
Safety and Immunogenicity of Betuvax-CoV-2, an RBD-Fc-Based SARS-CoV-2 Recombinant Vaccine: Preliminary Results of the First-in-Human, Randomized, Double-Blind, Placebo-Controlled Phase I/II Clinical Trial

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Methods
2.1. Study Design and Participants
2.2. Randomization and Masking
2.3. Formulations and Dosages of the Test Drug
2.4. Procedures
2.5. Cell Immunity Assessment
2.6. Primary Outcomes
2.7. Secondary Outcomes
2.7.1. Safety Outcomes
2.7.2. Immunogenicity Outcomes
2.8. Statistical Analysis
2.9. Role of the Funding Source
3. Results
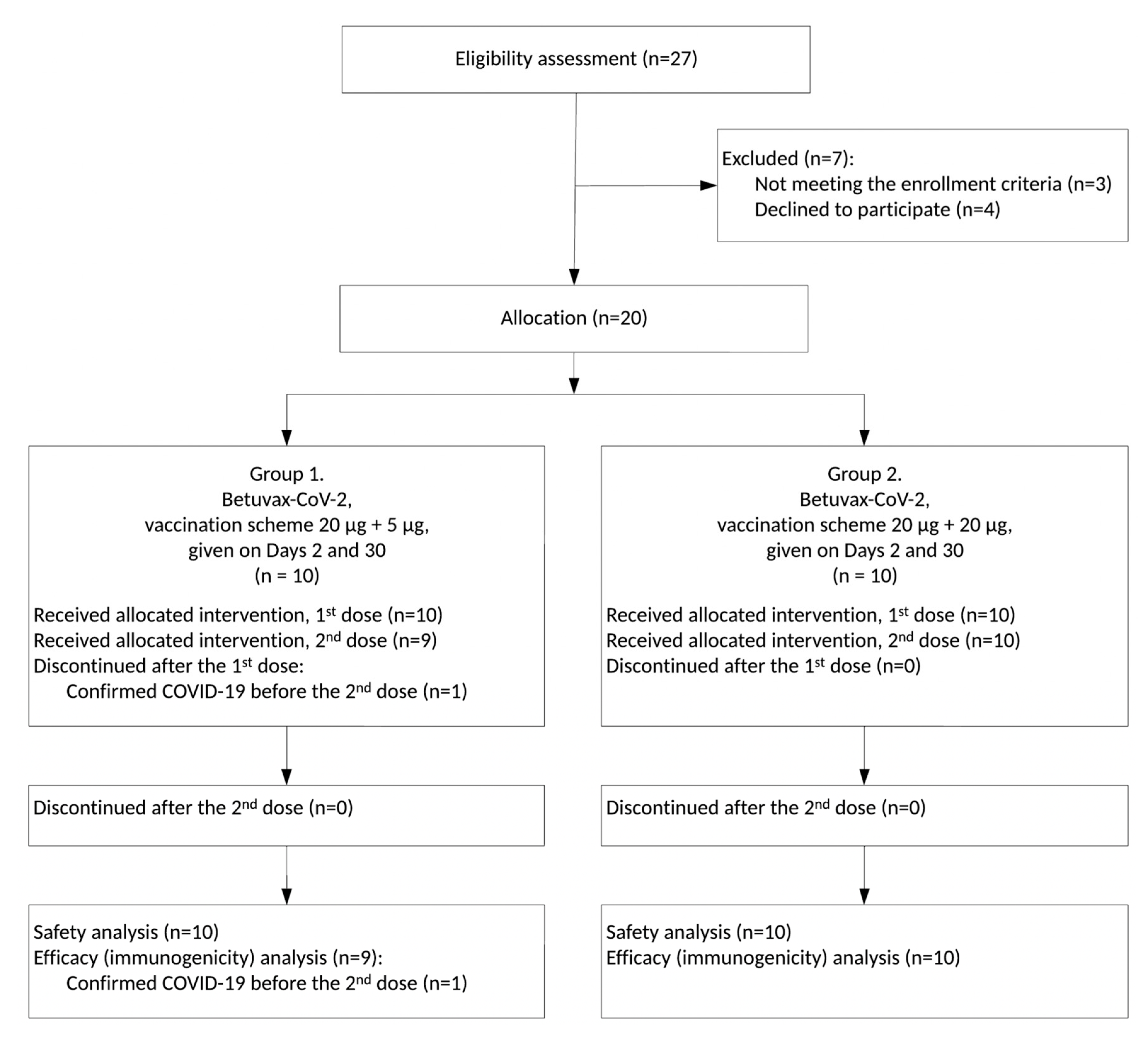
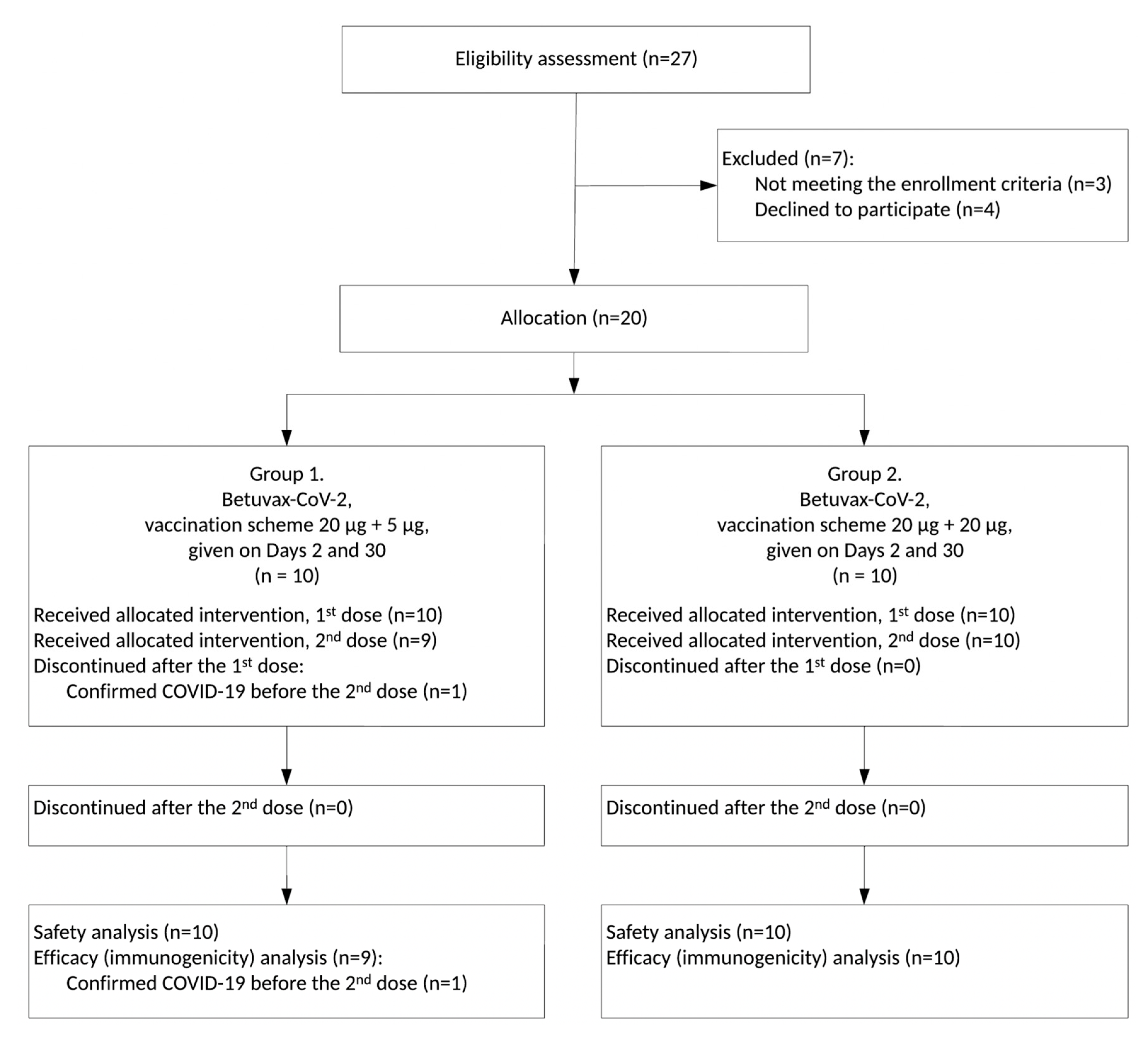
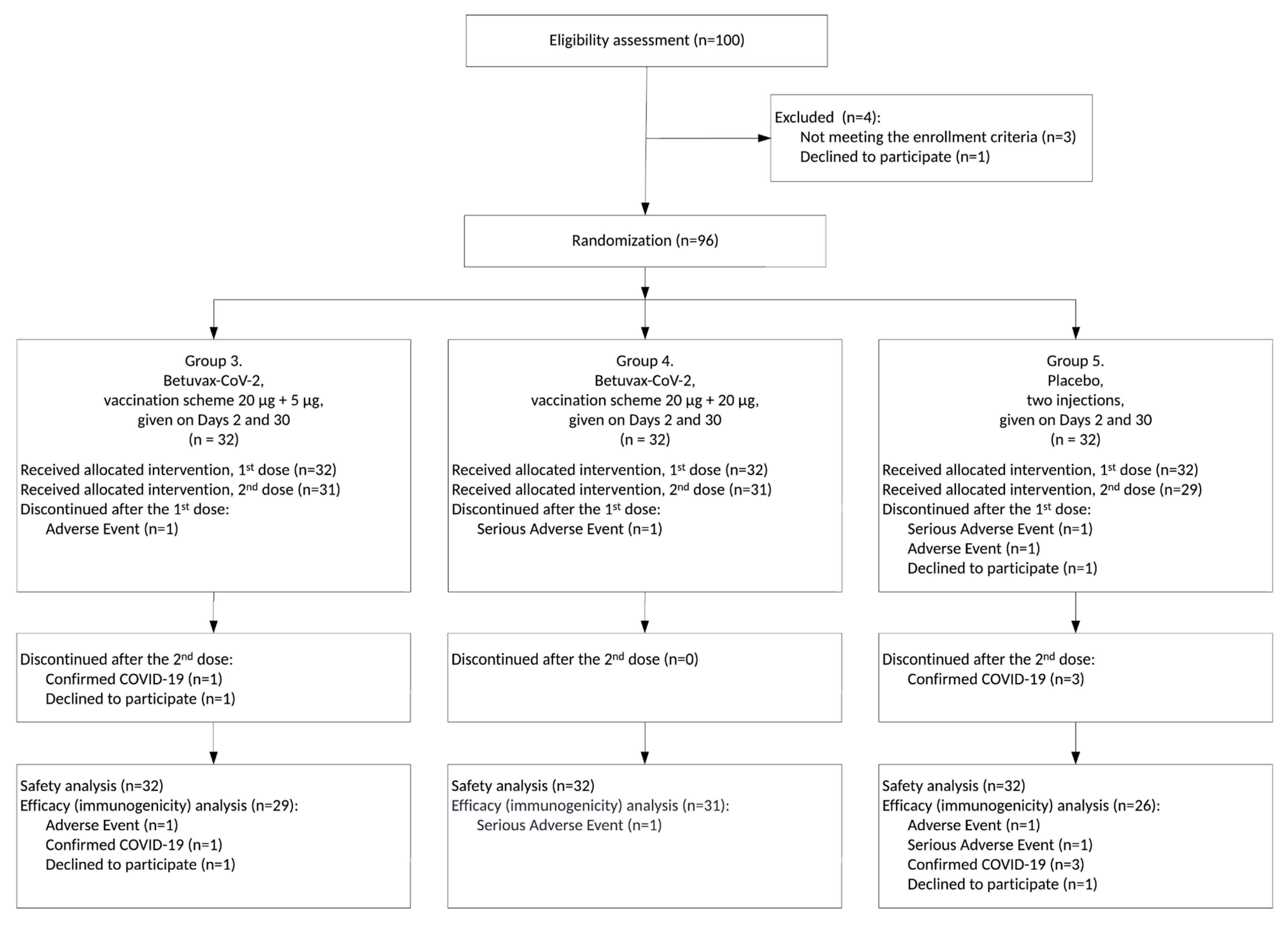
3.1. Study Design
3.2. Safety and Tolerability
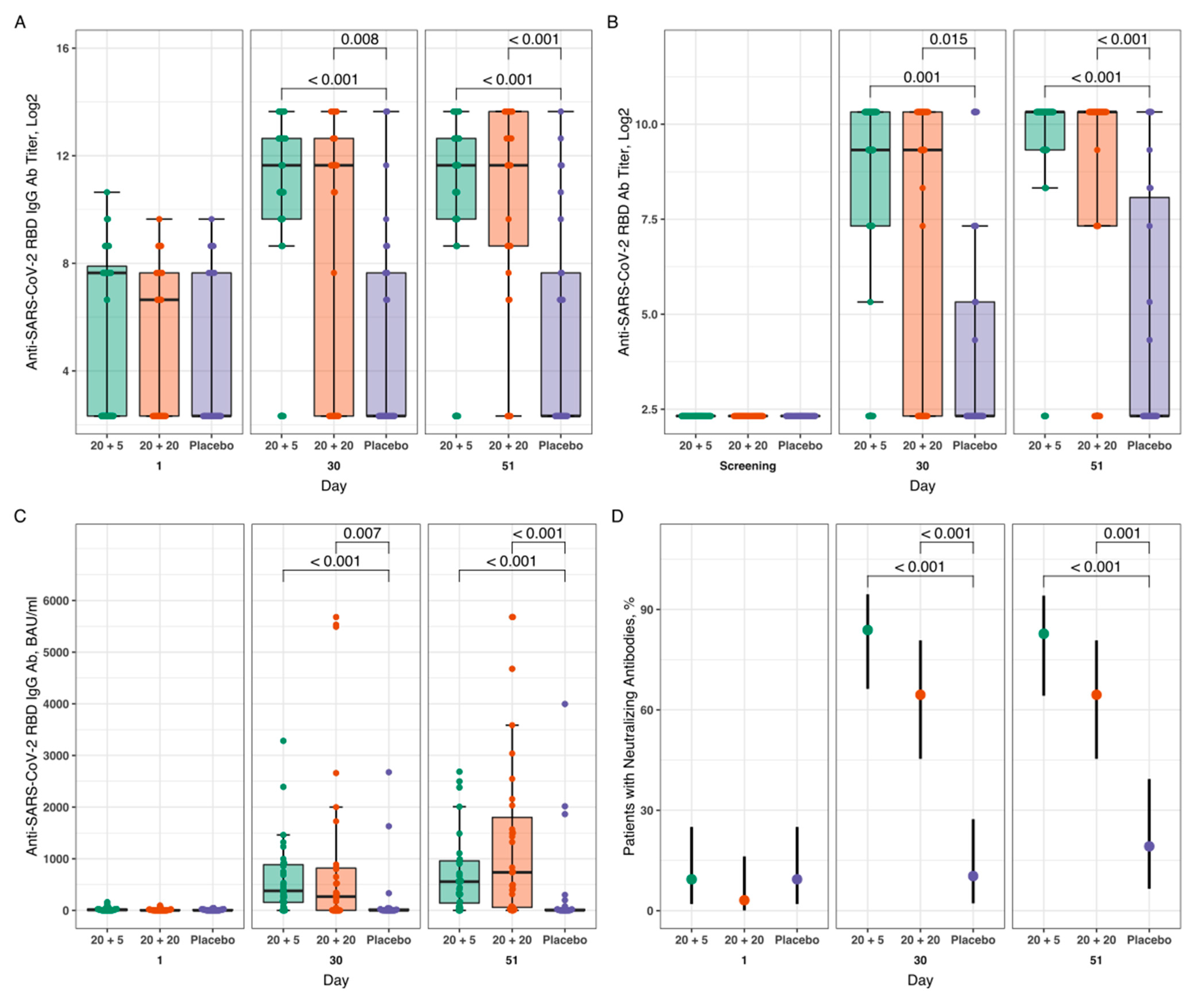
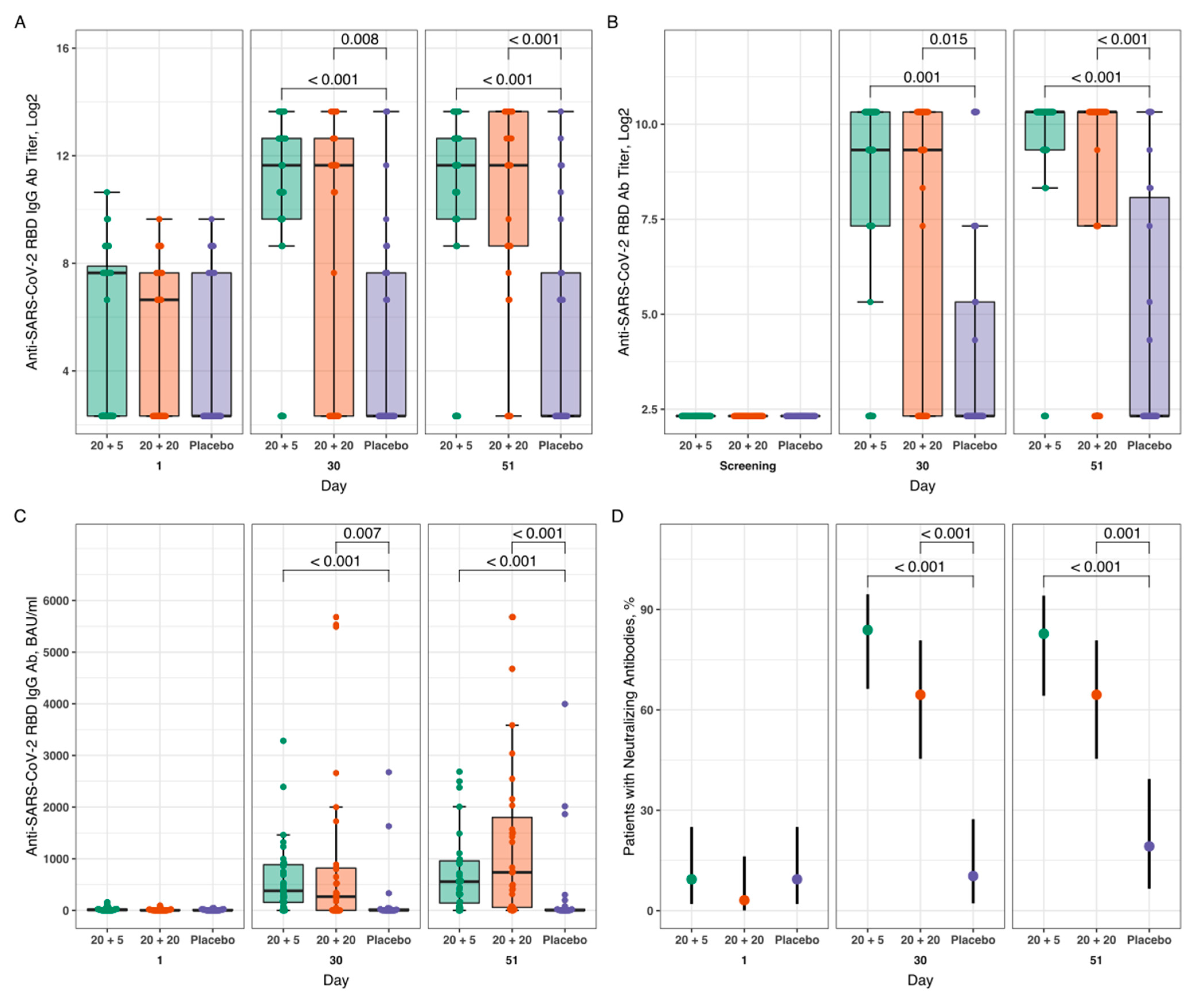
3.3. Humoral Immune Response
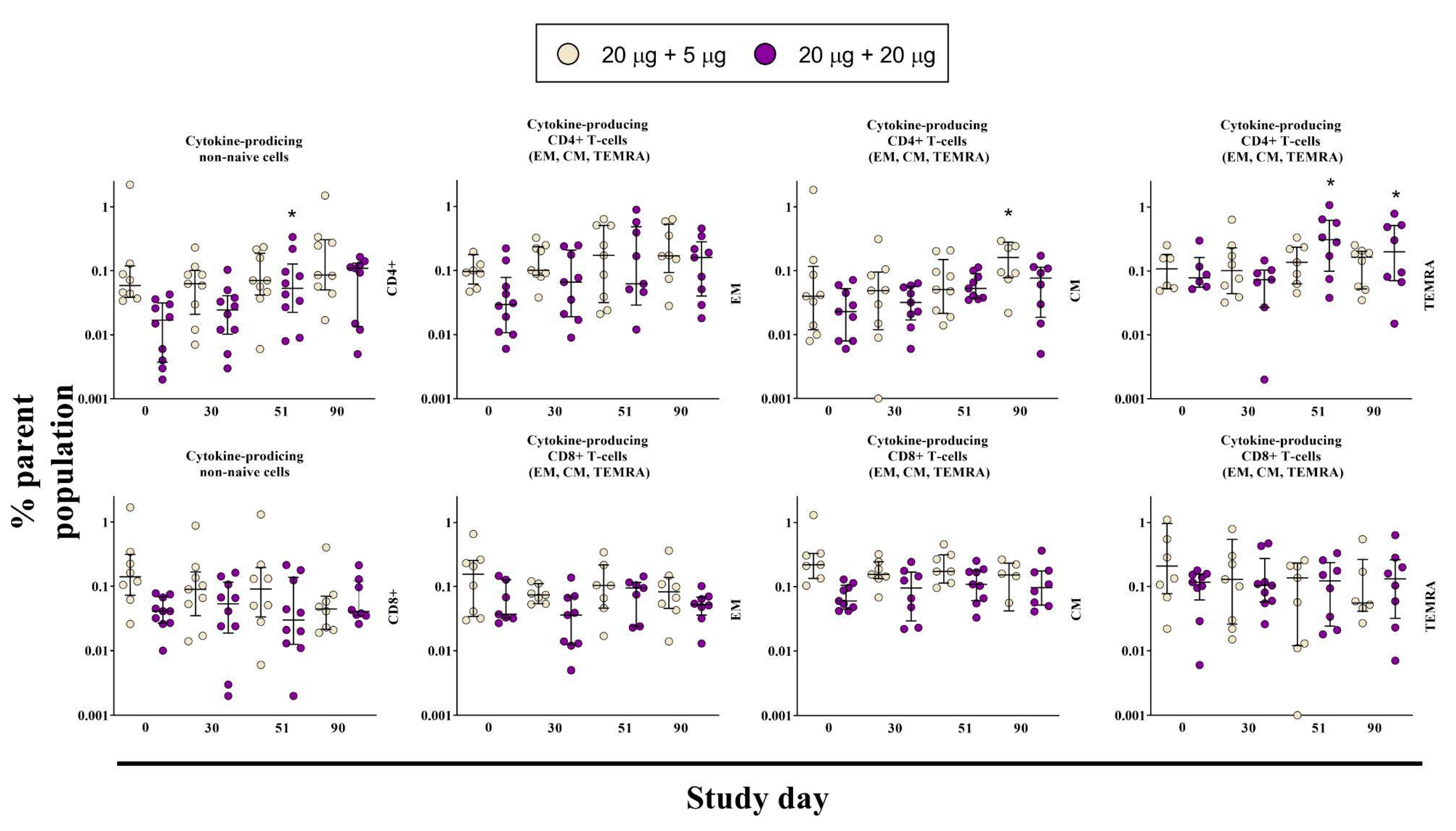
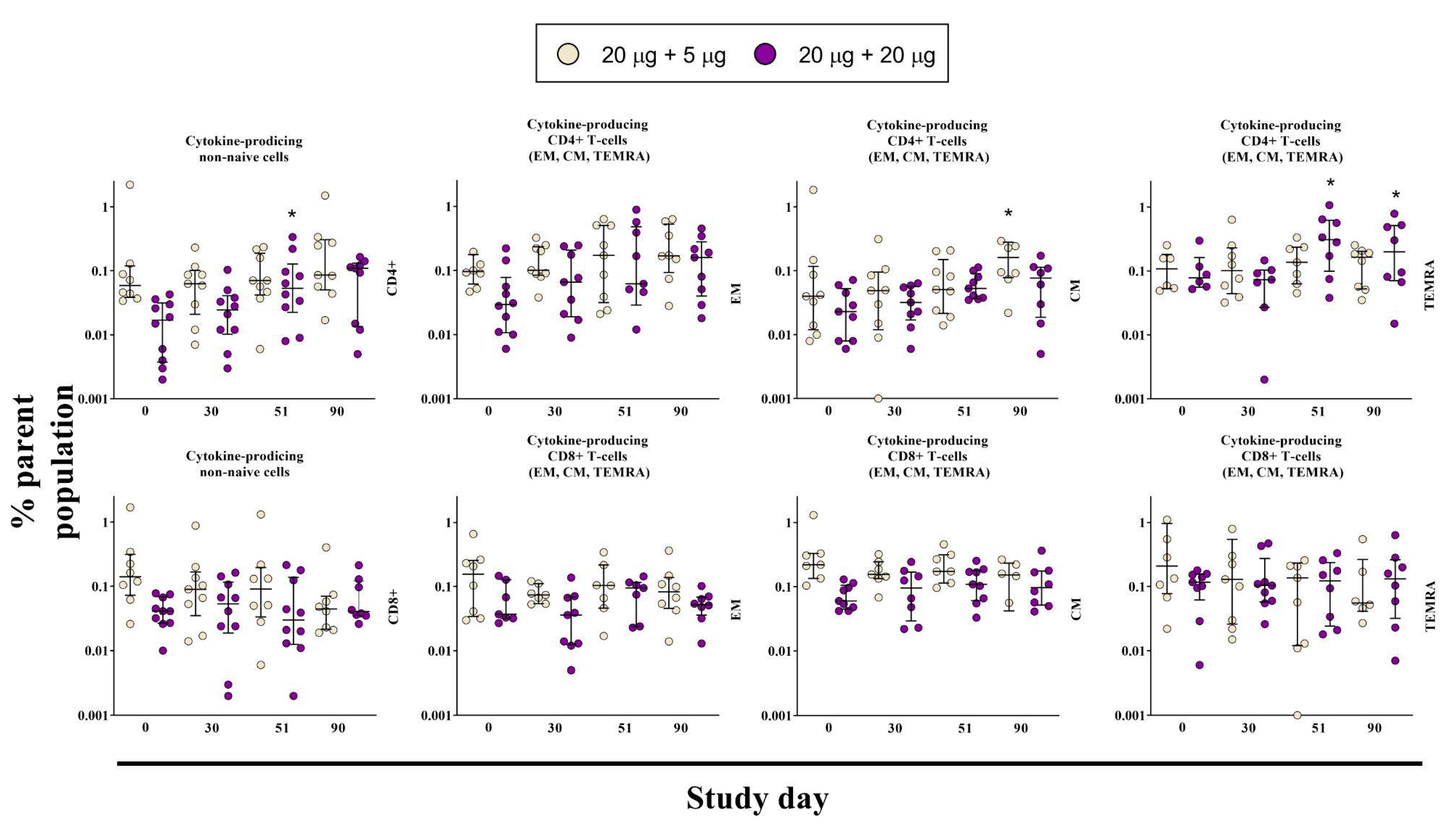
3.4. Cell-Mediated Immune Response
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. WHO Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int/ (accessed on 19 October 2022).
- Kudriavtsev, A.V.; Vakhrusheva, A.V.; Novoseletsky, V.N.; Bozdaganyan, M.E.; Shaitan, K.V.; Kirpichnikov, M.P.; Sokolova, O.S. Immune Escape Associated with RBD Omicron Mutations and SARS-CoV-2 Evolution Dynamics. Viruses 2022, 14, 1603. [Google Scholar] [CrossRef] [PubMed]
- Krasilnikov, I.V.; Kudriavtsev, A.V.; Vakhrusheva, A.V.; Frolova, M.E.; Ivanov, A.V.; Stukova, M.A.; Romanovskaya-Romanko, E.A.; Vasilyev, K.A.; Mushenkova, N.V.; Isaev, A.A. Design and Immunological Properties of the Novel Subunit Virus-like Vaccine against SARS-CoV-2. Vaccines 2022, 10, 69. [Google Scholar] [CrossRef] [PubMed]
- Vakhrusheva, A.V.; Kudriavtsev, A.V.; Kryuchkov, N.A.; Deev, R.V.; Frolova, M.E.; Blagodatskikh, K.A.; Djonovic, M.; Nedorubov, A.A.; Odintsova, E.; Ivanov, A.V.; et al. SARS-CoV-2 Subunit Virus-like Vaccine Demonstrates High Safety Profile and Protective Efficacy: Preclinical Study. Vaccines 2022, 10, 1290. [Google Scholar] [CrossRef]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Pérez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA COVID-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef]
- El Sahly, H.M.; Baden, L.R.; Essink, B.; Doblecki-Lewis, S.; Martin, J.M.; Anderson, E.J.; Campbell, T.B.; Clark, J.; Jackson, L.A.; Fichtenbaum, C.J.; et al. Efficacy of the mRNA-1273 SARS-CoV-2 Vaccine at Completion of Blinded Phase. N. Engl. J. Med. 2021, 385, 1774–1785. [Google Scholar] [CrossRef] [PubMed]
- Voysey, M.; Clemens, S.A.C.; Madhi, S.A.; Weckx, L.Y.; Folegatti, P.M.; Aley, P.K.; Angus, B.; Baillie, V.L.; Barnabas, S.L.; Bhorat, Q.E.; et al. Safety and efficacy of the ChAdOx1 nCoV-19 vaccine (AZD1222) against SARS-CoV-2: An interim analysis of four randomized controlled trials in Brazil, South Africa, and the UK. Lancet 2021, 397, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Sadoff, J.; Gray, G.; Vandebosch, A.; Cárdenas, V.; Shukarev, G.; Grinsztejn, B.; Goepfert, P.A.; Truyers, C.; Fennema, H.; Spiessens, B.; et al. Safety and Efficacy of Single-Dose Ad26.COV2.S Vaccine against COVID-19. N. Engl. J. Med. 2021, 384, 2187–2201. [Google Scholar] [CrossRef]
- Kitagawa, H.; Kaiki, Y.; Sugiyama, A.; Nagashima, S.; Kurisu, A.; Nomura, T.; Omori, K.; Akita, T.; Shigemoto, N.; Tanaka, J.; et al. Adverse reactions to the BNT162b2 and mRNA-1273 mRNA COVID-19 vaccines in Japan. J. Infect. Chemother. 2022, 28, 576–581. [Google Scholar] [CrossRef]
- Hernández, A.F.; Calina, D.; Poulas, K.; Docea, A.O.; Tsatsakis, A.M. Safety of COVID-19 vaccines administered in the EU: Should we be concerned? Toxicol. Rep. 2021, 1, 871–879. [Google Scholar] [CrossRef]
- Paul-Ehrlich-Institut. Safety Report; Paul-Ehrlich-Institut: Langen, Germany, 2022; pp. 1–26. [Google Scholar]
- Greinacher, A.; Thiele, T.; Warkentin, T.E.; Weisser, K.; Kyrle, P.A.; Eichinger, S. Thrombotic Thrombocytopenia after ChAdOx1 nCov-19 Vaccination. N. Engl. J. Med. 2021, 384, 2092–2101. [Google Scholar] [CrossRef]
- Anand, P.; Stahel, V.P. Review the safety of COVID-19 mRNA vaccines: A review. Patient Saf. Surg. 2021, 15, 20. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, N.; Grossegesse, M.; Neumann, M.; Schaade, L.; Nitsche, A. Evaluation of a commercial ELISA as alternative to plaque reduction neutralization test to detect neutralizing antibodies against SARS-CoV-2. Sci. Rep. 2022, 12, 3549. [Google Scholar] [CrossRef] [PubMed]
- Suntronwong, N.; Assawakosri, S.; Kanokudom, S.; Yorsaeng, R.; Auphimai, C.; Thongmee, T.; Vichaiwattana, P.; Duangchinda, T.; Chantima, W.; Pakchotanon, P.; et al. Strong Correlations between the Binding Antibodies against Wild-Type and Neutralizing Antibodies against Omicron BA.1 and BA.2 Variants of SARS-CoV-2 in Individuals Following Booster (Third-Dose) Vaccination. Diagnostics 2022, 12, 1781. [Google Scholar] [CrossRef] [PubMed]
- Perkmann, T.; Mucher, P.; Perkmann-Nagele, N.; Radakovics, A.; Repl, M.; Koller, T.; Schmetterer, K.G.; Bigenzahn, J.W.; Leitner, F.; Jordakieva, G.; et al. The Comparability of Anti-Spike SARS-CoV-2 Antibody Tests is Time-Dependent: A Prospective Observational Study. Microbiol. Spectr. 2022, 10, e0140221. [Google Scholar] [CrossRef] [PubMed]
- Naaber, P.; Tserel, L.; Kangro, K.; Sepp, E.; Jürjenson, V.; Adamson, A.; Haljasmägi, L.; Rumm, A.P.; Maruste, R.; Kärner, J.; et al. Dynamics of antibody response to BNT162b2 vaccine after six months: A longitudinal prospective study. Lancet Reg. Heal. Eur. 2021, 10, 100208. [Google Scholar] [CrossRef]
- Lau, C.S.; Phua, S.K.; Liang, Y.L.; Oh, M.L.H.; Aw, T.-C. BNT162b2 mRNA COVID-19 Vaccine Elicited Antibody Responses in COVID-19-naïve Subjects. J. Exp. Pathol. 2021, 2, 117–127. [Google Scholar]
- Damerau, L.; Mühlenbruch, G.; Evenschor-Ascheid, A.; Fussen, C.; Nienhaus, A.; Terschüren, C.; Herold, R.; Harth, V. Coronavirus Vaccination: Spike Antibody Levels in Health Workers after Six Months—A Cross-Sectional Study. Int. J. Environ. Res. Public Health. 2022, 19, 11422. [Google Scholar] [CrossRef]
- Mishra, S.K.; Pradhan, S.K.; Pati, S.; Sahu, S.; Nanda, R.K. Waning of Anti-spike Antibodies in AZD1222 (ChAdOx1) Vaccinated Healthcare Providers: A Prospective Longitudinal Study. Cureus 2021, 13, e19879. [Google Scholar] [CrossRef]
- Santonato, D.; Malvicini, M.A.; Novau, A.; Torres, S.F.; Siaba Serrate, A.; Romano, M.V.; Brenzoni, P.G.; Fabbro, L.; Paulosky, L.; Cornistein, W. Seroprevalence in Argentinian healthcare workers after vaccination with Sputnik V. Int. J. Infect. Control. 2022, 18, 21791. [Google Scholar] [CrossRef]
- Bayram, A.; Demirbakan, H.; Günel Karadeniz, P.; Erdoğan, M.; Koçer, I. Quantitation of antibodies against SARS-CoV-2 spike protein after two doses of CoronaVac in healthcare workers. J. Med. Virol. 2021, 93, 5560–5567. [Google Scholar] [CrossRef]
- Gilbert, P.B.; Montefiori, D.C.; McDermott, A.B.; Fong, Y.; Benkeser, D.; Deng, W.; Zhou, H.; Houchens, C.R.; Martins, K.; Jayashankar, L.; et al. Immune correlates analysis of the mRNA-1273 COVID-19 vaccine efficacy clinical trial. Science 2022, 375, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Dimeglio, C.; Herin, F.; Martin-Blondel, G.; Miedougé, M.; Izopet, J. Antibody titers and protection against a SARS-CoV-2 infection. J. Infect. 2022, 84, 248–288. [Google Scholar] [CrossRef]
- Feng, S.; Phillips, D.J.; White, T.; Sayal, H.; Aley, P.K.; Bibi, S.; Dold, C.; Fuskova, M.; Gilbert, S.C.; Hirsch, I.; et al. Correlates of protection against symptomatic and asymptomatic SARS-CoV-2 infection. Nat. Med. 2021, 27, 2032–2040. [Google Scholar] [CrossRef] [PubMed]
- Dimeglio, C.; Migueres, M.; Bouzid, N.; Chapuy-Regaud, S.; Gernigon, C.; Da-Silva, I.; Porcheron, M.; Martin-Blondel, G.; Herin, F.; Izopet, J. Antibody Titers and Protection against Omicron (BA.1 and BA.2) SARS-CoV-2 Infection. Vaccines 2022, 10, 1548. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | N | M ± SD | 95% CI | Min–Max | Me | IQR | p | |
|---|---|---|---|---|---|---|---|---|
| Weight, kg | G1 | 10 | 64.2 ± 10.6 | 56.6–71.8 | 48.0–79.0 | 63.0 | 13.0 | 0.571 2 |
| G2 | 10 | 67.3 ± 13.3 | 57.8–76.8 | 50.0–90.0 | 65.0 | 20.0 | ||
| Age, years | G1 | 10 | - | - | 26.1–57.5 | 32.2 | 6.9 | 0.853 1 |
| G2 | 10 | - | - | 18.3–51.4 | 35.6 | 16.2 | ||
| BMI, kg/m | G1 | 10 | - | - | 18.8–29.4 | 20.6 | 5.2 | 0.853 1 |
| G2 | 10 | - | - | 18.6–29.8 | 20.4 | 4.6 | ||
| Height, cm | G1 | 10 | 170.5 ± 7.8 | 164.9–176.1 | 160.0–186.0 | 170.0 | 12.0 | 0.375 2 |
| G2 | 10 | 174.1 ± 9.8 | 167.1–181.1 | 160.0–188.0 | 173.5 | 17.0 |
| Group | N | M ± SD | 95% CI | Min–Max | Me | IQR | p | |
|---|---|---|---|---|---|---|---|---|
| Weight, kg | G3 | 32 | - | - | 55.3–96.0 | 71.9 | 15.1 | 0.157 1 |
| G4 | 32 | - | - | 50.0–100.1 | 65.1 | 12.0 | ||
| G5 | 32 | - | - | 52.0–92.3 | 67.1 | 16.7 | ||
| Age, years | G3 | 32 | - | - | 19.6–53.2 | 31.8 | 12.9 | 0.709 1 |
| G4 | 32 | - | - | 19.4–58.1 | 28.7 | 14.0 | ||
| G5 | 32 | - | - | 20.8–58.7 | 29.9 | 11.2 | ||
| BMI, kg/m | G3 | 32 | 23.6 ± 2.3 | 24.5–22.8 | 18.8–28.3 | 23.4 | 3.2 | 0.660 2 |
| G4 | 32 | 23.7 ± 2.4 | 24.6–22.9 | 19.6–28.8 | 23.8 | 4.1 | ||
| G5 | 32 | 23.2 ± 3.0 | 24.2–22.1 | 18.6–29.3 | 22.7 | 4.3 | ||
| Height, cm | G3 | 32 | - | - | 155.0–190.0 | 174.0 | 9.5 | 0.590 1 |
| G4 | 32 | - | - | 153.0–195.0 | 167.5 | 12.0 | ||
| G5 | 32 | - | - | 158.0–190.0 | 169.5 | 13.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kudriavtsev, A.V.; Vakhrusheva, A.V.; Kryuchkov, N.A.; Frolova, M.E.; Blagodatskikh, K.A.; Ivanishin, T.V.; Djonovic, M.; Romanovskaya-Romanko, E.A.; Kovalenko, A.N.; Lioznov, D.A.; et al. Safety and Immunogenicity of Betuvax-CoV-2, an RBD-Fc-Based SARS-CoV-2 Recombinant Vaccine: Preliminary Results of the First-in-Human, Randomized, Double-Blind, Placebo-Controlled Phase I/II Clinical Trial. Vaccines 2023, 11, 326. https://doi.org/10.3390/vaccines11020326
Kudriavtsev AV, Vakhrusheva AV, Kryuchkov NA, Frolova ME, Blagodatskikh KA, Ivanishin TV, Djonovic M, Romanovskaya-Romanko EA, Kovalenko AN, Lioznov DA, et al. Safety and Immunogenicity of Betuvax-CoV-2, an RBD-Fc-Based SARS-CoV-2 Recombinant Vaccine: Preliminary Results of the First-in-Human, Randomized, Double-Blind, Placebo-Controlled Phase I/II Clinical Trial. Vaccines. 2023; 11(2):326. https://doi.org/10.3390/vaccines11020326
Chicago/Turabian StyleKudriavtsev, Aleksandr V., Anna V. Vakhrusheva, Nickolay A. Kryuchkov, Maria E. Frolova, Konstantin A. Blagodatskikh, Taras V. Ivanishin, Milana Djonovic, Ekaterina A. Romanovskaya-Romanko, Anton N. Kovalenko, Dmitry A. Lioznov, and et al. 2023. "Safety and Immunogenicity of Betuvax-CoV-2, an RBD-Fc-Based SARS-CoV-2 Recombinant Vaccine: Preliminary Results of the First-in-Human, Randomized, Double-Blind, Placebo-Controlled Phase I/II Clinical Trial" Vaccines 11, no. 2: 326. https://doi.org/10.3390/vaccines11020326
APA StyleKudriavtsev, A. V., Vakhrusheva, A. V., Kryuchkov, N. A., Frolova, M. E., Blagodatskikh, K. A., Ivanishin, T. V., Djonovic, M., Romanovskaya-Romanko, E. A., Kovalenko, A. N., Lioznov, D. A., Zubkova, T. G., Teplykh, S. V., Oseshnyuk, R. A., Stukova, M. A., Isaev, A. A., & Krasilnikov, I. V. (2023). Safety and Immunogenicity of Betuvax-CoV-2, an RBD-Fc-Based SARS-CoV-2 Recombinant Vaccine: Preliminary Results of the First-in-Human, Randomized, Double-Blind, Placebo-Controlled Phase I/II Clinical Trial. Vaccines, 11(2), 326. https://doi.org/10.3390/vaccines11020326