
In Vitro Transcribed RNA-Based Platform Vaccines: Past, Present, and Future



Abstract
:1. Introduction
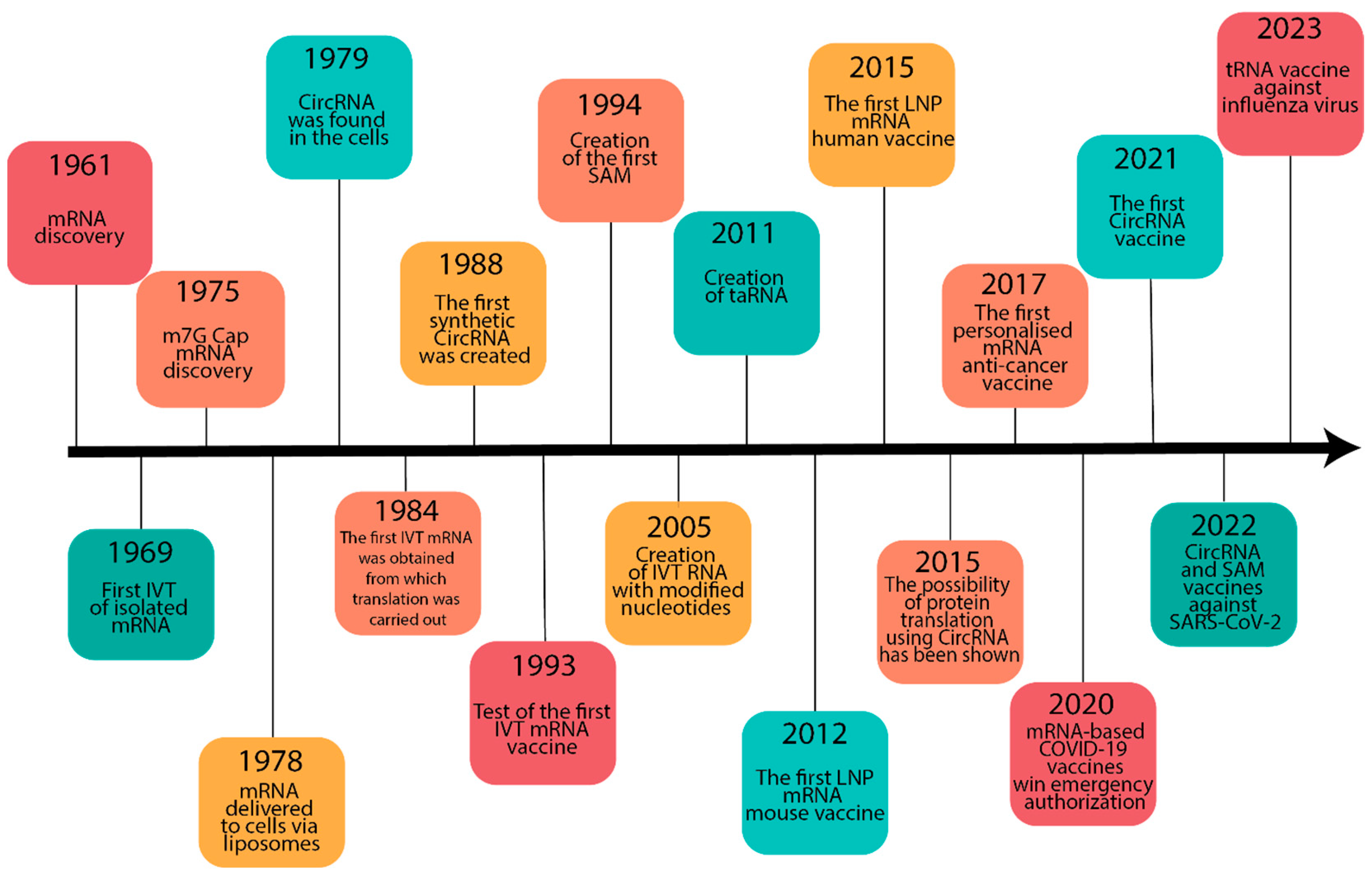
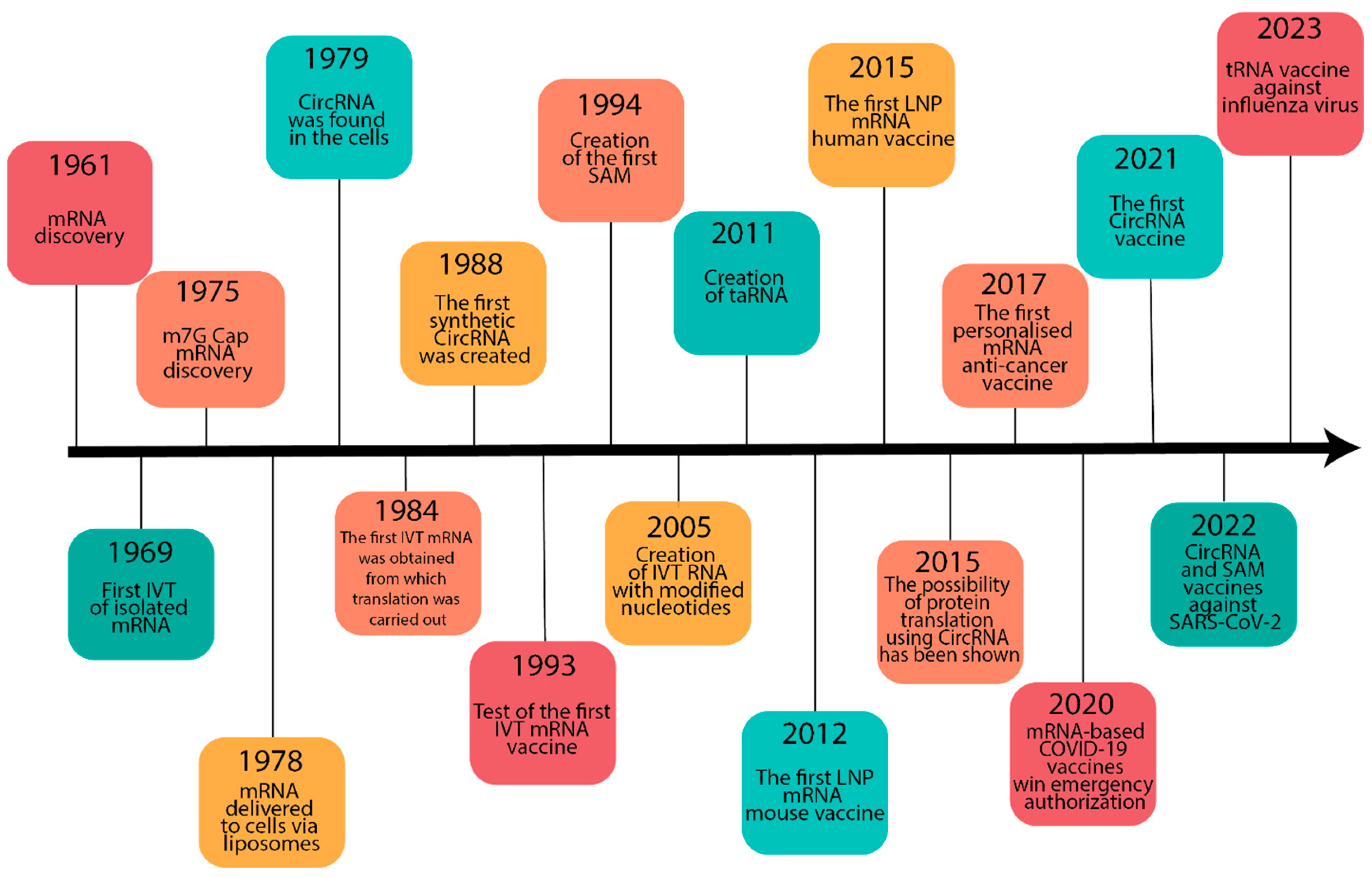
2. Historical Background
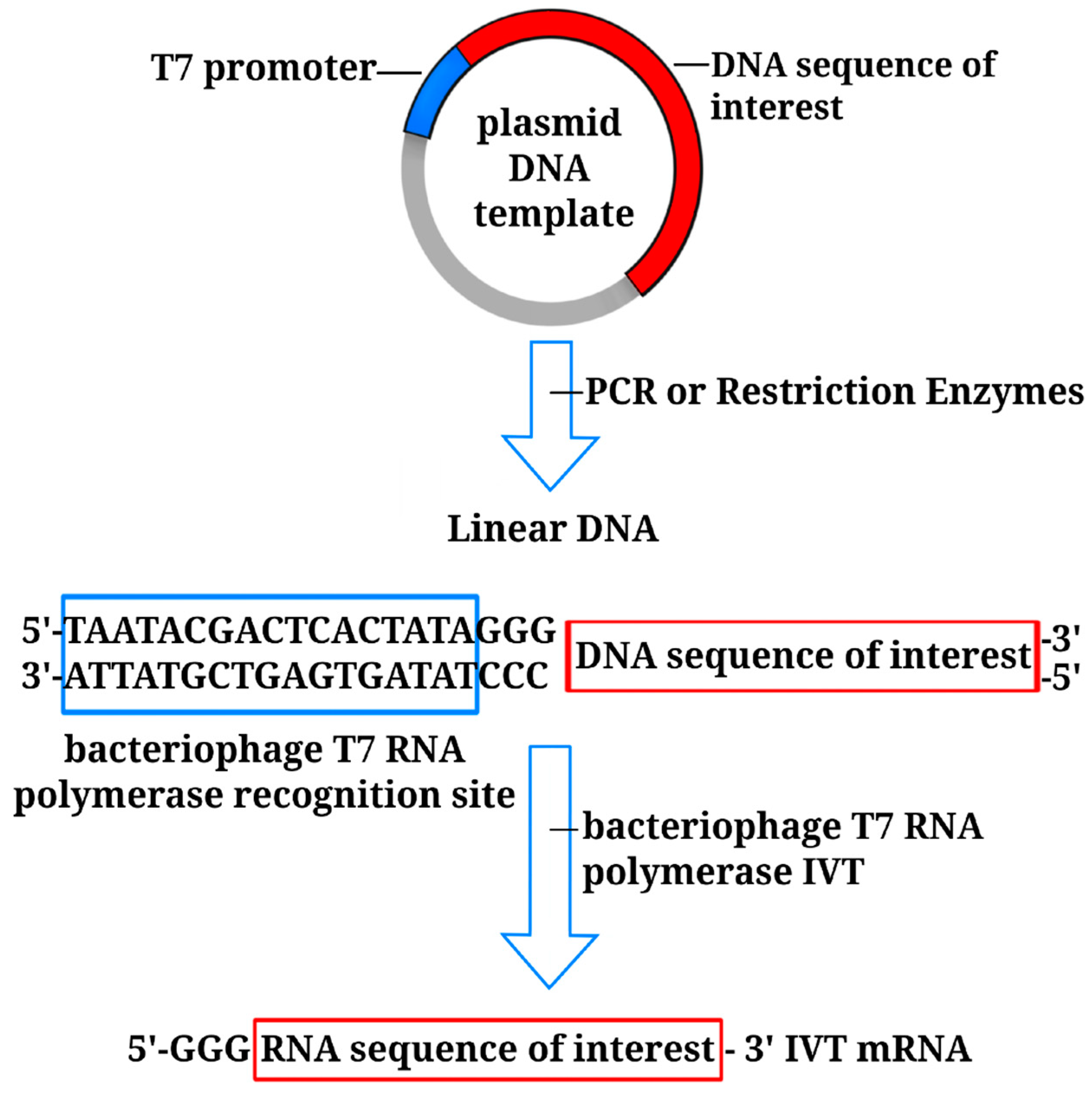
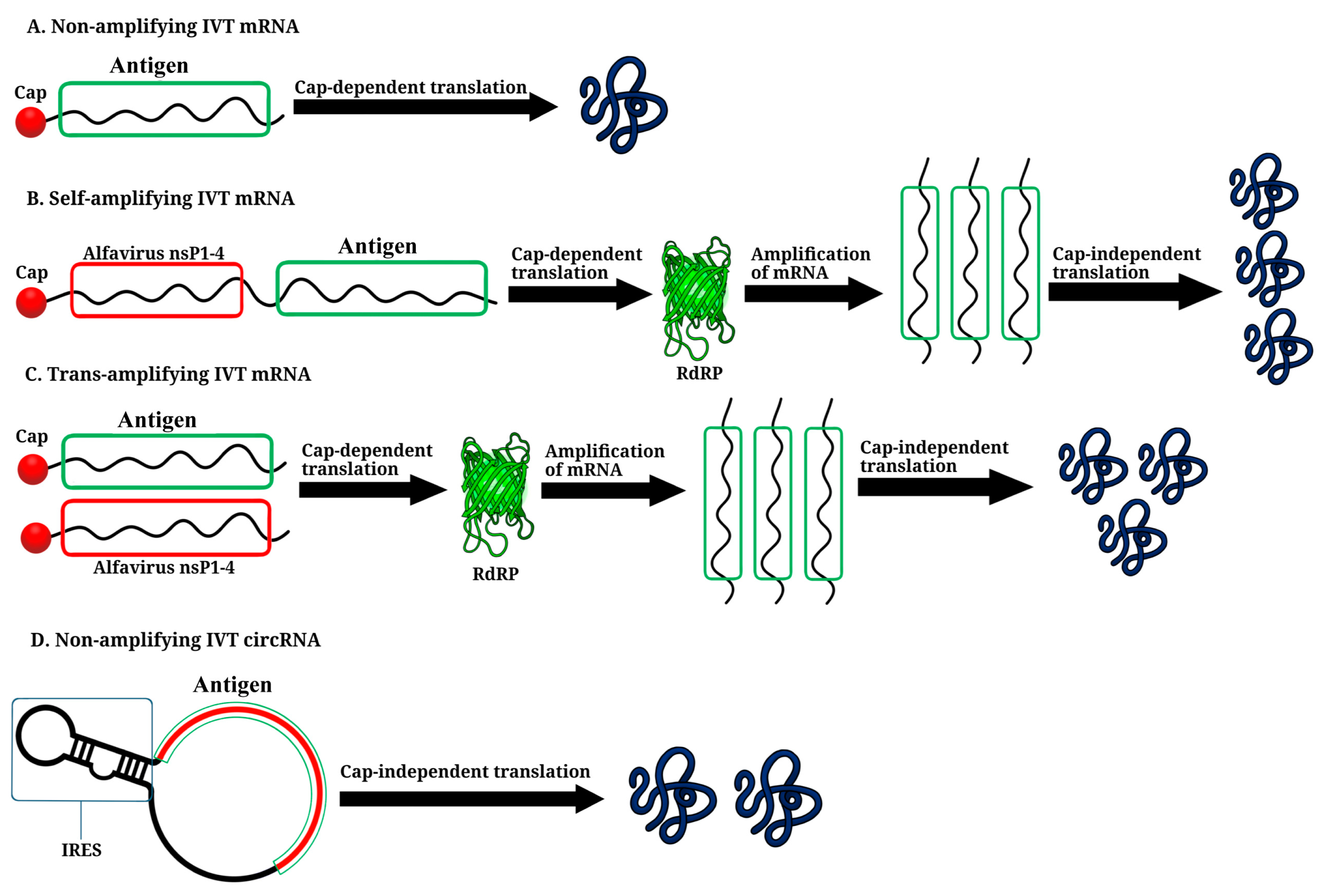
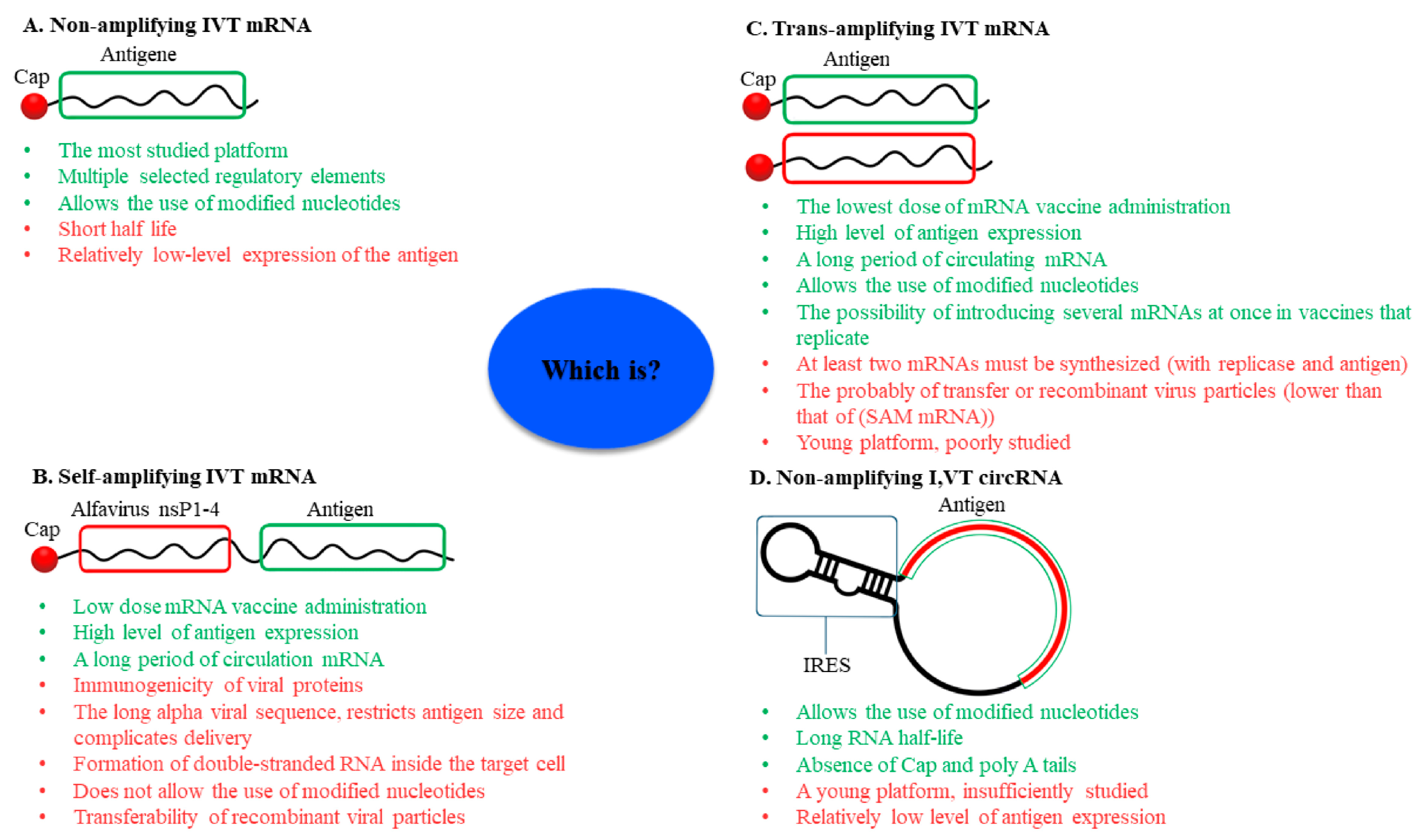
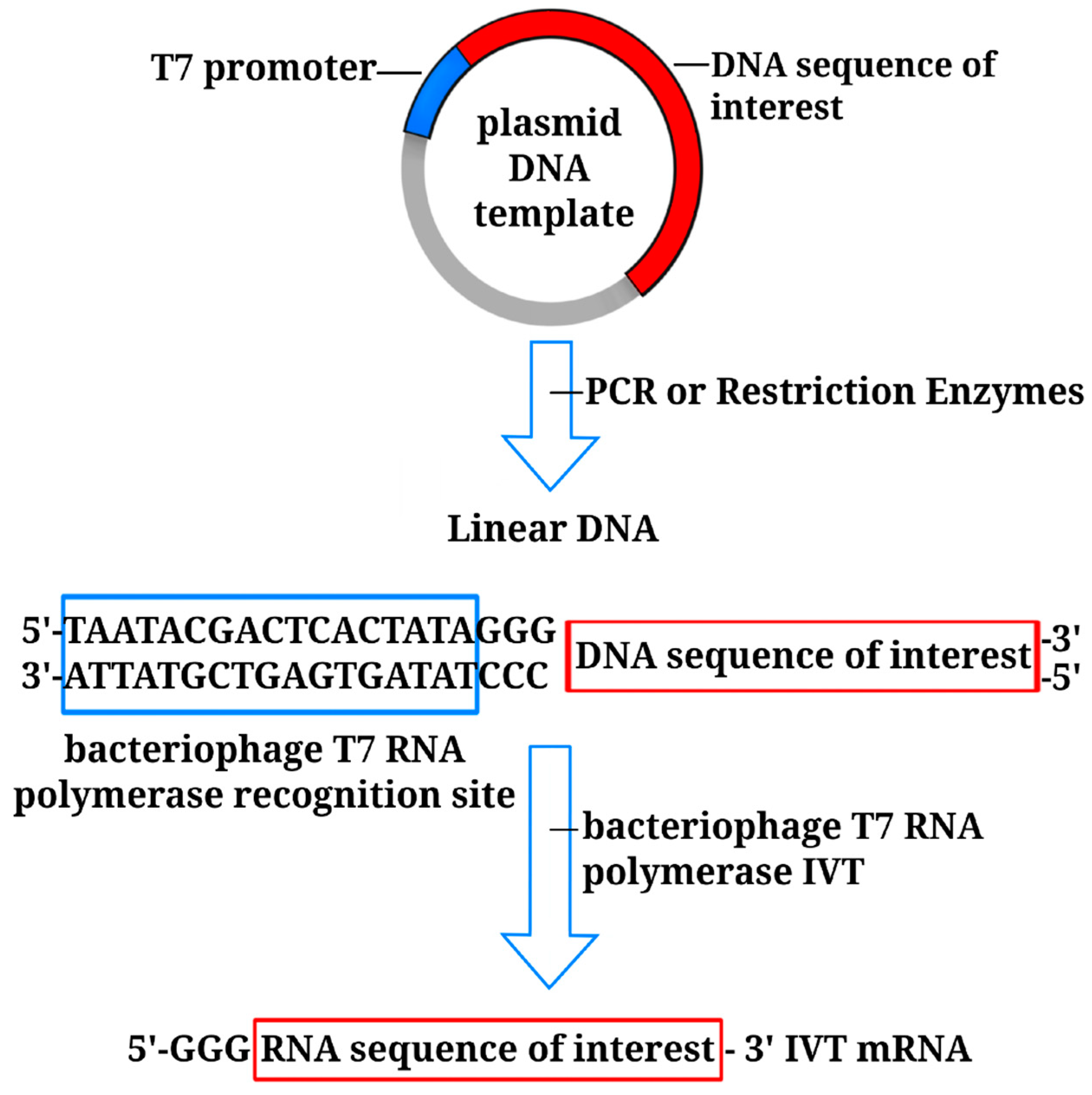
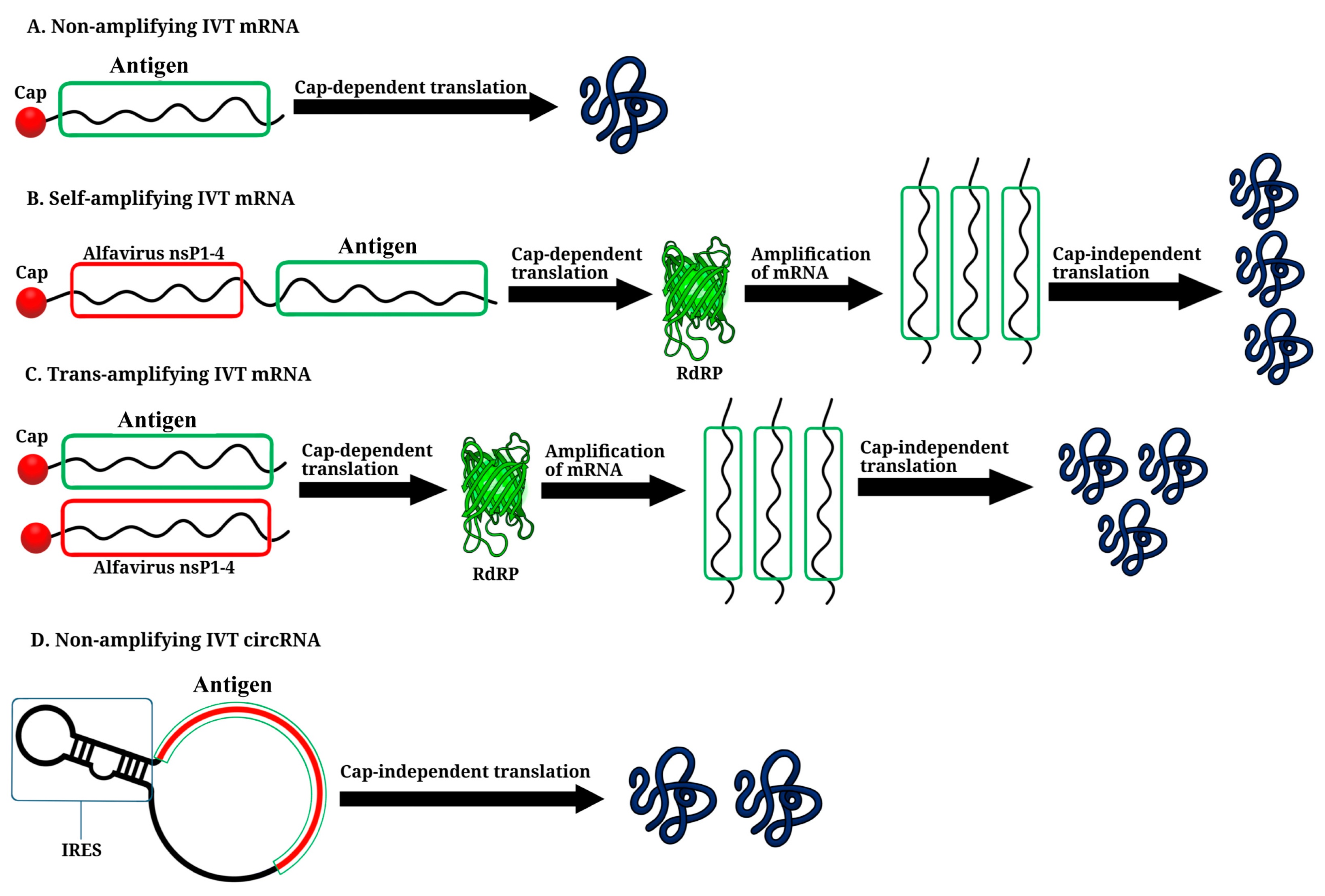
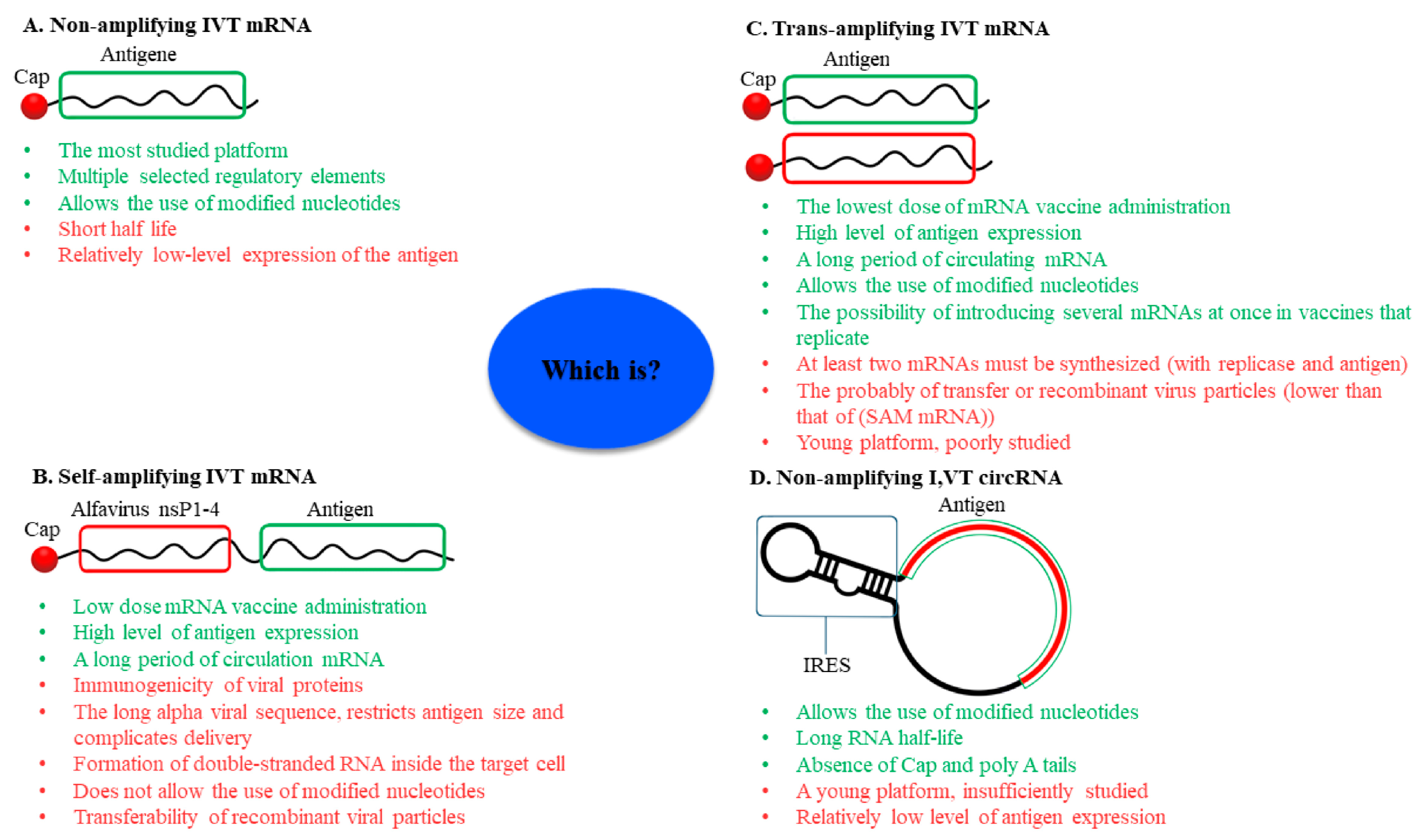
3. Non-Amplifying In Vitro-Transcribed mRNA Platform
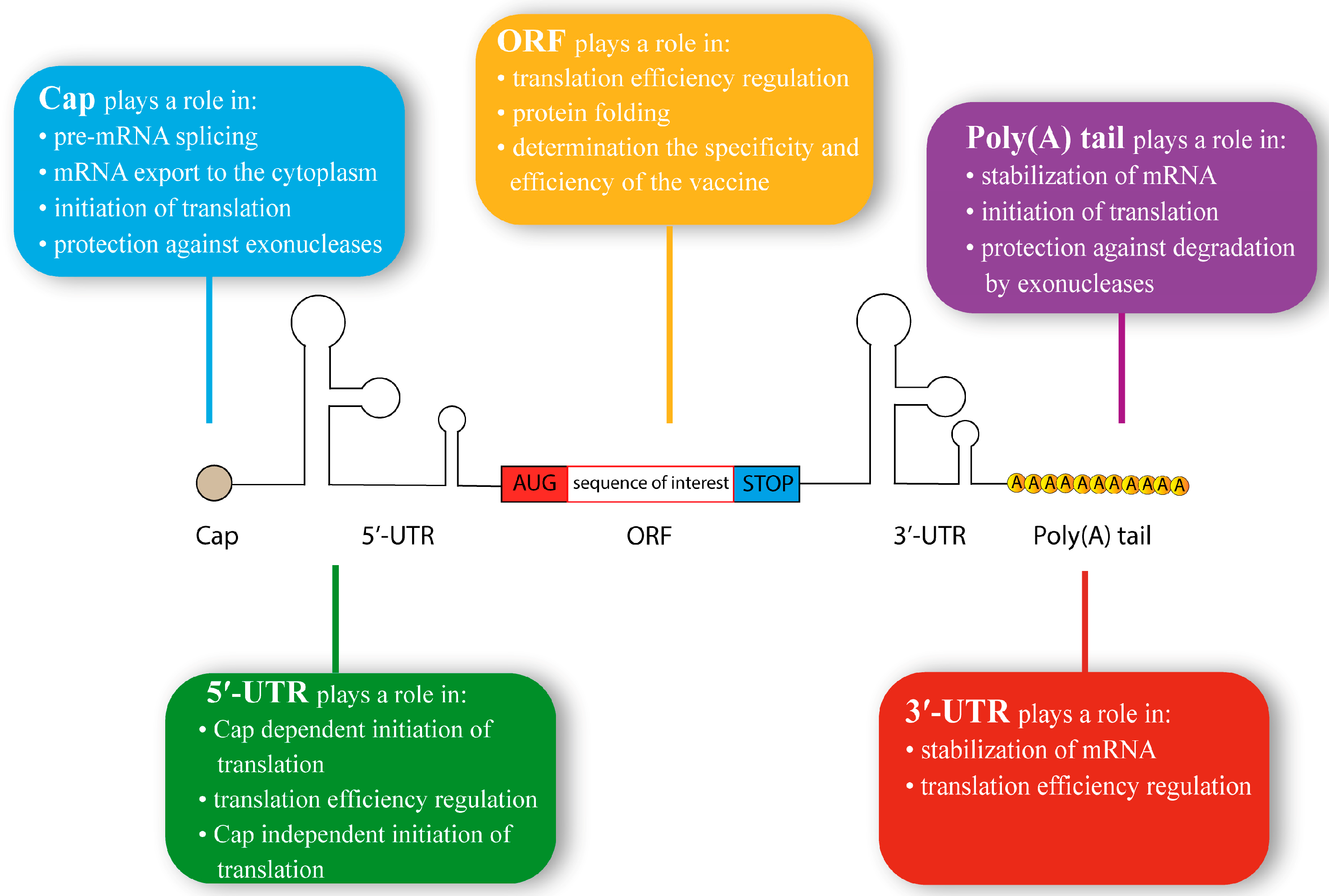
3.1. 5′ Cap Structure
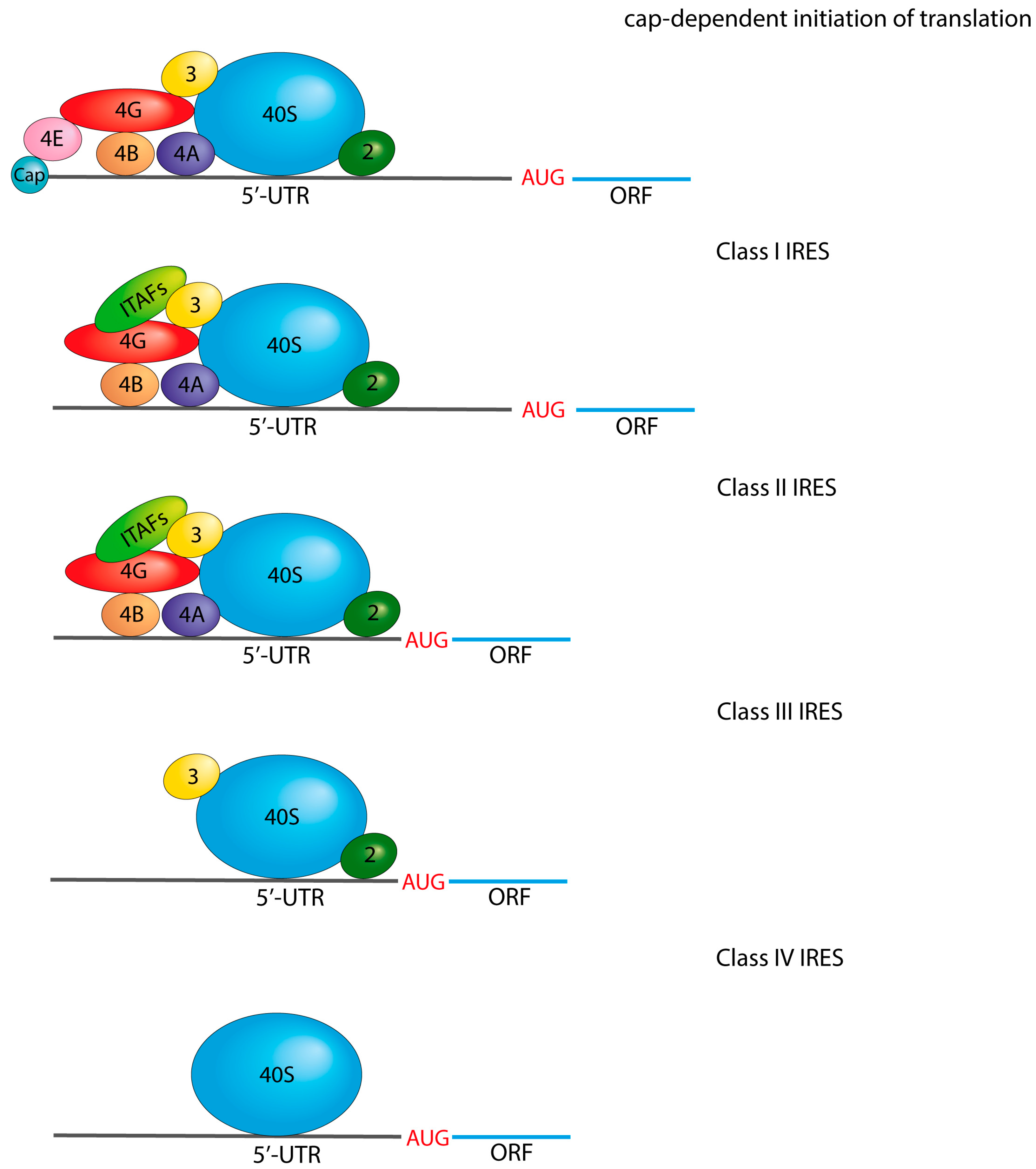
3.2. 5′-Untranslated Region Structure
3.3. Open Reading Frame Structure
3.4. 3′-Untranslated Region Structure
3.5. Poly(A) Tail Structure
4. Self-Amplifying IVT mRNA Platform
5. Trans-Amplifying IVT mRNA Platform
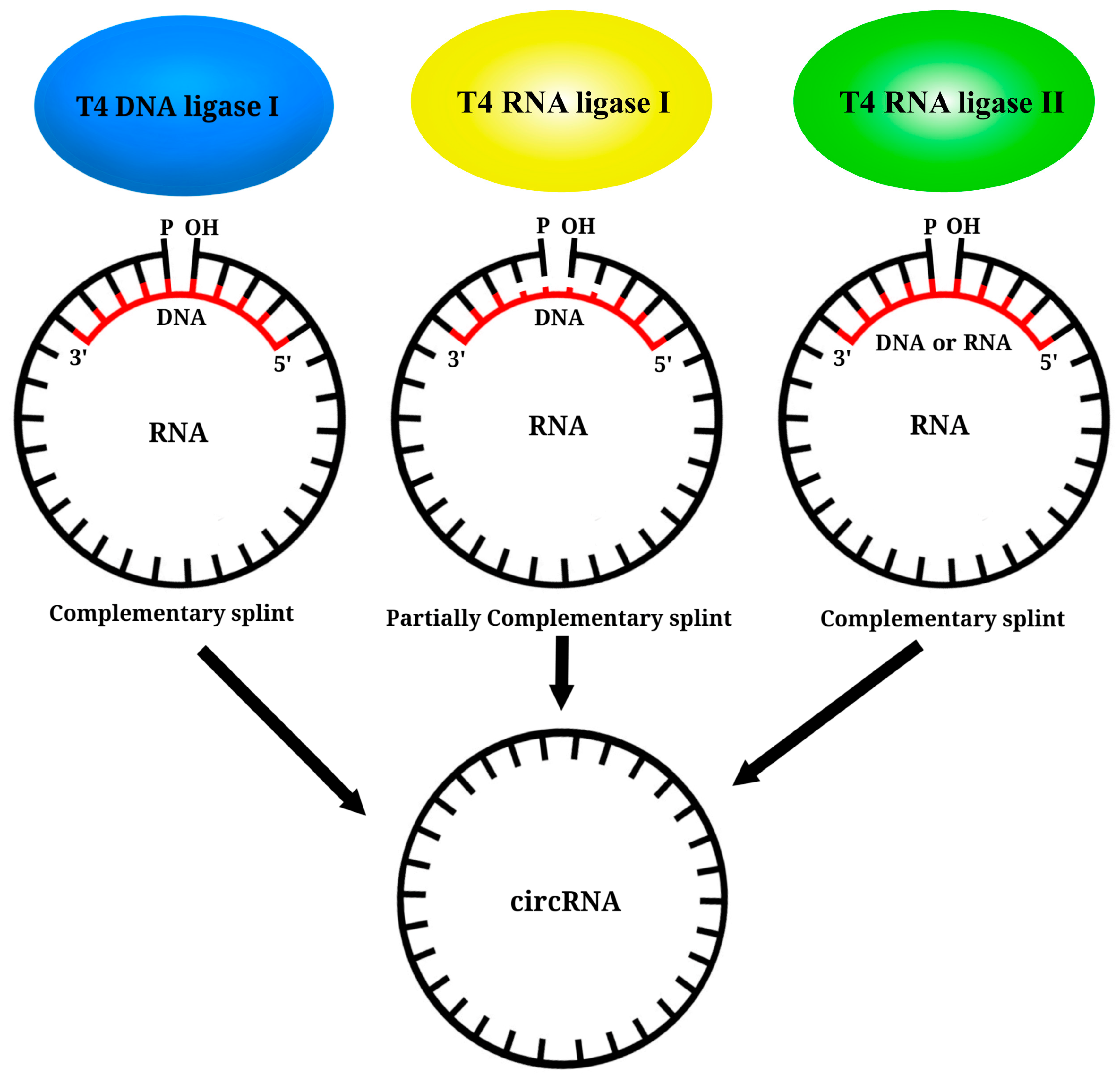
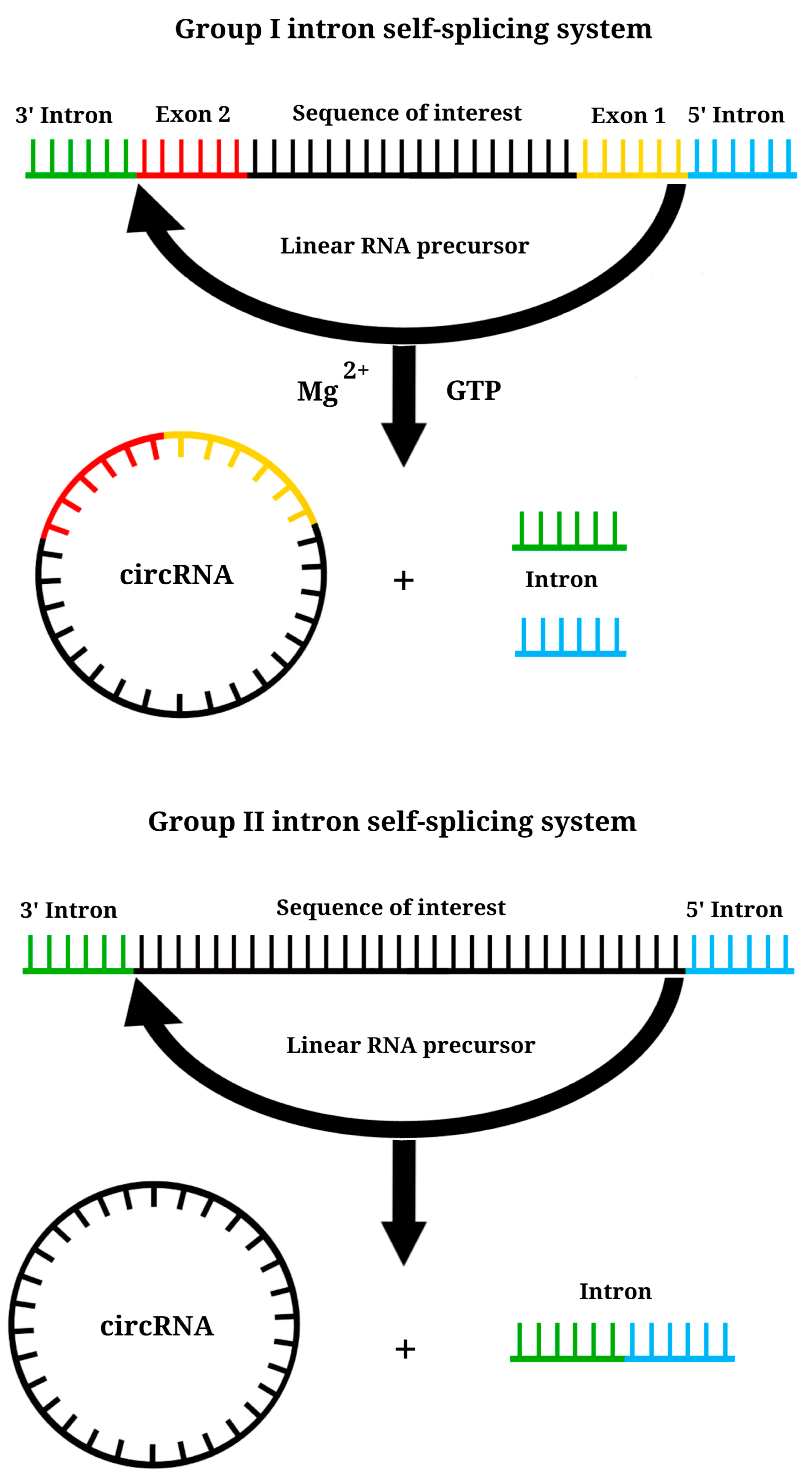
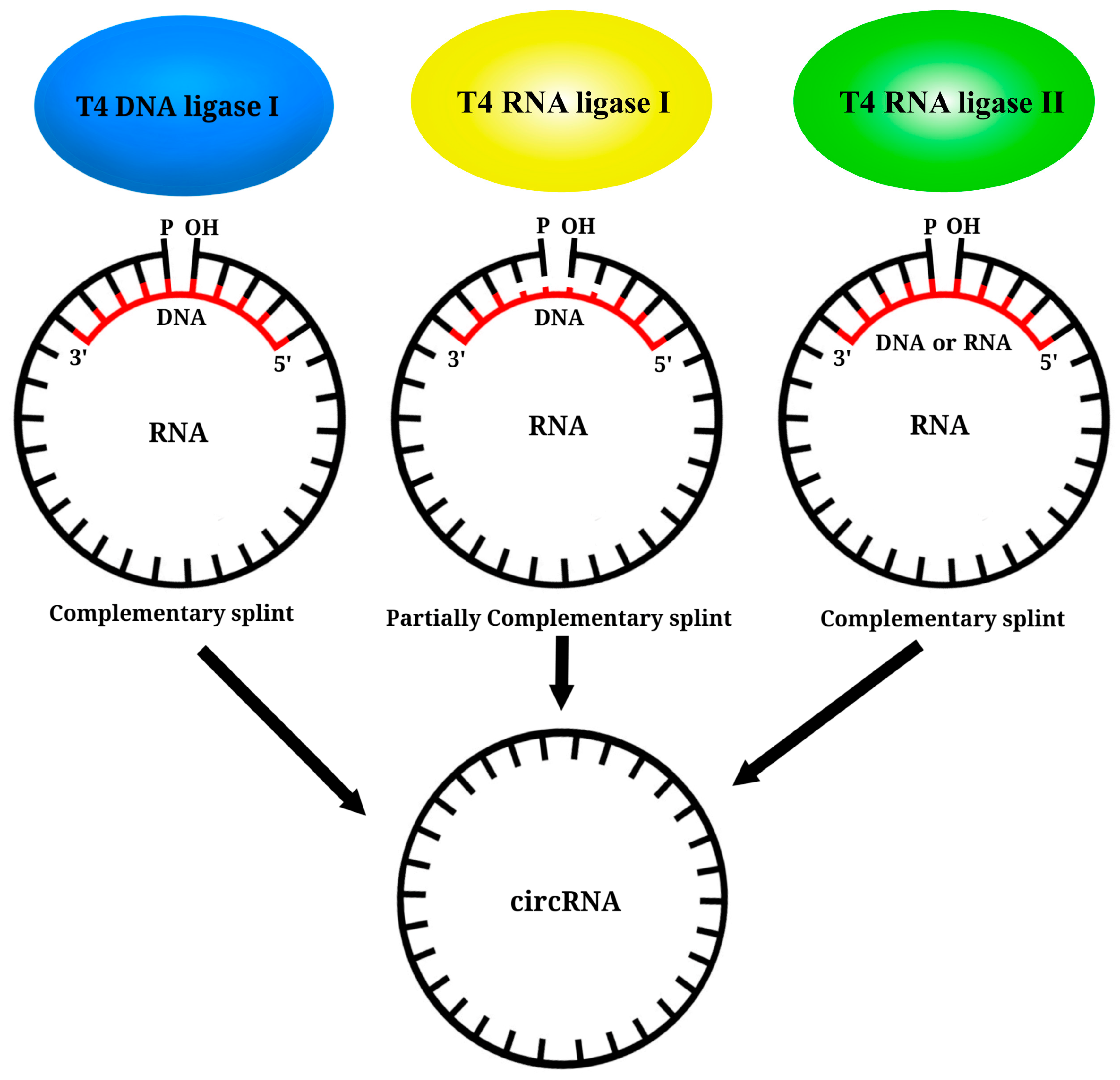
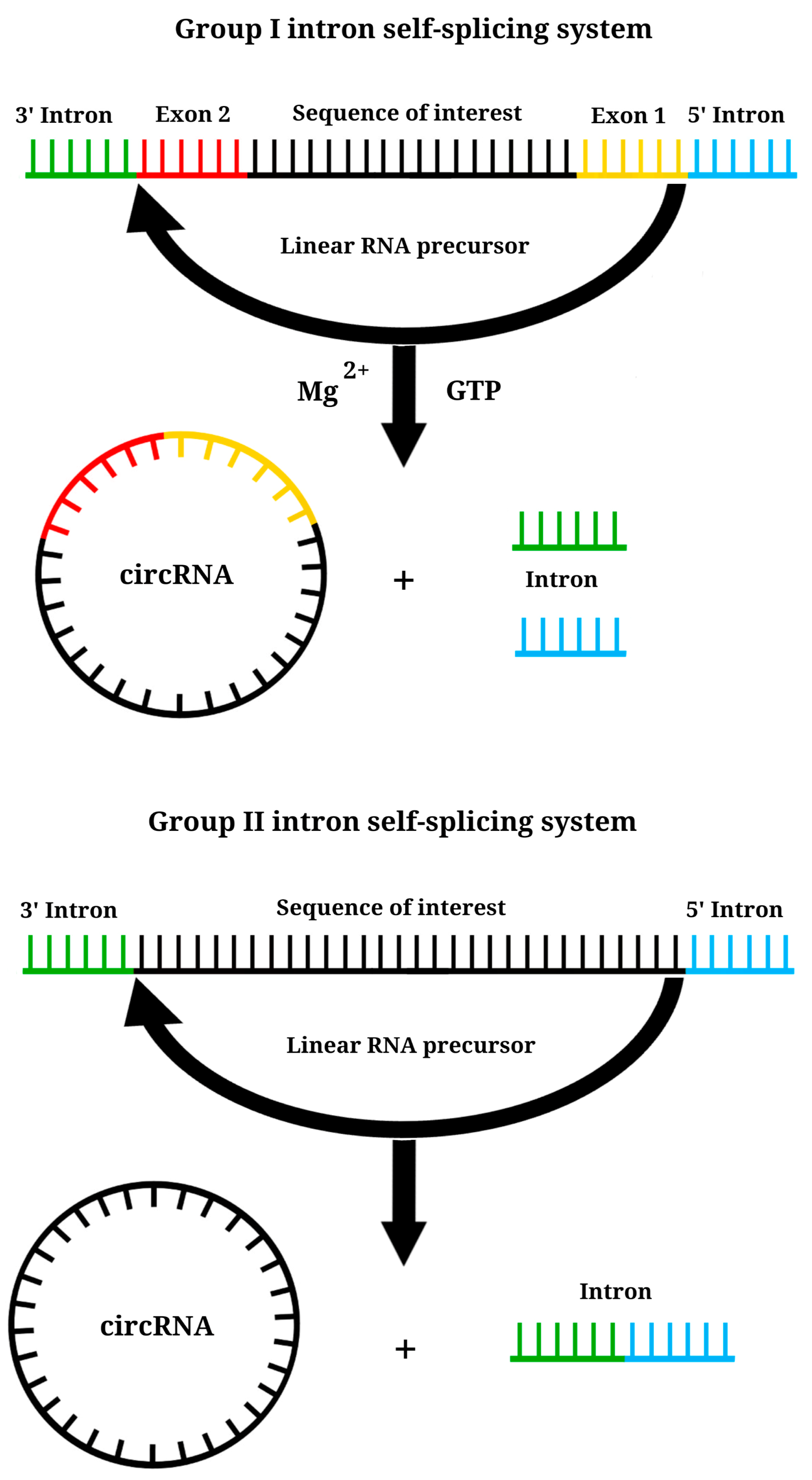
6. Circular RNA Platform
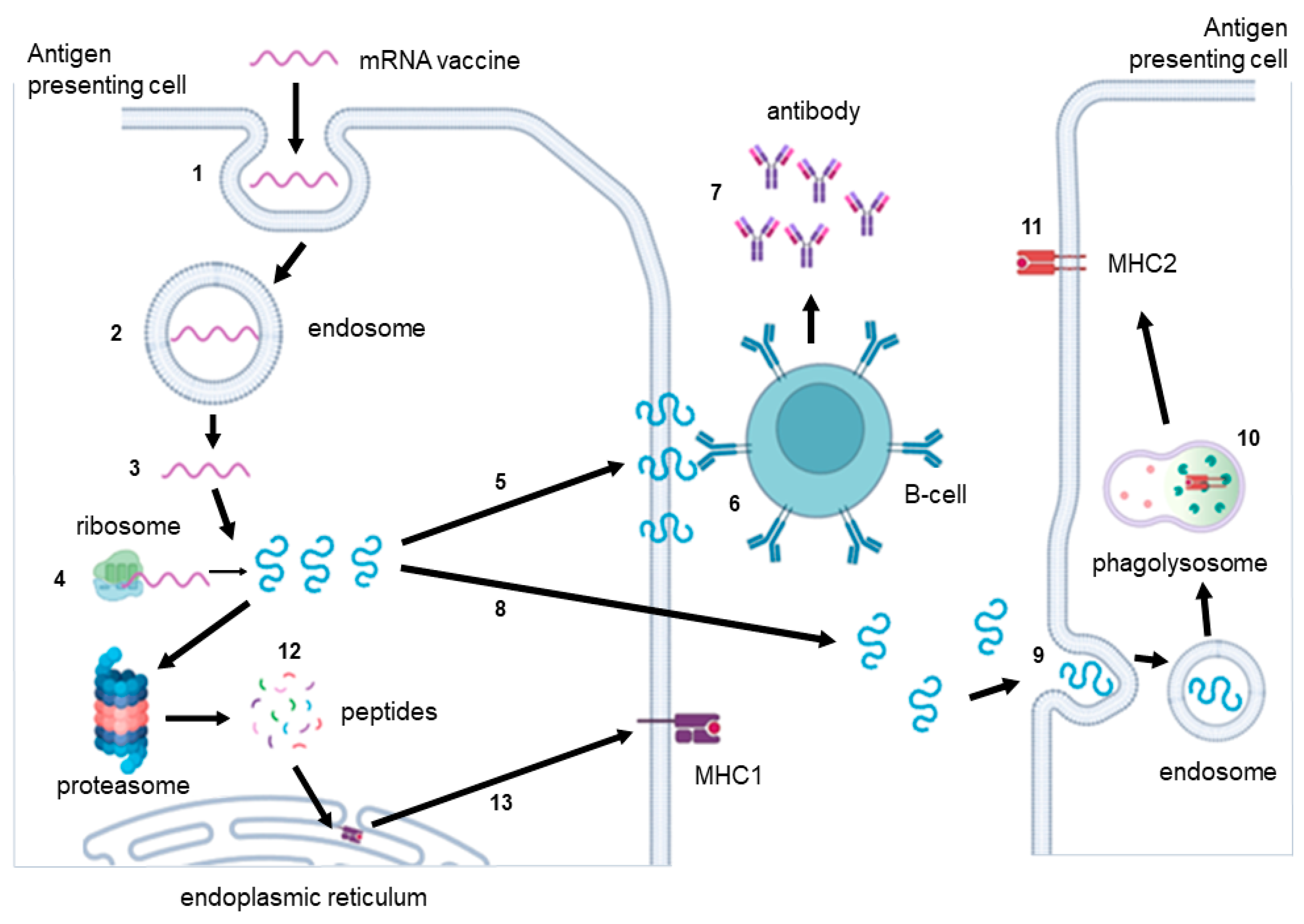
7. Immunogenicity of RNA Vaccines
8. Future of mRNA-Based Platform Vaccine
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Plotkin, S.A. Vaccines: The Fourth Century. Clin. Vaccine Immunol. 2009, 16, 1709–1719. [Google Scholar] [CrossRef] [PubMed]
- Probst, J.; Weide, B.; Scheel, B.; Pichler, B.J.; Hoerr, I.; Rammensee, H.G.; Pascolo, S. Spontaneous cellular uptake of exogenous messenger RNA in vivo is nucleic acid-specific, saturable and ion dependent. Gene Ther. 2007, 14, 1175–1180. [Google Scholar] [CrossRef] [PubMed]
- Ahammad, I.; Lira, S.S. Designing a novel mRNA vaccine against SARS-CoV-2: An immunoinformatics approach. Int. J. Biol. Macromol. 2020, 162, 820–837. [Google Scholar] [CrossRef]
- Li, W.; Joshi, M.D.; Singhania, S.; Ramsey, K.H.; Murthy, A.K. Peptide Vaccine: Progress and Challenges. Vaccines 2014, 2, 515–536. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.T.; Foged, C.; Korsholm, K.S.; Rades, T.; Christensen, D. Liposome-Based Adjuvants for Subunit Vaccines: Formulation Strategies for Subunit Antigens and Immunostimulators. Pharmaceutics 2016, 8, 7. [Google Scholar] [CrossRef] [PubMed]
- Schlake, T.; Thess, A.; Fotin-Mleczek, M.; Kallen, K.J. Developing mRNA-vaccine technologies. RNA Biol. 2012, 9, 1319–1330. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef]
- Chen, N.; Xia, P.; Li, S.; Zhang, T.; Wang, T.T.; Zhu, J. RNA sensors of the innate immune system and their detection of pathogens. IUBMB Life 2017, 69, 297–304. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Pelc, R.S.; Muramatsu, H.; Andersen, H.; DeMaso, C.R.; Dowd, K.A.; Sutherland, L.L.; Scearce, R.M.; Parks, R.; et al. Zika virus protection by a single low-dose nucleoside-modified mRNA vaccination. Nature 2017, 543, 248–251. [Google Scholar] [CrossRef]
- Cobb, M. Who discovered messenger RNA? Curr. Biol. 2015, 25, 526–532. [Google Scholar] [CrossRef]
- Zahednezhad, F.; Saadat, M.; Valizadeh, H.; Zakeri-Milani, P.; Baradaran, B. Liposome and immune system interplay: Challenges and potentials. J. Control. Release 2019, 10, 194–209. [Google Scholar] [CrossRef] [PubMed]
- Sanger, H.L.; Klotz, G.; Riesner, D.; Gross, H.J.; Kleinschmidt, A.K. Viroids are single-stranded covalently closed circular RNA molecules existing as highly base-paired rod-like structures. Proc. Natl. Acad. Sci. USA 1976, 73, 3852–3856. [Google Scholar] [CrossRef] [PubMed]
- Krieg, P.A.; Melton, D.A. In vitro RNA synthesis with SP6 RNA polymerase. Methods Enzymol. 1987, 155, 397–415. [Google Scholar] [PubMed]
- Dolgin, E. The tangled history of mRNA vaccines. Nature 2021, 597, 318–324. [Google Scholar] [CrossRef]
- Zhou, X.; Berglund, P.; Rhodes, G.; Parker, S.E.; Jondal, M.; Liljeström, P. Self-replicating Semliki Forest virus RNA as recombinant vaccine. Vaccine 1994, 12, 1510–1514. [Google Scholar] [CrossRef]
- Spuul, P.; Balistrer, G.; Hellstrom, K.; Golubtsov, A.V.; Jokitalo, E.; Ahola, T. Assembly of alphavirus replication complexes from RNA and protein components in a novel trans-replication system in mammalian cells. J. Virol. 2011, 85, 4739–4751. [Google Scholar] [CrossRef]
- Hou, X.; Zaks, T.; Langer, R.; Youn, H.; Im, H.J. Lipid nanoparticles for mRNA delivery. Nat. Rev. Mater. 2021, 6, 1078–1094. [Google Scholar] [CrossRef]
- Albertsen, C.H.; Kulkarni, J.A.; Witzigmann, D.; Lind, M.; Petersson, K.; Simonsen, J.B. The role of lipid components in lipid nanoparticles for vaccines and gene therapy. Adv. Drug Deliv. Rev. 2022, 188, 114416. [Google Scholar] [CrossRef]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.P.; Simon, P.; Löwer, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrörs, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef]
- Pascolo, S. Synthetic Messenger RNA-Based Vaccines: From Scorn to Hype. Viruses 2021, 13, 270. [Google Scholar] [CrossRef]
- Pardi, N.; Muramatsu, H.; Weissman, D.; Kariko, K. In vitro transcription of long RNA containing modified nucleosides. Methods Mol. Biol. 2013, 969, 29–42. [Google Scholar] [CrossRef]
- Rio, D.C. Expression and purification of active recombinant T7 RNA polymerase from E. coli. In RNA: A Laboratory Manual; CSHL Press: Cold Spring Harbor, NY, USA, 2013. [Google Scholar] [CrossRef]
- Deng, Z.; Tian, Y.; Song, J.; An, G.; Yang, P. mRNA Vaccines: The Dawn of a New Era of Cancer Immunotherapy. Front. Immunol. 2022, 13, 887125. [Google Scholar] [CrossRef] [PubMed]
- Klocker, N.; Weissenboeck, F.P.; Dulmen, M.; Spacek, P.; Huwel, S.; Rentmeister, A. Photocaged 5′ cap analogues for optical control of mRNA translation in cells. Nat. Chem. 2022, 14, 905–913. [Google Scholar] [CrossRef] [PubMed]
- To, K.K.; Cho, W.C. An overview of rational design of mRNA-based therapeutics and vaccines. Expert Opin. Drug Discov. 2021, 16, 1307–1317. [Google Scholar] [CrossRef]
- Ramanathan, A.; Robb, G.B.; Chan, S.-H. mRNA capping: Biological functions and applications. Nucleic Acids Res. 2016, 44, 7511–7526. [Google Scholar] [CrossRef]
- Mauer, J.; Luo, X.; Blanjoie, A.; Jiao, X.; Grozhik, A.V.; Patil, D.P.; Linder, B.; Pickering, B.F.; Vasseur, J.J.; Chen, Q.; et al. Reversible methylation of m6Am in the 5′ cap controls mRNA stability. Nature 2017, 541, 371–375. [Google Scholar] [CrossRef]
- Tat, T.T.; Kiss, D.L. From m6A to Cap-Adjacent m6Am and their Effects on mRNAs. Epitranscriptomics 2021, 12, 325–351. [Google Scholar] [CrossRef]
- Drazkowska, K.; Tomecki, R.; Warminski, M.; Baran, N.; Cysewski, D.; Depaix, A.; Kasprzyk, R.; Kowalska, J.; Jemielity, J.; Sikorski, P.J. 2′-O-Methylation of the second transcribed nucleotide within the mRNA 5′ cap impacts the protein production level in a cell-specific manner and contributes to RNA immune evasion. Nucleic Acids Res. 2022, 50, 9051–9071. [Google Scholar] [CrossRef] [PubMed]
- Linares-Fernandez, S.; Lacroix, C.; Exposito, J.-Y.; Verrier, B. Tailoring mRNA Vaccine to Balance Innate/Adaptive Immune Response. Trends Mol. Med. 2020, 26, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Pardi, N.; Hogan, M.J.; Weissman, D. Recent advances in mRNA vaccine technology. Curr. Opin. Immunol. 2020, 65, 14–20. [Google Scholar] [CrossRef]
- Stepinski, J.; Waddell, C.; Stolarski, R.; Darzynkiewicz, E.; Rhoads, R.E. Synthesis and properties of mRNAs containing the novel “anti-reverse” cap analogs 7-methyl (3′-O-methyl) GpppG and 7-methyl (3′-deoxy) GpppG. RNA 2001, 7, 1486–1495. [Google Scholar] [PubMed]
- Jemielity, J.; Fowler, T.; Zuberek, J.; Stepinski, J.; Lewdorowicz, M.; Niedzwiecka, A.; Stolarski, R.; Darzynkiewicz, E.; Rhoads, R.E. Novel “anti-reverse” cap analogs with superior translational properties. RNA 2003, 9, 1108–1122. [Google Scholar] [CrossRef] [PubMed]
- Henderson, J.M.; Ujita, A.; Hill, E.; Yousif-Rosales, S.; Smith, C.; Ko, N.; McReynolds, T.; Cabral, C.R.; Escamilla-Powers, J.R.; Houston, M.E. Cap 1 messenger RNA synthesis with co-transcriptional CleanCap® analog by in vitro transcription. Curr. Protoc. 2021, 1, e39. [Google Scholar] [CrossRef] [PubMed]
- TriLink BioTechnologies. Cap Analogs. Available online: https://www.trilinkbiotech.com/products-services/nucleoside-triphosphates-nucleotides.html?cap_analogs=1878 (accessed on 10 September 2023).
- Hornblower, B.; Robb, G.B.; Tzertzinis, G. Minding your caps and tails—Considerations for functional mRNA synthesis. DNA 2018, 5, 3. [Google Scholar]
- TriLink BioTechnologies. CleanCap. Available online: https://www.trilinkbiotech.com/cleancap-old21-3 (accessed on 10 September 2023).
- Moradian, H.; Roch, T.; Anthofer, L.; Lendlein, A.; Gossen, M. Chemical modification of uridine modulates mRNA-mediated proinflammatory and antiviral response in primary human macrophages. Mol. Ther. Nucleic Acids 2022, 27, 854–869. [Google Scholar] [CrossRef]
- Niederer, R.O.; Rojas-Duran, M.F.; Zinshteyn, B.; Gilbert, W.V. Direct analysis of ribosome targeting illuminates thousand-fold regulation of translation initiation. Cell Syst. 2022, 13, 256–264. [Google Scholar] [CrossRef]
- Leppek, K.; Das, R.; Barna, M. Functional 5′ UTR mRNA structures in eukaryotic translation regulation and how to find them. Nat. Rev. Mol. Cell Biol. 2018, 19, 158–174. [Google Scholar] [CrossRef]
- Hinnebusch, A.G.; Ivanov, I.P.; Sonenberg, N. Translational control by 5′-untranslated regions of eukaryotic mRNAs. Science 2016, 352, 1413–1416. [Google Scholar] [CrossRef]
- Haizel, S.A.; Bhardwaj, U.; Gonzalez, R.L., Jr.; Mitra, S.; Goss, D.J. 5′-UTR recruitment of the translation initiation factor eIF4GI or DAP5 drives cap-independent translation of a subset of human mRNAs. J. Biol. Chem. 2020, 295, 11693–11706. [Google Scholar] [CrossRef]
- Hinnebusch, A.G. Molecular mechanism of scanning and start codon selection in eukaryotes. Microbiol. Mol. Biol. Rev. 2011, 75, 434–467. [Google Scholar] [CrossRef]
- Weingarten-Gabbay, S.; Elias-Kirma, S.; Nir, R.; Gritsenko, A.A.; Stern-Ginossar, N.; Yakhini, Z.; Weinberger, A.; Segal, E. Comparative genetics. Systematic discovery of cap-independent translation sequences in human and viral genomes. Science 2016, 351, aad4939. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, R.; Tang, L.; Yang, L. Nonviral Delivery Systems of mRNA Vaccines for Cancer Gene Therapy. Pharmaceutics 2022, 14, 512. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Mao, Y.; Ji, Q.; Dersh, D.; Yewdell, J.W.; Qian, S.B. Decoding mRNA translatability and stability from the 5′ UTR. Nat. Struct. Mol. Biol. 2020, 27, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Linares-Fernández, S.; Moreno, J.; Lambert, E.; Mercier-Gouy, P.; Vachez, L.; Verrier, B.; Exposito, J.Y. Combining an optimized mRNA template with a double purification process allows strong expression of in vitro transcribed mRNA. Mol. Ther. Nucleic Acids 2021, 26, 945–956. [Google Scholar] [CrossRef]
- Sample, P.J.; Wang, B.; Reid, D.W.; Presnyak, V.; McFadyen, I.J.; Morris, D.R.; Seelig, G. Human 5′ UTR design and variant effect prediction from a massively parallel translation assay. Nat. Biotechnol. 2019, 37, 803–809. [Google Scholar] [CrossRef]
- Trepotec, Z.; Aneja, M.K.; Geiger, J.; Hasenpusch, G.; Plank, C.; Rudolph, C. Maximizing the Translational Yield of mRNA Therapeutics by Minimizing 5′-UTRs. Tissue Eng. Part A 2019, 25, 69–79. [Google Scholar] [CrossRef]
- Ko, H.L.; Park, H.-J.; Kim, J.; Kim, H.; Youn, H.; Nam, J.H. Development of an RNA Expression Platform Controlled by Viral Internal Ribosome Entry Sites. J. Microbiol. Biotechnol. 2019, 29, 127–140. [Google Scholar] [CrossRef]
- Deviatkin, A.A.; Simonov, R.A.; Trutneva, K.A.; Maznina, A.A.; Soroka, A.B.; Kogan, A.A.; Feoktistova, S.G.; Khavina, E.M.; Mityaeva, O.N.; Volchkov, P.Y. Cap-Independent Circular mRNA Translation Efficiency. Vaccines 2023, 11, 238. [Google Scholar] [CrossRef]
- Svitkin, Y.V.; Siddiqui, N.; Sonenberg, N. Protein Synthesis Initiation in Eukaryotes: IRES-mediated Internal Initiation. eLS 2015, 1–11. [Google Scholar] [CrossRef]
- Trainor, B.M.; Shcherbik, N. Short and Sweet: Viral 5′-UTR as a Canonical and Non-Canonical Translation Initiation Switch. J. Cell. Immunol. 2021, 3, 296–304. [Google Scholar] [CrossRef]
- Kreiter, S.; Selmi, A.; Diken, M.; Sebastian, M.; Osterloh, P.; Schild, H.; Huber, C.; Tureci, O.; Sahin, U. Increased antigen presentation efficiency by coupling antigens to MHC class I trafficking signals. J. Immunol. 2008, 180, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Mediani, L.; Guillen-Boixet, J.; Vinet, J.; Franzmann, T.M.; Bigi, I.; Mateju, D.; Carrà, A.D.; Morelli, F.F.; Tiago, T.; Poser, I.; et al. Defective ribosomal products challenge nuclear function by impairing nuclear condensate dynamics and immobilizing ubiquitin. EMBO J. 2019, 38, e101341. [Google Scholar] [CrossRef] [PubMed]
- Weissman, D. mRNA transcript therapy. Expert Rev. Vaccines 2015, 14, 265–281. [Google Scholar] [CrossRef]
- Bullock, T.N.J. CD40 stimulation as a molecular adjuvant for cancer vaccines and other immunotherapies. Cell. Mol. Immunol. 2022, 19, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, S.L.; Bai, A.; Bailey, D.; Ichikawa, K.; Zielinski, J.; Karp, R.; Apte, A.; Arnold, K.; Zacharek, S.J.; Iliou, M.S.; et al. Durable anticancer immunity from intratumoral administration of IL-23, IL-36γ, and OX40L mRNAs. Sci. Transl. Med. 2019, 11, eaat9143. [Google Scholar] [CrossRef]
- Fan, N.; Chen, K.; Zhu, R.; Zhang, Z.; Huang, H.; Qin, S.; Zheng, Q.; He, Z.; He, X.; Xiao, W.; et al. Manganese-coordinated mRNA vaccines with enhanced mRNA expression and immunogenicity induce robust immune responses against SARS-CoV-2 variants. Sci. Adv. 2022, 8, eabq3500. [Google Scholar] [CrossRef]
- Tanida-Miyake, E.; Koike, M.; Uchiyama, Y.; Tanida, I. Optimization of mNeonGreen for Homo sapiens increases its fluorescent intensity in mammalian cells. PLoS ONE 2018, 13, e0191108. [Google Scholar] [CrossRef]
- Zhong, F.; Cao, W.; Chan, E.; Tay, P.N.; Cahya, F.F.; Zhang, H.; Lu, J. Deviation from major codons in the Toll-like receptor genes is associated with low Toll-like receptor expression. Immunology 2005, 114, 83–93. [Google Scholar] [CrossRef]
- Kimchi-Sarfaty, C.; Oh, J.M.; Kim, I.-W.; Sauna, Z.E.; Calcagno, A.M.; Ambudkar, S.V.; Gottesman, M.M. A “silent” polymorphism in the MDR1 gene changes substrate specificity. Science 2007, 315, 525–528. [Google Scholar] [CrossRef]
- Kanampalliwar, A.M.; Soni, R.; Girdhar, A.; Tiwari, A. Reverse Vaccinology: Basics and Applications. Methods Mol. Biol. 2020, 2131, 1–16. [Google Scholar] [CrossRef]
- Yang, Z.; Bogdan, P.; Nazarian, S. An in silico deep learning approach to multi-epitope vaccine design: A SARS-CoV-2 case study. Sci. Rep. 2021, 11, 3238. [Google Scholar] [CrossRef]
- Serruto, D.; Rappuoli, R. Post-genomic vaccine development. FEBS Lett. 2006, 580, 2985–2992. [Google Scholar] [CrossRef] [PubMed]
- Fadilah, F.; Paramita, R.I.; Erlina, L.; Khaerunissa, A.I.; Puspita, E.W.; Aryo, T. Linker Optimization in Breast Cancer Multiepitope Peptide Vaccine Design Based on Molecular Study. ICOLIB 2023, 27, 528–538. [Google Scholar] [CrossRef]
- Suhrbier, A. Multi-epitope DNA vaccines. Immunol. Cell Biol. 1997, 75, 402–408. [Google Scholar] [CrossRef]
- Chen, X.; Zaro, J.L.; Shen, W.-C. Fusion protein linkers: Property, design and functionality. Adv. Drug Deliv. Rev. 2013, 65, 1357–1369. [Google Scholar] [CrossRef]
- Ali, M.; Pandey, R.K.; Khatoon, N.; Narula, A.; Mishra, A.K.; Prajapati, V.K. Exploring dengue genome to construct a multi-epitope based subunit vaccine by utilizing immunoinformatics approach to battle against dengue infection. Sci. Rep. 2017, 7, 9232. [Google Scholar] [CrossRef] [PubMed]
- Sanami, S.; Azadegan-Dehkordi, F.; Rafieian-Kopaei, M.; Salehi, M.; Ghasemi-Dehnoo, M.; Mahooti, M.; Alizadeh, M.; Bagheri, N. Design of a multi-epitope vaccine against cervical cancer using immunoinformatics approaches. Sci. Rep. 2021, 11, 12397. [Google Scholar] [CrossRef]
- Banerjee, S.; Majumder, K.; Gutierrez, G.J.; Gupta, D.; Mittal, B. Immuno-informatics approach for multi-epitope vaccine designing against SARS-CoV-2. BioRxiv 2020. BioRxiv:2020-07. [Google Scholar] [CrossRef]
- Mittal, A.; Sasidharan, S.; Raj, S.; Balaji, S.N.; Saudagar, P. Exploring the Zika Genome to Design a Potential Multiepitope Vaccine Using an Immunoinformatics Approach. Int. J. Pept. Res. Ther. 2020, 26, 2231–2240. [Google Scholar] [CrossRef]
- Mayr, C. What Are 3′ UTRs Doing? Cold Spring Harb. Perspect. Biol. 2019, 11, a034728. [Google Scholar] [CrossRef]
- Siepel, A.; Bejerano, G.; Pedersen, J.S.; Hinrichs, A.S.; Hou, M.; Rosenbloom, K.; Clawson, H.; Spieth, J.; Hillier, L.W.; Richards, S.; et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005, 15, 1034–1050. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Lu, J.; Kulbokas, E.J.; Golub, T.R.; Mootha, V.; Lindblad-Toh, K.; Lander, E.S.; Kellis, M. Systematic discovery of regulatory motifs in human promoters and 3′ UTRs by comparison of several mammals. Nature 2005, 434, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Orlandini von Niessen, A.G.; Poleganov, M.A.; Rechner, C.; Plaschke, A.; Kranz, L.M.; Fesser, S.; Diken, M.; Löwer, M.; Vallazza, B.; Beissert, T.; et al. Improving mRNA-Based Therapeutic Gene Delivery by Expression-Augmenting 3′ UTRs Identified by Cellular Library Screening. Mol. Ther. 2019, 27, 824–836. [Google Scholar] [CrossRef]
- Holtkamp, S.; Kreiter, S.; Selmi, A.; Simon, P.; Koslowski, M.; Huber, C.; Türeci, O.; Sahin, U. Modification of antigen-encoding RNA increases stability, translational efficacy, and T-cell stimulatory capacity of dendritic cells. Blood 2006, 108, 4009–4017. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.; Liu, X.; Li, M.; Zhang, Z.; Song, L.; Zhu, B.; Wu, X.; Liu, J.; Zhao, D.; Li, Y. Advances in COVID-19 mRNA vaccine development. Signal Transduct. Target. Ther. 2022, 7, 94. [Google Scholar] [CrossRef]
- Kontoyiannis, D.; Pasparakis, M.; Pizarro, T.T.; Cominelli, F.; Kollias, G. Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: Implications for joint and gut-associated immunopathologies. Immunity 1999, 10, 387–398. [Google Scholar] [CrossRef]
- Trepotec, Z.; Geiger, J.; Plank, C.; Aneja, M.K.; Rudolph, C. Segmented poly(A) tails significantly reduce recombination of plasmid DNA without affecting mRNA translation efficiency or half-life. RNA 2019, 25, 507–518. [Google Scholar] [CrossRef]
- Pardi, N.; Parkhouse, K.; Kirkpatrick, E.; McMahon, M.; Zost, S.J.; Mui, B.L.; Tam, Y.K.; Karikó, K.; Barbosa, C.J.; Madden, T.D.; et al. Nucleoside-Modified mRNA Immunization Elicits Influenza Virus Hemagglutinin Stalk-Specific Antibodies. Nat. Commun. 2018, 9, 3361. [Google Scholar] [CrossRef]
- Grier, A.; Burleigh, S.; Sahni, J.; Clough, C.A.; Cardot, V.; Choe, D.C.; Krutein, M.C.; Rawlings, D.J.; Jensen, M.C.; Scharenberg, A.M.; et al. pEVL: A Linear Plasmid for Generating mRNA IVT Templates with Extended Encoded Poly(A) Sequences. Mol. Ther. Nucleic Acids 2016, 5, e306. [Google Scholar] [CrossRef]
- Park, J.-E.; Yi, H.; Kim, Y.; Chang, H.; Kim, V.N. Regulation of Poly(A) Tail and Translation during the Somatic Cell Cycle. Mol. Cell 2016, 62, 462–471. [Google Scholar] [CrossRef]
- Qin, S.; Tang, X.; Chen, Y.; Chen, K.; Fan, N.; Xiao, W.; Zheng, Q.; Li, G.; Teng, Y.; Wu, M.; et al. mRNA-based therapeutics: Powerful and versatile tools to combat diseases. Signal Transduct. Target. Ther. 2022, 7, 166. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Kariko, K.; Tureci, O. mRNA-based therapeutics-developing a new class of drugs. Nat. Rev. Drug Discov. 2014, 13, 759–780. [Google Scholar] [CrossRef]
- Voet, D.; Voet, J.G. Biochemistry, 4th ed.; John Wiley & Sons: New York, NY, USA, 2011; pp. 123–126. [Google Scholar]
- Preiss, T.; Muckenthaler, M.; Hentze, M.W. Poly(A)-tail-promoted translation in yeast: Implications for translational control. RNA 1998, 4, 1321–1331. [Google Scholar] [CrossRef]
- Vogel, A.B.; Kanevsky, I.; Che, Y.; Swanson, K.A.; Muik, A.; Vormehr, M.; Kranz, L.M.; Walzer, K.C.; Hein, S.; Güler, A.; et al. A prefusion SARS-CoV-2 spike RNA vaccine is highly immunogenic and prevents lung infection in non-human primates. Biorxiv 2020. BioRxiv:2020-09. [Google Scholar] [CrossRef]
- Xia, X. Detailed dissection and critical evaluation of the Pfizer/BioNTech and Moderna mRNA vaccines. Vaccines 2021, 9, 734. [Google Scholar] [CrossRef] [PubMed]
- Nance, K.D.; Meier, J.L. Modifications in an Emergency: The Role of N1-Methylpseudouridine in COVID-19 Vaccines. ACS Cent. Sci. 2021, 7, 748–756. [Google Scholar] [CrossRef]
- Jeeva, S.; Kim, K.-H.; Shin, C.H.; Wang, B.Z.; Kang, S.M. An Update on mRNA-Based Viral Vaccines. Vaccines 2021, 9, 965. [Google Scholar] [CrossRef]
- Lima, S.A.; Chipman, L.; Nicholson, A.; Chen, Y.H.; Yee, B.A.; Yeo, G.W.; Coller, J.; Pasquinelli, A.E. Short poly(A) tails are a conserved feature of highly expressed genes. Nat. Struct. Mol. Biol. 2017, 24, 1057–1063. [Google Scholar] [CrossRef]
- Liu, Y.; Nie, H.; Sun, R.; Wang, J.; Lu, F. Enhancement of synthetic mRNA translation efficiency through engineered poly(A) tails. BioRxiv 2021. [Google Scholar] [CrossRef]
- Bhattacharya, M.; Sharma, A.R.; Ghosh, P.; Patra, P.; Patra, B.C.; Lee, S.S.; Chakraborty, C. Bioengineering of Novel Non-Replicating mRNA (NRM) and Self-Amplifying mRNA (SAM) Vaccine Candidates Against SARS-CoV-2 Using Immunoinformatics Approach. Mol. Biotechnol. 2022, 64, 510–525. [Google Scholar] [CrossRef]
- Brito, L.A.; Kommareddy, S.; Maione, D.; Uematsu, Y.; Giovani, C.; Berlanda Scorza, F.; Otten, G.R.; Yu, D.; Mandl, C.W.; Mason, P.W.; et al. Self-amplifying mRNA vaccines. Adv. Genet. 2015, 89, 179–233. [Google Scholar] [CrossRef]
- Tan, Y.B.; Yien Law, M.C.; Luo, D. Targeting the alphavirus virus replication process for antiviral development. Antivir. Res. 2023, 210, 105494. [Google Scholar] [CrossRef] [PubMed]
- Bakar, F.A.; Ng, L.F.P. Nonstructural Proteins of Alphavirus-Potential Targets for Drug Development. Viruses 2018, 10, 71. [Google Scholar] [CrossRef] [PubMed]
- Gotte, B.; Liu, L.; McInerney, G.M. The enigmatic alphavirus non-structural protein 3 (nsP3) revealing its secrets at last. Viruses 2018, 10, 105. [Google Scholar] [CrossRef] [PubMed]
- Forrester, N.L.; Guerbois, M.; Seymour, R.L.; Spratt, H.; Weaver, S.C. Vector-borne transmission imposes a severe bottleneck on an RNA virus population. PLoS Pathog. 2012, 8, e1002897. [Google Scholar] [CrossRef]
- Kim, J.; Eygeris, Y.; Gupta, M.; Sahay, G. Self-assembled mRNA vaccines. Adv. Drug Deliv. Rev. 2021, 170, 83–112. [Google Scholar] [CrossRef]
- Blakney, A.K.; McKay, P.F.; Shattock, R.J. Structural Components for Amplification of Positive and Negative Strand VEEV Splitzicons. Front. Mol. Biosci. 2018, 5, 71. [Google Scholar] [CrossRef]
- Bloom, K.; Berg, F.; Arbuthnot, P. Self-amplifying RNA vaccines for infectious diseases. Gene Ther. 2021, 28, 117–129. [Google Scholar] [CrossRef]
- Pollock, K.M.; Cheeseman, H.M.; Szubert, A.J.; Libri, V.; Boffito, M.; Owen, D.; Bern, H.; O’Hara, J.; McFarlane, L.R.; Lemm, N.M.; et al. Safety and Immunogenicity of a Self-Amplifying RNA Vaccine Against COVID-19: COVAC1, a Phase I, Dose-Ranging Trial. EClinicalMedicine 2022, 44, 101262. [Google Scholar] [CrossRef]
- Maruggi, G.; Zhang, C.; Li, J.; Ulmer, J.B.; Yu, D. mRNA as a transformative technology for vaccine development to control infectious diseases. Mol. Ther. 2019, 27, 757–772. [Google Scholar] [CrossRef]
- Carpentier, K.S.; Morrison, T.E. Innate immune control of alphavirus infection. Curr. Opin. Virol. 2018, 28, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Beissert, T.; Perkovic, M.; Vogel, A.; Erbar, S.; Walzer, K.C.; Hempel, T.; Brill, S.; Haefner, E.; Becker, R.; Türeci, Ö.; et al. A Trans-amplifying RNA Vaccine Strategy for Induction of potent protective immunity. Mol. Ther. 2020, 28, 119–128. [Google Scholar] [CrossRef]
- Schmidt, C.; Haefner, E.; Gerbeth, J.; Beissert, T.; Sahin, U.; Perkovic, M.; Schnierle, B.S. A taRNA vaccine candidate induces a specific immune response that protects mice against Chikungunya virus infections. Mol. Ther. Nucleic Acids 2022, 28, 743–754. [Google Scholar] [CrossRef] [PubMed]
- Palladino, G.; Chang, C.; Lee, C.; Music, N.; De Souza, I.; Nolasco, J.; Amoah, S.; Suphaphiphat, P.; Otten, G.R.; Settembre, E.C.; et al. Self-amplifying mRNA SARS-CoV-2 vaccines raise cross-reactive immune response to variants and prevent infection in animal models. Mol. Ther. Methods Clin. Dev. 2022, 25, 225–235. [Google Scholar] [CrossRef] [PubMed]
- McCafferty, S.; Haque, A.K.M.A.; Vandierendonck, A.; Weidensee, B.; Plovyt, M.; Stuchlíková, M.; François, N.; Valembois, S.; Heyndrickx, L.; Michiels, J.; et al. A dual-antigen self-amplifying RNA SARS-CoV-2 vaccine induces potent humoral and cellular immune responses and protects against SARS-CoV-2 variants through T cell-mediated immunity. Mol. Ther. 2022, 30, 2968–2983. [Google Scholar] [CrossRef]
- Chang, C.; Music, N.; Cheung, M.; Rossignol, E.; Bedi, S.; Patel, H.; Safari, M.; Lee, C.; Otten, G.R.; Settembre, E.C.; et al. Self-amplifying mRNA bicistronic influenza vaccines raise cross-reactive immune responses in mice and prevent infection in ferrets. Mol. Ther. Methods Clin. Dev. 2022, 27, 195–205. [Google Scholar] [CrossRef]
- Stokes, A.; Pion, J.; Binazon, O.; Laffont, B.; Bigras, M.; Dubois, G.; Blouin, K.; Young, J.K.; Ringenberg, M.A.; Ben Abdeljelil, N.; et al. Nonclinical safety assessment of repeated administration and biodistribution of a novel rabies self-amplifying mRNA vaccine in rats. Regul. Toxicol. Pharmacol. 2020, 113, 104648. [Google Scholar] [CrossRef]
- Erasmus, J.H.; Khandhar, A.P.; O’Connor, M.A.; Walls, A.C.; Hemann, E.A.; Murapa, P.; Archer, J.; Leventhal, S.; Fuller, J.T.; Lewis, T.; et al. An Alphavirus-derived replicon RNA vaccine induces SARS-CoV-2 neutralizing antibody and T cell responses in mice and nonhuman primates. Sci. Transl. Med. 2020, 12, eabc9396. [Google Scholar] [CrossRef]
- Luisi, K.; Morabito, K.M.; Burgomaster, K.E.; Sharma, M.; Kong, W.P.; Foreman, B.M.; Patel, S.; Fisher, B.; Aleshnick, M.A.; Laliberte, J.; et al. Development of a potent Zika virus vaccine using self-amplifying messenger RNA. Sci. Adv. 2020, 6, eaba5068. [Google Scholar] [CrossRef]
- Chahal, J.S.; Khan, O.F.; Cooper, C.L.; McPartlan, J.S.; Tsosie, J.K.; Tilley, L.D.; Sidik, S.M.; Lourido, S.; Langer, R.; Bavari, S.; et al. Dendrimer-RNA nanoparticles generate protective immunity against lethal Ebola, H1N1 influenza, and Toxoplasma gondii challenges with a single dose. Proc. Natl. Acad. Sci. USA 2016, 113, E4133–E4142. [Google Scholar] [CrossRef]
- Samsa, M.M.; Dupuy, L.C.; Beard, C.W.; Six, C.M.; Schmaljohn, C.S.; Mason, P.W.; Geall, A.J.; Ulmer, J.B.; Yu, D. Self-Amplifying RNA Vaccines for Venezuelan Equine Encephalitis Virus Induce Robust Protective Immunogenicity in Mice. Mol. Ther. 2019, 27, 850–865. [Google Scholar] [CrossRef] [PubMed]
- Moyo, N.; Vogel, A.B.; Buus, S.; Erbar, S.; Wee, E.G.; Sahin, U.; Hanke, T. Efficient Induction of T Cells against Conserved HIV-1 Regions by Mosaic Vaccines Delivered as Self-Amplifying mRNA. Mol. Ther. Methods Clin. Dev. 2019, 12, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Teague, B.; Zhang, Y.; Su, Z.; Porter, E.; Dobosh, B.; Wagner, T.; Irvine, D.J.; Weiss, R. In vitro evolution of enhanced RNA replicons for immunotherapy. Sci. Rep. 2019, 9, 6932. [Google Scholar] [CrossRef]
- Fuller, D.H.; Berglund, P. Amplifying RNA vaccine development. N. Engl. J. Med. 2020, 382, 2469–2471. [Google Scholar] [CrossRef] [PubMed]
- Perkovic, M.; Gawletta, S.; Hempel, T.; Brill, S.; Nett, E.; Sahin, U.; Beissert, T. A trans-amplifying RNA simplified to essential elements is highly replicative and robustly immunogenic in mice. Mol. Ther. 2023, 31, 1636–1646. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, C.; Schnierle, B.S. Self-Amplifying RNA Vaccine Candidates: Alternative Platforms for mRNA Vaccine Development. Pathogens 2023, 12, 138. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, C.; Hastert, F.D.; Gerbeth, J.; Beissert, T.; Sahin, U.; Perkovic, M.; Schnierle, B.S. A Bivalent Trans-Amplifying RNA Vaccine Candidate Induces Potent Chikungunya and Ross River Virus Specific Immune Responses. Vaccines 2022, 10, 1374. [Google Scholar] [CrossRef]
- Hsu, M.T.; Coca-Prados, M. Electron microscopic evidence for the circular form of RNA in the cytoplasm of eukaryotic cells. Nature 1979, 280, 339–340. [Google Scholar] [CrossRef]
- Nigro, J.M.; Cho, K.R.; Fearon, E.R.; Kern, S.E.; Ruppert, J.M.; Oliner, J.D.; Kinzler, K.W.; Vogelstein, B. Scrambled exons. Cell 1991, 64, 607–613. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Z. Efficient backsplicing produces translatable circular mRNAs. RNA 2015, 21, 172–179. [Google Scholar] [CrossRef]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Jeck, W.R.; Sorrentino, J.A.; Wang, K.; Slevin, M.K.; Burd, C.E.; Liu, J.; Marzluff, W.F.; Sharpless, N.E. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 2013, 19, 141–157. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Liu, D.; He, Q.; Liu, J.; Mao, Q.; Liang, Z. Research progress on circular RNA vaccines. Front. Immunol. 2022, 13, 1091797. [Google Scholar] [CrossRef]
- Pamudurti, N.R.; Bartok, O.; Jens, M.; Ashwal-Fluss, R.; Stottmeister, C.; Ruhe, L.; Hanan, M.; Wyler, E.; Perez-Hernandez, D.; Ramberger, E.; et al. Translation of CircRNAs. Mol. Cell 2017, 66, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.-C.; Wong, C.-W.; Liang, P.-P.; Shi, M.; Cao, Y.; Rao, S.T.; Tsui, S.K.; Waye, M.M.; Zhang, Q.; Fu, W.M. Translation of the circular RNA circβ-catenin promotes liver cancer cell growth through activation of the Wnt pathway. Genome Biol. 2019, 20, 84. [Google Scholar] [CrossRef]
- Chen, C.; Sarnow, P. Initiation of protein synthesis by the eukaryotic translational apparatus on circular RNAs. Science 1995, 268, 415–417. [Google Scholar] [CrossRef]
- Wesselhoeft, R.A.; Kowalski, P.S.; Anderson, D.G. Engineering circular RNA for potent and stable translation in eukaryotic cells. Nat. Commun. 2018, 9, 2629. [Google Scholar] [CrossRef]
- Qu, L.; Yi, Z.; Shen, Y.; Lin, L.; Chen, F.; Xu, Y.; Wu, Z.; Tang, H.; Zhang, X.; Tian, F. Circular RNA vaccines against SARS-CoV-2 and emerging variants. Cell 2022, 185, 1728–1744.e16. [Google Scholar] [CrossRef]
- Chen, R.; Wang, S.K.; Belk, J.A.; Amaya, L.; Li, Z.; Cardenas, A.; Abe, B.T.; Chen, C.K.; Wender, P.A.; Chang, H.Y. Engineering circular RNA for enhanced protein production. Nat. Biotechnol. 2023, 41, 262–272. [Google Scholar] [CrossRef]
- Chen, X.; Lu, Y. Circular RNA: Biosynthesis in vitro. Front. Bioeng. Biotechnol. 2021, 9, 787881. [Google Scholar] [CrossRef]
- Obi, P.; Chen, Y.G. The Design and Synthesis of Circular RNAs. Methods 2021, 196, 85–103. [Google Scholar] [CrossRef]
- Moore, M.J. Joining RNA molecules with T4 DNA ligase. Methods Mol. Biol. 1999, 118, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Abe, N.; Kodama, A.; Abe, H. Preparation of Circular RNA In Vitro. Methods Mol. Biol. 2018, 1724, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Deana, A.; Celesnik, H.; Belasco, J.G. The bacterial enzyme RppH triggers messenger RNA degradation by 5′ pyrophosphate removal. Nature 2008, 451, 355–358. [Google Scholar] [CrossRef] [PubMed]
- Petkovic, S.; Müller, S. Synthesis and Engineering of Circular RNAs. Methods Mol. Biol. 2018, 1724, 167–180. [Google Scholar] [CrossRef]
- Lohman, G.J.S.; Tabor, S.; Nichols, N.M. Current Protocols in Molecular Biology—DNA ligases. Curr. Protoc. Mol. Biol. 2011, 94, 3.14.1–3.14.7. [Google Scholar] [CrossRef]
- Chen, Y.G.; Kim, M.V.; Chen, X.; Batista, P.J.; Aoyama, S.; Wilusz, J.E.; Iwasaki, A.; Chang, H.Y. Sensing Self and Foreign Circular RNAs by Intron Identity. Mol. Cell 2017, 67, 228–238. [Google Scholar] [CrossRef]
- Kershaw, C.J.; O’Keefe, R.T. Splint ligation of RNA with T4 DNA ligase—Recombinant and in vitro RNA synthesis, methods and protocols. Methods Mol. Biol. 2012, 941, 257–269. [Google Scholar] [CrossRef]
- Uhlenbeck, O.C.; Gumport, R.I. T4 RNA ligase. Enzyme 1982, 15, 31–58. [Google Scholar]
- Muller, S.; Appel, B. In Vitro circularization of RNA. RNA Biol. 2017, 14, 1018–1027. [Google Scholar] [CrossRef]
- Petkovic, S.; Muller, S. RNA Circularization Strategies In Vivo and In Vitro. Nucleic Acids Res. 2015, 43, 2454–2465. [Google Scholar] [CrossRef]
- Wang, L.; Ruffner, D.E. Oligoribonucleotide circularization by ‘template-mediated’ ligation with T4 RNA ligase: Synthesis of circular hammerhead ribozymes. Nucleic Acids Res. 1998, 26, 2502–2504. [Google Scholar] [CrossRef] [PubMed]
- Costello, A.; Lao, N.T.; Barron, N.; Clynes, M. Reinventing the Wheel: Synthetic Circular RNAs for Mammalian Cell Engineering. Trends Biotechnol. 2020, 38, 217–230. [Google Scholar] [CrossRef] [PubMed]
- Breuer, J.; Rossbach, O. Production and purification of artificial circular RNA sponges for application in molecular biology and medicine. Methods Protoc. 2020, 3, 42. [Google Scholar] [CrossRef]
- Carmona, E. Circular RNA: Design Criteria for Optimal Therapeutical Utility; Harvard University: Cambridge, UK, 2019. [Google Scholar]
- Bullard, D.R.; Bowater, R.P. Direct Comparison of Nick-Joining Activity of the Nucleic Acid Ligases from Bacteriophage T4. Biochem. J. 2006, 398, 135–144. [Google Scholar] [CrossRef]
- Chen, H.; Cheng, K.; Liu, X.; An, R.; Komiyama, M.; Liang, X. Preferential production of RNA rings by T4 RNA ligase 2 without any splint through rational design of precursor strand. Nucleic Acids Res. 2020, 48, e54. [Google Scholar] [CrossRef] [PubMed]
- Puttaraju, M.; Been, M.D. Group I permuted intron-exon (PIE) sequences self-splice to produce circular exons. Nucleic Acids Res. 1992, 20, 5357–5364. [Google Scholar] [CrossRef]
- Ford, E.; Ares, M., Jr. Synthesis of circular RNA in bacteria and yeast using RNA cyclase ribozymes derived from a group I intron of phage T4. Proc. Natl. Acad. Sci. USA 1994, 91, 3117–3121. [Google Scholar] [CrossRef]
- Rausch, J.W.; Hein, W.F.; Payea, M.J.; Sherpa, C.; Gorospe, M.; Le Grice, S.F.J. Characterizing and circumventing sequence restrictions for synthesis of circular RNA in vitro. Nucleic Acids Res. 2021, 49, e35. [Google Scholar] [CrossRef]
- Lee, K.H.; Kim, S.; Song, J.; Han, S.R.; Kim, J.H.; Lee, S.W. Efficient circular RNA engineering by end-to-end self-targeting and splicing reaction using Tetrahymena group I intron ribozyme. Nucleic Acids Res. 2023, 51, e78. [Google Scholar] [CrossRef]
- Rostain, W.; Shen, S.; Cordero, T.; Rodrigo, G.; Jaramillo, A. Engineering a circular riboregulator in Escherichia coli. Biodesign Res. 2020, 1–9. [Google Scholar] [CrossRef]
- Mikheeva, S.; Hakim-Zarga, M.; Carlson, D.; Jarrell, K. Use of an engineered ribozyme to produce a circular human exon. Nucleic Acids Res. 1997, 25, 5085–5094. [Google Scholar] [CrossRef] [PubMed]
- Lambowitz, A.M.; Zimmerly, S. Group II introns: Mobile ribozymes that invade DNA. Cold Spring Harb. Perspect. Biol. 2011, 3, a003616. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wei, H.; Zhang, K.; Li, Z.; Wei, T.; Tang, C.; Yang, Y.; Wang, Z. A flexible, efficient, and scalable platform to produce circular RNAs as new therapeutics. BioRxiv 2022. [Google Scholar] [CrossRef]
- Murray, H.L.; Mikheeva, S.; Coljee, V.W.; Turczyk, B.M.; Donahue, W.F.; Bar-Shalom, A.; Jarrell, K.A. Excision of group II introns as circles. Mol. Cell 2001, 8, 201–211. [Google Scholar] [CrossRef]
- Diegelman, A.M.; Kool, E.T. Generation of circular RNAs and trans—Cleaving catalytic RNAs by rolling transcription of circular DNA oligonucleotides encoding hairpin ribozymes. Nucleic Acids Res. 1998, 26, 3235–3241. [Google Scholar] [CrossRef]
- Petkovic, S.; Muller, S. RNA Self-Processing: Formation of Cyclic Species and Concatemers from a Small Engineered RNA. FEBS Lett. 2013, 587, 2435–2440. [Google Scholar] [CrossRef]
- Dallas, A.; Balatskaya, S.V.; Kuo, T.-C.; Ilves, H.; Vlassov, A.V.; Kaspar, R.L.; Kisich, K.O.; Kazakov, S.A.; Johnston, B.H. Hairpin Ribozyme-Antisense RNA Constructs Can Act as Molecular Lassos. Nucleic Acids Res. 2008, 36, 6752–6766. [Google Scholar] [CrossRef]
- Hieronymus, R.; Muller, S. Engineering of Hairpin Ribozyme Variants for RNA Recombination and Splicing. Ann. N. Y. Acad. Sci. 2019, 1447, 135–143. [Google Scholar] [CrossRef]
- Fisher, A.J.; Beal, P.A. Structural basis for eukaryotic mRNA modification. Curr. Opin. Struct. Biol. 2018, 53, 59–68. [Google Scholar] [CrossRef]
- Kariko, K.; Muramatsu, H.; Ludwig, J.; Weissman, D. Generating the optimal mRNA for therapy: HPLC purification eliminates immune activation and improves translation of nucleoside-modified, protein-encoding mRNA. Nucleic Acids Res. 2011, 39, e142. [Google Scholar] [CrossRef] [PubMed]
- Vaidyanathan, S.; Azizian, K.T.; Haque, A.A.; Henderson, J.M.; Hendel, A.; Shore, S.; Antony, J.S.; Hogrefe, R.I.; Kormann, M.S.D.; Porteus, M.H.; et al. Uridine depletion and chemical modification increase Cas9 mRNA activity and reduce immunogenicity without HPLC purification. Mol. Ther. Nucleic Acids 2018, 12, 530–542. [Google Scholar] [CrossRef]
- Hornung, V.; Ellegast, J.; Kim, S.; Brzózka, K.; Jung, A.; Kato, H.; Poeck, H.; Akira, S.; Conzelmann, K.K.; Schlee, M.; et al. Hartmann, G. 5′-Triphosphate RNA is the ligand for RIG-I. Science 2006, 314, 994–997. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Takeuchi, O.; Mikamo-Satoh, E.; Hirai, R.; Kawai, T.; Matsushita, K.; Hiiragi, A.; Dermody, T.S.; Fujita, T.; Akira, S. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J. Exp. Med. 2008, 205, 1601–1610. [Google Scholar] [CrossRef] [PubMed]
- Pulit-Penaloza, J.A.; Scherbik, S.V.; Brinton, M.A. Activation of Oas1a gene expression by type I IFN requires both STAT1 and STAT2 while only STAT2 is required for Oas1b activation. Virology 2012, 425, 71–81. [Google Scholar] [CrossRef]
- Xu, S.; Yang, K.; Li, R.; Zhang, L. mRNA Vaccine Era-Mechanisms, Drug Platform and Clinical Prospection. Int. J. Mol. Sci. 2020, 21, 6582. [Google Scholar] [CrossRef] [PubMed]
- Bidram, M.; Zhao, Y.; Shebardina, N.G.; Baldin, A.V.; Bazhin, A.V.; Ganjalikhany, M.R.; Zamyatnin, A.A., Jr.; Ganjalikhani-Hakemi, M. mRNA-Based Cancer Vaccines: A Therapeutic Strategy for the Treatment of Melanoma Patients. Vaccines 2021, 9, 1060. [Google Scholar] [CrossRef]
- Freund, I.; Eigenbrod, T.; Helm, M.; Dalpke, A. RNA Modifications Modulate Activation of Innate Toll-Like Receptors. Genes 2019, 10, 92. [Google Scholar] [CrossRef]
- Rouf, N.Z.; Biswas, S.; Tarannum, N.; Oishee, L.M.; Muna, M.M. Demystifying mRNA vaccines: An emerging platform at the forefront of cryptic diseases. RNA Biol. 2022, 19, 386–410. [Google Scholar] [CrossRef]
- Boccaletto, P.; Machnicka, M.A.; Purta, E.; Feder, M.; Grosjean, H.; Bujnicki, J.M. MODOMICS: A database of RNA modification pathways. Nucleic Acids Res. 2018, 46, D303–D307. [Google Scholar] [CrossRef]
- Kariko, K.; Buckstein, M.; Ni, H.; Weissman, D. Suppression of RNA recognition by Toll-like receptors: The impact of nucleoside modification and the evolutionary origin of RNA. Immunity 2005, 23, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Gao, F.; Chang, Y.; Zhao, Q.; He, X. Advances of mRNA vaccine in tumor: A maze of opportunities and challenges. Biomark. Res. 2023, 11, 6. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Zhang, Q.; Feng, X.H.; Liu, J. Synthetic modified messenger RNA for therapeutic applications. Acta Biomater. 2021, 131, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.; Kim, M.; Seo, Y.; Moon, Y.S.; Lee, H.J.; Lee, K.; Lee, H. Emergence of synthetic mRNA: In vitro synthesis of mRNA and its applications in regenerative medicine. Biomaterials 2018, 156, 172–193. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Yi, C. Chemical Modifications to RNA: A New Layer of Gene Expression Regulation. ACS Chem. Biol. 2017, 12, 316–325. [Google Scholar] [CrossRef]
- Durbin, A.F.; Wang, C.; Marcotrigiano, J.; Gehrke, L. RNAs Containing Modified Nucleotides Fail To Trigger RIG-I Conformational Changes for Innate Immune Signaling. mBio 2016, 7, e00833-16. [Google Scholar] [CrossRef]
- Gote, V.; Bolla, P.K.; Kommineni, N.; Butreddy, A.; Nukala, P.K.; Palakurthi, S.S.; Khan, W. A Comprehensive Review of mRNA Vaccines. Int. J. Mol. Sci. 2023, 24, 2700. [Google Scholar] [CrossRef]
- Mu, X.; Hur, S. Immunogenicity of In Vitro-Transcribed RNA. Acc. Chem. Res. 2021, 54, 4012–4023. [Google Scholar] [CrossRef]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Salmon-Divon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M.; et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012, 485, 201–206. [Google Scholar] [CrossRef]
- Meyer, K.D.; Saletore, Y.; Zumbo, P.; Elemento, O.; Mason, C.E.; Jaffrey, S.R. Comprehensive Analysis of mRNA Methylation Reveals Enrichment in 3′ UTRs and Near Stop Codons. Cell 2012, 149, 1635–1646. [Google Scholar] [CrossRef]
- Chen, Y.G.; Chen, R.; Ahmad, S.; Verma, R.; Kasturi, S.P.; Amaya, L.; Broughton, J.P.; Kim, J.; Cadena, C.; Pulendran, B.; et al. N6-Methyladenosine Modification Controls Circular RNA Immunity. Mol. Cell 2019, 76, 96–109.e9. [Google Scholar] [CrossRef]
- Kostyusheva, A.; Brezgin, S.; Glebe, D.; Kostyushev, D.; Chulanov, V. Host-cell interactions in HBV infection and pathogenesis: The emerging role of m6A modification. Emerg. Microbes Infect. 2021, 10, 2264–2275. [Google Scholar] [CrossRef]
- Yang, Y.; Fan, X.; Mao, M.; Song, X.; Wu, P.; Zhang, Y.; Jin, Y.; Yang, Y.; Chen, L.L.; Wang, Y.; et al. Extensive translation of circular RNAs driven by N6-methyladenosine. Cell Res. 2017, 27, 626–641. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Wang, J.Q.; Tan, Y.; Yuan, R.; Chen, Z.S.; Zou, C. RNA methylation and cancer treatment. Pharmacol. Res. 2021, 174, 105937. [Google Scholar] [CrossRef] [PubMed]
- Wesselhoeft, R.A.; Kowalski, P.S.; Parker-Hale, F.C.; Huang, Y.; Bisaria, N.; Anderson, D.G. RNA Circularization Diminishes Immunogenicity and Can Extend Translation Duration In Vivo. Mol. Cell 2019, 74, 508–520.e4. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.X.; Guo, S.K.; Nan, F.; Xu, Y.F.; Yang, L.; Chen, L.L. RNA circles with minimized immunogenicity as potent PKR inhibitors. Mol. Cell 2022, 82, 420–434.e6. [Google Scholar] [CrossRef]
- Tai, J.; Chen, Y.G. Differences in the immunogenicity of engineered circular RNAs. J. Mol. Cell Biol. 2023, 15, mjad002. [Google Scholar] [CrossRef]
- Rigby, R.E.; Webb, L.M.; Mackenzie, K.J.; Li, Y.; Leitch, A.; Reijns, M.A.; Lundie, R.J.; Revuelta, A.; Davidson, D.J.; Diebold, S.; et al. RNA:DNA hybrids are a novel molecular pattern sensed by TLR9. EMBO J. 2014, 33, 542–558. [Google Scholar] [CrossRef]
- Roers, A.; Hiller, B.; Hornung, V. Recognition of Endogenous Nucleic Acids by the Innate Immune System. Immunity 2016, 44, 739–754. [Google Scholar] [CrossRef]
- Lee, J.H.; Chiang, C.; Gack, M.U. Endogenous Nucleic Acid Recognition by RIG-I-Like Receptors and cGAS. J. Interferon Cytokine Res. Off. J. Int. Soc. Interferon Cytokine Res. 2019, 39, 450–458. [Google Scholar] [CrossRef]
- Wang, L.; Sun, L.; Byrd, K.M.; Ko, C.C.; Zhao, Z.; Fang, J. AIM2 Inflammasome’s First Decade of Discovery: Focus on Oral Diseases. Front. Immunol. 2020, 11, 1487. [Google Scholar] [CrossRef] [PubMed]
- Mu, X.; Greenwald, E.; Ahmad, S.; Hur, S. An origin of the immunogenicity of in vitro transcribed RNA. Nucleic Acids Res. 2018, 46, 5239–5249. [Google Scholar] [CrossRef] [PubMed]
- Xiao, M.-S.; Wilusz, J.E. An improved method for circular RNA purification using RNase R that efficiently removes linear RNAs containing G-quadruplexes or structured 3′ ends. Nucleic Acids Res. 2019, 47, 8755–8769. [Google Scholar] [CrossRef]
- Weissman, D.; Pardi, N.; Muramatsu, H.; Karikó, K. Synthetic messenger RNA and cell metabolism modulation, methods and protocols. Methods Mol. Biol. 2012, 969, 43–54. [Google Scholar] [CrossRef]
- Baiersdorfer, M.; Boros, G.; Muramatsu, H.; Mahiny, A.; Vlatkovic, I.; Sahin, U.; Karikó, K. A Facile Method for the Removal of dsRNA Contaminant from In Vitro-Transcribed mRNA. Mol. Therapy. Nucleic Acids 2019, 15, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Rosa, S.S.; Prazeres, D.M.F.; Azevedo, A.M.; Marques, M.P.C. mRNA vaccines manufacturing: Challenges and bottlenecks. Vaccine 2021, 39, 2190–2200. [Google Scholar] [CrossRef]
- Rzymski, P.; Szuster-Ciesielska, A.; Dzieciątkowski, T.; Gwenzi, W.; Fal, A. mRNA vaccines: The future of prevention of viral infections? J. Med. Virol. 2023, 95, e28572. [Google Scholar] [CrossRef]
- Al Fayez, N.; Nassar, M.S.; Alshehri, A.A.; Alnefaie, M.K.; Almughem, F.A.; Alshehri, B.Y.; Alawad, A.O.; Tawfik, E.A. Recent Advancement in mRNA Vaccine Development and Applications. Pharmaceutics 2023, 15, 1972. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Cap | Structure | Capping Enzymes | Capping Stage |
|---|---|---|---|
| Cap 0 | m7G-ppp-N-p | Vaccinia Capping Enzyme (VCE) | Separate stage |
| Cap 1 | m7G-ppp-N2’-Me-Op | Vaccinia Capping Enzyme (VCE) and mRNA cap 2′O-methyltransferase (MTase) | Two separate stages |
| Anti-Reverse Cap Analog (ARCA) | 3′-O-Me-m7G5′-ppp-5′G | RNA polymerases of bacteriophages T3, T7 or SP6 | Сo-transcriptionally |
| CleanCap® Reagent AG 3′OMe | m7G3′-O-Me-ppp-A2′-O-Me-p-G | RNA polymerases of bacteriophages T3, T7 or SP6 | Сo-transcriptionally |
| CleanCap® Reagent AG | m7G-ppp-A2′-O-Me-p-G | RNA polymerases of bacteriophages T3, T7 or SP6 | Сo-transcriptionally |
| CleanCap® Reagent AU | m7G-ppp-A2′-O-Me-p-U | RNA polymerases of bacteriophages T3, T7 or SP6 | Сo-transcriptionally |
| Nucleic Acid | PRR | Effects |
|---|---|---|
| ssRNA | TLR7 | Inhibition of protein translation; Synthesis of pro-inflammatory cytokines |
| TLR8 | Synthesis of pro-inflammatory cytokines | |
| NOD2 | Synthesis of pro-inflammatory cytokines | |
| dsRNA | TLR3 | Inhibition of protein translation; Synthesis of pro-inflammatory cytokines |
| RIG-I | Inhibition of protein translation; Synthesis of pro-inflammatory cytokines | |
| MDA-5 | Inhibition of protein translation; Synthesis of pro-inflammatory cytokines | |
| NLRP3 | Synthesis of pro-inflammatory cytokines | |
| PKR | Stop of translation | |
| OAS | RNA degradation | |
| CircRNA | RIG-I | Inhibition of protein translation; Synthesis of pro-inflammatory cytokines |
| PKR | Stop of translation | |
| dsDNA, RNA/DNA | TLR9 | Synthesis of pro-inflammatory cytokines |
| cGAS | Synthesis of pro-inflammatory cytokines | |
| AIM2 | Synthesis of pro-inflammatory cytokines |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perenkov, A.D.; Sergeeva, A.D.; Vedunova, M.V.; Krysko, D.V. In Vitro Transcribed RNA-Based Platform Vaccines: Past, Present, and Future. Vaccines 2023, 11, 1600. https://doi.org/10.3390/vaccines11101600
Perenkov AD, Sergeeva AD, Vedunova MV, Krysko DV. In Vitro Transcribed RNA-Based Platform Vaccines: Past, Present, and Future. Vaccines. 2023; 11(10):1600. https://doi.org/10.3390/vaccines11101600
Chicago/Turabian StylePerenkov, Alexey D., Alena D. Sergeeva, Maria V. Vedunova, and Dmitri V. Krysko. 2023. "In Vitro Transcribed RNA-Based Platform Vaccines: Past, Present, and Future" Vaccines 11, no. 10: 1600. https://doi.org/10.3390/vaccines11101600
APA StylePerenkov, A. D., Sergeeva, A. D., Vedunova, M. V., & Krysko, D. V. (2023). In Vitro Transcribed RNA-Based Platform Vaccines: Past, Present, and Future. Vaccines, 11(10), 1600. https://doi.org/10.3390/vaccines11101600