
Integration of Bulk and Single-Cell RNA-Seq Data to Construct a Prognostic Model of Membrane Tension-Related Genes for Colon Cancer


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Acquisition
2.2. Construction and Validation of the MTRG Prognostic Model
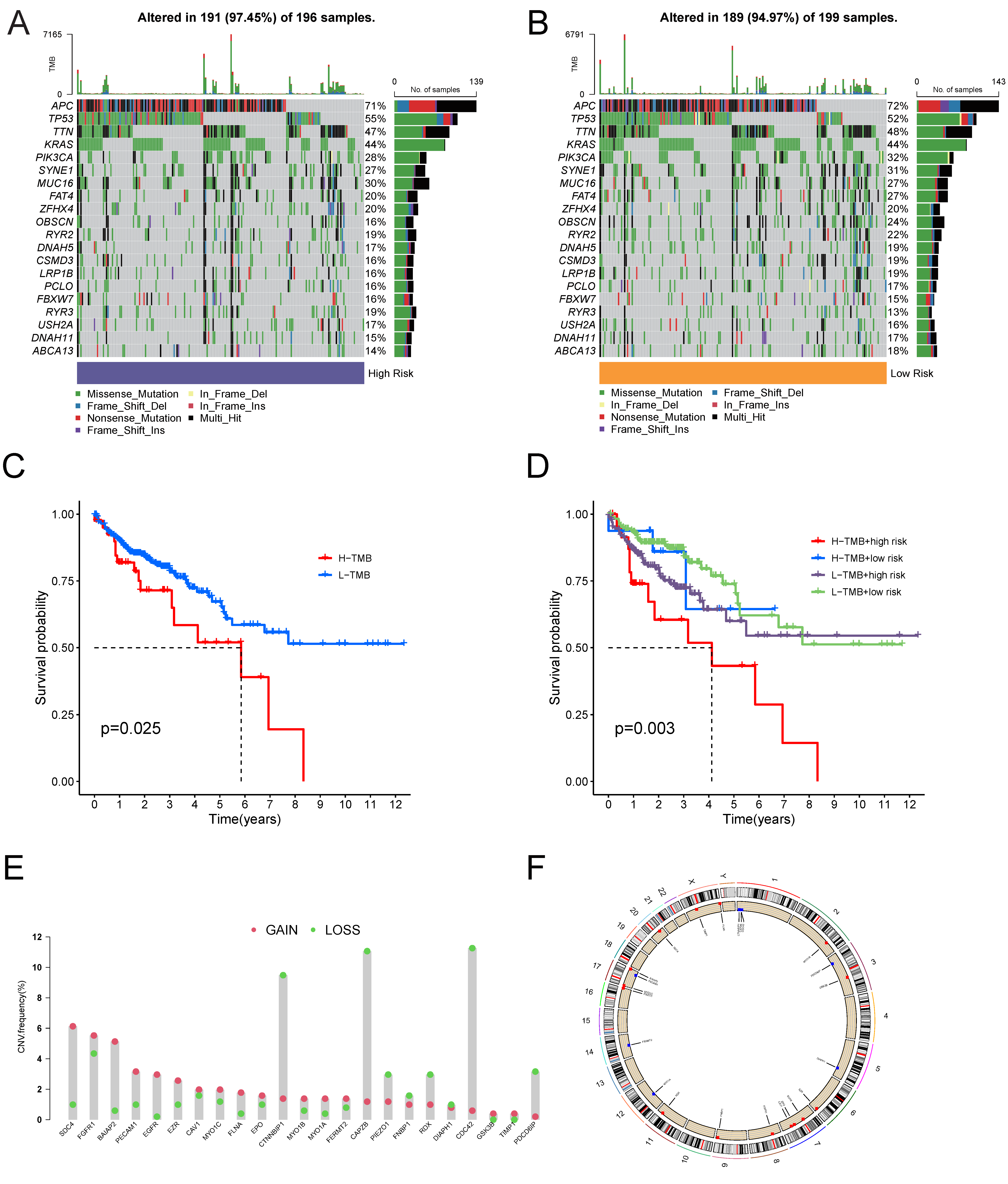
2.3. Visualization of Mutation and CNV Data
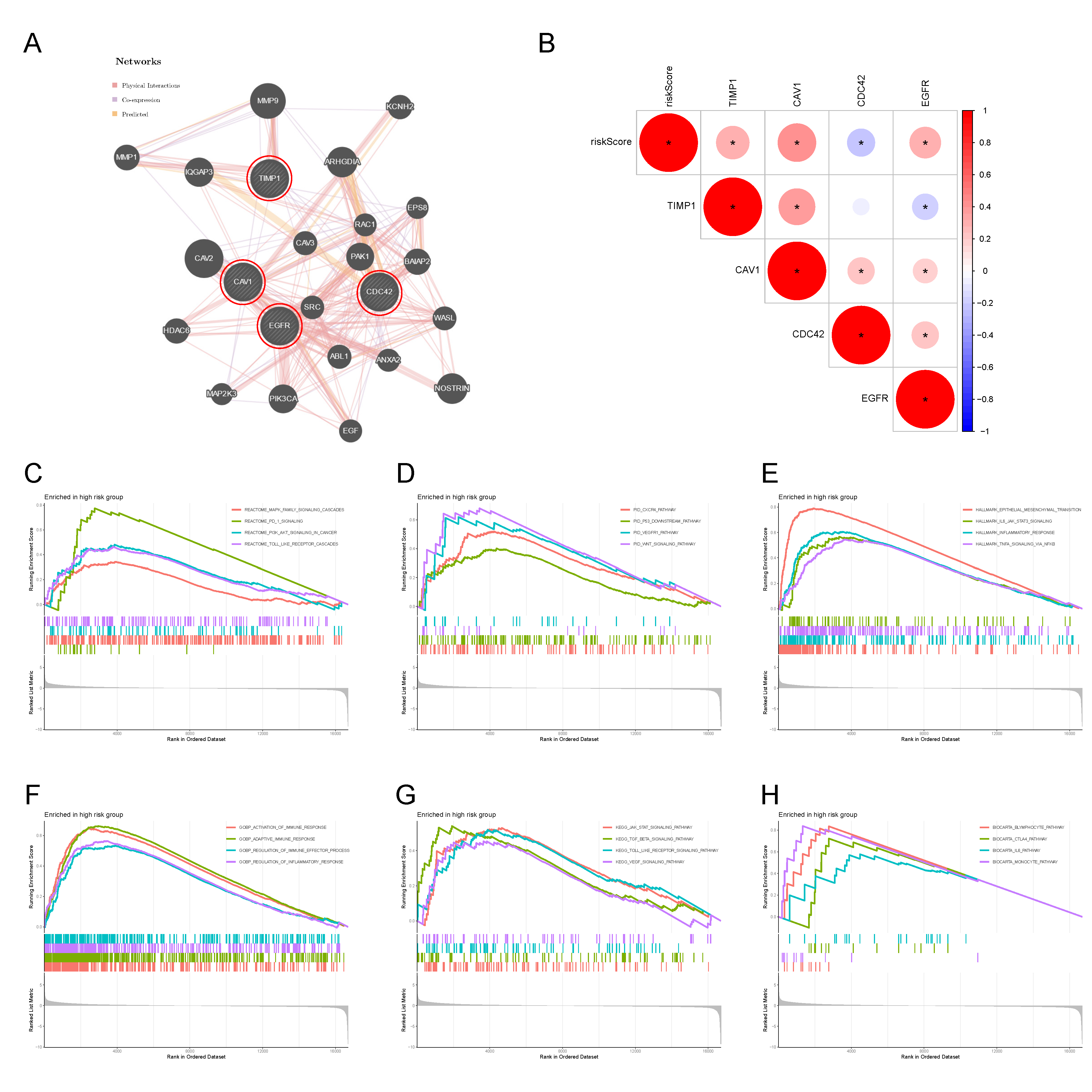
2.4. Protein Interaction Network Construction and GSEA Analysis
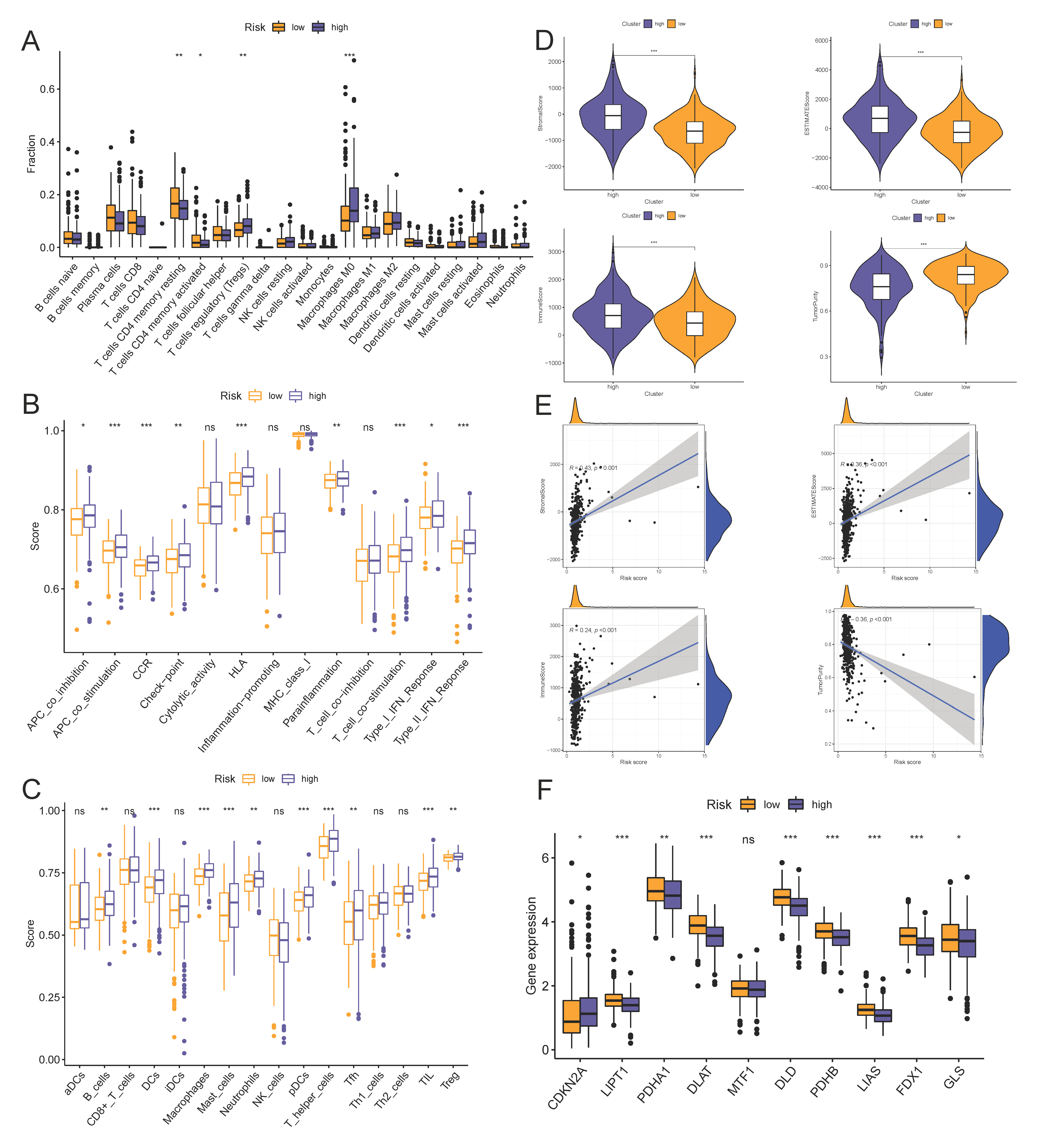
2.5. Immune Cell Infiltration, Gene Set Enrichment Analysis, and ICI-Related Gene Expression between the High- and Low-Risk Groups
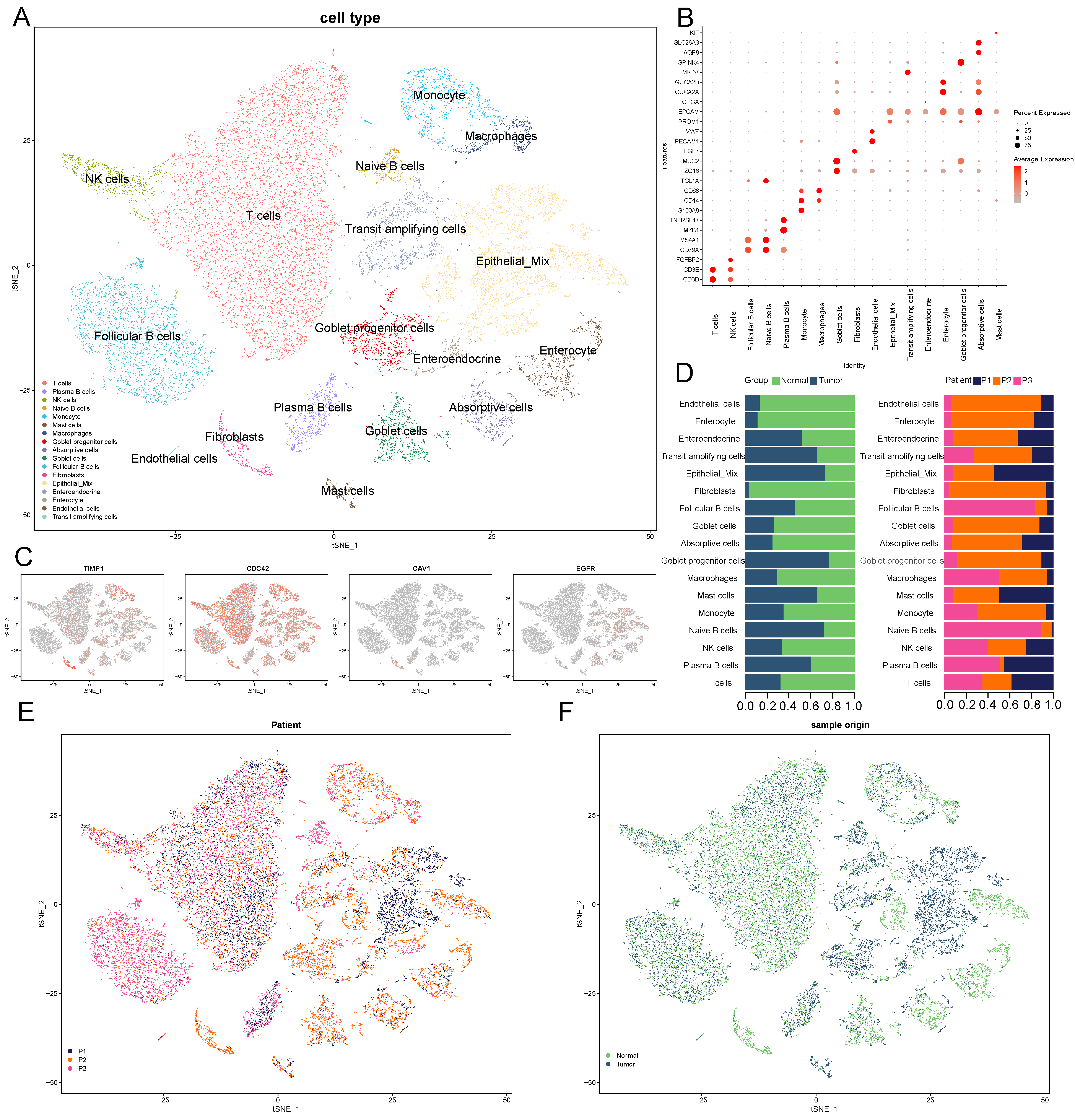
2.6. Dimensionality Reduction, Clustering, and Annotation of scRNA-Seq Data
2.7. Subclusters of Major Cell Types and Pseudo-Time Analysis
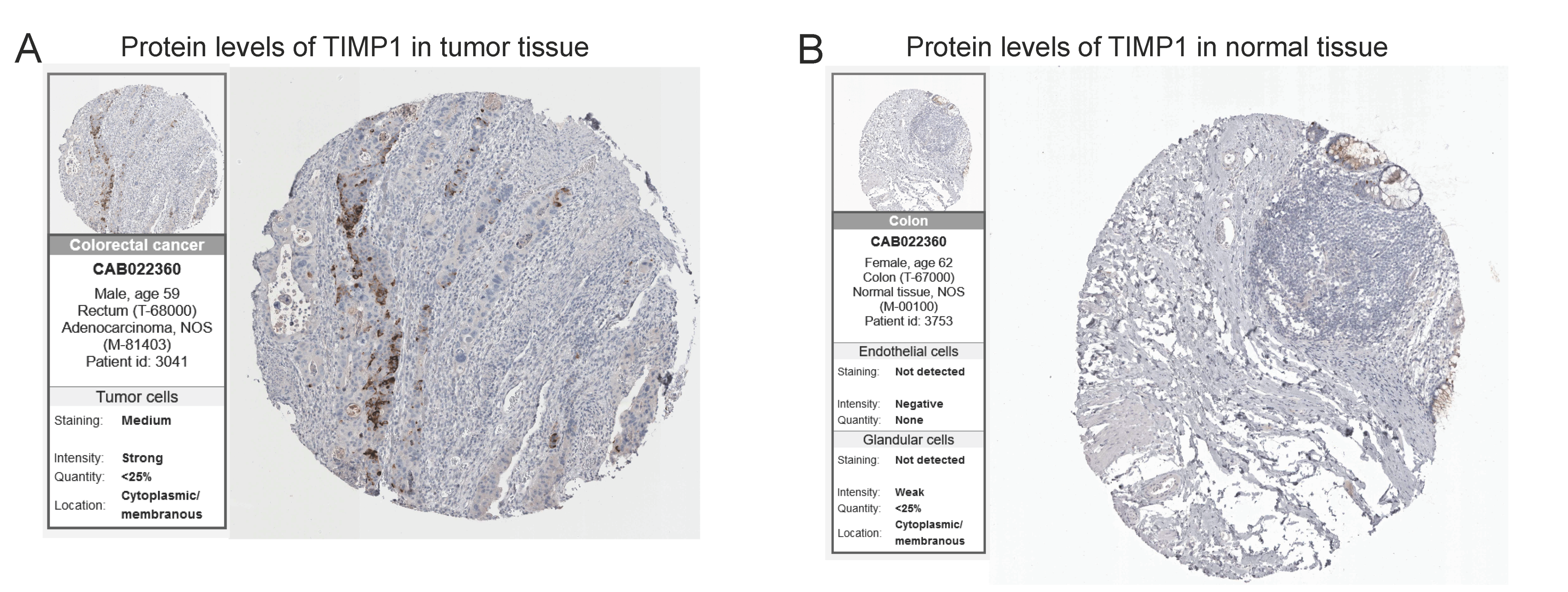
2.8. Validation of Protein Expression by the HPA Database
2.9. Molecular Docking
2.10. Statistical Analysis
3. Results
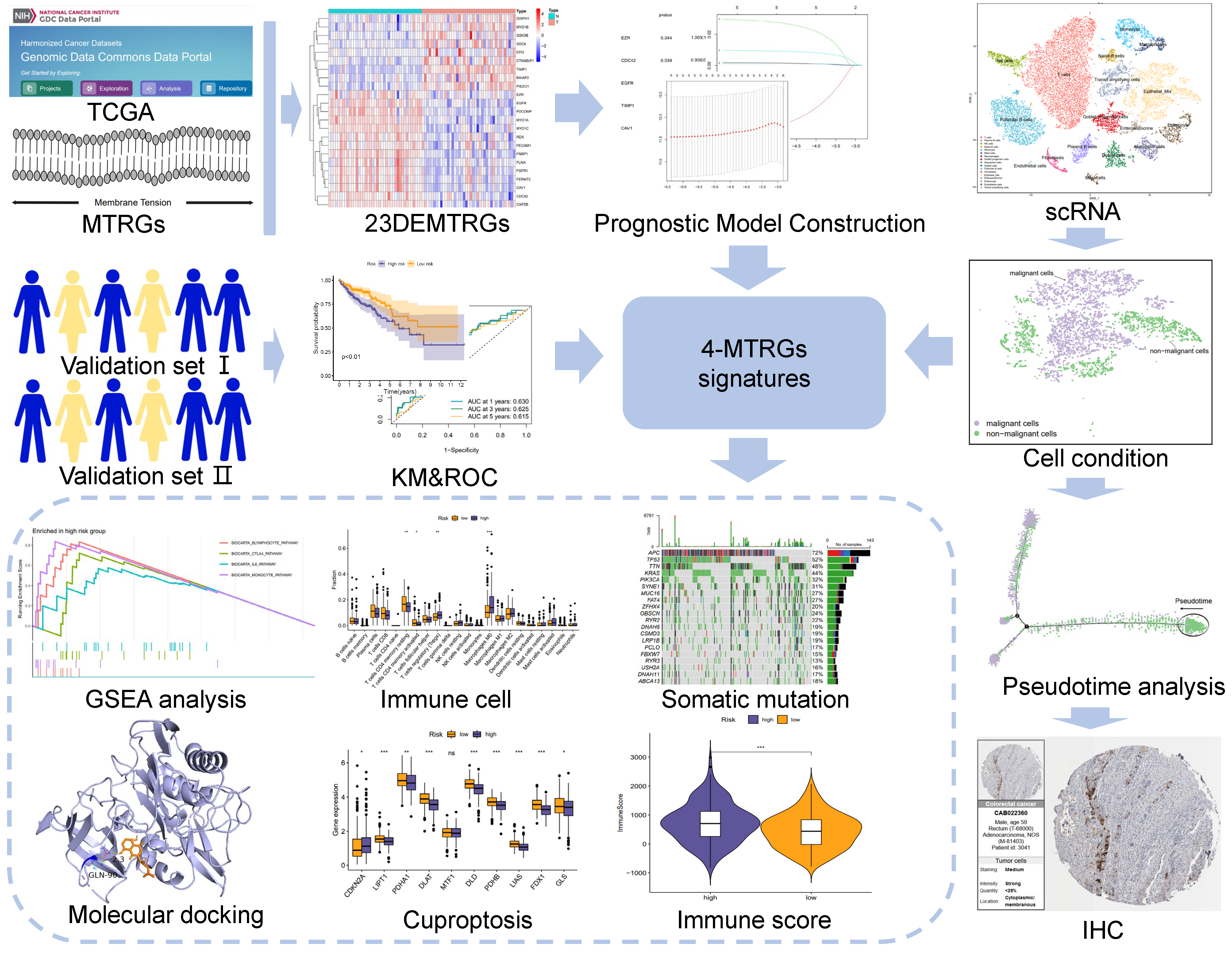
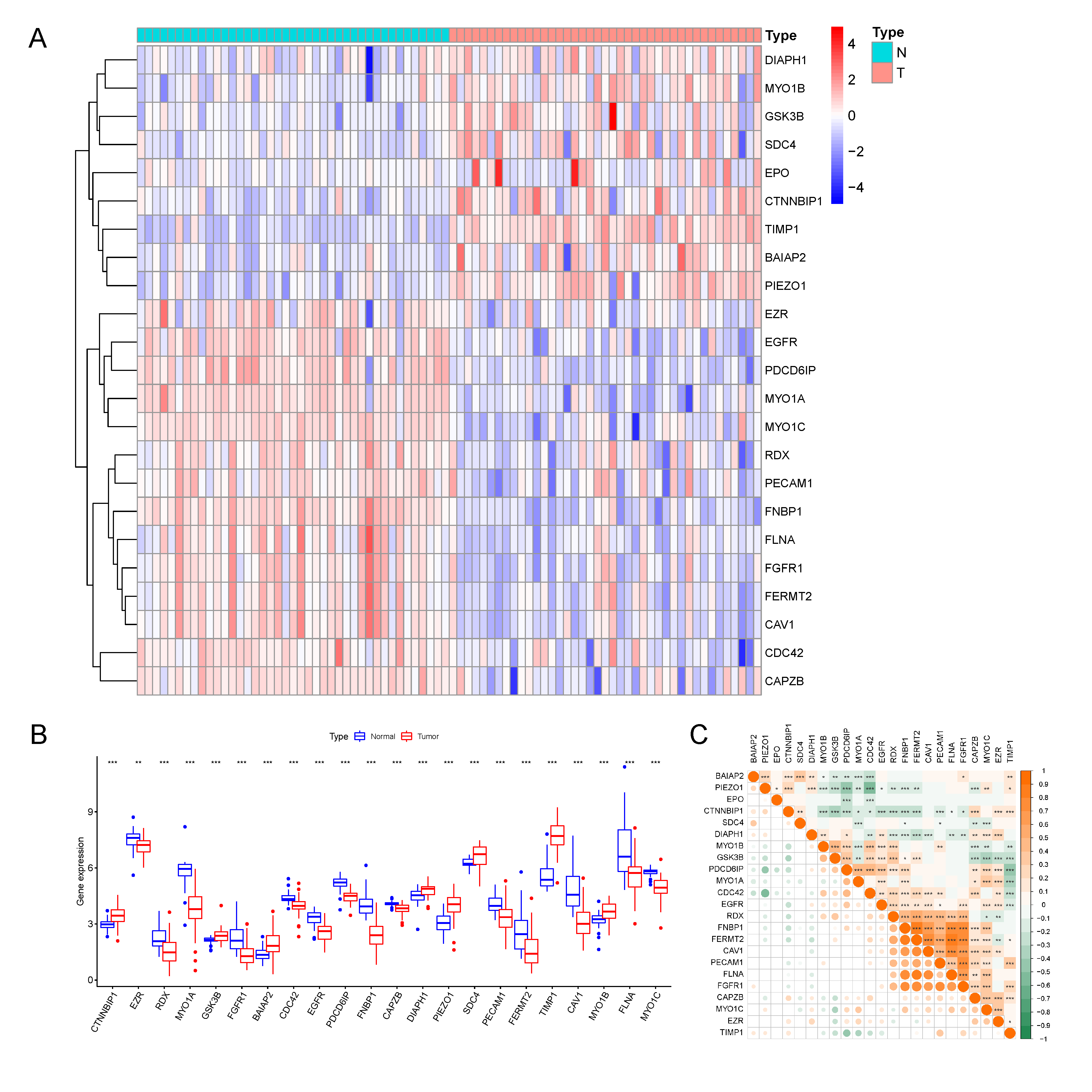
3.1. Identification of 23 Differentially Expressed MTRGs
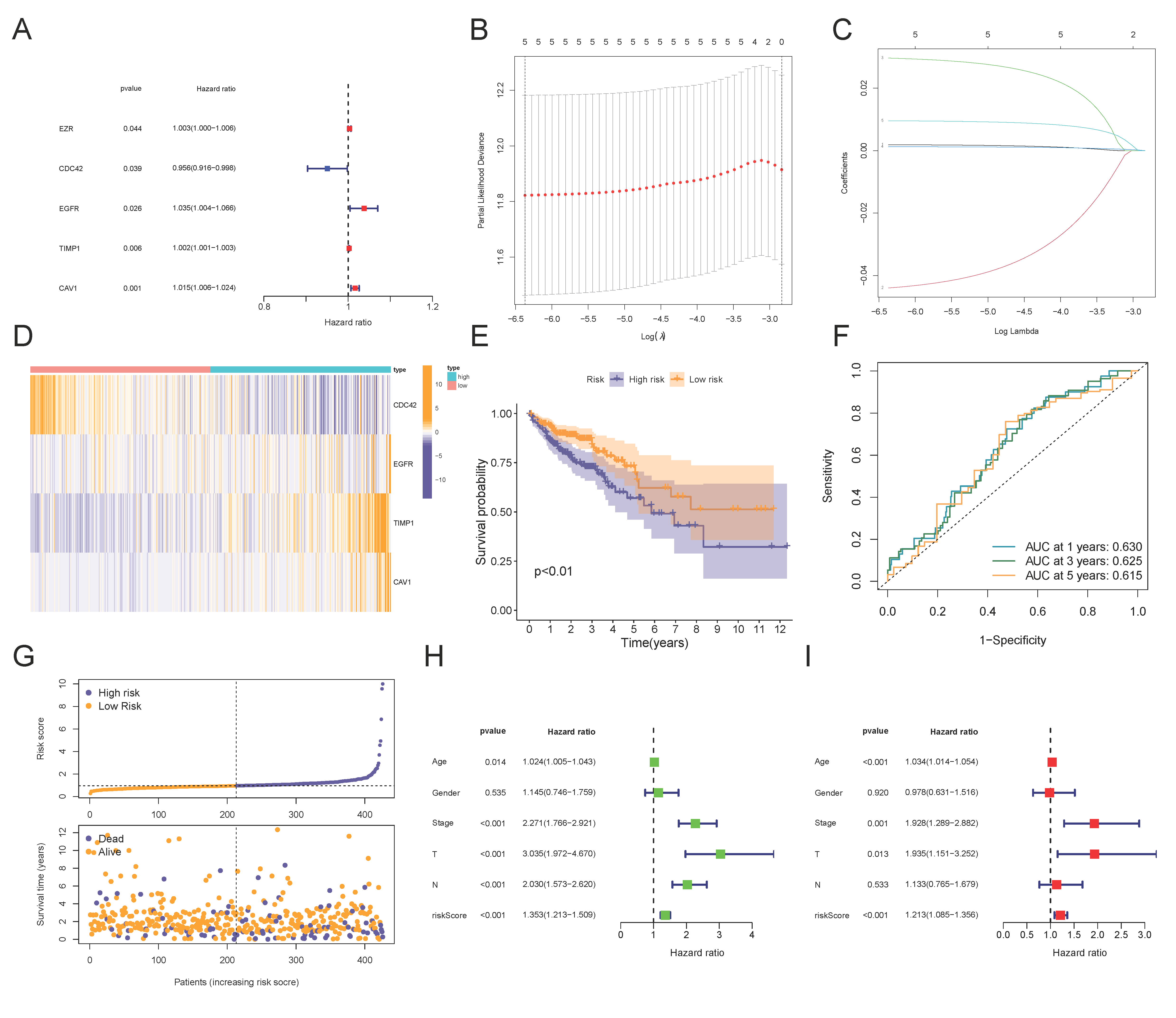
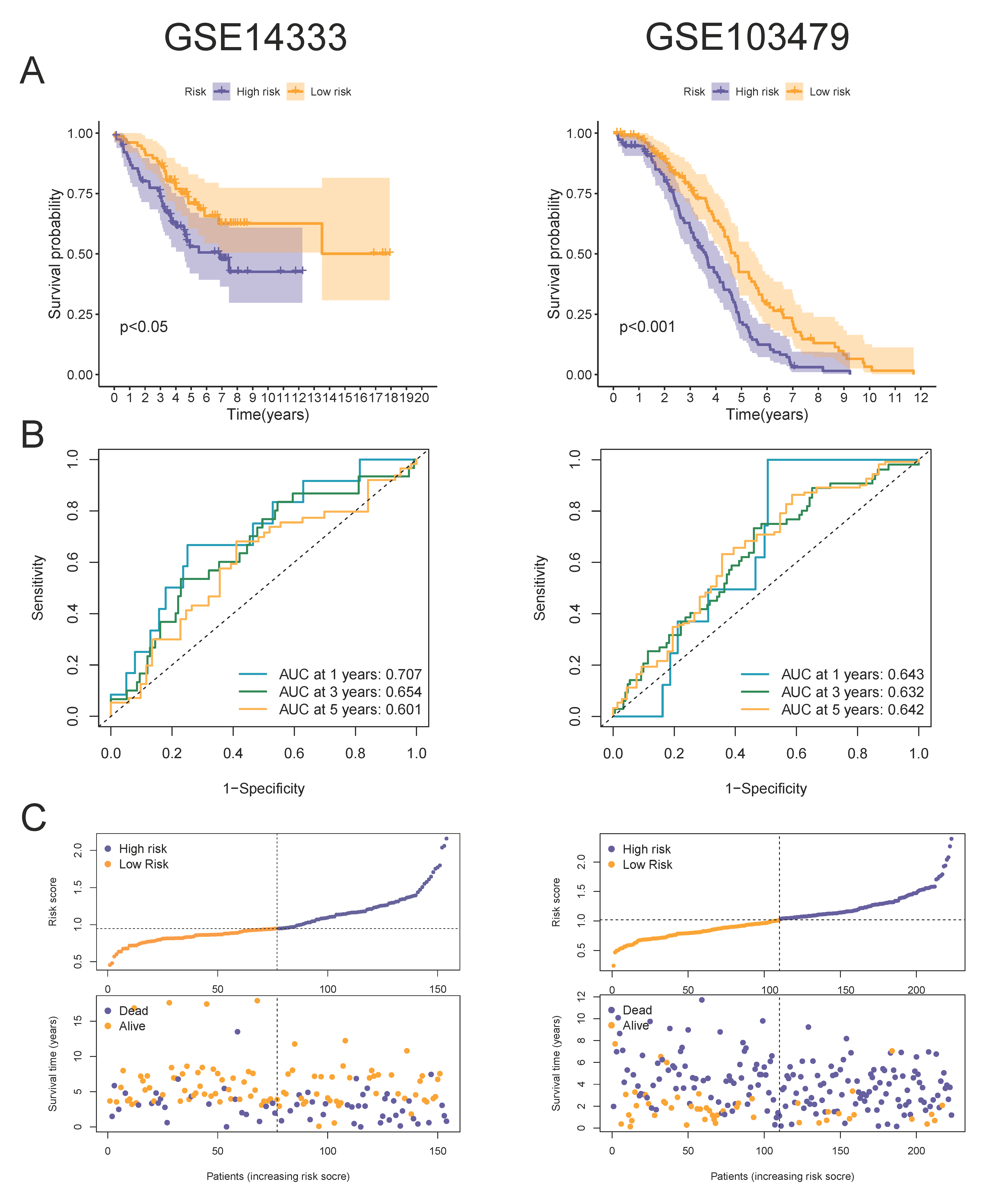
3.2. Construction and Validation of a 4-MTRG Prognostic Model
3.3. Somatic Mutation Profile of the Tumor Samples
3.4. Protein Interaction Network of the 4-MTRG Signature and GSEA Analysis
3.5. Risk Scores Related to Different Immune Cell Infiltration, Immune Status, and ICIs
3.6. scRNA-Seq and Cell Type Characteristics of Normal and COAD Samples
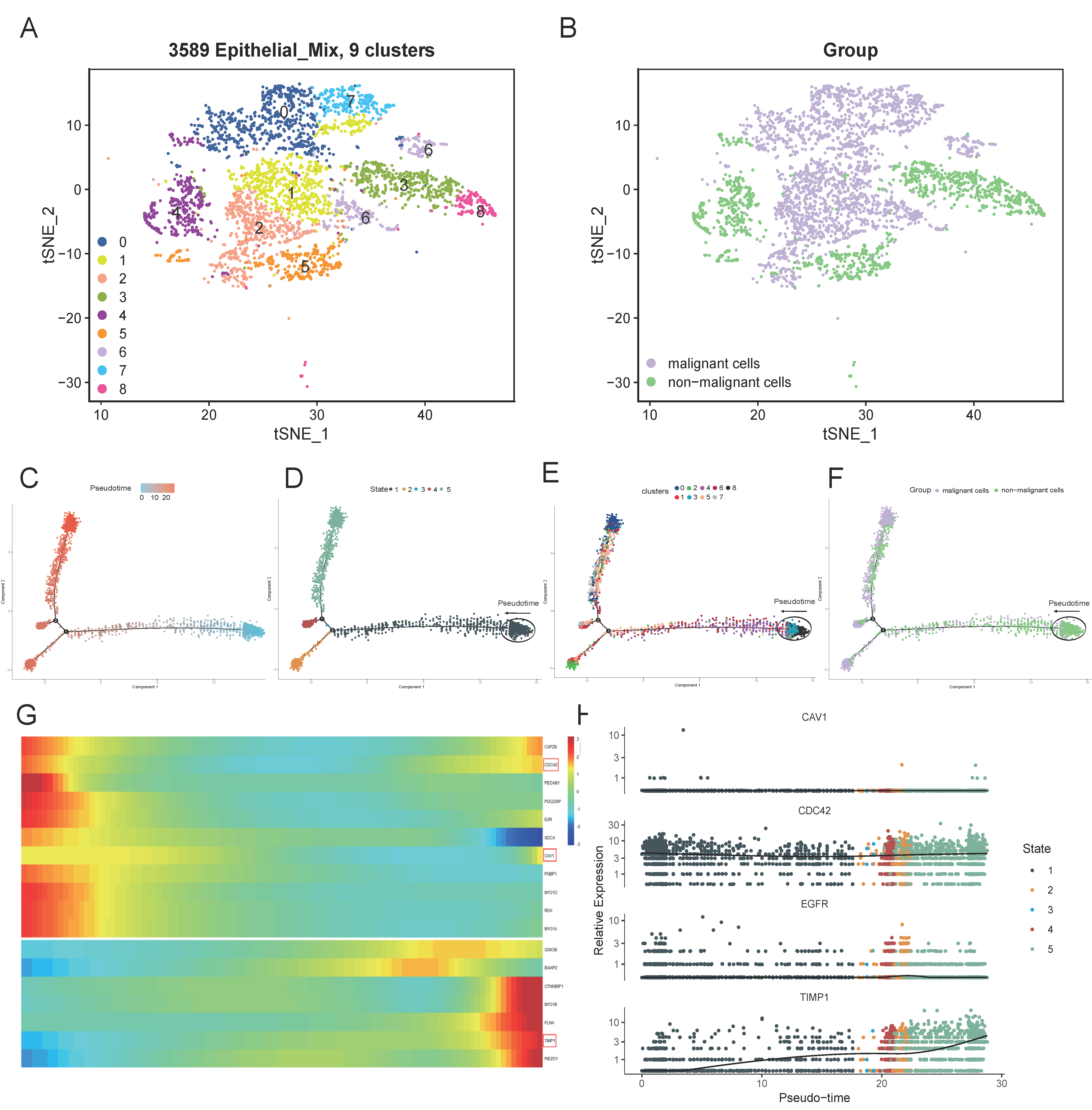
3.7. Subclusters of the Epithelial Cell and Pseudo-Time analysis
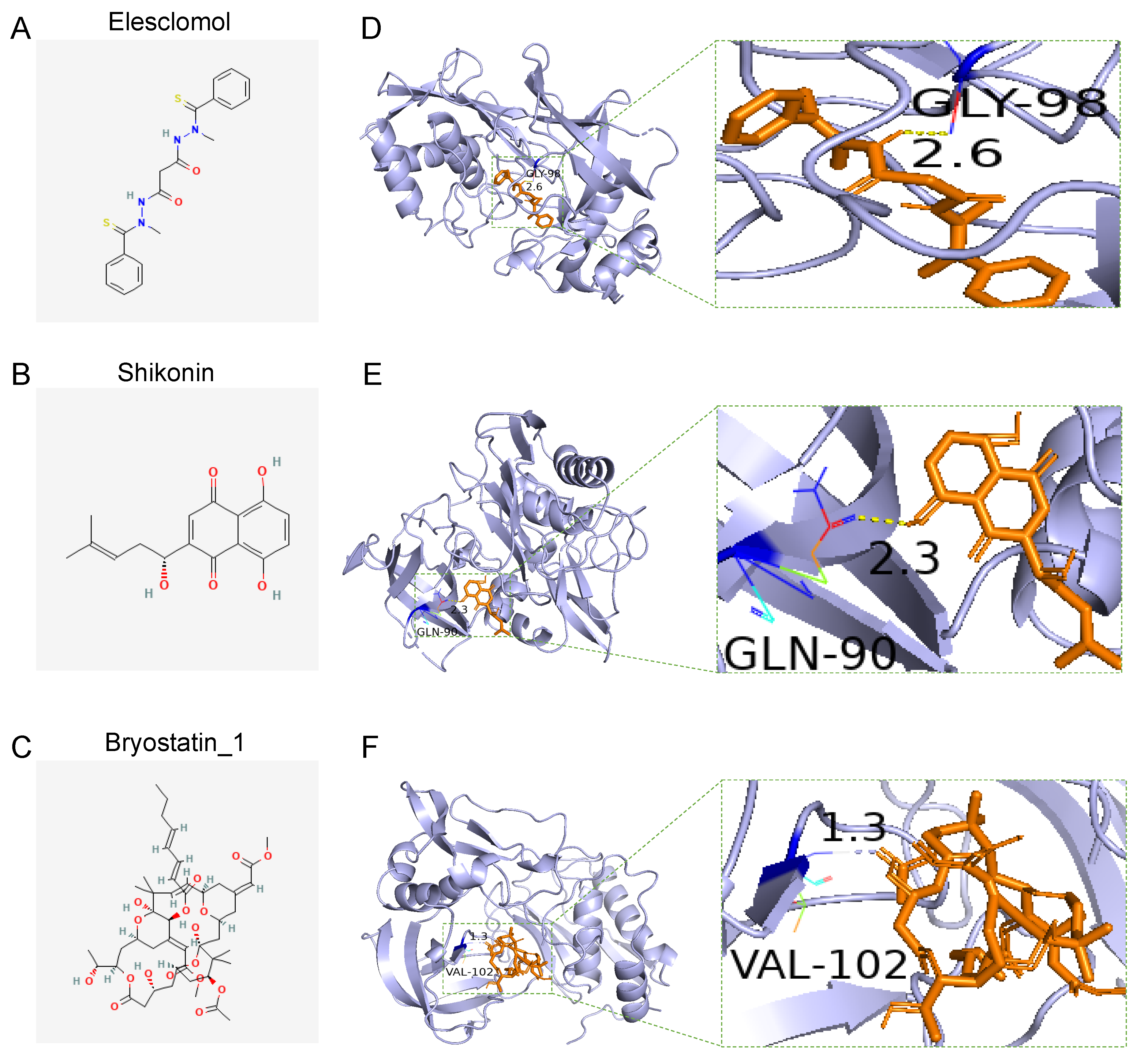
3.8. Drug Sensitivity of the Two Groups and Docking of Drug Candidates to TIMP1
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Ahmed, M. Colon Cancer: A Clinician’s Perspective in 2019. Gastroenterol. Res. 2020, 13, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Benson, A.B.; Venook, A.P.; Al-Hawary, M.M.; Arain, M.A.; Chen, Y.-J.; Ciombor, K.K.; Cohen, S.; Cooper, H.S.; Deming, D.; Farkas, L.; et al. Colon Cancer, Version 2.2021, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2021, 19, 329–359. [Google Scholar] [CrossRef] [PubMed]
- Bretou, M.; Jouannot, O.; Fanget, I.; Pierobon, P.; Larochette, N.; Gestraud, P.; Guillon, M.; Emiliani, V.; Gasman, S.; Desnos, C.; et al. Cdc42 controls the dilation of the exocytotic fusion pore by regulating membrane tension. Mol. Biol. Cell 2014, 25, 3195–3209. [Google Scholar] [CrossRef]
- Chen, B.; Khodadoust, M.S.; Liu, C.L.; Newman, A.M.; Alizadeh, A.A. Profiling Tumor Infiltrating Immune Cells with CIBERSORT. In Cancer Systems Biology, Methods in Molecular Biology; Von Stechow, L., Ed.; Springer: New York, NY, USA, 2018; pp. 243–259. [Google Scholar] [CrossRef]
- Cheng, W.-L.; Feng, P.-H.; Lee, K.-Y.; Chen, K.-Y.; Sun, W.-L.; Van Hiep, N.; Luo, C.-S.; Wu, S.-M. The Role of EREG/EGFR Pathway in Tumor Progression. Int. J. Mol. Sci. 2021, 22, 12828. [Google Scholar] [CrossRef]
- Chronopoulos, A.; Thorpe, S.D.; Cortes, E.; Lachowski, D.; Rice, A.J.; Mykuliak, V.V.; Róg, T.; Lee, D.A.; Hytönen, V.P.; Del Río Hernández, A.E. Syndecan-4 tunes cell mechanics by activating the kindlin-integrin-RhoA pathway. Nat. Mater 2020, 19, 669–678. [Google Scholar] [CrossRef] [PubMed]
- Chugh, P.; Clark, A.G.; Smith, M.B.; Cassani, D.A.D.; Dierkes, K.; Ragab, A.; Roux, P.P.; Charras, G.; Salbreux, G.; Paluch, E. Actin cortex architecture regulates cell surface tension. Nat. Cell Biol. 2017, 19, 689–697. [Google Scholar] [CrossRef]
- De Belly, H.; Stubb, A.; Yanagida, A.; Labouesse, C.; Jones, P.H.; Paluch, E.K.; Chalut, K.J. Membrane Tension Gates ERK-Mediated Regulation of Pluripotent Cell Fate. Cell Stem Cell 2021, 28, 273–284.e6. [Google Scholar] [CrossRef]
- Deng, X.; Lin, D.; Zhang, X.; Shen, X.; Yang, Z.; Yang, L.; Lu, X.; Yu, L.; Zhang, N.; Lin, J. Profiles of immune-related genes and immune cell infiltration in the tumor microenvironment of diffuse lower-grade gliomas. J. Cell. Physiol. 2020, 235, 7321–7331. [Google Scholar] [CrossRef]
- Diz-Muñoz, A.; Fletcher, D.A.; Weiner, O.D. Use the force: Membrane tension as an organizer of cell shape and motility. Trends Cell Biol. 2013, 23, 47–53. [Google Scholar] [CrossRef]
- Du, W.; Frankel, T.L.; Green, M.; Zou, W. IFNγ signaling integrity in colorectal cancer immunity and immunotherapy. Cell. Mol. Immunol. 2022, 19, 23–32. [Google Scholar] [CrossRef]
- Fu, Z.Y.; Lv, J.H.; Ma, C.Y.; Yang, D.P.; Wang, T. Tissue inhibitor of metalloproteinase-1 decreased chemosensitivity of MDA-435 breast cancer cells to chemotherapeutic drugs through the PI3K/AKT/NF-kB pathway. Biomed. Pharmacother. 2011, 65, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Huang, Z.; Duan, J.; Nice, E.C.; Lin, J.; Huang, C. Elesclomol induces copper-dependent ferroptosis in colorectal cancer cells via degradation of ATP7A. Mol. Oncol. 2021, 15, 3527–3544. [Google Scholar] [CrossRef] [PubMed]
- Giannopoulou, E.; Antonacopoulou, A.; Floratou, K.; Papavassiliou, A.G.; Kalofonos, H.P. Dual targeting of EGFR and HER-2 in colon cancer cell lines. Cancer Chemother. Pharmacol. 2009, 63, 973–981. [Google Scholar] [CrossRef] [PubMed]
- Del Pulgar, T.G.; Valdes-Mora, F.; Bandrés, E.; Pérez-Palacios, R.; Espina, C.; Cejas, P.; García-Cabezas, M.A.; Nistal, M.; Casado, E.; González-Barón, M.; et al. Cdc42 is highly expressed in colorectal adenocarcinoma and downregulates ID4 through an epigenetic mechanism. Int. J. Oncol. 2008, 33, 185–193. [Google Scholar] [CrossRef]
- Gong, Y.; Scott, E.; Lu, R.; Xu, Y.; Oh, W.K.; Yu, Q. TIMP-1 Promotes Accumulation of Cancer Associated Fibroblasts and Cancer Progression. PLoS ONE 2013, 8, e77366. [Google Scholar] [CrossRef] [PubMed]
- Ha, T.-K.; Chi, S.-G. CAV1/caveolin 1 enhances aerobic glycolysis in colon cancer cells via activation of SLC2A3/GLUT3 transcription. Autophagy 2012, 8, 1684–1685. [Google Scholar] [CrossRef] [PubMed]
- Hardbower, D.M.; Coburn, L.A.; Asim, M.; Singh, K.; Sierra, J.C.; Barry, D.P.; Gobert, A.P.; Piazuelo, M.B.; Washington, M.K.; Wilson, K.T. EGFR-mediated macrophage activation promotes colitis-associated tumorigenesis. Oncogene 2017, 36, 3807–3819. [Google Scholar] [CrossRef]
- Issa, I.A.; Noureddine, M. Colorectal cancer screening: An updated review of the available options. World J. Gastroenterol. 2017, 23, 5086–5096. [Google Scholar] [CrossRef]
- Keren, K. Membrane tension leads the way. Proc. Natl. Acad. Sci. USA 2011, 108, 14379–14380. [Google Scholar] [CrossRef]
- Kryczek, I.; Lin, Y.; Nagarsheth, N.; Peng, D.; Zhao, L.; Zhao, E.; Vatan, L.; Szeliga, W.; Dou, Y.; Owens, S.; et al. IL-22+CD4+ T cells promote colorectal cancer stemness via STAT3 transcription factor activation and induction of the methyltransferase DOT1L. Immunity 2014, 40, 772–784. [Google Scholar] [CrossRef]
- Lachowski, D.; Matellan, C.; Gopal, S.; Cortes, E.; Robinson, B.K.; Saiani, A.; Miller, A.F.; Stevens, M.M.; Hernández, A.E.D.R. Substrate Stiffness-Driven Membrane Tension Modulates Vesicular Trafficking via Caveolin-1. ACS Nano 2022, 16, 4322–4337. [Google Scholar] [CrossRef] [PubMed]
- Long, A.G.; Lundsmith, E.T.; Hamilton, K.E. Inflammation and Colorectal Cancer. Curr. Colorectal Cancer Rep. 2017, 13, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Magee, M.S.; Abraham, T.S.; Baybutt, T.R.; Flickinger, J.C.; Ridge, N.A.; Marszalowicz, G.P.; Prajapati, P.; Hersperger, A.R.; Waldman, S.A.; Snook, A.E. Human GUCY2C-Targeted Chimeric Antigen Receptor (CAR)-Expressing T Cells Eliminate Colorectal Cancer Metastases. Cancer Immunol. Res. 2018, 6, 509–516. [Google Scholar] [CrossRef] [PubMed]
- del Maldonado, M.M.; Medina, J.I.; Velazquez, L.; Dharmawardhane, S. Targeting Rac and Cdc42 GEFs in Metastatic Cancer. Front. Cell Dev. Biol. 2020, 8. [Google Scholar] [CrossRef]
- Mayakonda, A.; Lin, D.-C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef]
- Mellman, I.; Yarden, Y. Endocytosis and Cancer. Cold Spring Harb. Perspect. Biol. 2013, 5, a016949. [Google Scholar] [CrossRef]
- MMercier, V.; Larios, J.; Molinard, G.; Goujon, A.; Matile, S.; Gruenberg, J.; Roux, A. Endosomal membrane tension regulates ESCRT-III-dependent intra-lumenal vesicle formation. Nat. Cell Biol. 2020, 22, 947–959. [Google Scholar] [CrossRef]
- Nagasu, S.; Sudo, T.; Kinugasa, T.; Yomoda, T.; Fujiyoshi, K.; Shigaki, T.; Akagi, Y. Y-box-binding protein 1 inhibits apoptosis and upregulates EGFR in colon cancer. Oncol. Rep. 2019, 41, 2889–2896. [Google Scholar] [CrossRef]
- Osmani, N.; Peglion, F.; Chavrier, P.; Etienne-Manneville, S. Cdc42 localization and cell polarity depend on membrane traffic. J. Cell Biol. 2010, 191, 1261–1269. [Google Scholar] [CrossRef]
- Park, J.S.; Burckhardt, C.J.; Lazcano, R.; Solis, L.M.; Isogai, T.; Li, L.; Chen, C.; Gao, B.; Minna, J.D.; Bachoo, R.; et al. Mechanical regulation of glycolysis via cytoskeleton architecture. Nature 2020, 578, 621–626. [Google Scholar] [CrossRef]
- Pontes, B.; Monzo, P.; Gauthier, N.C. Membrane tension: A challenging but universal physical parameter in cell biology. Semin. Cell Dev. Biol. 2017, 71, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Price, B.; Dennison, C.; Tschesche, H.; Elliott, E. Neutrophil Tissue Inhibitor of Matrix Metalloproteinases-1 Occurs in Novel Vesicles That Do Not Fuse with the Phagosome. J. Biol. Chem. 2000, 275, 28308–28315. [Google Scholar] [CrossRef] [PubMed]
- Riggi, M.; Niewola-Staszkowska, K.; Chiaruttini, N.; Colom, A.; Kusmider, B.; Mercier, V.; Soleimanpour, S.; Stahl, M.; Matile, S.; Roux, A.; et al. Decrease in plasma membrane tension triggers PtdIns(4,5)P2 phase separation to inactivate TORC2. Nat. Cell Biol. 2018, 20, 1043–1051. [Google Scholar] [CrossRef] [PubMed]
- Shihata, W.A.; Michell, D.L.; Andrews, K.L.; Chin-Dusting, J.P.F. Caveolae: A Role in Endothelial Inflammation and Mechanotransduction? Front. Physiol. 2016, 7, 628. [Google Scholar] [CrossRef]
- Sobrero, A.F.; Puccini, A.; Shi, Q.; Grothey, A.; Andrè, T.; Shields, A.F.; Souglakos, I.; Yoshino, T.; Iveson, T.; Ceppi, M.; et al. A new prognostic and predictive tool for shared decision making in stage III colon cancer. Eur. J. Cancer 2020, 138, 182–188. [Google Scholar] [CrossRef]
- Song, G.; Xu, S.; Zhang, H.; Wang, Y.; Xiao, C.; Jiang, T.; Wu, L.; Zhang, T.; Sun, X.; Zhong, L.; et al. TIMP1 is a prognostic marker for the progression and metastasis of colon cancer through FAK-PI3K/AKT and MAPK pathway. J. Exp. Clin. Cancer Res. 2016, 35, 148. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics, 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Thottacherry, J.J.; Kosmalska, A.J.; Kumar, A.; Vishen, A.S.; Elosegui-Artola, A.; Pradhan, S.; Sharma, S.; Singh, P.P.; Guadamillas, M.C.; Chaudhary, N.; et al. Mechanochemical feedback control of dynamin independent endocytosis modulates membrane tension in adherent cells. Nat. Commun. 2018, 9, 4217. [Google Scholar] [CrossRef]
- Tsujita, K.; Satow, R.; Asada, S.; Nakamura, Y.; Arnes, L.; Sako, K.; Fujita, Y.; Fukami, K.; Itoh, T. Homeostatic membrane tension constrains cancer cell dissemination by counteracting BAR protein assembly. Nat. Commun. 2021, 12, 5930. [Google Scholar] [CrossRef]
- Tsvetkov, P.; Coy, S.; Petrova, B.; Dreishpoon, M.; Verma, A.; Abdusamad, M.; Rossen, J.; Joesch-Cohen, L.; Humeidi, R.; Spangler, R.D.; et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science 2022, 375, 1254–1261. [Google Scholar] [CrossRef]
- Warde-Farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.T.; et al. The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010, 38, W214–W220. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Lu, Z.; Li, L.; Ma, M.; Long, F.; Wu, R.; Huang, L.; Chou, J.; Yang, K.; Zhang, Y.; et al. Identification and Validation of Ferroptosis-Related LncRNA Signatures as a Novel Prognostic Model for Colon Cancer. Front. Immunol. 2022, 12, 783362. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Lin, C.; Chen, X.; Li, S.; Li, X.; Xiao, B. Structure deformation and curvature sensing of PIEZO1 in lipid membranes. Nature 2022, 604, 377–383. [Google Scholar] [CrossRef]
- Zhang, N.; Peng, F.; Wang, Y.; Yang, L.; Wu, F.; Wang, X.; Ye, C.; Han, B.; He, G. Shikonin induces colorectal carcinoma cells apoptosis and autophagy by targeting galectin-1/JNK signaling axis. Int. J Biol. Sci. 2020, 16, 147–161. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Kelaria, S.; Kerstetter, J.; Wang, J. The functional and prognostic implications of regulatory T cells in colorectal carcinoma. J. Gastrointest. Oncol. 2015, 6, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Zhang, J.; Zeng, D.; Sun, H.; Rong, X.; Shi, M.; Bin, J.; Liao, Y.; Liao, W. Immune cell infiltration as a biomarker for the diagnosis and prognosis of stage I-III colon cancer. Cancer Immunol. Immunother. 2019, 68, 433–442. [Google Scholar] [CrossRef]
- Zhu, J.; Kong, W.; Xie, Z. Expression and Prognostic Characteristics of Ferroptosis-Related Genes in Colon Cancer. Int. J. Mol. Sci. 2021, 22, 5652. [Google Scholar] [CrossRef]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Fu, Y.; Zhang, K.; Li, Y. Integration of Bulk and Single-Cell RNA-Seq Data to Construct a Prognostic Model of Membrane Tension-Related Genes for Colon Cancer. Vaccines 2022, 10, 1562. https://doi.org/10.3390/vaccines10091562
Li J, Fu Y, Zhang K, Li Y. Integration of Bulk and Single-Cell RNA-Seq Data to Construct a Prognostic Model of Membrane Tension-Related Genes for Colon Cancer. Vaccines. 2022; 10(9):1562. https://doi.org/10.3390/vaccines10091562
Chicago/Turabian StyleLi, Jiacheng, Yugang Fu, Kehui Zhang, and Yong Li. 2022. "Integration of Bulk and Single-Cell RNA-Seq Data to Construct a Prognostic Model of Membrane Tension-Related Genes for Colon Cancer" Vaccines 10, no. 9: 1562. https://doi.org/10.3390/vaccines10091562
APA StyleLi, J., Fu, Y., Zhang, K., & Li, Y. (2022). Integration of Bulk and Single-Cell RNA-Seq Data to Construct a Prognostic Model of Membrane Tension-Related Genes for Colon Cancer. Vaccines, 10(9), 1562. https://doi.org/10.3390/vaccines10091562