
Delving into Molecular Pathways: Analyzing the Mechanisms of Action of Monoclonal Antibodies Integrated in IMGT/mAb-DB for Myasthenia Gravis




Abstract
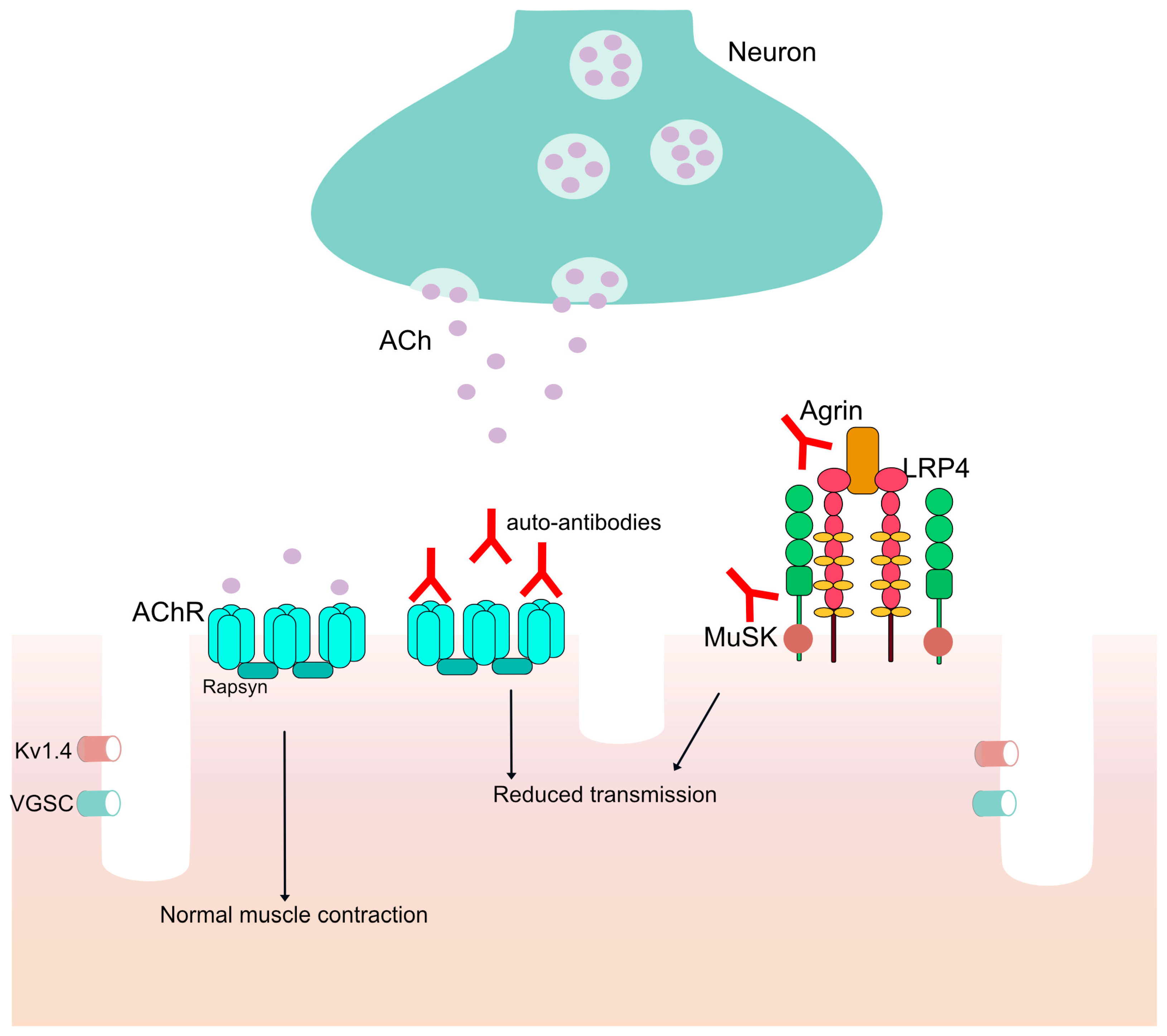
:1. Introduction
2. Methods
3. Results
3.1. Monoclonal Antibodies for Complement Inhibition
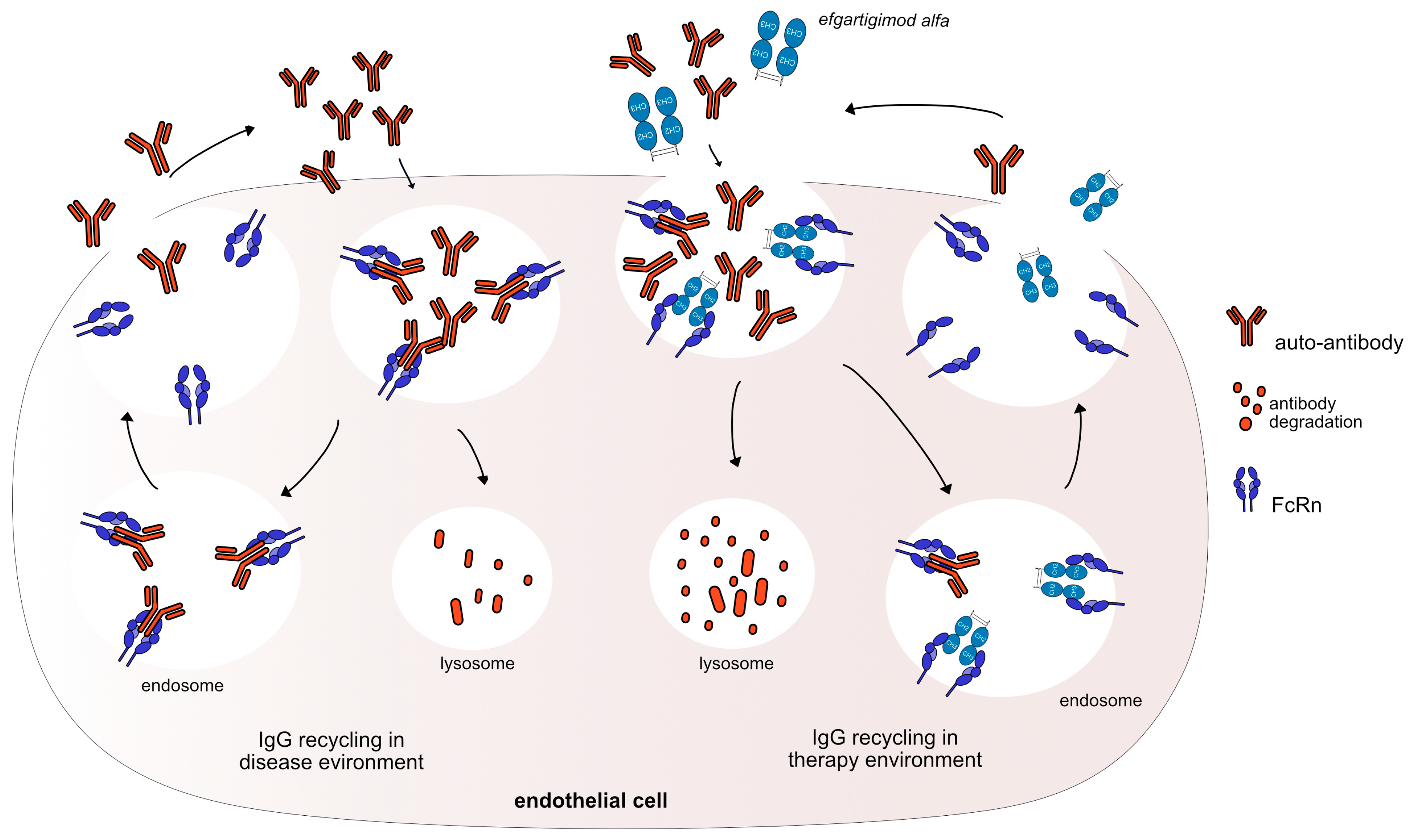
3.2. Monoclonal Antibodies as FcRn Antagonists
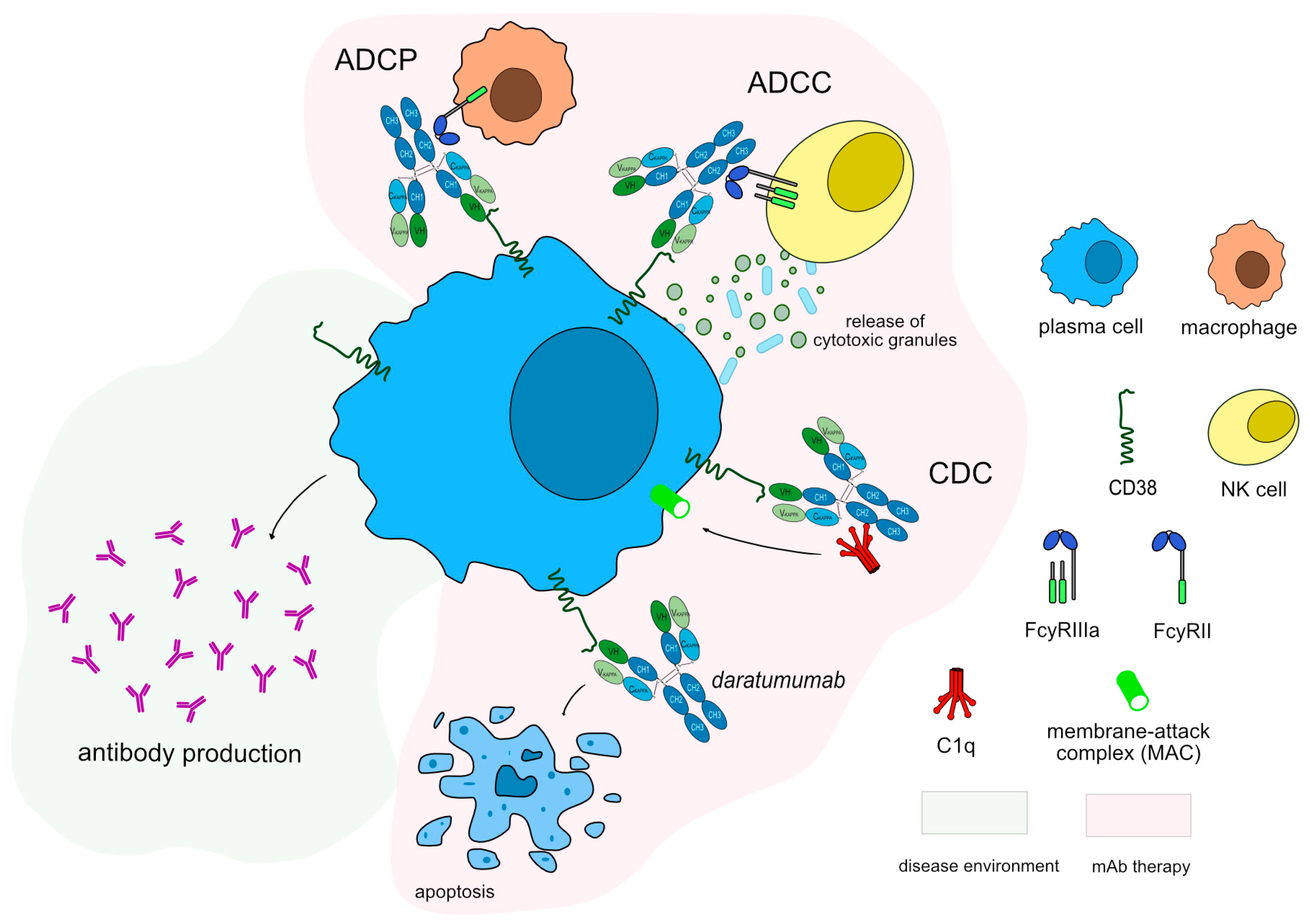
3.3. Monoclonal Antibodies for B Cell Depletion
3.4. Therapies Directed to Plasma Cell

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| INN mAbs | Target | Receptor Identification | IMGT MOA ** | Clinical Indication | IMGT Variants | FDA/EMA Approval |
|---|---|---|---|---|---|---|
| eculizumab * (mAbID 37) | C5 | IgG2–G4-kappa | Blocking ** Complement inhibitor | Dermatomyositis, Nephritis, Paroxysmal nocturnal hemoglobinuria (PNH), Psoriasis, Rheumatoid Arthritis (RA), Atypical hemolytic uremic syndrome (aHUS), Neuromyelitis optica (NMO), Myasthenia Gravis (MG), Kidney transplantation treatment of antibody-mediated rejection (AMR), Kidney transplantation prevention of delayed graft function (DGF) | EMA: October 2003 FDA: March 2007 | |
| ravulizumab * (mAbID 674) | IgG2–G4-kappa | PNH|Atypical hemolytic uremic syndrome|MG | G4v24 CH3 L107, S114 Half-life extension | EMA: May 216 FDA: December 2018 | ||
| crovalimab (mAbID 783) | IgG1-kappa | PNH | G1v94 CH2 R1.2, R1.1, K3, G110, S115, S116 ADCC and CDC reduction G1v100 CH3 R118, E120 decrease Rheumatoid factor (RF) binding Fc variants with enhanced FcRn binding G1v85 CH3 L107, A114 half life extensions | |||
| gefurulimab *, *** (mAbID 1253) | VH–VH’ | Complement component deficiency|MG | ||||
| tesidolumab (mAbID 535) | IgG1-lambda2 | Age-related macular degeneration (AMD)|Choroiditis | G1v14 CH2 A1.3, A1.2 ADCC and CDC reduction | |||
| vilobelimab *** (mAbID 1038) | IgG4-kappa | Hidradenitis suppurativa | Inflammation | ||||
| olendalizumab (mAbID 585) | IgG2–G4-kappa | Graft-versus-host disease (GvHD)|Antiphospholipid syndrome (APS) | ||||
| pozelimab * (mAbID 898) | IgG4-kappa | PNH|MG | G4v5 h P10 Half-IG exchange reduction | |||
| efgartigimod alfa * (mAbID 731) | FCGRT | Fc-gamma1 | Neutralizing ** FcRn inhibitor | MG|Primary immune thrombocytopenia (ITP)|Chronic inflammatory demyelinating polyneuropathy (CIDP) | G1v96 CH2 Y15.1, T16, E18; CH3 K113, F114 Half-life extension without pH dependency | FDA: December 2021 |
| batoclimab * (mAbID 943) | IgG1-lambda2 | Autoimmune diseases|MG | G1v14 CH2 A1.3, A1.2 ADCC and CDC reduction | |||
| rozanolixizumab * (mAbID 943) | IgG4-kappa | MG|Thrombocytopenia/Immune thrombocytopenia (ITP) | G4v5 h P10 Half-IG exchange reduction | FDA: April 2018 | ||
| nipocalimab * (mAbID 1020) | IgG1-lambda3 | Autoimmune diseases|MG | G1v29 CH2 A84.4 No N-glycosylation site ADCC reduction | |||
| orilanolimab (mAbID 854) | IgG4-kappa | Autoimmune diseases| Pemphigus vulgaris (PV)| Warm antibody autoimmune hemolytic anemia | G4v5 h P10 Half-IG exchange reduction | |||
| iscalimab *,*** (mAbID 799) | CD40 | IgG1-kappa | Blocking ** Immunosuppressant | Psoriasis|Kindey transplant rejection|MG|Sjögren’s syndrome (SjS)|Graves’ orbitopathy (GO) | G1v29 CH2 A84.4 No N-glycosylation site ADCC reduction | |
| bleselumab (mAbID 563) | IgG4-kappa | Psoriasis|Organ transplant immunological rejection suppression | G4v5 h P10 Half-IG exchange reduction G4v3 CH2 E1.2 ADCC and CDC reduction | |||
| ravagalimab (mAbID 806) | IgG1-kappa | Crohn’s disease (CD) | G1v14 CH2 A1.3, A1.2 ADCC and CDC reduction G1v42 CH2 Q14; CH3 L107 Half-life extension | |||
| inebilizumab *,*** (mAbID 553) | CD19 | IgG1-kappa | Blocking ** Immunosuppressant, Fc-effector function | MG|Multiple sclerosis (MS)|NMO|MG Scleroderma| Neuromyelitis optica spectrum disorder| Chronic lymphocytic leukemia (CLL)| Lymphoma diffuse large B cell (DLBCL) | FDA: February 2016 | |
| obexelimab (mAbID 518) | IgG1-kappa | Autoimmune diseases| RA|Systemic lupus erythematosus (SLE)| IgG4-related disease (IgG4-RD) | G1v25 CH2 E29, F113 B cell inhibition | |||
| rituximab * (mAbID 161) | MS4A1 (CD20) | IgG1-kappa | Blocking ** Immunosuppressant, Fc-effector function | CLL (CD20-positive, in combination with fludaraline and cyclophosphamide (FC))| Solid organ transplantation| RA|MG DLBCL (in combination with hyaluronidase)| Wegener’s Granulomatosis (WG) and Microscopic Polygamiitis (MPA), in combination with glucocorticoids| Non-Hodgkin’s lymphoma (NHL), follicular CD20 positive, relapsed or refractory low grade|PV| Chronic focal encephalitis (CFE)|Waldenstrom macroglobulinemia (WM) | E EMA: June 1998 FDA: November 1997 | |
| ofatumumab * (mAbID 194) | IgG1-kappa | CLL|NHL|RA|MS (relapsing remitting)| Lymphoma follicular (LF)|NMO|PV|MG | EMA: April 2010 (withdrawn) FDA: October 2009 | |||
| ublituximab (mAbID 372) | IgG1-kappa | CLL|DLBCL|NMO|MS, relapsing-remitting | FDA: August 2016 | |||
| ocrelizumab (mAbID 227) | IgG1-kappa | RA|SLE|MS| Lupus Nephritis| Primary progressive multiple sclerosis (PPMS) | FDA: March 2017 | |||
| divozilimab (mAbID 1060) | IgG1-kappa | MS, relapsing-remitting|Systemic Scleroderma|NMOSD | ||||
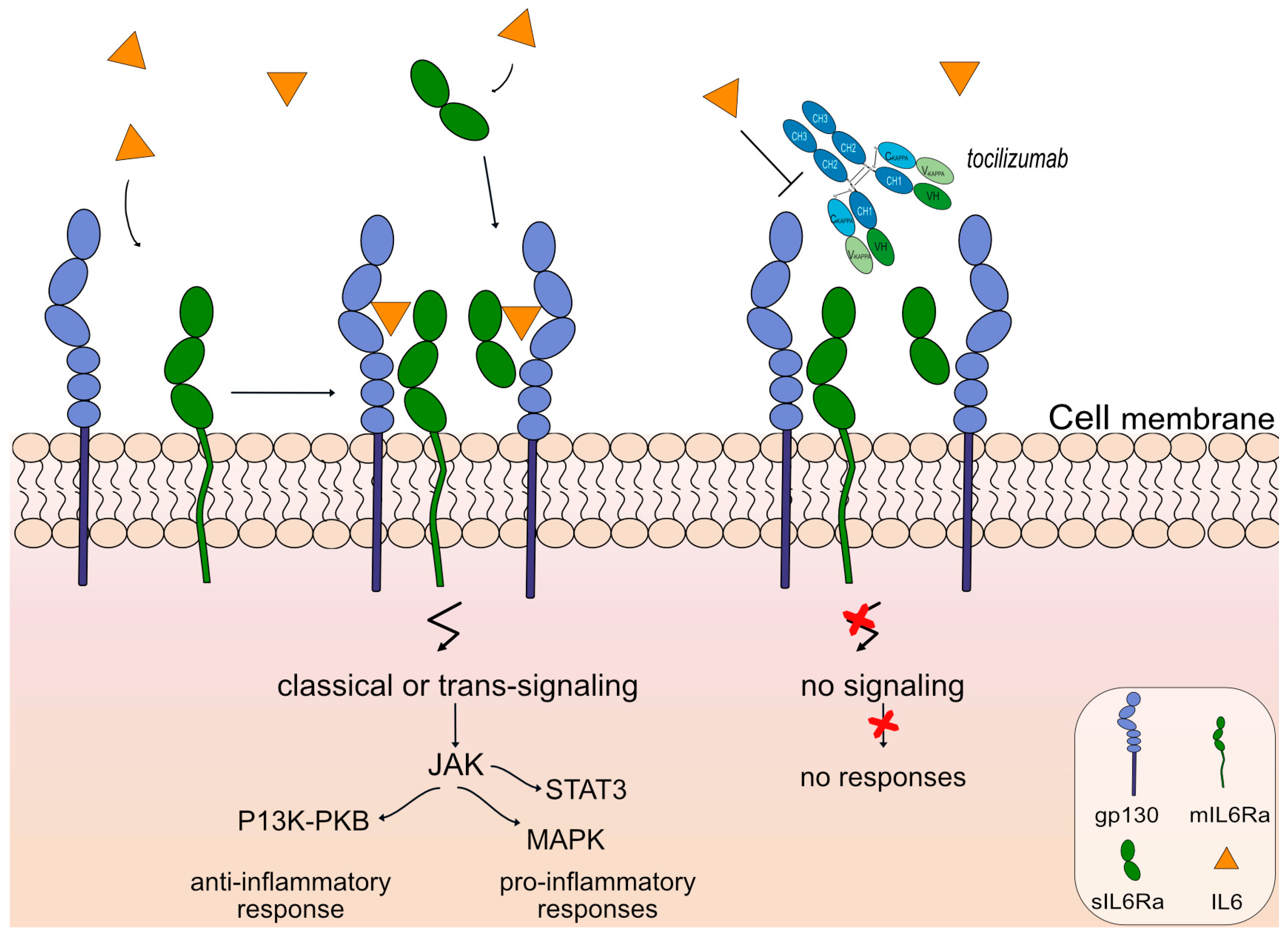
| tocilizumab * (mAbID 96) | IL6R | IgG1-kappa | Blocking ** Immunosuppressant | Lymphoproliferative disorder giant lymph node hyperplasia (Castleman’s disease)|Multiple myeloma (MM)|RA|Systemic juvenile idiopathic arthritis (SJIA)|Systemic sclerosis| Cytokine Release Syndrome (CRS)|NMO|Large-vessel vasculitis Giant cell arteritis| Polyarticular Juvenile Idiopathic Arthritis (PJIA)|Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection (COVID-19)|MG | EMA: January 2009 FDA: January 2010 | |
| satralizumab (mAbID 586) | IgG2-kappa | NMO | EMA: June 2016 FDA: August 2020 | |||
| levilimab *** (mAbID 887) | IgG1-kappa | RA | G1v76 CH2 P1.4, V1.3, A1.2 ADCC and CDC reduction G1v21 CH2 Y15.1, T16, E18 Half-life extension | |||
| sarilumab (mAbID 400) | IgG1-kappa | RA|Ankylosing spondylitis (AS)|Uveitis Polymyalgia Rheumatica (PMR) | FDA: May 2017 | |||
| vobarilizumab (mAbID 523) | VH–VH’ | RA|SLE| Inflammatory conditions | ||||
| clazakizumab (mAbID 414) | IL6 | IgG1-kappa | Blocking ** Immunosuppressant | CD|RA|Psoriatic arthritis (PSA)|AMR|Cancers, lung | G1v29 CH2 A84.4 No-glycosylation site ADCC reduction | FDA: August 2019 |
| olokizumab *** (mAbID 353) | IgG4-kappa | Autoimmune diseases|CD|RA | G4v5 h P10 Half-IG exchange reduction | |||
| siltuximab (mAbID 297) | IgG1-kappa | MM|Multicentric Castleman’s disease (MCD)|Renal cell carcinoma (RCC)|Neoplasms | EMA: May 2014 FDA: April 2014 | |||
| sirukumab *** (mAbID 384) | IgG1-kappa | RA (Despite Methotrexate Therapy)| Lupus nephritis|Juvenile Idiopathic Arthitis (JIA), pediatric|Giant cell arteritis|PMR | FDA: July 2017 | |||
| ziltivekimab *** (mAbID 979) | IgG1-kappa | Anemia | G1v21 CH2 Y15.1, T16, E18 Half-life extension | |||
| mezagitamab * (mAbID 882) | CD38 | IgG1-lambda | Blocking ** Immunosuppressant, Fc-effector function | MM|SLE|MG | FDA: January 2019 | |
| daratumumab * (mAbID 301) | IgG1-kappa | MM|AL amyloidosis|Myeloma, multiple (MM), recurrent or refractory (in combination with lenalidomide and dexamethasone or bortezomib and dexamethasone) | FDA: November 2015 | |||
| felzartamab (mAbID 1011) | IgG1-lambda2 | MM | ||||
| isatuximab (mAbID 539) | IgG1-kappa | MM|Hematologic malignancies | FDA: May 2014 |
3.5. Monoclonal Antibodies to Inactivate T-Cell Functions
4. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AChEI | acetylcholinesterase inhibitor |
| AChR | acetylcholine receptor |
| ADCC | antibody-dependent cytotoxicity |
| ADCP | antibody-dependent cellular phagocytosis |
| aHUS | atypical hemolytic uremic syndrome |
| C5 | complement 5 |
| CD19 | cluster of differentiation 19 |
| CD38 | cluster of differentiation 38 |
| CD40 | cluster of differentiation 40 |
| CDC | complement-dependent cytotoxicity |
| EAMG | experimental |
| EMA | European Medicines Agency |
| EOMG | early onset Myasthenia Gravis |
| FcRn | fragment crystallizable neonatal receptor |
| FcγR | Fc gamma (γ) receptors |
| FDA | U.S. Food and Drug Administration |
| gMG | generalized Myasthenia Gravis |
| HGNC | HUGO Gene Nomenclature Committee |
| Ig | immunoglobulin |
| IL6 | interleukin 6 |
| IL6ST | interleukin 6 signal transducer |
| IMGT® | the international ImMunoGeneTics information system® |
| INN | international nonproprietary name |
| LOMG | late-onset myasthenia gravis |
| LRP4 | lipoprotein related protein 4 |
| mAbs | monoclonal antibodies |
| MAC | membrane attack complex |
| MG | Myasthenia Gravis |
| MOA | mechanism of action |
| MS | multiple sclerosis |
| MS4A1 | Membrane Spanning 4-Domains A1 |
| MuSK | muscle-specific kinase |
| NMJ | neuromuscular junction |
| NMO | neuromyelitis optica |
| NMOSD | neuromyelitis optica spectrum disorder |
| PNH | paroxysmal nocturnal hemoglobinuria |
| RA | rheumatoid arthritis |
| TCC | terminal complement complex |
| WHO | world health organization |
References
- Verschuuren, J.J.; Huijbers, M.G.; Plomp, J.J.; Niks, E.H.; Molenaar, P.C.; Martinez-Martinez, P.; Gomez, A.M.; De Baets, M.H.; Losen, M. Pathophysiology of myasthenia gravis with antibodies to the acetylcholine receptor, muscle-specific kinase and low-density lipoprotein receptor-related protein 4. Autoimmun. Rev. 2013, 12, 918–923. [Google Scholar] [CrossRef] [PubMed]
- Paz, M.L.; Barrantes, F.J. Autoimmune Attack of the Neuromuscular Junction in Myasthenia Gravis: Nicotinic Acetylcholine Receptors and Other Targets. ACS Chem. Neurosci. 2019, 10, 2186–2194. [Google Scholar] [CrossRef] [PubMed]
- Garcia Estevez, D.A.; Pardo Fernandez, J. Myasthenia gravis. Update on diagnosis and therapy. Med. Clin. 2023, 161, 119–127. [Google Scholar] [CrossRef]
- Pechlivanidou, M.; Ninou, E.; Karagiorgou, K.; Tsantila, A.; Mantegazza, R.; Francesca, A.; Furlan, R.; Dudeck, L.; Steiner, J.; Tzartos, J.; et al. Autoimmunity to neuronal nicotinic acetylcholine receptors. Pharmacol. Res. 2023, 192, 106790. [Google Scholar] [CrossRef] [PubMed]
- Gasperi, C.; Melms, A.; Schoser, B.; Zhang, Y.; Meltoranta, J.; Risson, V.; Schaeffer, L.; Schalke, B.; Kröger, S. Anti-agrin autoantibodies in myasthenia gravis. Neurology 2014, 82, 1976–1983. [Google Scholar] [CrossRef] [PubMed]
- Gomez, A.M.; Broeck, J.V.D.; Vrolix, K.; Janssen, S.P.; Lemmens, M.A.M.; Van Der Esch, E.; Duimel, H.; Frederik, P.; Molenaar, P.C.; Martínez-Martínez, P.; et al. Antibody effector mechanisms in myasthenia gravis-pathogenesis at the neuromuscular junction. Autoimmunity 2010, 43, 353–370. [Google Scholar] [CrossRef] [PubMed]
- Berrih-Aknin, S.; Le Panse, R. Myasthenia gravis: A comprehensive review of immune dysregulation and etiological mechanisms. J. Autoimmun. 2014, 52, 90–100. [Google Scholar] [CrossRef]
- Golfinopoulou, R.; Papageorgiou, L.; Efthimiadou, A.; Bacopoulou, F.; Chrousos, G.P.; Eliopoulos, E.; Vlachakis, D. Clinical Genomic, phenotype and epigenetic insights into the pathology, autoimmunity and weight management of patients with Myasthenia Gravis (Review). Mol. Med. Rep. 2021, 24, 512. [Google Scholar] [CrossRef]
- Tsiamalos, P.; Kordas, G.; Kokla, A.; Poulas, K.; Tzartos, S.J. Epidemiological and immunological profile of muscle-specific kinase myasthenia gravis in Greece. Eur. J. Neurol. 2009, 16, 925–930. [Google Scholar] [CrossRef]
- Li, Y.; Peng, Y.; Yang, H. Serological diagnosis of myasthenia gravis and its clinical significance. Ann. Transl. Med. 2023, 11, 290. [Google Scholar] [CrossRef]
- Zisimopoulou, P.; Evangelakou, P.; Tzartos, J.; Lazaridis, K.; Zouvelou, V.; Mantegazza, R.; Antozzi, C.; Andreetta, F.; Evoli, A.; Deymeer, F.; et al. A comprehensive analysis of the epidemiology and clinical characteristics of anti-LRP4 in myasthenia gravis. J. Autoimmun. 2014, 52, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Tzartos, J.S.; Zhang, B.; Belimezi, M.; Ragheb, S.; Bealmear, B.; Lewis, R.A.; Xiong, W.-C.; Lisak, R.P.; Tzartos, S.J.; Mei, L. Autoantibodies to lipoprotein-related protein 4 in patients with double-seronegative myasthenia gravis. Arch. Neurol. 2012, 69, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Phillips, L.H. The epidemiology of myasthenia gravis. Semin. Neurol. 2004, 24, 17–20. [Google Scholar] [CrossRef] [PubMed]
- Hughes, B.W.; Moro De Casillas, M.L.; Kaminski, H.J. Pathophysiology of myasthenia gravis. Semin. Neurol. 2004, 24, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Sanders, D.B.; Wolfe, G.I.; Narayanaswami, P. International consensus guidance for management of myasthenia gravis: Executive summary. Neurology 2016, 87, 419–425. [Google Scholar] [CrossRef]
- Seybold, M.E.; Drachman, D.B. Gradually increasing doses of prednisone in myasthenia gravis. Reducing the hazards of treatment. N. Engl. J. Med. 1974, 290, 81–84. [Google Scholar] [CrossRef]
- Morren, J.; Li, Y. Maintenance immunosuppression in myasthenia gravis, an update. J. Neurol. Sci. 2020, 410, 116648. [Google Scholar] [CrossRef] [PubMed]
- Alhaidar, M.K.; Abumurad, S.; Soliven, B.; Rezania, K. Current Treatment of Myasthenia Gravis. J. Clin. Med. 2022, 11, 1597. [Google Scholar] [CrossRef]
- Howard, J.F., Jr.; Barohn, R.J.; Cutter, G.R.; Freimer, M.; Juel, V.C.; Mozaffar, T.; Mellion, M.L.; Benatar, M.G.; Farrugia, M.E.; Wang, J.J.; et al. A randomized, double-blind, placebo-controlled phase II study of eculizumab in patients with refractory generalized myasthenia gravis. Muscle Nerve 2013, 48, 76–84. [Google Scholar] [CrossRef]
- Legendre, C.M.; Licht, C.; Muus, P.; Greenbaum, L.A.; Babu, S.; Bedrosian, C.; Bingham, C.; Cohen, D.J.; Delmas, Y.; Douglas, K.; et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N. Engl. J. Med. 2013, 368, 2169–2181. [Google Scholar] [CrossRef]
- Dmytrijuk, A.; Robie-Suh, K.; Cohen, M.H.; Rieves, D.; Weiss, K.; Pazdur, R. FDA report: Eculizumab (Soliris) for the treatment of patients with paroxysmal nocturnal hemoglobinuria. Oncologist 2008, 13, 993–1000. [Google Scholar] [CrossRef]
- Pittock, S.J.; Berthele, A.; Fujihara, K.; Kim, H.J.; Levy, M.; Palace, J.; Nakashima, I.; Terzi, M.; Totolyan, N.; Viswanathan, S.; et al. Eculizumab in Aquaporin-4-Positive Neuromyelitis Optica Spectrum Disorder. N. Engl. J. Med. 2019, 381, 614–625. [Google Scholar] [CrossRef]
- Manso, T.; Folch, G.; Giudicelli, V.; Jabado-Michaloud, J.; Kushwaha, A.; Ngoune, V.N.; Georga, M.; Papadaki, A.; Debbagh, C.; Pégorier, P.; et al. IMGT® databases, related tools and web resources through three main axes of research and development. Nucleic Acids Res. 2021, 50, D1262–D1272. [Google Scholar] [CrossRef]
- Manso, T.; Kushwaha, A.; Abdollahi, N.; Duroux, P.; Giudicelli, V.; Kossida, S. Mechanisms of action of monoclonal antibodies in oncology integrated in IMGT/mAb-DB. Front. Immunol. 2023, 14, 1129323. [Google Scholar] [CrossRef]
- Ehrenmann, F.; Kaas, Q.; Lefranc, M.P. IMGT/3Dstructure-DB and IMGT/DomainGapAlign: A database and a tool for immunoglobulins or antibodies, T cell receptors, MHC, IgSF and MhcSF. Nucleic Acids Res. 2010, 38, D301–D307. [Google Scholar] [CrossRef]
- Lefranc, M.P.; Lefranc, G. IMGT ((R)) Nomenclature of Engineered IGHG Variants Involved in Antibody Effector Properties and Formats. Antibodies 2022, 11, 65. [Google Scholar] [CrossRef]
- Engel, A.G.; Lambert, E.H.; Howard, F.M. Immune complexes (IgG and C3) at the motor end-plate in myasthenia gravis: Ultrastructural and light microscopic localization and electrophysiologic correlations. Mayo Clin. Proc. 1977, 52, 267–280. [Google Scholar]
- Romi, F.; Kristoffersen, E.K.; Aarli, J.A.; Gilhus, N.E. The role of complement in myasthenia gravis: Serological evidence of complement consumption in vivo. J. Neuroimmunol. 2005, 158, 191–194. [Google Scholar] [CrossRef]
- Kaminski, H.J.; Kusner, L.L.; Richmonds, C.; Medof, M.E.; Lin, F. Deficiency of decay accelerating factor and CD59 leads to crisis in experimental myasthenia. Exp. Neurol. 2006, 202, 287–293. [Google Scholar] [CrossRef]
- Howard, J.F., Jr. Myasthenia gravis: The role of complement at the neuromuscular junction. Ann. N. Y. Acad. Sci. 2018, 1412, 113–128. [Google Scholar] [CrossRef]
- Mantegazza, R.; Vanoli, F.; Frangiamore, R.; Cavalcante, P. Complement Inhibition for the Treatment of Myasthenia Gravis. Immunotargets Ther. 2020, 9, 317–331. [Google Scholar] [CrossRef]
- Rother, R.P.; Rollins, S.A.; Mojcik, C.F.; Brodsky, R.A.; Bell, L. Discovery and development of the complement inhibitor eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria. Nat. Biotechnol. 2007, 25, 1256–1264. [Google Scholar] [CrossRef] [PubMed]
- Nicholas, S.R. Sanderson, Complement and myasthenia gravis. Mol. Immunol. 2022, 151, 11–18. [Google Scholar] [CrossRef]
- Stern, R.M.; Connell, N.T. Ravulizumab: A novel C5 inhibitor for the treatment of paroxysmal nocturnal hemoglobinuria. Ther. Adv. Hematol. 2019, 10, 2040620719874728. [Google Scholar] [CrossRef]
- Vu, T.; Ortiz, S.; Katsuno, M.; Annane, D.; Mantegazza, R.; Beasley, K.N.; Aguzzi, R.; Howard, J.F. Ravulizumab pharmacokinetics and pharmacodynamics in patients with generalized myasthenia gravis. J. Neurol. 2023, 270, 3129–3137. [Google Scholar] [CrossRef]
- Kulasekararaj, A.G.; Hill, A.; Rottinghaus, S.T.; Langemeijer, S.; Wells, R.; Gonzalez-Fernandez, F.A.; Gaya, A.; Lee, J.W.; Gutierrez, E.O.; Piatek, C.I.; et al. Ravulizumab (ALXN1210) vs eculizumab in C5-inhibitor-experienced adult patients with PNH: The 302 study. Blood 2019, 133, 540–549. [Google Scholar] [CrossRef]
- Jang, J.H.; Weyne, J.; Chaudhari, U.; Harari, O.; Miller, J.; Dain, B.; Meagher, K.A.; Rodgers, M.L.; Perlee, L.; Morton, L.; et al. Pozelimab, a Human Monoclonal Antibody Against Complement Factor C5, Provided Inhibition of Intravascular Hemolysis in Patients with Paroxysmal Nocturnal Hemoglobinuria. Blood 2021, 138, 1128. [Google Scholar] [CrossRef]
- Latuszek, A.; Liu, Y.; Olsen, O.; Foster, R.; Cao, M.; Lovric, I.; Yuan, M.; Liu, N.; Chen, H.; Zhang, Q.; et al. Inhibition of complement pathway activation with Pozelimab, a fully human antibody to complement component C5. PLoS ONE 2020, 15, e0231892. [Google Scholar] [CrossRef]
- Pyzik, M.; Sand, K.M.; Hubbard, J.J.; Andersen, J.T.; Sandlie, I.; Blumberg, R.S. The Neonatal Fc Receptor (FcRn): A Misnomer? Front. Immunol. 2019, 10, 1540. [Google Scholar] [CrossRef]
- Dalakas, M.C.; Spaeth, P.J. The importance of FcRn in neuro-immunotherapies: From IgG catabolism, FCGRT gene polymorphisms, IVIg dosing and efficiency to specific FcRn inhibitors. Ther. Adv. Neurol. Disord. 2021, 14, 1756286421997381. [Google Scholar] [CrossRef]
- Gable, K.L.; Guptill, J.T. Antagonism of the Neonatal Fc Receptor as an Emerging Treatment for Myasthenia Gravis. Front. Immunol. 2019, 10, 3052. [Google Scholar] [CrossRef] [PubMed]
- Bril, V.; Drużdż, A.; Grosskreutz, J.; Habib, A.A.; Mantegazza, R.; Sacconi, S.; Utsugisawa, K.; Vissing, J.; Vu, T.; Boehnlein, M.; et al. Safety and efficacy of rozanolixizumab in patients with generalised myasthenia gravis (MycarinG): A randomised, double-blind, placebo-controlled, adaptive phase 3 study. Lancet Neurol. 2023, 22, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Iorio, R.; Damato, V.; Alboini, P.E.; Evoli, A. Efficacy and safety of rituximab for myasthenia gravis: A systematic review and meta-analysis. J. Neurol. 2015, 262, 1115–1119. [Google Scholar] [CrossRef] [PubMed]
- Tannemaat, M.R.; Verschuuren, J. Emerging therapies for autoimmune myasthenia gravis: Towards treatment without corticosteroids. Neuromuscul. Disord. 2020, 30, 111–119. [Google Scholar] [CrossRef]
- Saccà, F.; Pane, C.; Espinosa, P.E.; Sormani, M.P.; Signori, A. Efficacy of innovative therapies in myasthenia gravis: A systematic review, meta-analysis and network meta-analysis. Eur. J. Neurol 2023, 30, 3854–3867. [Google Scholar] [CrossRef] [PubMed]
- Hehir, M.K., 2nd; Li, Y. Diagnosis and Management of Myasthenia Gravis. Continuum 2022, 28, 1615–1642. [Google Scholar] [CrossRef]
- Zouvelou, V.; Psimenou, E. Double Seropositive Myasthenia Gravis Successfully Treated with Rituximab. J. Clin. Neuromuscul. Dis. 2022, 24, 116–117. [Google Scholar] [CrossRef]
- Forsthuber, T.G.; Cimbora, D.M.; Ratchford, J.N.; Katz, E.; Stüve, O. B cell-based therapies in CNS autoimmunity: Differentiating CD19 and CD20 as therapeutic targets. Ther. Adv. Neurol. Disord. 2018, 11, 175628641876169. [Google Scholar] [CrossRef]
- Nair, S.S.; Jacob, S. Novel Immunotherapies for Myasthenia Gravis. Immunotargets Ther. 2023, 12, 25–45. [Google Scholar] [CrossRef]
- Tedder, T.F.; Klejman, G.; Schlossman, S.F.; Saito, H. Structure of the gene encoding the human B lymphocyte differentiation antigen CD20 (B1). J. Immunol. 1989, 142, 2560–2568. [Google Scholar] [CrossRef]
- Tedder, T.F.; Boyd, A.W.; Freedman, A.S.; Nadler, L.M.; Schlossman, S.F. The B cell surface molecule B1 is functionally linked with B cell activation and differentiation. J. Immunol. 1985, 135, 973–979. [Google Scholar] [CrossRef]
- Léveillé, C.; Al-Daccak, R.; Mourad, W. CD20 is physically and functionally coupled to MHC class II and CD40 on human B cell lines. Eur. J. Immunol. 1999, 29, 65–74. [Google Scholar] [CrossRef]
- Boyles, J.S.; Sadowski, D.; Potter, S.; Vukojicic, A.; Parker, J.; Chang, W.Y.; Ma, Y.L.; Chambers, M.G.; Nelson, J.; Barmettler, B.; et al. A nondepleting anti-CD19 antibody impairs B cell function and inhibits autoimmune diseases. JCI Insight 2023, 8, e166137. [Google Scholar] [CrossRef]
- Tandan, R.; Hehir, M.K.; Waheed, W.; Howard, D.B. Rituximab treatment of myasthenia gravis: A systematic review. Muscle Nerve 2017, 56, 185–196. [Google Scholar] [CrossRef]
- Narayanaswami, P.; Sanders, D.B.; Wolfe, G.; Benatar, M.; Cea, G.; Evoli, A.; Gilhus, N.E.; Illa, I.; Kuntz, N.L.; Massey, J.; et al. International Consensus Guidance for Management of Myasthenia Gravis: 2020 Update. Neurology 2021, 96, 114–122. [Google Scholar] [CrossRef]
- Łosińska, K.; Korkosz, M.; Pripp, A.H.; Haugeberg, G. Real-world experience of rituximab biosimilar GP2013 in rheumatoid arthritis patients naive to or switched from reference rituximab. Rheumatol. Int. 2023, 43, 881–888. [Google Scholar] [CrossRef]
- Chmielewska, N.; Szyndler, J. Targeting CD20 in multiple sclerosis—Review of current treatment strategies. Neurol. Neurochir. Pol. 2023, 57, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Rual, C.; Biotti, D.; Lepine, Z.; Delourme, A.; Le Berre, J.; Treiner, E.; Ciron, J. 2 grams versus 1 gram rituximab as maintenance schedule in multiple sclerosis, neuromyelitis optica spectrum disorders and related diseases: What B-cell repopulation data tell us. Mult. Scler. Relat. Disord. 2023, 71, 104563. [Google Scholar] [CrossRef] [PubMed]
- Waters, M.J.; Field, D.; Ravindran, J. Refractory myasthenia gravis successfully treated with ofatumumab. Muscle Nerve 2019, 60, E45–E47. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Tejerina, D.; Sotoca, J.; Llaurado, A.; López-Diego, V.; Juntas-Morales, R.; Salvado, M. New Targeted Agents in Myasthenia Gravis and Future Therapeutic Strategies. J. Clin. Med. 2022, 11, 6394. [Google Scholar] [CrossRef]
- Menon, D.; Bril, V. Pharmacotherapy of Generalized Myasthenia Gravis with Special Emphasis on Newer Biologicals. Drugs 2022, 82, 865–887. [Google Scholar] [CrossRef]
- Frampton, J.E. Inebilizumab: First Approval. Drugs 2020, 80, 1259–1264. [Google Scholar] [CrossRef]
- Viela Bio. A Randomized, Double-Blind, Multicenter, Placebocontrolled Phase 3 Study with Open-Label Period to Evaluate the Efficacy and Safety of Inebilizumab in Adults with Myasthenia Gravis; Viela Bio: Gaithersburg, MD, USA, 2021. [Google Scholar]
- Gallagher, S.; Turman, S.; Yusuf, I.; Akhgar, A.; Wu, Y.; Roskos, L.K.; Herbst, R.; Wang, Y. Pharmacological profile of MEDI-551, a novel anti-CD19 antibody, in human CD19 transgenic mice. Int. Immunopharmacol. 2016, 36, 205–212. [Google Scholar] [CrossRef]
- Chen, D.; Gallagher, S.; Monson, N.L.; Herbst, R.; Wang, Y. Inebilizumab, a B Cell-Depleting Anti-CD19 Antibody for the Treatment of Autoimmune Neurological Diseases: Insights from Preclinical Studies. J. Clin. Med. 2016, 5, 107. [Google Scholar] [CrossRef]
- Tenca, C.; Merlo, A.; Zarcone, D.; Saverino, D.; Bruno, S.; De Santanna, A.; Ramarli, D.; Fabbi, M.; Pesce, C.; Deaglio, S.; et al. Death of T cell precursors in the human thymus: A role for CD38. Int. Immunol. 2003, 15, 1105–1116. [Google Scholar] [CrossRef] [PubMed]
- Scheibe, F.; Ostendorf, L.; Prüss, H.; Radbruch, H.; Aschman, T.; Hoffmann, S.; Blau, I.; Meisel, C.; Alexander, T.; Meisel, A. Daratumumab for treatment-refractory antibody-mediated diseases in neurology. Eur. J. Neurol. 2022, 29, 1847–1854. [Google Scholar] [CrossRef] [PubMed]
- Field-Smith, A.; Morgan, G.J.; Davies, F.E. Bortezomib (Velcadetrade mark) in the Treatment of Multiple Myeloma. Ther. Clin. Risk Manag. 2006, 2, 271–279. [Google Scholar] [CrossRef]
- Gomez, A.M. Proteasome inhibition with bortezomib depletes plasma cells and autoantibodies in experimental autoimmune myasthenia gravis. J. Immunol. 2011, 186, 2503–2513. [Google Scholar] [CrossRef]
- Clark, E.A. CD40: A cytokine receptor in search of a ligand. Tissue Antigens 1990, 36, 33–36. [Google Scholar] [CrossRef]
- van Kooten, C.; Banchereau, J. CD40-CD40 ligand. J. Leukoc. Biol. 2000, 67, 2–17. [Google Scholar] [CrossRef]
- Huda, R.; Tuzun, E.; Christadoss, P. Targeting complement system to treat myasthenia gravis. Rev. Neurosci. 2014, 25, 575–583. [Google Scholar] [CrossRef]
- Hunter, C.A.; Jones, S.A. IL-6 as a keystone cytokine in health and disease. Nat. Immunol. 2015, 16, 448–457. [Google Scholar] [CrossRef]
- Kang, S.; Tanaka, T.; Narazaki, M.; Kishimoto, T. Targeting Interleukin-6 Signaling in Clinic. Immunity 2019, 50, 1007–1023. [Google Scholar] [CrossRef]
- Spencer, S.; Köstel Bal, S.; Egner, W.; Lango Allen, H.; Raza, S.I.; Ma, C.A.; Gürel, M.; Zhang, Y.; Sun, G.; Sabroe, R.A.; et al. Loss of the interleukin-6 receptor causes immunodeficiency, atopy, and abnormal inflammatory responses. J. Exp. Med. 2019, 216, 1986–1998. [Google Scholar] [CrossRef]
- Garbers, C.; Thaiss, W.; Jones, G.W.; Waetzig, G.H.; Lorenzen, I.; Guilhot, F.; Lissilaa, R.; Ferlin, W.G.; Grötzinger, J.; Jones, S.A.; et al. Inhibition of classic signaling is a novel function of soluble glycoprotein 130 (sgp130), which is controlled by the ratio of interleukin 6 and soluble interleukin 6 receptor. J. Biol. Chem. 2011, 286, 42959–42970. [Google Scholar] [CrossRef]
- Hirano, T. IL-6 in inflammation, autoimmunity and cancer. Int. Immunol. 2021, 33, 127–148. [Google Scholar] [CrossRef]
- Biggioggero, M.; Crotti, C.; Becciolini, A.; Favalli, E.G. Tocilizumab in the treatment of rheumatoid arthritis: An evidence-based review and patient selection. Drug Des. Dev. Ther. 2019, 13, 57–70. [Google Scholar] [CrossRef]
- Yan, X.; Tang, W.; Zhang, Z.; Zhang, Y.; Luo, C.; Tang, X. Tocilizumab in Systemic Juvenile Idiopathic Arthritis: Response Differs by Disease Duration at Medication Initiation and by Phenotype of Disease. Front. Pediatr. 2021, 9, 735846. [Google Scholar] [CrossRef]
- Khanna, D.; Lin, C.J.F.; Furst, D.E.; Goldin, J.; Kim, G.; Kuwana, M.; Allanore, Y.; Matucci-Cerinic, M.; Distler, O.; Shima, Y.; et al. Tocilizumab in systemic sclerosis: A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Respir. Med. 2020, 8, 963–974. [Google Scholar] [CrossRef] [PubMed]
- Traboulsee, A.; Greenberg, B.M.; Bennett, J.L.; Szczechowski, L.; Fox, E.; Shkrobot, S.; Yamamura, T.; Terada, Y.; Kawata, Y.; Wright, P.; et al. Safety and efficacy of satralizumab monotherapy in neuromyelitis optica spectrum disorder: A randomised, double-blind, multicentre, placebo-controlled phase 3 trial. Lancet Neurol. 2020, 19, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Kusner, L.L.; Kaminski, H.J.; Soltys, J. Effect of complement and its regulation on myasthenia gravis pathogenesis. Expert Rev. Clin. Immunol. 2008, 4, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Alabbad, S.; AlGaeed, M.; Sikorski, P.; Kaminski, H.J. Monoclonal Antibody-Based Therapies for Myasthenia Gravis. BioDrugs 2020, 34, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Zhang, J.; Meng, J.; Jiang, G.; Yan, Z.; Yang, Y.; Chen, Z.; You, W.; Wang, Z.; Chen, G. Different Monoclonal Antibodies in Myasthenia Gravis: A Bayesian Network Meta-Analysis. Front. Pharmacol. 2021, 12, 790834. [Google Scholar] [CrossRef]
- Marshall, H.T.; Djamgoz, M.B.A. Immuno-Oncology: Emerging Targets and Combination Therapies. Front. Oncol. 2018, 8, 315. [Google Scholar] [CrossRef]




| INN mAbs | Receptor Identification | IMGT Variants | IMGT MOA * | Clinical Trials for MG |
|---|---|---|---|---|
| eculizumab | IgG2–G4-kappa | - | Blocking * Complement Inhibitor | Phase III (NCT03759366|NCT02301624)|NCT01997229)| Phase II (NCT00727194 Observational (NCT04202341) |
| ravulizumab | IgG2–G4-kappa | G4v24 CH3 L107, S114 Half-life extension | Phase III (NCT05644561|NCT03920293) Observational (NCT04202341) | |
| gefurulimab | VH-VH’ | - | Phase III (NCT05556096) | |
| pozelimab | IgG4-kappa | G4v5 h P10 Half-IG exchange reduction | Phase III (NCT05070858) |
| INN mAbs | Receptor Identification | IMGT Variants | IMGT MOA * | Clinical Trials for MG |
|---|---|---|---|---|
| efgartigimod alfa | IgG1-Fc fragment | G1v46 CH3 K113, F114 increase FcRn binding | Neutralizing * FcRn inhibitor | Phase III (NCT04980495) |
| batoclimab | IgG1 | G1v14 CH2 A1.3, A1.2 ADCC and CDC reduction | Phase III (NCT05403541) | |
| rozanolixizumab | IgG4-kappa | G4v5 h P10 Half-IG exchange reduction | Phase III (NCT03971422|NCT05681715|NCT04124965) | |
| nipocalimab | IgG1-lambda3 | G1v29 A84.4 No N-glycosylation site ADCC reduction | Phase III (NCT05265273|NCT04951622) | |
| orilanolimab | IgG4-kappa | G4v5 h P10 Half-IG exchange reduction | Phase I (discontinued) |
| Target | INN mAbs | Receptor Identification | IMGT MOA * | Clinical Trials for MG |
|---|---|---|---|---|
| MS4A1 | rituximab | IgG1-kappa | Blocking * Immunosuppressant, Fc-effector function | Phase III NCT05868837|NCT05332587 |
| ofatumumab | IgG1-kappa | - | ||
| CD19 | inebilizumab | IgG1-kappa | Phase III NCT04524273 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Golfinopoulou, R.; Giudicelli, V.; Manso, T.; Kossida, S. Delving into Molecular Pathways: Analyzing the Mechanisms of Action of Monoclonal Antibodies Integrated in IMGT/mAb-DB for Myasthenia Gravis. Vaccines 2023, 11, 1756. https://doi.org/10.3390/vaccines11121756
Golfinopoulou R, Giudicelli V, Manso T, Kossida S. Delving into Molecular Pathways: Analyzing the Mechanisms of Action of Monoclonal Antibodies Integrated in IMGT/mAb-DB for Myasthenia Gravis. Vaccines. 2023; 11(12):1756. https://doi.org/10.3390/vaccines11121756
Chicago/Turabian StyleGolfinopoulou, Rebecca, Véronique Giudicelli, Taciana Manso, and Sofia Kossida. 2023. "Delving into Molecular Pathways: Analyzing the Mechanisms of Action of Monoclonal Antibodies Integrated in IMGT/mAb-DB for Myasthenia Gravis" Vaccines 11, no. 12: 1756. https://doi.org/10.3390/vaccines11121756
APA StyleGolfinopoulou, R., Giudicelli, V., Manso, T., & Kossida, S. (2023). Delving into Molecular Pathways: Analyzing the Mechanisms of Action of Monoclonal Antibodies Integrated in IMGT/mAb-DB for Myasthenia Gravis. Vaccines, 11(12), 1756. https://doi.org/10.3390/vaccines11121756