H.; Rodell, T.C.; Laccabue, D.; et al. Combined GS-4774 and Tenofovir Therapy Can Improve HBV-Specific T-Cell Responses in Patients with Chronic Hepatitis. Gastroenterology 2019, 157, 227–241.e7. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.S.; Mutimer, D.; Heo, J.; Tak, W.Y.; Rosenberg, W.; KukJang, B.; Kim, Y.; Forton, D.; Tasker, S.; Georges, B. A phase 1b evaluation of HepTcell HBV- specific immunotherapy in nuc- controlled, eAg negative chronic HBV infection [abstract]. J. Hepatol. 2019, 70, e50. [Google Scholar] [CrossRef]
- Bourgine, M.; Crabe, S.; Lobaina, Y.; Guillen, G.; Aguilar, J.C.; Michel, M.L. Nasal route favors the induction of CD4+ T cell responses in the liver of HBV-carrier mice immunized with a recombinant hepatitis B surface-and core-based therapeutic vaccine. Antivir. Res. 2018, 153, 23–32. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Dose |
|---|---|
| PBS | 150 μL |
| GM-CSF + IFN-α + rHBVvac (GMI-HBVac) | 10 μg + 10,000 IU + 1 μg |
| CTX | 15 mg/kg |
| CTX + GMI-HBVac | 15 mg/kg + 10 μg + 10,000 IU + 1 μg |
| MAF | 2 μg/mL |
| MAF + GMI-HBVac | 2 μg/mL + 10 μg + 10,000 IU + 1 μg |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, Q.; Zhong, Y.; Wang, B. Mafosfamide Boosts GMI-HBVac against HBV via Treg Depletion in HBV-Infected Mice. Vaccines 2023, 11, 1026. https://doi.org/10.3390/vaccines11061026
Lin Q, Zhong Y, Wang B. Mafosfamide Boosts GMI-HBVac against HBV via Treg Depletion in HBV-Infected Mice. Vaccines. 2023; 11(6):1026. https://doi.org/10.3390/vaccines11061026
Chicago/Turabian StyleLin, Qin, Yiwei Zhong, and Bin Wang. 2023. "Mafosfamide Boosts GMI-HBVac against HBV via Treg Depletion in HBV-Infected Mice" Vaccines 11, no. 6: 1026. https://doi.org/10.3390/vaccines11061026
APA StyleLin, Q., Zhong, Y., & Wang, B. (2023). Mafosfamide Boosts GMI-HBVac against HBV via Treg Depletion in HBV-Infected Mice. Vaccines, 11(6), 1026. https://doi.org/10.3390/vaccines11061026