
Mouse Models for Unravelling Immunology of Blood Stage Malaria


Abstract
1. The Immune Response to Plasmodium Asexual Blood Stages Dictates Malarial Disease

{kind=link}
| Disease Manifestation | Species of Plasmodium | Probable Mechanism | Severity | References |
|---|---|---|---|---|
| Fever | All species | Schizogony-induced inflammation from 24–72 h depending on parasite species. Mediated by endogenous pyrogens induced during schizogeny such as TNF-α, IL-6, IL-1β and prostaglandin E2). | Not generally lethal | [14] |
| Anemia | All species | Erythrophagocytosis. Dyserythropoiesis. RBC destruction by parasite replication. | Can be lethal | [15] |
| Cerebral malaria | P. falciparum (children) P. falciparum and P. vivax (Adults) | Vascular activation via parasite sequestration on the brain endothelium, followed by breakdown of the blood brain barrier, vasogenic odema and herniation. | 20% mortality | [16,17,18,19] |
| Malaria-associated acute respiratory distress and Malaria-associated Acute Lung Injury | P. falciparum, P. vivax P. knowlsei P. ovale | Vascular activation via parasite sequestration on the pulmonary endothelial followed by pulmonary leak. | 40% mortality | [20,21,22] |
| Hepatomegaly/Liver fibrosis | P. falciparum P. vivax P. ovale | Jaundice and hepatic dysfunction due to infiltration of iRBCs and sequestration of iRBCs in the liver. Results in activation of hepatic stellate cells to become myofibroblasts. | Normally an indicator of severe malaria | [23,24] |
| Acute Kidney Injury (AKI) | P. falciparum P. vivax P. malariae | Glomerulonephritis, acute tubular necrosis and acute interstitial nephritis due to hemodynamic dysfunction and inflammation. Results in proteinuria, microalbuminuria and urinary casts along with hemolytic-uremic syndrome. Contributes to metabolic acidosis and can be exacerbated by liver damage. | Normally an indicator of severe malaria and found in around 40% of those with severe disease | [25] |
| Lactic acidosis | P. falciparum | Tissue hypoperfusion and hypoxia resulting from capillary obstruction with sequestered iRBCs and anemia Production of lactate by iRBCs. Impaired lactate clearance by the liver and kidney. | Normally an indicator of severe malaria | [26] |
| Hypoglycemia | P. falciparum P. vivax | Illness-induced fasting and inhibition of gluconeogenesis. | An indicator of severe malaria and more common in children than adults. Predicts mortality in malaria | [27] |
2. Utility of Rodent Plasmodium Species in the Investigation of Blood Stage Immunology
| Species | Clone | RBC Preference | Phenotypes | References |
|---|---|---|---|---|
| P. berghei | ANKA | Reticulocyte preference but will invade normocytes | Asynchronous life cycle, sequesters in the liver, lung and brain. Evidence of weight loss and anemia normally present at the time of death. C57BL/6J: lethal infection with breakdown of the BBB and death between day 7–10 p.i. Pronounced pulmonary pathology. BALB/c: Death from hyperparasitemia. No discernible cerebral complications. Less extensive lung pathology. DBA/2J: No discernible cerebral complications. some pulmonary pathology but less pronounced than BALB/c mice. Death ~day 20 p.i from hyperparasitemia and anemia. Pet shop mice: resistant to death by cerebral malaria. Death ~day 20 p.i. from hyperparasitemia. | [40,41,42,43] |
| NK65 New York (NY) | Reticulocyte preference | Accumulates mostly in the lung, with very little accumulation in the brain. Causes anemia over the course of infection. C57BL/6: death in ~20 days from respiratory distress. BALB/c: no development of MA-ARDS. | [44,45] | |
| NK65 Edinburgh (E) | Normocytes and Reticulocytes | Accumulates in the lung but not the brain with some evidence of anemia. C57BL/6: death in 7–10 days from respiratory distress. Early increase in peripheral parasitemia. BALB/c: Resistant to respiratory distress upon infection. | [44,45] | |
| K173 | Reticulocyte preference | Very little parasite accumulation/sequestration in the brain. Does not produce gametocytes due to laboratory adaptation from passaging. C57BL/6: Used as model for cerebral malaria. Early death after infection due to cerebral pathology accompanied with very high parasitemia. Causes lung pathology with increased pulmonary oedema. | [46,47] | |
| P. yoelii | XL (also known as 17XL) | Normocytes and reticulocytes | C57BL/6: Lethal within ~10 days p.i. due to hyperparasitemia and severe anemia. BALB/c: Lethal within ~10 days p.i. DBA/2: Non-lethal infection. | [48] |
| XNL (also known as 17XNL) | Strong reticulocyte preference | C57BL/6: Resolving non-lethal infection accompanied by anemia. BALB/c: Resolving non-lethal infection. | [48,49] | |
| YM | Normocytes and reticulocytes | C57BL/6: Derivative of the XL line. Lethal within ~10 days p.i. due to hyperparasitemia and severe anemia. DBA/2: Lethal infection in ~10 days p.i. B10: Non-lethal infection. | [50,51,52,53] | |
| nigeriensis N67 | Normocytes and reticulocytes | C57BL/6: Lethal at ~15–20 days p.i. due to hyperparasitemia. | [50,52,54] | |
| nigeriensis N67C | Normocytes and reticulocytes | C57BL/6: Lethal within 7 days p.i. | [50,54,55] | |
| P. chabaudi | chabaudi AS | Normocytes and reticulocytes | Synchronous life cycle. Sequesters predominantly in the lung and liver. C57BL/6: Resolving non-lethal infection accompanied by anemia, thrombocytopenia, hypoglycemia, weight loss and hypothermia. Recrudescent infections and sub-patent for up to 3 months. BALB/c: More severe infection than in C57BL/6J mice but generally non-lethal in most BALB/c lines. A/J mice: Lethal anemia due to poor control of iRBCs and hyperparasitemia. | [31,56] |
| chabaudi BC | Normocytes and reticulocytes | C57BL/6: similar symptoms to P. chabaudi AS but more severe. | [57] | |
| chabaudi CB | Normocytes and reticulocytes | C57BL/6: similar symptoms to P. chabaudi AS but more severe. | [58] | |
| chabaudi ER | Normocytes and reticulocytes | C57BL/6: Similar symptoms to those of P. chabaudi AS. Recrudescent infections occurs at 20 to 25 days p.i. | [31,57,59] | |
| chabaudi adami | Preference for younger normocytes over reticulocytes | BALB/c mice: Non-lethal infection with single peak of infection around 10 days p.i. A/J mice: Non-lethal resolving infection. C57BL//6 mice: Non-lethal resolving infection. | [60,61,62,63,64] | |
| P. vinckei | vinckei CY | Normocytes Not thought to invade reticulocytes | CBA: Lethal infection by 6 days p.i. with hyperparasitemia. | [65] |
| vinckei ATCC 30091 | Normocytes Not thought to invade reticulocytes | ICR outbred mice: Lethal infection within 8–10 p.i. | [66] | |
| petteri AS | Normocytes Not thought to invade reticulocytes | AKR mice: Lethal in 5 days p.i. due to fast growing parasites. | [67,68] | |
| petteri BS | Normocytes Not thought to invade reticulocytes | ICR outbred mice: Non-lethal infection with a peak of parasitemia at 9 days p.i. CBA: Non-lethal infection with a peak of parasitemia at 9 days p.i. | [65,66] | |
| petteri AR | Normocytes Not thought to invade reticulocytes | AKR mice: Non-lethal with patent parasitemia not detectable by 22 days p.i. | [67,68] | |
| petteri CR | Normocytes Not thought to invade reticulocytes | BALB/c: single peak of non-lethal infection. CBA: single peak at 6 days p.i.; non-lethal infection. | [65,69] | |
| petteri HW | Normocytes Not thought to invade reticulocytes | C57BL/6: Lethal infection at 8–10 days p.i. from hyperparasitemia. | [70] |
| Rodent Species | Model Uses | Main Features | References | |
|---|---|---|---|---|
| Plasmodium chabaudi | Innate immune responses | Control of iRBCs by monocytes and γδ T cells, but not neutrophils or NK cells. Direct activation of DC for activation of T cell responses. | [85,86,87,88,89,90,91,92] | |
| T cell responses | Requires T cells for control of iRBCs. | [59] | ||
| Immune regulation | IL-10 is required for clinical immunity. TGF-β provides some protection against pathogenesis. | [93,94] | ||
| Generation of humoral immunity | Participation of both IgG and IgM in control of iRBCs. Does not require antibodies for control of acute infection with iRBCs. Antibodies contribute to control of iRBCs during chronic infection. | [11,95,96,97] | ||
| T and B cell memory responses | Generates memory T and B cell responses that expand upon secondary challenge infection. | [98,99,100] | ||
| Immune basis of clonal virulence | Clonal virulence is associated with differences in the immune response induction. | [101,102] | ||
| Host genetic basis of immune resistance to infection by iRBCs | Genetic control of host immune responses mediates immunological control of iRBCs and level of clinical immunity. | [103,104] | ||
| Plasmodium yoelii | Innate immune responses | Macrophages are protective against iRBCs. Minimal contribution of neutrophils,γδ-T cells or NK cells to control of iRBCs. | [105,106,107] | |
| T cell responses | Requires T cell for control of iRBCs. | [108] | ||
| Immune regulation | IL-10 and TGF-β are required for clinical immunity. | [93,109] | ||
| Generation of humoral immunity | Requires antibodies for control of iRBCs during acute infection. | [110] | ||
| T and B cell memory responses | Generates memory T and B cell responses that expand upon secondary challenge infection. | [111,112] | ||
| Immune correlates of lethal vs. non-lethal infection | Lethality is correlated with faster parasite growth and an early burst of TGF-β. | [113] | ||
| Plasmodium berghei | ANKA | CD8 T cell induced vascular leak | [114,115] | |
| Pulmonary vascular leakage and leukocyte infiltration | [116] | |||
| Clonal differences in the induction of experimental cerebral malaria | [117] | |||
| NK65 Edinburgh | Pulmonary vascular leakage and leukocyte infiltration | [45,118] | ||
| NK65 New York | Pulmonary vascular leakage and leukocyte infiltration | [45] | ||
| Transgene | Functionality | Transgenic Parasites | Properties of the Transgenic Parasite | References |
|---|---|---|---|---|
| Luciferase | In vivo and ex vivo visualization of organ-specific parasite sequestration. Can be imaged after injection of D-luciferin using an IVIS imager | P. berghei ANKA | Expression of luciferase under the eEF1α promoter (constitutive) or the AMA-1 promoter (schizont-specific). | [119] |
| P. berghei NK65 Edinburgh | Expression of luciferase under the AMA-1 promoter (schizont-specific). | [118] | ||
| P. chabaudi AS | Expression of luciferase under the eEF1α promoter (constitutive). | [56] | ||
| Allelic replacement of rodent Plasmodium proteins with human Plasmodium proteins | Study of immune responses to human Plasmodium parasite vaccine targets | P. berghei ANKA | Express P. falciparum merozoite protein-119 (MSP-119). | [120] |
| P. berghei ANKA | Express P. falciparum apical membrane antigen-1 (AMA-1). | [121] | ||
| P. berghei ANKA P. berghei NK65 | Express P. vivax merozoite protein-119 (MSP-119). | [122] | ||
| Fluorescent proteins | Imaging of parasites ex-vivo by microscopy. Assessment of phagocytosis by flow cytometry or Imagestream. | P. chabaudi AS | Expression of GFP under the eEF1α promoter. | [85] |
| P. chabaudi AJ | Expression of RFP under the eEF1α promoter. | [83] | ||
| P. yoelii XNL | Expression of GFP under the eEF1α promoter. | [123] | ||
| P. berghei ANKA | Expression of GFP under the eEF1α promoter. | [124] | ||
| P. berghei NK65E | Expression of GFP under the AMA-1 promoter (schizont-specific). | [118] | ||
| P. berghei NK65NY | Expression of GFP under the AMA-1 promoter (schizont-specific). | [45] | ||
| Insertion of peptide epitopes recognized by available TCR transgenic mice | Quantification of antigen-specific Plasmodium-induced T cells responses | P. yoelii XNL | Express a CD4 and CD8 immunodominant epitope from the glycoprotein LCMV. Can be tracked using SMARTA or P14 TCR transgenic mice. | [112] |
| P. yoelii XNL | Express a CD4 and CD8 immunodominant epitope from the model antigen chicken ovalbumin. Can be tracked using OVA TCR transgenic mice (OT-I and OT-II). | [125] | ||
| P. berghei ANKA | Express a CD4 and CD8 immunodominant epitope from the model antigen chicken ovalbumin. Can be tracked using OVA TCR transgenic mice (OT-I and OT-II). | [126] | ||
| Endogenous parasite target epitope | Parasite species | Transgenic Mouse line | Properties of the transgenic mouse line | References |
| RPL6120–127 | P. berghei ANKA | Pb-I H2-Kb C57BL/6 | Transgenic Pb-I CD8 T cells with Vα8.3/Vβ10 TCRs that recognize PbRPL6120–127 in P. berghei ANKA liver stages but cross-react with blood stage antigens in P. berghei ANKA, PcRPLL6130–137 (P. chabaudi) and PyRPL6123–130 (P. yoelii). | [127,128] |
| HSP90484–496 | P. berghei ANKA | Pb-II I-Ab C57BL/6 | Transgenic Pb-II CD4 T cells with Vα2/Vβ12 TCRs that react cross-react to P. berghei NK65, P. chabaudi AS and P. yoelii XNL. | [129,130] |
| 37/39 kDa fragment of MSP-11157–1171 | P. chabaudi AS | B5 I-Ed BALB/c | Contains B5 MSP-1-reactive CD4 T cells with Vα2/Vβ8 TCRs reactive to MSP-11157–1171. | [131] |
| 21 kDa fragment of MSP-1 | P. chabaudi AS | IghNIMP23/+ C57BL/6 | B cells express the NIMP23 Ig heavy chain and harbor B cells that react to the 21 kDa fragment of P. chabaudi AS MSP-1. | [132] |
| Cell Type | Mouse Strain | Parasite Strain | Sequencing Type | Database Access Code | References |
|---|---|---|---|---|---|
| Whole Blood and spleen | C57BL/6 | P. chabaudi chabaudi AS and P. chabaudi chabaudi CB | Microarray | GSE93631 | [133] |
| Whole Blood and spleen | C57BL6 | P. chabaudi chabaudi AS | Microarray | GSE123391 | [133] |
| GSE145781 | [134] | ||||
| Bone marrow | C57BL/6J and C57BL/6J Elf4−/− mice | P. yoelii XNL | RNAseq | GSE121035 | [135] |
| Monocyte derived dendritic cells | C57BL/6 | P. berghei ANKA | RNAseq | GSE126381 | [136] |
| Splenic Macrophages | C57BL/6 | P. berghei ANKA | Microarray | GSE111593 | [137] |
| Macrophages | BALB/c | P. yoelii XNL-Luc | RNAseq | GSE115906 | [138] |
| Red pulp macrophages | C57BL/6 | P. chabaudi chabaudi AS | Microarray | GSE23565 | [139] |
| NK cells | C57BL/6 | P. chabaudi chabaudi AS | Microarray | GSE12727 | [140] |
| γδ T cells | C57BL/6J | P. chabaudi chabaudi AJ | Single cell TCR sequencing and RNAseq | GSE108478 | [89] |
| CD4 T cells | C57BL/6 | P. berghei ANKA | microarray | GSE24903 | [141] |
| C57BL//6J and Uba3fl/fl-Lck Cre+ (KO) Uba3fl/fl and Uba3ΔT mice on a C57BL/6J background | P. yoelii XNL | RNAseq | GSE111066 | [142] | |
| C57BL/6 | P. chabaudi chabaudi AS | Microarray Single-cell RNAseq | GSE81196 GSE81197 | [143] | |
| C57BL/6 | P. yoelii XNL | Microarray | GSE85896 | [144] | |
| Regulatory T cells | BALB/c | P. yoelii XNL | Microarray | GSE34621 | [49] |
| B cells | Tbx21fl/flCd23Cre and CD23Cre+ mice on a C57BL/6J background | P. berghei ANKA | RNAseq ATACseq | GSE120729 GSE120727 | [145] |
| IFN-γR1−/− and C57BL/6J | P. chabaudi chabaudi AS | RNAseq | GSE85205 | [146] | |
| C57BL/6 | P. yoelii XNL | RNAseq | GSE134548 | [110] | |
| C57BL/6 | P. chabaudi chabaudi AS | RNAseq | GSE115155 | [132] | |
| Microglia | C57BL/6 wildtype and IFNAR−/− | P. berghei ANKA | Microarray | GSE119650 GSE86082 | [147] |
3. Genetic Control of the Immune Responses to Plasmodium Infection
4. Modelling the Influence of Environmental Factors on Immune Responses to Plasmodium Infection
5. Mechanisms of Anti-Parasite Immunity: What Have We Learnt about Control of iRBCs from Mouse Models of Plasmodium Infection?
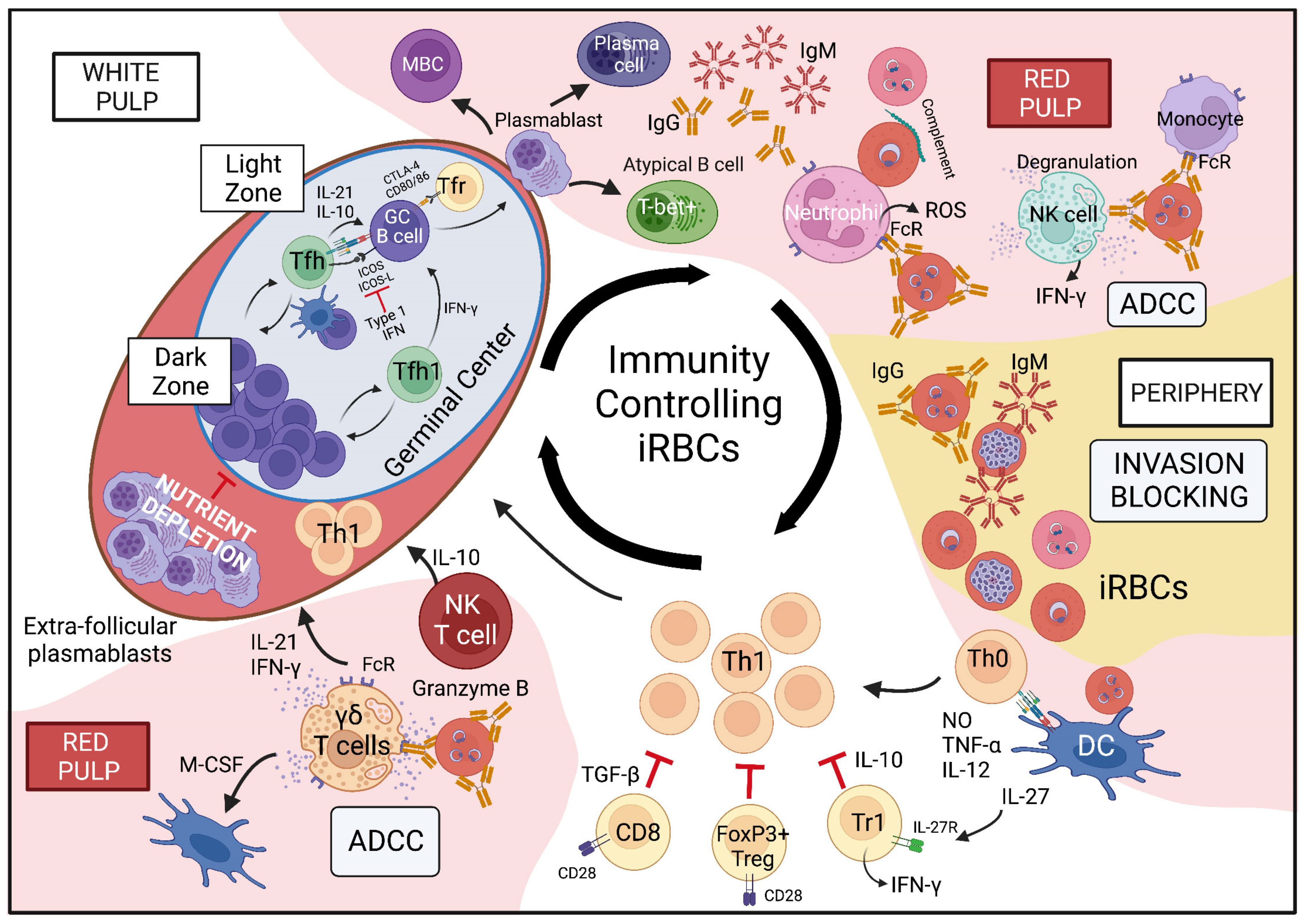
5.1. Invasion Blocking Is a Key Mechanism of Anti-Parasite Antibodies for the Control of iRBCs
5.2. Plasmodium Blood Stage Infection Leads to the Development of Memory B Cells That Respond during Secondary Challenge Infection
5.3. Development of Functional Anti-Plasmodium Blood Stage GC Responses
5.4. The Importance of Innate Immune Cells in Control of iRBCs
6. Immunopathogenesis of Malaria and Clinical Immunity
6.1. Organ-Specific Pathologies
6.2. Mechanisms of Inflammation-Induced SMA
6.3. T Cell-Mediated Breakdown of the Blood-Brain Barrier in Cerebral Malaria
6.4. Mechanisms of Malaria-Associated Acute Lung Injury (MA-ALI) in Malaria-Associated Acute Respiratory Distress Syndrome (MA-ARDS)
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- WHO. World Malaria Report; WHO: Geneva, Switzerland, 2021. [Google Scholar]
- Olotu, A.; Fegan, G.; Wambua, J.; Nyangweso, G.; Leach, A.; Lievens, M.; Kaslow, D.C.; Njuguna, P.; Marsh, K.; Bejon, P. Seven-year efficacy of RTS, S/AS01 malaria vaccine among young African children. N. Engl. J. Med. 2016, 374, 2519–2529. [Google Scholar] [CrossRef] [PubMed]
- Tinto, H.; Otieno, W.; Gesase, S.; Sorgho, H.; Otieno, L.; Liheluka, E.; Valéa, I.; Sing’oei, V.; Malabeja, A.; Valia, D. Long-term incidence of severe malaria following RTS, S/AS01 vaccination in children and infants in Africa: An open-label 3-year extension study of a phase 3 randomised controlled trial. Lancet Infect. Dis. 2019, 19, 821–832. [Google Scholar] [CrossRef]
- Datoo, M.S.; Natama, M.H.; Some, A.; Traore, O.; Rouamba, T.; Bellamy, D.; Yameogo, P.; Valia, D.; Tegneri, M.; Ouedraogo, F.; et al. Efficacy of a low-dose candidate malaria vaccine, R21 in adjuvant Matrix-M, with seasonal administration to children in Burkina Faso: A randomised controlled trial. Lancet 2021, 397, 1809–1818. [Google Scholar] [CrossRef]
- Vaughan, A.M.; Kappe, S.H.I. Malaria Parasite Liver Infection and Exoerythrocytic Biology. Cold Spring Harb. Perspect. Med. 2017, 7, a025486. [Google Scholar] [CrossRef]
- WHO Severe Malaria. 2014. Available online: https://onlinelibrary.wiley.com/doi/full/10.1111/tmi.12313_2 (accessed on 8 September 2022).
- Ndila, C.M.; Uyoga, S.; Macharia, A.W.; Nyutu, G.; Peshu, N.; Ojal, J.; Shebe, M.; Awuondo, K.O.; Mturi, N.; Tsofa, B. Human candidate gene polymorphisms and risk of severe malaria in children in Kilifi, Kenya: A case-control association study. Lancet Haematol. 2018, 5, e333–e345. [Google Scholar] [CrossRef]
- Marquet, S.; Conte, I.; Poudiougou, B.; Argiro, L.; Dessein, H.; Couturier, C.; Burté, F.; Oumar, A.A.; Brown, B.J.; Traore, A. A functional IL22 polymorphism (rs2227473) is associated with predisposition to childhood cerebral malaria. Sci. Rep. 2017, 7, 41636. [Google Scholar] [CrossRef]
- Jenkins, N.E.; Mwangi, T.W.; Kortok, M.; Marsh, K.; Craig, A.G.; Williams, T.N. A polymorphism of intercellular adhesion molecule-1 is associated with a reduced incidence of nonmalarial febrile illness in Kenyan children. Clin. Infect. Dis. 2005, 41, 1817–1819. [Google Scholar] [CrossRef]
- Villarino, N.F.; LeCleir, G.R.; Denny, J.E.; Dearth, S.P.; Harding, C.L.; Sloan, S.S.; Gribble, J.L.; Campagna, S.R.; Wilhelm, S.W.; Schmidt, N.W. Composition of the gut microbiota modulates the severity of malaria. Proc. Natl. Acad. Sci. USA 2016, 113, 2235–2240. [Google Scholar] [CrossRef]
- Matar, C.G.; Anthony, N.R.; O’Flaherty, B.M.; Jacobs, N.T.; Priyamvada, L.; Engwerda, C.R.; Speck, S.H.; Lamb, T.J. Gammaherpesvirus Co-Infection with Malaria Suppresses Anti-Parasitic Humoral Immunity. PLoS Pathog. 2015, 11, e1004858. [Google Scholar] [CrossRef]
- Weiss, G.E.; Traore, B.; Kayentao, K.; Ongoiba, A.; Doumbo, S.; Doumtabe, D.; Kone, Y.; Dia, S.; Guindo, A.; Traore, A.; et al. The Plasmodium falciparum-specific human memory B cell compartment expands gradually with repeated malaria infections. PLoS Pathog. 2010, 6, e1000912. [Google Scholar] [CrossRef]
- Wipasa, J.; Suphavilai, C.; Okell, L.C.; Cook, J.; Corran, P.H.; Thaikla, K.; Liewsaree, W.; Riley, E.M.; Hafalla, J.C. Long-lived antibody and B Cell memory responses to the human malaria parasites, Plasmodium falciparum and Plasmodium vivax. PLoS Pathog. 2010, 6, e1000770. [Google Scholar] [CrossRef]
- Oakley, M.S.; Gerald, N.; McCutchan, T.F.; Aravind, L.; Kumar, S. Clinical and molecular aspects of malaria fever. Trends Parasitol. 2011, 27, 442–449. [Google Scholar] [CrossRef]
- Perkins, D.J.; Were, T.; Davenport, G.C.; Kempaiah, P.; Hittner, J.B.; Ong’echa, J.M. Severe malarial anemia: Innate immunity and pathogenesis. Int. J. Biol. Sci. 2011, 7, 1427–1442. [Google Scholar] [CrossRef]
- Dvorin, J.D. Getting Your Head around Cerebral Malaria. Cell Host Microbe 2017, 22, 586–588. [Google Scholar] [CrossRef]
- Storm, J.; Jespersen, J.S.; Seydel, K.B.; Szestak, T.; Mbewe, M.; Chisala, N.V.; Phula, P.; Wang, C.W.; Taylor, T.E.; Moxon, C.A. Cerebral malaria is associated with differential cytoadherence to brain endothelial cells. EMBO Mol. Med. 2019, 11, e9164. [Google Scholar] [CrossRef]
- Kessler, A.; Dankwa, S.; Bernabeu, M.; Harawa, V.; Danziger, S.A.; Duffy, F.; Kampondeni, S.D.; Potchen, M.J.; Dambrauskas, N.; Vigdorovich, V. Linking EPCR-binding PfEMP1 to brain swelling in pediatric cerebral malaria. Cell Host Microbe 2017, 22, 601–614.e5. [Google Scholar] [CrossRef]
- Ponsford, M.J.; Medana, I.M.; Prapansilp, P.; Hien, T.T.; Lee, S.J.; Dondorp, A.M.; Esiri, M.M.; Day, N.P.; White, N.J.; Turner, G.D. Sequestration and microvascular congestion are associated with coma in human cerebral malaria. J. Infect. Dis. 2012, 205, 663–671. [Google Scholar] [CrossRef]
- Koh, Y. Update in acute respiratory distress syndrome. J. Intensive Care 2014, 2, 2. [Google Scholar] [CrossRef]
- Punsawad, C.; Viriyavejakul, P.; Setthapramote, C.; Palipoch, S. Enhanced expression of Fas and FasL modulates apoptosis in the lungs of severe P. falciparum malaria patients with pulmonary edema. Int. J. Clin. Exp. Pathol. 2015, 8, 10002. [Google Scholar]
- Maknitikul, S.; Luplertlop, N.; Grau, G.E.; Ampawong, S. Dysregulation of pulmonary endothelial protein C receptor and thrombomodulin in severe falciparum malaria-associated ARDS relevant to hemozoin. PLoS ONE 2017, 12, e0181674. [Google Scholar] [CrossRef]
- Blumberg, L.; Lee, R.P.; Lipman, J.; Beards, S. Predictors of mortality in severe malaria: A two year experience in a non-endemic area. Anaesth. Intensive Care 1996, 24, 217–223. [Google Scholar] [CrossRef]
- Viriyavejakul, P.; Khachonsaksumet, V.; Punsawad, C. Liver changes in severe Plasmodium falciparum malaria: Histopathology, apoptosis and nuclear factor kappa B expression. Malar. J. 2014, 13, 106. [Google Scholar] [CrossRef]
- Silva, G.B.D.J.; Pinto, J.R.; Barros, E.J.G.; Farias, G.M.N.; Daher, E.F. Kidney involvement in malaria: An update. Rev. Inst. Med. Trop. Sao Paulo 2017, 59, e53. [Google Scholar]
- Possemiers, H.; Vandermosten, L.; Van den Steen, P.E. Etiology of lactic acidosis in malaria. PLoS Pathog. 2021, 17, e1009122. [Google Scholar] [CrossRef]
- Thien, H.V.; Kager, P.A.; Sauerwein, H.P. Hypoglycemia in falciparum malaria: Is fasting an unrecognized and insufficiently emphasized risk factor? Trends Parasitol. 2006, 22, 410–415. [Google Scholar] [CrossRef]
- Chen, Q.; Amaladoss, A.; Ye, W.; Liu, M.; Dummler, S.; Kong, F.; Wong, L.H.; Loo, H.L.; Loh, E.; Tan, S.Q.; et al. Human natural killer cells control Plasmodium falciparum infection by eliminating infected red blood cells. Proc. Natl. Acad. Sci. USA 2014, 111, 1479–1484. [Google Scholar] [CrossRef]
- Zhang, L.L.; Li, J.L.; Ji, M.X.; Tian, D.; Wang, L.Y.; Chen, C.; Tian, M. Attenuated P. falciparum Parasite Shows Cytokine Variations in Humanized Mice. Front. Immunol. 2020, 11, 1801. [Google Scholar] [CrossRef]
- Killick-Kendrick, R.; Peters, W. Rodent Malaria; Academic Press: London, UK, 1978. [Google Scholar]
- Stephens, R.; Culleton, R.L.; Lamb, T.J. The contribution of Plasmodium chabaudi to our understanding of malaria. Trends Parasitol. 2012, 28, 73–82. [Google Scholar] [CrossRef]
- Smith, J.D.; Chitnis, C.E.; Craig, A.G.; Roberts, D.J.; Hudson-Taylor, D.E.; Peterson, D.S.; Pinches, R.; Newbold, C.I.; Miller, L.H. Switches in expression of Plasmodium falciparum var genes correlate with changes in antigenic and cytoadherent phenotypes of infected erythrocytes. Cell 1995, 82, 101–110. [Google Scholar] [CrossRef]
- Su, X.Z.; Heatwole, V.M.; Wertheimer, S.P.; Guinet, F.; Herrfeldt, J.A.; Peterson, D.S.; Ravetch, J.A.; Wellems, T.E. The large diverse gene family var encodes proteins involved in cytoadherence and antigenic variation of Plasmodium falciparum-infected erythrocytes. Cell 1995, 82, 89–100. [Google Scholar] [CrossRef]
- Niang, M.; Bei, A.K.; Madnani, K.G.; Pelly, S.; Dankwa, S.; Kanjee, U.; Gunalan, K.; Amaladoss, A.; Yeo, K.P.; Bob, N.S.; et al. STEVOR is a Plasmodium falciparum erythrocyte binding protein that mediates merozoite invasion and rosetting. Cell Host Microbe 2014, 16, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Goel, S.; Palmkvist, M.; Moll, K.; Joannin, N.; Lara, P.; Akhouri, R.R.; Moradi, N.; Ojemalm, K.; Westman, M.; Angeletti, D.; et al. RIFINs are adhesins implicated in severe Plasmodium falciparum malaria. Nat. Med. 2015, 21, 314–317. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, D.A.; Jarra, W.; Koernig, S.; Fonager, J.; Fernandez-Reyes, D.; Blythe, J.E.; Waller, C.; Preiser, P.R.; Langhorne, J. Host immunity modulates transcriptional changes in a multigene family (yir) of rodent malaria. Mol. Microbiol. 2005, 58, 636–647. [Google Scholar] [CrossRef] [PubMed]
- Fonager, J.; Cunningham, D.; Jarra, W.; Koernig, S.; Henneman, A.A.; Langhorne, J.; Preiser, P. Transcription and alternative splicing in the yir multigene family of the malaria parasite Plasmodium y. yoelii: Identification of motifs suggesting epigenetic and post-transcriptional control of RNA expression. Mol. Biochem. Parasitol. 2007, 156, 1–11. [Google Scholar] [CrossRef]
- Lawton, J.; Brugat, T.; Yan, Y.X.; Reid, A.J.; Bohme, U.; Otto, T.D.; Pain, A.; Jackson, A.; Berriman, M.; Cunningham, D.; et al. Characterization and gene expression analysis of the cir multi-gene family of Plasmodium chabaudi chabaudi (AS). BMC Genom. 2012, 13, 125. [Google Scholar] [CrossRef]
- Yam, X.Y.; Brugat, T.; Siau, A.; Lawton, J.; Wong, D.S.; Farah, A.; Twang, J.S.; Gao, X.; Langhorne, J.; Preiser, P.R. Characterization of the Plasmodium Interspersed Repeats (PIR) proteins of Plasmodium chabaudi indicates functional diversity. Sci. Rep. 2016, 6, 23449. [Google Scholar] [CrossRef]
- Darling, T.K.; Mimche, P.N.; Bray, C.; Umaru, B.; Brady, L.M.; Stone, C.; Eboumbou Moukoko, C.E.; Lane, T.E.; Ayong, L.S.; Lamb, T.J. EphA2 contributes to disruption of the blood-brain barrier in cerebral malaria. PLoS Pathog. 2020, 16, e1008261. [Google Scholar] [CrossRef]
- Darling, T.K.; Schenk, M.P.; Zhou, C.C.; Maloba, F.M.; Mimche, P.N.; Gibbins, J.M.; Jobe, S.M.; Lamb, T.J. Platelet alpha-granules contribute to organ-specific pathologies in a mouse model of severe malaria. Blood Adv. 2020, 4, 1–8. [Google Scholar] [CrossRef]
- Epiphanio, S.; Campos, M.G.; Pamplona, A.; Carapau, D.; Pena, A.C.; Ataide, R.; Monteiro, C.A.; Felix, N.; Costa-Silva, A.; Marinho, C.R.; et al. VEGF promotes malaria-associated acute lung injury in mice. PLoS Pathog. 2010, 6, e1000916. [Google Scholar] [CrossRef]
- Hanum, P.S.; Hayano, M.; Kojima, S. Cytokine and chemokine responses in a cerebral malaria-susceptible or -resistant strain of mice to Plasmodium berghei ANKA infection: Early chemokine expression in the brain. Int. Immunol. 2003, 15, 633–640. [Google Scholar] [CrossRef]
- Niikura, M.; Kamiya, S.; Kita, K.; Kobayashi, F. Coinfection with nonlethal murine malaria parasites suppresses pathogenesis caused by Plasmodium berghei NK65. J. Immunol. 2008, 180, 6877–6884. [Google Scholar] [CrossRef]
- Vandermosten, L.; Pham, T.T.; Possemiers, H.; Knoops, S.; Van Herck, E.; Deckers, J.; Franke-Fayard, B.; Lamb, T.J.; Janse, C.J.; Opdenakker, G.; et al. Experimental malaria-associated acute respiratory distress syndrome is dependent on the parasite-host combination and coincides with normocyte invasion. Malar. J. 2018, 17, 102. [Google Scholar] [CrossRef]
- Hee, L.; Dinudom, A.; Mitchell, A.J.; Grau, G.E.; Cook, D.I.; Hunt, N.H.; Ball, H.J. Reduced activity of the epithelial sodium channel in malaria-induced pulmonary oedema in mice. Int. J. Parasitol. 2011, 41, 81–88. [Google Scholar] [CrossRef]
- Janse, C.J.; Boorsma, E.G.; Ramesar, J.; Grobbee, M.J.; Mons, B. Host cell specificity and schizogony of Plasmodium berghei under different in vitro conditions. Int. J. Parasitol. 1989, 19, 509–514. [Google Scholar] [CrossRef]
- Hojo-Souza, N.S.; de Azevedo, P.O.; de Castro, J.T.; Teixeira-Carvalho, A.; Lieberman, J.; Junqueira, C.; Gazzinelli, R.T. Contributions of IFN-gamma and granulysin to the clearance of Plasmodium yoelii blood stage. PLoS Pathog. 2020, 16, e1008840. [Google Scholar] [CrossRef]
- Abel, S.; Luckheide, N.; Westendorf, A.M.; Geffers, R.; Roers, A.; Muller, W.; Sparwasser, T.; Matuschewski, K.; Buer, J.; Hansen, W. Strong impact of CD4+ Foxp3+ regulatory T cells and limited effect of T cell-derived IL-10 on pathogen clearance during Plasmodium yoelii infection. J. Immunol. 2012, 188, 5467–5477. [Google Scholar] [CrossRef]
- Xia, L.; Wu, J.; Pattaradilokrat, S.; Tumas, K.; He, X.; Peng, Y.C.; Huang, R.; Myers, T.G.; Long, C.A.; Wang, R.; et al. Detection of host pathways universally inhibited after Plasmodium yoelii infection for immune intervention. Sci. Rep. 2018, 8, 15280. [Google Scholar] [CrossRef]
- Yoeli, M.; Hargreaves, B.; Carter, R.; Walliker, D. Sudden increase in virulence in a strain of Plasmodium berghei yoelii. Ann. Trop. Med. Parasitol. 1975, 69, 173–178. [Google Scholar] [CrossRef]
- Nair, S.C.; Xu, R.; Pattaradilokrat, S.; Wu, J.; Qi, Y.; Zilversmit, M.; Ganesan, S.; Nagarajan, V.; Eastman, R.T.; Orandle, M.S.; et al. A Plasmodium yoelii HECT-like E3 ubiquitin ligase regulates parasite growth and virulence. Nat. Commun. 2017, 8, 223. [Google Scholar] [CrossRef]
- Sayles, P.C.; Wassom, D.L. Immunoregulation in murine malaria. Susceptibility of inbred mice to infection with Plasmodium yoelii depends on the dynamic interplay of host and parasite genes. J. Immunol. 1988, 141, 241–248. [Google Scholar]
- Wu, J.; Tian, L.; Yu, X.; Pattaradilokrat, S.; Li, J.; Wang, M.; Yu, W.; Qi, Y.; Zeituni, A.E.; Nair, S.C.; et al. Strain-specific innate immune signaling pathways determine malaria parasitemia dynamics and host mortality. Proc. Natl. Acad. Sci. USA 2014, 111, E511–E520. [Google Scholar] [CrossRef]
- Lacerda-Queiroz, N.; Riteau, N.; Eastman, R.T.; Bock, K.W.; Orandle, M.S.; Moore, I.N.; Sher, A.; Long, C.A.; Jankovic, D.; Su, X.Z. Mechanism of splenic cell death and host mortality in a Plasmodium yoelii malaria model. Sci. Rep. 2017, 7, 10438. [Google Scholar] [CrossRef]
- Brugat, T.; Cunningham, D.; Sodenkamp, J.; Coomes, S.; Wilson, M.; Spence, P.J.; Jarra, W.; Thompson, J.; Scudamore, C.; Langhorne, J. Sequestration and histopathology in Plasmodium chabaudi malaria are influenced by the immune response in an organ-specific manner. Cell. Microbiol. 2014, 16, 687–700. [Google Scholar] [CrossRef]
- Mackinnon, M.J.; Read, A.F. Genetic Relationships between Parasite Virulence and Transmission in the Rodent Malaria Plasmodium Chabaudi. Evolution 1999, 53, 689–703. [Google Scholar] [CrossRef]
- Lamb, T.J.; Langhorne, J. The severity of malarial anaemia in Plasmodium chabaudi infections of BALB/c mice is determined independently of the number of circulating parasites. Malar. J. 2008, 7, 68. [Google Scholar] [CrossRef]
- Fonseca, L.; Seixas, E.; Butcher, G.; Langhorne, J. Cytokine responses of CD4+ T cells during a Plasmodium chabaudi chabaudi (ER) blood-stage infection in mice initiated by the natural route of infection. Malar. J. 2007, 6, 77. [Google Scholar] [CrossRef]
- Yadava, A.; Kumar, S.; Dvorak, J.A.; Milon, G.; Miller, L.H. Trafficking of Plasmodium chabaudi adami-infected erythrocytes within the mouse spleen. Proc. Natl. Acad. Sci. USA 1996, 93, 4595–4599. [Google Scholar] [CrossRef]
- Scorza, T.; Grubb, K.; Cambos, M.; Santamaria, C.; Tshikudi Malu, D.; Spithill, T.W. Vaccination with a Plasmodium chabaudi adami multivalent DNA vaccine cross-protects A/J mice against challenge with P. c. adami DK and virulent Plasmodium chabaudi chabaudi AS parasites. Int. J. Parasitol. 2008, 38, 819–827. [Google Scholar] [CrossRef]
- Gaudreault, V.; Wirbel, J.; Jardim, A.; Rohrbach, P.; Scorza, T. Red Blood Cells Preconditioned with Hemin Are Less Permissive to Plasmodium Invasion In Vivo and In Vitro. PLoS ONE 2015, 10, e0140805. [Google Scholar]
- Cavacini, L.A.; Guidotti, M.; Parke, L.A.; Melancon-Kaplan, J.; Weidanz, W.P. Reassessment of the role of splenic leukocyte oxidative activity and macrophage activation in expression of immunity to malaria. Infect. Immun. 1989, 57, 3677–3682. [Google Scholar] [CrossRef]
- Van der Heyde, H.C.; Elloso, M.M.; Roopenian, D.C.; Manning, D.D.; Weidanz, W.P. Expansion of the CD4-, CD8- gamma delta T cell subset in the spleens of mice during non-lethal blood-stage malaria. Eur. J. Immunol. 1993, 23, 1846–1850. [Google Scholar] [CrossRef] [PubMed]
- Ramaprasad, A.; Klaus, S.; Douvropoulou, O.; Culleton, R.; Pain, A. Plasmodium vinckei genomes provide insights into the pan-genome and evolution of rodent malaria parasites. BMC Biol. 2021, 19, 69. [Google Scholar] [CrossRef] [PubMed]
- LaCrue, A.N.; Scheel, M.; Kennedy, K.; Kumar, N.; Kyle, D.E. Effects of artesunate on parasite recrudescence and dormancy in the rodent malaria model Plasmodium vinckei. PLoS ONE 2011, 6, e26689. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, J.; Siddiqui, A.J.; Goyal, M.; Prakash, K.; Soni, A.; Puri, S.K.; Srivastava, M. Host immune response is severely compromised during lethal Plasmodium vinckei infection. Parasitol. Res. 2015, 114, 3445–3457. [Google Scholar] [CrossRef]
- Viens, P.; Chevalier, J.L.; Sonea, S.; Yoeli, M. The effect of reticulocytosis on Plasmodium vinckei infection in white mice. Action of phenylhydrazine and of repeated bleedings. Can. J. Microbiol. 1971, 17, 257–261. [Google Scholar] [CrossRef]
- Cavacini, L.A.; Parke, L.A.; Weidanz, W.P. Resolution of acute malarial infections by T cell-dependent non-antibody-mediated mechanisms of immunity. Infect. Immun. 1990, 58, 2946–2950. [Google Scholar] [CrossRef]
- Vigario, A.M.; Belnoue, E.; Cumano, A.; Marussig, M.; Miltgen, F.; Landau, I.; Mazier, D.; Gresser, I.; Renia, L. Inhibition of Plasmodium yoelii blood-stage malaria by interferon alpha through the inhibition of the production of its target cell, the reticulocyte. Blood 2001, 97, 3966–3971. [Google Scholar] [CrossRef]
- Masopust, D.; Sivula, C.P.; Jameson, S.C. Of mice, dirty mice, and men: Using mice to understand human immunology. J. Immunol. 2017, 199, 383–388. [Google Scholar] [CrossRef]
- Mestas, J.; Hughes, C.C. Of mice and not men: Differences between mouse and human immunology. J. Immunol. 2004, 172, 2731–2738. [Google Scholar] [CrossRef]
- Steiniger, B.S. Human spleen microanatomy: Why mice do not suffice. Immunology 2015, 145, 334–346. [Google Scholar] [CrossRef]
- Weiss, L. The spleen in malaria: The role of barrier cells. Immunol. Lett. 1990, 25, 165–172. [Google Scholar] [CrossRef]
- Drenckhahn, D.; Wagner, J. Stress fibers in the splenic sinus endothelium in situ: Molecular structure, relationship to the extracellular matrix, and contractility. J. Cell Biol. 1986, 102, 1738–1747. [Google Scholar] [CrossRef]
- Hommel, M.; David, P.H.; Oligino, L.D. Surface alterations of erythrocytes in Plasmodium falciparum malaria. Antigenic variation, antigenic diversity, and the role of the spleen. J. Exp. Med. 1983, 157, 1137–1148. [Google Scholar] [CrossRef]
- Gilks, C.F.; Walliker, D.; Newbold, C.I. Relationships between sequestration, antigenic variation and chronic parasitism in Plasmodium chabaudi chabaudi—A rodent malaria model. Parasite Immunol. 1990, 12, 45–64. [Google Scholar] [CrossRef]
- Urban, B.C.; Hien, T.T.; Day, N.P.; Phu, N.H.; Roberts, R.; Pongponratn, E.; Jones, M.; Mai, N.T.; Bethell, D.; Turner, G.D.; et al. Fatal Plasmodium falciparum malaria causes specific patterns of splenic architectural disorganization. Infect. Immun. 2005, 73, 1986–1994. [Google Scholar] [CrossRef]
- Achtman, A.H.; Khan, M.; MacLennan, I.C.; Langhorne, J. Plasmodium chabaudi chabaudi infection in mice induces strong B cell responses and striking but temporary changes in splenic cell distribution. J. Immunol. 2003, 171, 317–324. [Google Scholar] [CrossRef]
- Cadman, E.T.; Abdallah, A.Y.; Voisine, C.; Sponaas, A.M.; Corran, P.; Lamb, T.; Brown, D.; Ndungu, F.; Langhorne, J. Alterations of splenic architecture in malaria are induced independently of Toll-like receptors 2, 4, and 9 or MyD88 and may affect antibody affinity. Infect. Immun. 2008, 76, 3924–3931. [Google Scholar] [CrossRef]
- Lanigan, T.M.; Kopera, H.C.; Saunders, T.L. Principles of genetic engineering. Genes 2020, 11, 291. [Google Scholar] [CrossRef]
- Clark, J.F.; Dinsmore, C.J.; Soriano, P. A most formidable arsenal: Genetic technologies for building a better mouse. Genes Dev. 2020, 34, 1256–1286. [Google Scholar] [CrossRef]
- Spence, P.J.; Cunningham, D.; Jarra, W.; Lawton, J.; Langhorne, J.; Thompson, J. Transformation of the rodent malaria parasite Plasmodium chabaudi. Nat. Protoc. 2011, 6, 553–561. [Google Scholar] [CrossRef]
- Janse, C.J.; Ramesar, J.; Waters, A.P. High-efficiency transfection and drug selection of genetically transformed blood stages of the rodent malaria parasite Plasmodium berghei. Nat. Protoc. 2006, 1, 346–356. [Google Scholar] [CrossRef]
- Sponaas, A.M.; Freitas do Rosario, A.P.; Voisine, C.; Mastelic, B.; Thompson, J.; Koernig, S.; Jarra, W.; Renia, L.; Mauduit, M.; Potocnik, A.J.; et al. Migrating monocytes recruited to the spleen play an important role in control of blood stage malaria. Blood 2009, 114, 5522–5531. [Google Scholar] [CrossRef]
- Knackstedt, S.L.; Georgiadou, A.; Apel, F.; Abu-Abed, U.; Moxon, C.A.; Cunnington, A.J.; Raupach, B.; Cunningham, D.; Langhorne, J.; Kruger, R.; et al. Neutrophil extracellular traps drive inflammatory pathogenesis in malaria. Sci. Immunol. 2019, 4, eaaw0336. [Google Scholar] [CrossRef]
- Rocha, B.C.; Marques, P.E.; Leoratti, F.M.S.; Junqueira, C.; Pereira, D.B.; Antonelli, L.; Menezes, G.B.; Golenbock, D.T.; Gazzinelli, R.T. Type I Interferon Transcriptional Signature in Neutrophils and Low-Density Granulocytes Are Associated with Tissue Damage in Malaria. Cell Rep. 2015, 13, 2829–2841. [Google Scholar] [CrossRef]
- Hernández-Castañeda, M.A.; Happ, K.; Cattalani, F.; Wallimann, A.; Blanchard, M.; Fellay, I.; Scolari, B.; Lannes, N.; Mbagwu, S.; Fellay, B. γδ T Cells Kill Plasmodium falciparum in a Granzyme-and Granulysin-Dependent Mechanism during the Late Blood Stage. J. Immunol. 2020, 204, 1798–1809. [Google Scholar] [CrossRef]
- Mamedov, M.R.; Scholzen, A.; Nair, R.V.; Cumnock, K.; Kenkel, J.A.; Oliveira, J.H.M.; Trujillo, D.L.; Saligrama, N.; Zhang, Y.; Rubelt, F. A macrophage colony-stimulating-factor-producing γδ T cell subset prevents malarial parasitemic recurrence. Immunity 2018, 48, 350–363.e7. [Google Scholar] [CrossRef]
- Deroost, K.; Langhorne, J. Gamma/Delta T Cells and Their Role in Protection against Malaria. Front. Immunol. 2018, 9, 2973. [Google Scholar] [CrossRef]
- Seixas, E.; Moura Nunes, J.F.; Matos, I.; Coutinho, A. The interaction between DC and Plasmodium berghei/chabaudi-infected erythrocytes in mice involves direct cell-to-cell contact, internalization and TLR. Eur. J. Immunol. 2009, 39, 1850–1863. [Google Scholar] [CrossRef]
- Voisine, C.; Mastelic, B.; Sponaas, A.M.; Langhorne, J. Classical CD11c+ dendritic cells, not plasmacytoid dendritic cells, induce T cell responses to Plasmodium chabaudi malaria. Int. J. Parasitol. 2010, 40, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Omer, F.M.; Riley, E.M. Transforming growth factor β production is inversely correlated with severity of murine malaria infection. J. Exp. Med. 1998, 188, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Sanni, L.A.; Omer, F.; Riley, E.; Langhorne, J. Pathology of Plasmodium chabaudi chabaudi infection and mortality in interleukin-10-deficient mice are ameliorated by anti-tumor necrosis factor alpha and exacerbated by anti-transforming growth factor beta antibodies. Infect. Immun. 2003, 71, 4850–4856. [Google Scholar] [CrossRef] [PubMed]
- Perez-Mazliah, D.; Nguyen, M.P.; Hosking, C.; McLaughlin, S.; Lewis, M.D.; Tumwine, I.; Levy, P.; Langhorne, J. Follicular Helper T Cells are Essential for the Elimination of Plasmodium Infection. EBioMedicine 2017, 24, 216–230. [Google Scholar] [CrossRef] [PubMed]
- Couper, K.N.; Phillips, R.S.; Brombacher, F.; Alexander, J. Parasite-specific IgM plays a significant role in the protective immune response to asexual erythrocytic stage Plasmodium chabaudi AS infection. Parasite Immunol. 2005, 27, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurty, A.T.; Thouvenel, C.D.; Portugal, S.; Keitany, G.J.; Kim, K.S.; Holder, A.; Crompton, P.D.; Rawlings, D.J.; Pepper, M. Somatically Hypermutated Plasmodium-Specific IgM(+) Memory B Cells Are Rapid, Plastic, Early Responders upon Malaria Rechallenge. Immunity 2016, 45, 402–414. [Google Scholar] [CrossRef]
- Carpio, V.H.; Opata, M.M.; Montanez, M.E.; Banerjee, P.P.; Dent, A.L.; Stephens, R. IFN-gamma and IL-21 Double Producing T Cells Are Bcl6-Independent and Survive into the Memory Phase in Plasmodium chabaudi Infection. PLoS ONE 2015, 10, e0144654. [Google Scholar] [CrossRef]
- Ndungu, F.M.; Cadman, E.T.; Coulcher, J.; Nduati, E.; Couper, E.; Macdonald, D.W.; Ng, D.; Langhorne, J. Functional memory B cells and long-lived plasma cells are generated after a single Plasmodium chabaudi infection in mice. PLoS Pathog. 2009, 5, e1000690. [Google Scholar] [CrossRef]
- Opata, M.M.; Carpio, V.H.; Ibitokou, S.A.; Dillon, B.E.; Obiero, J.M.; Stephens, R. Early effector cells survive the contraction phase in malaria infection and generate both central and effector memory T cells. J. Immunol. 2015, 194, 5346–5354. [Google Scholar] [CrossRef]
- Fairlie-Clarke, K.J.; Allen, J.E.; Read, A.F.; Graham, A.L. Quantifying variation in the potential for antibody-mediated apparent competition among nine genotypes of the rodent malaria parasite Plasmodium chabaudi. Infect. Genet. Evol. 2013, 20, 270–275. [Google Scholar] [CrossRef][Green Version]
- Long, G.H.; Chan, B.H.; Allen, J.E.; Read, A.F.; Graham, A.L. Blockade of TNF receptor 1 reduces disease severity but increases parasite transmission during Plasmodium chabaudi chabaudi infection. Int. J. Parasitol. 2008, 38, 1073–1081. [Google Scholar] [CrossRef]
- Fortin, A.; Stevenson, M.M.; Gros, P. Complex genetic control of susceptibility to malaria in mice. Genes Immun. 2002, 3, 177–186. [Google Scholar] [CrossRef]
- Huang, H.M.; McMorran, B.J.; Foote, S.J.; Burgio, G. Host genetics in malaria: Lessons from mouse studies. Mamm. Genome 2018, 29, 507–522. [Google Scholar] [CrossRef]
- Couper, K.N.; Blount, D.G.; Hafalla, J.C.; van Rooijen, N.; de Souza, J.B.; Riley, E.M. Macrophage-mediated but gamma interferon-independent innate immune responses control the primary wave of Plasmodium yoelii parasitemia. Infect. Immun. 2007, 75, 5806–5818. [Google Scholar] [CrossRef]
- Theess, W.; Sellau, J.; Steeg, C.; Klinke, A.; Baldus, S.; Cramer, J.P.; Jacobs, T. Myeloperoxidase Attenuates Pathogen Clearance during Plasmodium yoelii Nonlethal Infection. Infect. Immun. 2017, 85, e00475-16. [Google Scholar] [CrossRef]
- Miyakoda, M.; Bayarsaikhan, G.; Kimura, D.; Akbari, M.; Udono, H.; Yui, K. Metformin Promotes the Protection of Mice Infected with Plasmodium yoelii Independently of gammadelta T Cell Expansion. Front. Immunol. 2018, 9, 2942. [Google Scholar] [CrossRef]
- Mogil, R.J.; Patton, C.L.; Green, D.R. Cellular subsets involved in cell-mediated immunity to murine Plasmodium yoelii 17X malaria. J. Immunol. 1987, 138, 1933–1939. [Google Scholar]
- Couper, K.N.; Blount, D.G.; Wilson, M.S.; Hafalla, J.C.; Belkaid, Y.; Kamanaka, M.; Flavell, R.A.; de Souza, J.B.; Riley, E.M. IL-10 from CD4CD25Foxp3CD127 adaptive regulatory T cells modulates parasite clearance and pathology during malaria infection. PLoS Pathog. 2008, 4, e1000004. [Google Scholar] [CrossRef]
- Vijay, R.; Guthmiller, J.J.; Sturtz, A.J.; Surette, F.A.; Rogers, K.J.; Sompallae, R.R.; Li, F.; Pope, R.L.; Chan, J.A.; de Labastida Rivera, F.; et al. Infection-induced plasmablasts are a nutrient sink that impairs humoral immunity to malaria. Nat. Immunol. 2020, 21, 790–801. [Google Scholar] [CrossRef]
- Brown, S.L.; Bauer, J.J.; Lee, J.; Ntirandekura, E.; Stumhofer, J.S. IgM(+) and IgM(−) memory B cells represent heterogeneous populations capable of producing class-switched antibodies and germinal center B cells upon rechallenge with P. yoelii. J. Leukoc. Biol. 2022, in press. [Google Scholar] [CrossRef]
- Zander, R.A.; Vijay, R.; Pack, A.D.; Guthmiller, J.J.; Graham, A.C.; Lindner, S.E.; Vaughan, A.M.; Kappe, S.H.I.; Butler, N.S. Th1-like Plasmodium-Specific Memory CD4(+) T Cells Support Humoral Immunity. Cell Rep. 2017, 21, 1839–1852. [Google Scholar] [CrossRef]
- Omer, F.M.; de Souza, J.B.; Riley, E.M. Differential induction of TGF-beta regulates proinflammatory cytokine production and determines the outcome of lethal and nonlethal Plasmodium yoelii infections. J. Immunol. 2003, 171, 5430–5436. [Google Scholar] [CrossRef]
- McQuillan, J.A.; Mitchell, A.J.; Ho, Y.F.; Combes, V.; Ball, H.J.; Golenser, J.; Grau, G.E.; Hunt, N.H. Coincident parasite and CD8 T cell sequestration is required for development of experimental cerebral malaria. Int. J. Parasitol. 2011, 41, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Swanson, P.A., 2nd; Hart, G.T.; Russo, M.V.; Nayak, D.; Yazew, T.; Pena, M.; Khan, S.M.; Janse, C.J.; Pierce, S.K.; McGavern, D.B. CD8+ T Cells Induce Fatal Brainstem Pathology during Cerebral Malaria via Luminal Antigen-Specific Engagement of Brain Vasculature. PLoS Pathog. 2016, 12, e1006022. [Google Scholar] [CrossRef] [PubMed]
- Aitken, E.H.; Negri, E.M.; Barboza, R.; Lima, M.R.; Alvarez, J.M.; Marinho, C.R.; Caldini, E.G.; Epiphanio, S. Ultrastructure of the lung in a murine model of malaria-associated acute lung injury/acute respiratory distress syndrome. Malar. J. 2014, 13, 230. [Google Scholar] [CrossRef] [PubMed]
- Amani, V.; Boubou, M.I.; Pied, S.; Marussig, M.; Walliker, D.; Mazier, D.; Renia, L. Cloned lines of Plasmodium berghei ANKA differ in their abilities to induce experimental cerebral malaria. Infect. Immun. 1998, 66, 4093–4099. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.T.; Verheijen, M.; Vandermosten, L.; Deroost, K.; Knoops, S.; Van den Eynde, K.; Boon, L.; Janse, C.J.; Opdenakker, G.; Van den Steen, P.E. Pathogenic CD8(+) T Cells Cause Increased Levels of VEGF-A in Experimental Malaria-Associated Acute Respiratory Distress Syndrome, but Therapeutic VEGFR Inhibition Is Not Effective. Front. Cell. Infect. Microbiol. 2017, 7, 416. [Google Scholar] [CrossRef] [PubMed]
- Franke-Fayard, B.; Janse, C.J.; Cunha-Rodrigues, M.; Ramesar, J.; Buscher, P.; Que, I.; Lowik, C.; Voshol, P.J.; den Boer, M.A.; van Duinen, S.G.; et al. Murine malaria parasite sequestration: CD36 is the major receptor, but cerebral pathology is unlinked to sequestration. Proc. Natl. Acad. Sci. USA 2005, 102, 11468–11473. [Google Scholar] [CrossRef]
- De Koning-Ward, T.F.; O’Donnell, R.A.; Drew, D.R.; Thomson, R.; Speed, T.P.; Crabb, B.S. A new rodent model to assess blood stage immunity to the Plasmodium falciparum antigen merozoite surface protein 119 reveals a protective role for invasion inhibitory antibodies. J. Exp. Med. 2003, 198, 869–875. [Google Scholar] [CrossRef]
- Kocken, C.H.; van der Wel, A.M.; Dubbeld, M.A.; Narum, D.L.; van de Rijke, F.M.; van Gemert, G.J.; van der Linde, X.; Bannister, L.H.; Janse, C.; Waters, A.P.; et al. Precise timing of expression of a Plasmodium falciparum-derived transgene in Plasmodium berghei is a critical determinant of subsequent subcellular localization. J. Biol. Chem. 1998, 273, 15119–15124. [Google Scholar] [CrossRef]
- Dobrescu, I.; de Camargo, T.M.; Gimenez, A.M.; Murillo, O.; Amorim, K.; Marinho, C.R.F.; Soares, I.S.; Boscardin, S.B.; Bargieri, D.Y. Protective Immunity in Mice Immunized with P. vivax MSP119-Based Formulations and Challenged with P. berghei Expressing PvMSP119. Front. Immunol. 2020, 11, 28. [Google Scholar] [CrossRef]
- Ono, T.; Tadakuma, T.; Rodriguez, A. Plasmodium yoelii yoelii 17XNL constitutively expressing GFP throughout the life cycle. Exp. Parasitol. 2007, 115, 310–313. [Google Scholar] [CrossRef]
- Franke-Fayard, B.; Trueman, H.; Ramesar, J.; Mendoza, J.; van der Keur, M.; van der Linden, R.; Sinden, R.E.; Waters, A.P.; Janse, C.J. A Plasmodium berghei reference line that constitutively expresses GFP at a high level throughout the complete life cycle. Mol. Biochem. Parasitol. 2004, 137, 23–33. [Google Scholar] [CrossRef]
- Dookie, R.S.; Villegas-Mendez, A.; Kroeze, H.; Barrett, J.R.; Draper, S.J.; Franke-Fayard, B.M.; Janse, C.J.; MacDonald, A.S.; Couper, K.N. Combinatorial Tim-3 and PD-1 activity sustains antigen-specific Th1 cell numbers during blood-stage malaria. Parasite Immunol. 2020, 42, e12723. [Google Scholar] [CrossRef]
- Miyakoda, M.; Kimura, D.; Yuda, M.; Chinzei, Y.; Shibata, Y.; Honma, K.; Yui, K. Malaria-specific and nonspecific activation of CD8+ T cells during blood stage of Plasmodium berghei infection. J. Immunol. 2008, 181, 1420–1428. [Google Scholar] [CrossRef]
- Valencia-Hernandez, A.M.; Ng, W.Y.; Ghazanfari, N.; Ghilas, S.; de Menezes, M.N.; Holz, L.E.; Huang, C.; English, K.; Naung, M.; Tan, P.S.; et al. A Natural Peptide Antigen within the Plasmodium Ribosomal Protein RPL6 Confers Liver TRM Cell-Mediated Immunity against Malaria in Mice. Cell Host Microbe 2020, 27, 950–962.e7. [Google Scholar] [CrossRef]
- Lau, L.S.; Fernandez-Ruiz, D.; Mollard, V.; Sturm, A.; Neller, M.A.; Cozijnsen, A.; Gregory, J.L.; Davey, G.M.; Jones, C.M.; Lin, Y.H.; et al. CD8+ T cells from a novel T cell receptor transgenic mouse induce liver-stage immunity that can be boosted by blood-stage infection in rodent malaria. PLoS Pathog. 2014, 10, e1004135. [Google Scholar] [CrossRef]
- Enders, M.H.; Bayarsaikhan, G.; Ghilas, S.; Chua, Y.C.; May, R.; de Menezes, M.N.; Ge, Z.; Tan, P.S.; Cozijnsen, A.; Mollard, V.; et al. Plasmodium berghei Hsp90 contains a natural immunogenic I-A(b)-restricted antigen common to rodent and human Plasmodium species. Curr. Res. Immunol. 2021, 2, 79–92. [Google Scholar] [CrossRef]
- Fernandez-Ruiz, D.; Lau, L.S.; Ghazanfari, N.; Jones, C.M.; Ng, W.Y.; Davey, G.M.; Berthold, D.; Holz, L.; Kato, Y.; Enders, M.H.; et al. Development of a Novel CD4(+) TCR Transgenic Line That Reveals a Dominant Role for CD8(+) Dendritic Cells and CD40 Signaling in the Generation of Helper and CTL Responses to Blood-Stage Malaria. J. Immunol. 2017, 199, 4165–4179. [Google Scholar] [CrossRef]
- Stephens, R.; Albano, F.R.; Quin, S.; Pascal, B.J.; Harrison, V.; Stockinger, B.; Kioussis, D.; Weltzien, H.U.; Langhorne, J. Malaria-specific transgenic CD4(+) T cells protect immunodeficient mice from lethal infection and demonstrate requirement for a protective threshold of antibody production for parasite clearance. Blood 2005, 106, 1676–1684. [Google Scholar] [CrossRef]
- Perez-Mazliah, D.; Gardner, P.J.; Schweighoffer, E.; McLaughlin, S.; Hosking, C.; Tumwine, I.; Davis, R.S.; Potocnik, A.J.; Tybulewicz, V.L.; Langhorne, J. Plasmodium-specific atypical memory B cells are short-lived activated B cells. eLife 2018, 7, e39800. [Google Scholar] [CrossRef]
- Talavera-López, C.; Bediako, Y.; Lin, J.-W.; Valletta, J.J.; Recker, M.; Langhorne, J. Comparison of whole blood and spleen transcriptional signatures over the course of an experimental malaria infection. Sci. Rep. 2019, 9, 15853. [Google Scholar] [CrossRef]
- Zhao, Y.; Hosking, C.; Cunningham, D.; Langhorne, J.; Lin, J.-W. Transcriptome analysis of blood and spleen in virulent and avirulent mouse malaria infection. Sci. Data 2020, 7, 253. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, Z.; Cui, S.; Zhao, Y.; Craft, S.; Fikrig, E.; You, F. ELF4 facilitates innate host defenses against Plasmodium by activating transcription of Pf4 and Ppbp. J. Biol. Chem. 2019, 294, 7787–7796. [Google Scholar] [CrossRef]
- Pereira, L.M.; Assis, P.A.; de Araújo, N.M.; Durso, D.F.; Junqueira, C.; Ataíde, M.A.; Pereira, D.B.; Lien, E.; Fitzgerald, K.A.; Zamboni, D.S. Caspase-8 mediates inflammation and disease in rodent malaria. Nat. Commun. 2020, 11, 4596. [Google Scholar] [CrossRef]
- Oakley, M.S.; Chorazeczewski, J.K.; Aleshnick, M.; Anantharaman, V.; Majam, V.; Chawla, B.; Myers, T.G.; Su, Q.; Okoth, W.A.; Takeda, K. TCRβ-expressing macrophages induced by a pathogenic murine malaria correlate with parasite burden and enhanced phagocytic activity. PLoS ONE 2018, 13, e0201043. [Google Scholar] [CrossRef]
- Lai, S.M.; Sheng, J.; Gupta, P.; Renia, L.; Duan, K.; Zolezzi, F.; Karjalainen, K.; Newell, E.W.; Ruedl, C. Organ-specific fate, recruitment, and refilling dynamics of tissue-resident macrophages during blood-stage malaria. Cell Rep. 2018, 25, 3099–3109.e3. [Google Scholar] [CrossRef]
- Kim, C.C.; Nelson, C.S.; Wilson, E.B.; Hou, B.; DeFranco, A.L.; DeRisi, J.L. Splenic red pulp macrophages produce type I interferons as early sentinels of malaria infection but are dispensable for control. PLoS ONE 2012, 7, e48126. [Google Scholar]
- Kim, C.C.; Parikh, S.; Sun, J.C.; Myrick, A.; Lanier, L.L.; Rosenthal, P.J.; DeRisi, J.L. Experimental malaria infection triggers early expansion of natural killer cells. Infect. Immun. 2008, 76, 5873–5882. [Google Scholar] [CrossRef]
- Haque, A.; Best, S.E.; Ammerdorffer, A.; Desbarrieres, L.; de Oca, M.M.; Amante, F.H.; de Labastida Rivera, F.; Hertzog, P.; Boyle, G.M.; Hill, G.R.; et al. Type I interferons suppress CD4(+) T-cell-dependent parasite control during blood-stage Plasmodium infection. Eur. J. Immunol. 2011, 41, 2688–2698. [Google Scholar] [CrossRef]
- Cheng, Q.; Liu, J.; Pei, Y.; Zhang, Y.; Zhou, D.; Pan, W.; Zhang, J. Neddylation contributes to CD4+ T cell-mediated protective immunity against blood-stage Plasmodium infection. PLoS Pathog. 2018, 14, e1007440. [Google Scholar] [CrossRef]
- Fontana, M.F.; de Melo, G.L.; Anidi, C.; Hamburger, R.; Kim, C.Y.; Lee, S.Y.; Pham, J.; Kim, C.C. Macrophage colony stimulating factor derived from CD4+ T cells contributes to control of a blood-borne infection. PLoS Pathog. 2016, 12, e1006046. [Google Scholar] [CrossRef] [PubMed]
- Ames, R.Y.; Ting, L.-M.; Gendlina, I.; Kim, K.; Macian, F. The transcription factor NFAT1 participates in the induction of CD4+ T cell functional exhaustion during Plasmodium yoelii infection. Infect. Immun. 2017, 85, e00364-17. [Google Scholar] [CrossRef] [PubMed]
- Ly, A.; Liao, Y.; Pietrzak, H.; Ioannidis, L.J.; Sidwell, T.; Gloury, R.; Doerflinger, M.; Triglia, T.; Qin, R.Z.; Groom, J.R. Transcription factor T-bet in B cells modulates germinal center polarization and antibody affinity maturation in response to malaria. Cell Rep. 2019, 29, 2257–2269.e6. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.C.; Baccarella, A.M.; Bayat, A.; Pepper, M.; Fontana, M.F. FCRL5+ memory B cells exhibit robust recall responses. Cell Rep. 2019, 27, 1446–1460.e4. [Google Scholar] [CrossRef] [PubMed]
- Talavera-López, C.; Capuccini, B.; Mitter, R.; Lin, J.-W.; Langhorne, J. Transcriptomes of microglia in experimental cerebral malaria in mice in the presence and absence of Type I Interferon signaling. BMC Res. Notes 2018, 11, 913. [Google Scholar] [CrossRef]
- Friedman, M.J. Erythrocytic mechanism of sickle cell resistance to malaria. Proc. Natl. Acad. Sci. USA 1978, 75, 1994–1997. [Google Scholar] [CrossRef]
- Aidoo, M.; Terlouw, D.J.; Kolczak, M.S.; McElroy, P.D.; ter Kuile, F.O.; Kariuki, S.; Nahlen, B.L.; Lal, A.A.; Udhayakumar, V. Protective effects of the sickle cell gene against malaria morbidity and mortality. Lancet 2002, 359, 1311–1312. [Google Scholar] [CrossRef]
- Peters, A.L.; Noorden, C.J.V. Glucose-6-phosphate dehydrogenase deficiency and malaria: Cytochemical detection of heterozygous G6PD deficiency in women. J. Histochem. Cytochem. 2009, 57, 1003–1011. [Google Scholar] [CrossRef]
- Avalos, S.; Mejia, R.E.; Banegas, E.; Salinas, C.; Gutierrez, L.; Fajardo, M.; Galo, S.; Pinto, A.; Mejia, A.; Fontecha, G. G6PD deficiency, primaquine treatment, and risk of haemolysis in malaria-infected patients. Malar. J. 2018, 17, 415. [Google Scholar] [CrossRef]
- Mockenhaupt, F.P.; Ehrhardt, S.; Gellert, S.; Otchwemah, R.N.; Dietz, E.; Anemana, S.D.; Bienzle, U. α+-thalassemia protects African children from severe malaria. Blood 2004, 104, 2003–2006. [Google Scholar] [CrossRef]
- Hill, A.V.; Allsopp, C.E.; Kwiatkowski, D.; Anstey, N.M.; Twumasi, P.; Rowe, P.A.; Bennett, S.; Brewster, D.; McMichael, A.J.; Greenwood, B.M. Common west African HLA antigens are associated with protection from severe malaria. Nature 1991, 352, 595–600. [Google Scholar] [CrossRef]
- May, J.; Lell, B.; Luty, A.J.; Meyer, C.G.; Kremsner, P.G. HLA-DQB1*0501-restricted Th1 type immune responses to Plasmodium falciparum liver stage antigen 1 protect against malaria anemia and reinfections. J. Infect. Dis. 2001, 183, 168–172. [Google Scholar] [CrossRef]
- Troye-Blomberg, M.; Olerup, O.; Larsson, A.; Sjoberg, K.; Perlmann, H.; Riley, E.; Lepers, J.P.; Perlmann, P. Failure to detect MHC class II associations of the human immune response induced by repeated malaria infections to the Plasmodium falciparum antigen Pf155/RESA. Int. Immunol. 1991, 3, 1043–1051. [Google Scholar] [CrossRef]
- Gichohi-Wainaina, W.N.; Melse-Boonstra, A.; Feskens, E.J.; Demir, A.Y.; Veenemans, J.; Verhoef, H. Tumour necrosis factor allele variants and their association with the occurrence and severity of malaria in African children: A longitudinal study. Malar. J. 2015, 14, 249. [Google Scholar] [CrossRef][Green Version]
- McGuire, W.; Knight, J.C.; Hill, A.V.; Allsopp, C.E.; Greenwood, B.M.; Kwiatkowski, D. Severe malarial anemia and cerebral malaria are associated with different tumor necrosis factor promoter alleles. J. Infect. Dis. 1999, 179, 287–290. [Google Scholar] [CrossRef]
- McGuire, W.; Hill, A.V.; Allsopp, C.E.; Greenwood, B.M.; Kwiatkowski, D. Variation in the TNF-α promoter region associated with susceptibility to cerebral malaria. Nature 1994, 371, 508–511. [Google Scholar] [CrossRef]
- Clark, T.G.; Diakite, M.; Auburn, S.; Campino, S.; Fry, A.E.; Green, A.; Richardson, A.; Small, K.; Teo, Y.Y.; Wilson, J. Tumor necrosis factor and lymphotoxin-α polymorphisms and severe malaria in African populations. J. Infect. Dis. 2009, 199, 569–575. [Google Scholar] [CrossRef]
- Ravenhall, M.; Campino, S.; Sepulveda, N.; Manjurano, A.; Nadjm, B.; Mtove, G.; Wangai, H.; Maxwell, C.; Olomi, R.; Reyburn, H.; et al. MalariaGen, Novel genetic polymorphisms associated with severe malaria and under selective pressure in North-eastern Tanzania. PLoS Genet. 2018, 14, e1007172. [Google Scholar] [CrossRef]
- Malaria Genomic Epidemiology Network Writing Group; Band, G.; Rockett, K.A.; Spencer, C.C.; Kwiatkowski, D.P. A novel locus of resistance to severe malaria in a region of ancient balancing selection. Nature 2015, 526, 253–257. [Google Scholar]
- Timmann, C.; Thye, T.; Vens, M.; Evans, J.; May, J.; Ehmen, C.; Sievertsen, J.; Muntau, B.; Ruge, G.; Loag, W.; et al. Genome-wide association study indicates two novel resistance loci for severe malaria. Nature 2012, 489, 443–446. [Google Scholar] [CrossRef]
- Jallow, M.; Teo, Y.Y.; Small, K.S.; Rockett, K.A.; Deloukas, P.; Clark, T.G.; Kivinen, K.; Bojang, K.A.; Conway, D.J.; Pinder, M.; et al. Genome-wide and fine-resolution association analysis of malaria in West Africa. Nat. Genet. 2009, 41, 657–665. [Google Scholar] [CrossRef]
- Brisebarre, A.; Kumulungui, B.; Sawadogo, S.; Atkinson, A.; Garnier, S.; Fumoux, F.; Rihet, P. A genome scan for Plasmodium falciparum malaria identifies quantitative trait loci on chromosomes 5q31, 6p21.3, 17p12, and 19p13. Malar. J. 2014, 13, 198. [Google Scholar] [CrossRef]
- Maiga, B.; Dolo, A.; Touré, O.; Dara, V.; Tapily, A.; Campino, S.; Sepulveda, N.; Corran, P.; Rockett, K.; Clark, T. Fc gamma Receptor II a-H 131 R Polymorphism and Malaria Susceptibility in Sympatric Ethnic Groups, Fulani and Dogon of Mali. Scand. J. Immunol. 2014, 79, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Vafa, M.; Maiga, B.; Berzins, K.; Hayano, M.; Bereczky, S.; Dolo, A.; Daou, M.; Arama, C.; Kouriba, B.; Färnert, A. Associations between the IL-4-590 T allele and Plasmodium falciparum infection prevalence in asymptomatic Fulani of Mali. Microbes Infect. 2007, 9, 1043–1048. [Google Scholar] [CrossRef]
- Vafa, M.; Maiga, B.; Israelsson, E.; Dolo, A.; Doumbo, O.K.; Troye-Blomberg, M. Impact of the IL-4-590 C/T transition on the levels of Plasmodium falciparum specific IgE, IgG, IgG subclasses and total IgE in two sympatric ethnic groups living in Mali. Microbes Infect. 2009, 11, 779–784. [Google Scholar] [CrossRef]
- Dolo, A.; Modiano, D.; Maiga, B.; Daou, M.; Dolo, G.; Guindo, H.; Ba, M.; Maiga, H.; Coulibaly, D.; Perlman, H. Difference in susceptibility to malaria between two sympatric ethnic groups in Mali. Am. J. Trop. Med. Hyg. 2005, 72, 243–248. [Google Scholar] [CrossRef]
- Hedrick, P.W. Population genetics of malaria resistance in humans. Heredity 2011, 107, 283–304. [Google Scholar] [CrossRef]
- McCall, M.B.; Hopman, J.; Daou, M.; Maiga, B.; Dara, V.; Ploemen, I.; Nganou-Makamdop, K.; Niangaly, A.; Tolo, Y.; Arama, C. Early interferon-γ response against Plasmodium falciparum correlates with interethnic differences in susceptibility to parasitemia between sympatric Fulani and Dogon in Mali. J. Infect. Dis. 2010, 201, 142–152. [Google Scholar] [CrossRef]
- Laroque, A.; Min-Oo, G.; Tam, M.; Radovanovic, I.; Stevenson, M.M.; Gros, P. Genetic control of susceptibility to infection with Plasmodium chabaudi chabaudi AS in inbred mouse strains. Genes Immun. 2012, 13, 155–163. [Google Scholar] [CrossRef]
- Stevenson, M.; Lyanga, J.; Skamene, E. Murine malaria: Genetic control of resistance to Plasmodium chabaudi. Infect. Immun. 1982, 38, 80–88. [Google Scholar] [CrossRef]
- Bopp, S.E.; Ramachandran, V.; Henson, K.; Luzader, A.; Lindstrom, M.; Spooner, M.; Steffy, B.M.; Suzuki, O.; Janse, C.; Waters, A.P. Genome wide analysis of inbred mouse lines identifies a locus containing ppar-γ as contributing to enhanced malaria survival. PLoS ONE 2010, 5, e10903. [Google Scholar] [CrossRef]
- Burt, R.A.; Marshall, V.M.; Wagglen, J.; Rodda, F.R.; Senyschen, D.; Baldwin, T.M.; Buckingham, L.A.; Foote, S.J. Mice that are congenic for the char2 locus are susceptible to malaria. Infect. Immun. 2002, 70, 4750–4753. [Google Scholar] [CrossRef] [PubMed]
- Guénet, J.-L.; Benavides, F.; Panthier, J.-J.; Montagutelli, X. The different categories of genetically standardized populations of laboratory mice. In Genetics of the Mouse; Springer: Berlin/Heidelberg, Germany, 2015; pp. 319–359. [Google Scholar]
- Yang, X.; HayGlass, K.T.; Brunham, R.C. Genetically determined differences in IL-10 and IFN-gamma responses correlate with clearance of Chlamydia trachomatis mouse pneumonitis infection. J. Immunol. 1996, 156, 4338–4344. [Google Scholar] [PubMed]
- Cui, S.; Chesson, C.; Hope, R. Genetic variation within and between strains of outbred Swiss mice. Lab. Anim. 1993, 27, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Carothers, A.D.; Rudan, I.; Kolcic, I.; Polasek, O.; Hayward, C.; Wright, A.F.; Campbell, H.; Teague, P.; Hastie, N.D.; Weber, J.L. Estimating human inbreeding coefficients: Comparison of genealogical and marker heterozygosity approaches. Ann. Hum. Genet. 2006, 70 Pt 5, 666–676. [Google Scholar] [CrossRef]
- Lemes, R.B.; Nunes, K.; Carnavalli, J.E.P.; Kimura, L.; Mingroni-Netto, R.C.; Meyer, D.; Otto, P.A. Inbreeding estimates in human populations: Applying new approaches to an admixed Brazilian isolate. PLoS ONE 2018, 13, e0196360. [Google Scholar] [CrossRef]
- Abolins, S.; King, E.C.; Lazarou, L.; Weldon, L.; Hughes, L.; Drescher, P.; Raynes, J.G.; Hafalla, J.C.R.; Viney, M.E.; Riley, E.M. The comparative immunology of wild and laboratory mice, Mus musculus domesticus. Nat. Commun. 2017, 8, 14811. [Google Scholar] [CrossRef]
- Aylor, D.L.; Valdar, W.; Foulds-Mathes, W.; Buus, R.J.; Verdugo, R.A.; Baric, R.S.; Ferris, M.T.; Frelinger, J.A.; Heise, M.; Frieman, M.B.; et al. Genetic analysis of complex traits in the emerging Collaborative Cross. Genome Res. 2011, 21, 1213–1222. [Google Scholar] [CrossRef]
- Chesler, E.J. Out of the bottleneck: The Diversity Outcross and Collaborative Cross mouse populations in behavioral genetics research. Mamm. Genome 2014, 25, 3–11. [Google Scholar] [CrossRef]
- Collaborative Cross Consortium. The genome architecture of the Collaborative Cross mouse genetic reference population. Genetics 2012, 190, 389–401. [Google Scholar] [CrossRef]
- Threadgill, D.W.; Miller, D.R.; Churchill, G.A.; de Villena, F.P. The collaborative cross: A recombinant inbred mouse population for the systems genetic era. ILAR J. 2011, 52, 24–31. [Google Scholar] [CrossRef]
- Chesler, E.J.; Gatti, D.M.; Morgan, A.P.; Strobel, M.; Trepanier, L.; Oberbeck, D.; McWeeney, S.; Hitzemann, R.; Ferris, M.; McMullan, R.; et al. Diversity Outbred Mice at 21: Maintaining Allelic Variation in the Face of Selection. G3 Genes Genomes Genet. 2016, 6, 3893–3902. [Google Scholar] [CrossRef]
- Churchill, G.A.; Gatti, D.M.; Munger, S.C.; Svenson, K.L. The Diversity Outbred mouse population. Mamm. Genome 2012, 23, 713–718. [Google Scholar] [CrossRef]
- Bar, J.; Leung, J.M.; Hansen, C.; Loke, P.; Hall, A.R.; Conour, L.; Graham, A.L. Strong effects of lab-to-field environmental transitions on the bacterial intestinal microbiota of Mus musculus are modulated by Trichuris muris infection. FEMS Microbiol. Ecol. 2020, 96, fiaa167. [Google Scholar] [CrossRef]
- Lin, J.D.; Devlin, J.C.; Yeung, F.; McCauley, C.; Leung, J.M.; Chen, Y.H.; Cronkite, A.; Hansen, C.; Drake-Dunn, C.; Ruggles, K.V.; et al. Rewilding Nod2 and Atg16l1 Mutant Mice Uncovers Genetic and Environmental Contributions to Microbial Responses and Immune Cell Composition. Cell Host Microbe 2020, 27, 830–840.e4. [Google Scholar] [CrossRef]
- Yilmaz, B.; Portugal, S.; Tran, T.M.; Gozzelino, R.; Ramos, S.; Gomes, J.; Regalado, A.; Cowan, P.J.; d’Apice, A.J.; Chong, A.S.; et al. Gut microbiota elicits a protective immune response against malaria transmission. Cell 2014, 159, 1277–1289. [Google Scholar] [CrossRef]
- Yooseph, S.; Kirkness, E.F.; Tran, T.M.; Harkins, D.M.; Jones, M.B.; Torralba, M.G.; O’Connell, E.; Nutman, T.B.; Doumbo, S.; Doumbo, O.K.; et al. Stool microbiota composition is associated with the prospective risk of Plasmodium falciparum infection. BMC Genom. 2015, 16, 631. [Google Scholar] [CrossRef]
- Mandal, R.K.; Denny, J.E.; Namazzi, R.; Opoka, R.O.; Datta, D.; John, C.C.; Schmidt, N.W. Dynamic modulation of spleen germinal center reactions by gut bacteria during Plasmodium infection. Cell Rep. 2021, 35, 109094. [Google Scholar] [CrossRef]
- Mandal, R.K.; Crane, R.J.; Berkley, J.A.; Gumbi, W.; Wambua, J.; Ngoi, J.M.; Ndungu, F.M.; Schmidt, N.W. Longitudinal Analysis of Infant Stool Bacteria Communities before and after Acute Febrile Malaria and Artemether-Lumefantrine Treatment. J. Infect. Dis. 2019, 220, 687–698. [Google Scholar] [CrossRef]
- Stough, J.M.; Dearth, S.P.; Denny, J.E.; LeCleir, G.R.; Schmidt, N.W.; Campagna, S.R.; Wilhelm, S.W. Functional Characteristics of the Gut Microbiome in C57BL/6 Mice Differentially Susceptible to Plasmodium yoelii. Front. Microbiol. 2016, 7, 1520. [Google Scholar] [CrossRef]
- Waide, M.L.; Polidoro, R.; Powell, W.L.; Denny, J.E.; Kos, J.; Tieri, D.A.; Watson, C.T.; Schmidt, N.W. Gut Microbiota Composition Modulates the Magnitude and Quality of Germinal Centers during Plasmodium Infections. Cell Rep. 2020, 33, 108503. [Google Scholar] [CrossRef]
- Yeung, F.; Chen, Y.H.; Lin, J.D.; Leung, J.M.; McCauley, C.; Devlin, J.C.; Hansen, C.; Cronkite, A.; Stephens, Z.; Drake-Dunn, C.; et al. Altered Immunity of Laboratory Mice in the Natural Environment Is Associated with Fungal Colonization. Cell Host Microbe 2020, 27, 809–822.e6. [Google Scholar] [CrossRef]
- Beura, L.K.; Hamilton, S.E.; Bi, K.; Schenkel, J.M.; Odumade, O.A.; Casey, K.A.; Thompson, E.A.; Fraser, K.A.; Rosato, P.C.; Filali-Mouhim, A. Normalizing the environment recapitulates adult human immune traits in laboratory mice. Nature 2016, 532, 512–516. [Google Scholar] [CrossRef]
- Nahrendorf, W.; Scholzen, A.; Bijker, E.M.; Teirlinck, A.C.; Bastiaens, G.J.; Schats, R.; Hermsen, C.C.; Visser, L.G.; Langhorne, J.; Sauerwein, R.W. Memory B-cell and antibody responses induced by Plasmodium falciparum sporozoite immunization. J. Infect. Dis. 2014, 210, 1981–1990. [Google Scholar] [CrossRef]
- Coomes, S.M.; Pelly, V.S.; Kannan, Y.; Okoye, I.S.; Czieso, S.; Entwistle, L.J.; Perez-Lloret, J.; Nikolov, N.; Potocnik, A.J.; Biro, J.; et al. IFNgamma and IL-12 Restrict Th2 Responses during Helminth/Plasmodium Co-Infection and Promote IFNgamma from Th2 Cells. PLoS Pathog. 2015, 11, e1004994. [Google Scholar] [CrossRef]
- Schleicher, T.R.; Yang, J.; Freudzon, M.; Rembisz, A.; Craft, S.; Hamilton, M.; Graham, M.; Mlambo, G.; Tripathi, A.K.; Li, Y. A mosquito salivary gland protein partially inhibits Plasmodium sporozoite cell traversal and transmission. Nat. Commun. 2018, 9, 2908. [Google Scholar] [CrossRef]
- Isawa, H.; Yuda, M.; Orito, Y.; Chinzei, Y. A mosquito salivary protein inhibits activation of the plasma contact system by binding to factor XII and high molecular weight kininogen. J. Biol. Chem. 2002, 277, 27651–27658. [Google Scholar] [CrossRef]
- Depinay, N.; Hacini, F.; Beghdadi, W.; Peronet, R.; Mécheri, S. Mast cell-dependent down-regulation of antigen-specific immune responses by mosquito bites. J. Immunol. 2006, 176, 4141–4146. [Google Scholar] [CrossRef]
- Donovan, M.J.; Messmore, A.S.; Scrafford, D.A.; Sacks, D.L.; Kamhawi, S.; McDowell, M.A. Uninfected mosquito bites confer protection against infection with malaria parasites. Infect. Immun. 2007, 75, 2523–2530. [Google Scholar] [CrossRef]
- Bediako, Y.; Adams, R.; Reid, A.J.; Valletta, J.J.; Ndungu, F.M.; Sodenkamp, J.; Mwacharo, J.; Ngoi, J.M.; Kimani, D.; Kai, O.; et al. Repeated clinical malaria episodes are associated with modification of the immune system in children. BMC Med. 2019, 17, 60. [Google Scholar] [CrossRef]
- Coggeshall, L.; Kumm, H. Demonstration of passive immunity in experimental monkey malaria. J. Exp. Med. 1937, 66, 177–190. [Google Scholar] [CrossRef][Green Version]
- Cohen, S.; McGregor, I.; Carrington, S.J.N. Gamma-globulin and acquired immunity to human malaria. Nature 1961, 192, 733–737. [Google Scholar] [CrossRef] [PubMed]
- Sabchareon, A.; Burnouf, T.; Ouattara, D.; Attanath, P.; Bouharoun-Tayoun, H.; Chantavanich, P.; Foucault, C.; Chongsuphajaisiddhi, T.; Druilhe, P. Parasitologic and clinical human response to immunoglobulin administration in falciparum malaria. Am. J. Trop. Med. Hyg. 1991, 45, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Osier, F.H.; Feng, G.; Boyle, M.J.; Langer, C.; Zhou, J.; Richards, J.S.; McCallum, F.J.; Reiling, L.; Jaworowski, A.; Anders, R.F.; et al. Opsonic phagocytosis of Plasmodium falciparum merozoites: Mechanism in human immunity and a correlate of protection against malaria. BMC Med. 2014, 12, 108. [Google Scholar] [CrossRef] [PubMed]
- Turrini, F.; Ginsburg, H.; Bussolino, F.; Pescarmona, G.P.; Serra, M.V.; Arese, P. Phagocytosis of Plasmodium falciparum-infected human red blood cells by human monocytes: Involvement of immune and nonimmune determinants and dependence on parasite developmental stage. Blood 1992, 80, 801–808. [Google Scholar] [CrossRef]
- Yoneto, T.; Waki, S.; Takai, T.; Tagawa, Y.; Iwakura, Y.; Mizuguchi, J.; Nariuchi, H.; Yoshimoto, T. A critical role of Fc receptor-mediated antibody-dependent phagocytosis in the host resistance to blood-stage Plasmodium berghei XAT infection. J. Immunol. 2001, 166, 6236–6241. [Google Scholar] [CrossRef]
- Akter, J.; Khoury, D.S.; Aogo, R.; Lansink, L.I.; SheelaNair, A.; Thomas, B.S.; Laohamonthonkul, P.; Pernold, C.P.; Dixon, M.W.; Soon, M.S. Plasmodium-specific antibodies block in vivo parasite growth without clearing infected red blood cells. PLoS Pathog. 2019, 15, e1007599. [Google Scholar] [CrossRef]
- McCallum, F.J.; Persson, K.E.; Mugyenyi, C.K.; Fowkes, F.J.; Simpson, J.A.; Richards, J.S.; Williams, T.N.; Marsh, K.; Beeson, J.G. Acquisition of growth-inhibitory antibodies against blood-stage Plasmodium falciparum. PLoS ONE 2008, 3, e3571. [Google Scholar] [CrossRef]
- Osier, F.H.; Fegan, G.; Polley, S.D.; Murungi, L.; Verra, F.; Tetteh, K.K.; Lowe, B.; Mwangi, T.; Bull, P.C.; Thomas, A.W.; et al. Breadth and magnitude of antibody responses to multiple Plasmodium falciparum merozoite antigens are associated with protection from clinical malaria. Infect. Immun. 2008, 76, 2240–2248. [Google Scholar] [CrossRef]
- Rono, J.; Osier, F.H.; Olsson, D.; Montgomery, S.; Mhoja, L.; Rooth, I.; Marsh, K.; Färnert, A. Breadth of anti-merozoite antibody responses is associated with the genetic diversity of asymptomatic Plasmodium falciparum infections and protection against clinical malaria. Clin. Infect. Dis. 2013, 57, 1409–1416. [Google Scholar] [CrossRef]
- Chan, J.-A.; Howell, K.B.; Reiling, L.; Ataide, R.; Mackintosh, C.L.; Fowkes, F.J.; Petter, M.; Chesson, J.M.; Langer, C.; Warimwe, G.M. Targets of antibodies against Plasmodium falciparum-infected erythrocytes in malaria immunity. J. Clin. Investig. 2012, 122, 3227–3238. [Google Scholar] [CrossRef]
- Nogaro, S.I.; Hafalla, J.C.; Walther, B.; Remarque, E.J.; Tetteh, K.K.; Conway, D.J.; Riley, E.M.; Walther, M. The breadth, but not the magnitude, of circulating memory B cell responses to P. falciparum increases with age/exposure in an area of low transmission. PLoS ONE 2011, 6, e25582. [Google Scholar] [CrossRef]
- Rotman, H.L.; Daly, T.M.; Clynes, R.; Long, C.A. Fc receptors are not required for antibody-mediated protection against lethal malaria challenge in a mouse model. J. Immunol. 1998, 161, 1908–1912. [Google Scholar]
- Grun, J.; Weidanz, W. Antibody-independent immunity to reinfection malaria in B-cell-deficient mice. Infect. Immun. 1983, 41, 1197–1204. [Google Scholar] [CrossRef]
- Kubagawa, H.; Honjo, K.; Ohkura, N.; Sakaguchi, S.; Radbruch, A.; Melchers, F.; Jani, P.K. Functional Roles of the IgM Fc Receptor in the Immune System. Front. Immunol. 2019, 10, 945. [Google Scholar] [CrossRef]
- Kurtovic, L.; Behet, M.C.; Feng, G.; Reiling, L.; Chelimo, K.; Dent, A.E.; Mueller, I.; Kazura, J.W.; Sauerwein, R.W.; Fowkes, F.J.I.; et al. Human antibodies activate complement against Plasmodium falciparum sporozoites, and are associated with protection against malaria in children. BMC Med. 2018, 16, 61. [Google Scholar] [CrossRef]
- Boyle, M.J.; Reiling, L.; Feng, G.; Langer, C.; Osier, F.H.; Aspeling-Jones, H.; Cheng, Y.S.; Stubbs, J.; Tetteh, K.K.; Conway, D.J.; et al. Human antibodies fix complement to inhibit Plasmodium falciparum invasion of erythrocytes and are associated with protection against malaria. Immunity 2015, 42, 580–590. [Google Scholar] [CrossRef]
- Taylor, P.R.; Seixas, E.; Walport, M.J.; Langhorne, J.; Botto, M. Complement contributes to protective immunity against reinfection by Plasmodium chabaudi chabaudi parasites. Infect. Immun. 2001, 69, 3853–3859. [Google Scholar] [CrossRef]
- Grun, J.L.; Weidanz, W.P. Immunity to Plasmodium chabaudi adami in the B-cell-deficient mouse. Nature 1981, 290, 143–145. [Google Scholar] [CrossRef]
- Weidanz, W.P.; LaFleur, G.; Brown, A.; Burns, J.M.; Gramaglia, I.; van der Heyde, H.C. γδ T cells but not NK cells are essential for cell-mediated immunity against Plasmodium chabaudi malaria. Infect. Immun. 2010, 78, 4331–4340. [Google Scholar] [CrossRef]
- Kinyanjui, S.M.; Bejon, P.; Osier, F.H.; Bull, P.C.; Marsh, K. What you see is not what you get: Implications of the brevity of antibody responses to malaria antigens and transmission heterogeneity in longitudinal studies of malaria immunity. Malar. J. 2009, 8, 242. [Google Scholar] [CrossRef]
- Kinyanjui, S.M.; Conway, D.J.; Lanar, D.E.; Marsh, K. IgG antibody responses to Plasmodium falciparum merozoite antigens in Kenyan children have a short half-life. Malar. J. 2007, 6, 82. [Google Scholar] [CrossRef] [PubMed]
- Portugal, S.; Pierce, S.K.; Crompton, P.D. Young lives lost as B cells falter: What we are learning about antibody responses in malaria. J. Immunol. 2013, 190, 3039–3046. [Google Scholar] [CrossRef] [PubMed]
- Olatunde, A.C.; Hale, J.S.; Lamb, T.J. Cytokine-skewed Tfh cells: Functional consequences for B cell help. Trends Immunol. 2021, 42, 536–550. [Google Scholar] [CrossRef] [PubMed]
- Elsner, R.A.; Shlomchik, M.J. Germinal Center and Extrafollicular B Cell Responses in Vaccination, Immunity, and Autoimmunity. Immunity 2020, 53, 1136–1150. [Google Scholar] [CrossRef]
- Inamine, A.; Takahashi, Y.; Baba, N.; Miyake, K.; Tokuhisa, T.; Takemori, T.; Abe, R. Two waves of memory B-cell generation in the primary immune response. Int. Immunol. 2005, 17, 581–589. [Google Scholar] [CrossRef]
- Takemori, T.; Kaji, T.; Takahashi, Y.; Shimoda, M.; Rajewsky, K. Generation of memory B cells inside and outside germinal centers. Eur. J. Immunol. 2014, 44, 1258–1264. [Google Scholar] [CrossRef]
- Toyama, H.; Okada, S.; Hatano, M.; Takahashi, Y.; Takeda, N.; Ichii, H.; Takemori, T.; Kuroda, Y.; Tokuhisa, T. Memory B cells without somatic hypermutation are generated from Bcl6-deficient B cells. Immunity 2002, 17, 329–339. [Google Scholar] [CrossRef]
- Perez-Mazliah, D.; Ng, D.H.; Freitas do Rosario, A.P.; McLaughlin, S.; Mastelic-Gavillet, B.; Sodenkamp, J.; Kushinga, G.; Langhorne, J. Disruption of IL-21 signaling affects T cell-B cell interactions and abrogates protective humoral immunity to malaria. PLoS Pathog. 2015, 11, e1004715. [Google Scholar] [CrossRef]
- Sebina, I.; Fogg, L.G.; James, K.R.; Soon, M.S.F.; Akter, J.; Thomas, B.S.; Hill, G.R.; Engwerda, C.R.; Haque, A. IL-6 promotes CD4(+) T-cell and B-cell activation during Plasmodium infection. Parasite Immunol. 2017, 39, e12455. [Google Scholar] [CrossRef]
- Sebina, I.; James, K.R.; Soon, M.S.; Fogg, L.G.; Best, S.E.; Labastida Rivera, F.; Montes de Oca, M.; Amante, F.H.; Thomas, B.S.; Beattie, L.; et al. IFNAR1-Signalling Obstructs ICOS-Mediated Humoral Immunity during Non-Lethal Blood-Stage Plasmodium Infection. PLoS Pathog. 2016, 12, e1005999. [Google Scholar] [CrossRef]
- Hopp, C.S.; Sekar, P.; Diouf, A.; Miura, K.; Boswell, K.; Skinner, J.; Tipton, C.M.; Peterson, M.E.; Chambers, M.J.; Andrews, S.; et al. Plasmodium falciparum-specific IgM B cells dominate in children, expand with malaria, and produce functional IgM. J. Exp. Med. 2021, 218, e20200901. [Google Scholar] [CrossRef]
- Thouvenel, C.D.; Fontana, M.F.; Netland, J.; Krishnamurty, A.T.; Takehara, K.K.; Chen, Y.; Singh, S.; Miura, K.; Keitany, G.J.; Lynch, E.M.; et al. Multimeric antibodies from antigen-specific human IgM+ memory B cells restrict Plasmodium parasites. J. Exp. Med. 2021, 218, e20200942. [Google Scholar] [CrossRef]
- Pietrzak, H.M.; Ioannidis, L.J.; Hansen, D.S. IgM(+) memory B cells induced in response to Plasmodium berghei adopt a germinal centre B cell phenotype during secondary infection. Parasitology 2020, 147, 994–998. [Google Scholar] [CrossRef]
- Wykes, M.N.; Zhou, Y.-H.; Liu, X.Q.; Good, M.F. Plasmodium yoelii can ablate vaccine-induced long-term protection in mice. J. Immunol. 2005, 175, 2510–2516. [Google Scholar] [CrossRef]
- Obeng-Adjei, N.; Portugal, S.; Tran, T.M.; Yazew, T.B.; Skinner, J.; Li, S.; Jain, A.; Felgner, P.L.; Doumbo, O.K.; Kayentao, K.; et al. Circulating Th1-cell-type Tfh cells that exhibit impaired B cell help are preferentially activated during acute malaria in children. Cell Rep. 2015, 13, 425–439. [Google Scholar] [CrossRef]
- Arroyo, E.N.; Pepper, M. B cells are sufficient to prime the dominant CD4+ Tfh response to Plasmodium infection. J. Exp. Med. 2020, 217, e20190849. [Google Scholar] [CrossRef]
- Wikenheiser, D.J.; Ghosh, D.; Kennedy, B.; Stumhofer, J.S. The Costimulatory Molecule ICOS Regulates Host Th1 and Follicular Th Cell Differentiation in Response to Plasmodium chabaudi chabaudi AS Infection. J. Immunol. 2016, 196, 778–791. [Google Scholar] [CrossRef]
- O’Neal, K.A.; Latham, L.E.; Ntirandekura, E.; Foscue, C.L.; Stumhofer, J.S. ICOS Expression Is Required for Maintenance but Not the Formation of Germinal Centers in the Spleen in Response to Plasmodium yoelii Infection. Infect. Immun. 2022, 90, e0046821. [Google Scholar] [CrossRef]
- Butler, N.S.; Moebius, J.; Pewe, L.L.; Traore, B.; Doumbo, O.K.; Tygrett, L.T.; Waldschmidt, T.J.; Crompton, P.D.; Harty, J.T. Therapeutic blockade of PD-L1 and LAG-3 rapidly clears established blood-stage Plasmodium infection. Nat. Immunol. 2012, 13, 188. [Google Scholar] [CrossRef]
- Kurup, S.P.; Obeng-Adjei, N.; Anthony, S.M.; Traore, B.; Doumbo, O.K.; Butler, N.S.; Crompton, P.D.; Harty, J.T. Regulatory T cells impede acute and long-term immunity to blood-stage malaria through CTLA-4. Nat. Med. 2017, 23, 1220–1225. [Google Scholar] [CrossRef]
- Obeng-Adjei, N.; Portugal, S.; Holla, P.; Li, S.; Sohn, H.; Ambegaonkar, A.; Skinner, J.; Bowyer, G.; Doumbo, O.K.; Traore, B.; et al. Malaria-induced interferon-gamma drives the expansion of Tbethi atypical memory B cells. PLoS Pathog. 2017, 13, e1006576. [Google Scholar] [CrossRef]
- Hopp, C.S.; Skinner, J.; Anzick, S.L.; Tipton, C.M.; Peterson, M.E.; Li, S.; Doumbo, S.; Kayentao, K.; Ongoiba, A.; Martens, C.; et al. Atypical B cells up-regulate costimulatory molecules during malaria and secrete antibodies with T follicular helper cell support. Sci. Immunol. 2022, 7, eabn1250. [Google Scholar] [CrossRef]
- Guthmiller, J.J.; Graham, A.C.; Zander, R.A.; Pope, R.L.; Butler, N.S. Cutting Edge: IL-10 Is Essential for the Generation of Germinal Center B Cell Responses and Anti-Plasmodium Humoral Immunity. J. Immunol. 2017, 198, 617–622. [Google Scholar] [CrossRef]
- Ryg-Cornejo, V.; Ioannidis, L.J.; Ly, A.; Chiu, C.Y.; Tellier, J.; Hill, D.L.; Preston, S.P.; Pellegrini, M.; Yu, D.; Nutt, S.L.; et al. Severe Malaria Infections Impair Germinal Center Responses by Inhibiting T Follicular Helper Cell Differentiation. Cell Rep. 2016, 14, 68–81. [Google Scholar] [CrossRef]
- Zander, R.A.; Obeng-Adjei, N.; Guthmiller, J.J.; Kulu, D.I.; Li, J.; Ongoiba, A.; Traore, B.; Crompton, P.D.; Butler, N.S. PD-1 Co-inhibitory and OX40 Co-stimulatory Crosstalk Regulates Helper T Cell Differentiation and Anti-Plasmodium Humoral Immunity. Cell Host Microbe 2015, 17, 628–641. [Google Scholar] [CrossRef]
- James, K.R.; Soon, M.S.F.; Sebina, I.; Fernandez-Ruiz, D.; Davey, G.; Liligeto, U.N.; Nair, A.S.; Fogg, L.G.; Edwards, C.L.; Best, S.E.; et al. IFN Regulatory Factor 3 Balances Th1 and T Follicular Helper Immunity during Nonlethal Blood-Stage Plasmodium Infection. J. Immunol. 2018, 200, 1443–1456. [Google Scholar] [CrossRef]
- Carpio, V.H.; Aussenac, F.; Puebla-Clark, L.; Wilson, K.D.; Villarino, A.V.; Dent, A.L.; Stephens, R. T Helper Plasticity Is Orchestrated by STAT3, Bcl6, and Blimp-1 Balancing Pathology and Protection in Malaria. iScience 2020, 23, 101310. [Google Scholar] [CrossRef]
- Cope, A.; Le Friec, G.; Cardone, J.; Kemper, C. The Th1 life cycle: Molecular control of IFN-γ to IL-10 switching. Trends Immunol. 2011, 32, 278–286. [Google Scholar] [CrossRef]
- Surette, F.A.; Guthmiller, J.J.; Li, L.; Sturtz, A.J.; Vijay, R.; Pope, R.L.; McClellan, B.L.; Pack, A.D.; Zander, R.A.; Shao, P.; et al. Extrafollicular CD4 T cell-derived IL-10 functions rapidly and transiently to support anti-Plasmodium humoral immunity. PLoS Pathog. 2021, 17, e1009288. [Google Scholar] [CrossRef]
- Wikenheiser, D.J.; Brown, S.L.; Lee, J.; Stumhofer, J.S. NK1.1 Expression Defines a Population of CD4(+) Effector T Cells Displaying Th1 and Tfh Cell Properties That Support Early Antibody Production During Plasmodium yoelii Infection. Front. Immunol. 2018, 9, 2277. [Google Scholar] [CrossRef]
- Troye-Blomberg, M.; Arama, C.; Quin, J.; Bujila, I.; Ostlund Farrants, A.K. What will studies of Fulani individuals naturally exposed to malaria teach us about protective immunity to malaria? Scand. J. Immunol. 2020, 92, e12932. [Google Scholar] [CrossRef] [PubMed]
- Walther, M.; Woodruff, J.; Edele, F.; Jeffries, D.; Tongren, J.E.; King, E.; Andrews, L.; Bejon, P.; Gilbert, S.C.; De Souza, J.B.; et al. Innate immune responses to human malaria: Heterogeneous cytokine responses to blood-stage Plasmodium falciparum correlate with parasitological and clinical outcomes. J. Immunol. 2006, 177, 5736–5745. [Google Scholar] [CrossRef] [PubMed]
- Franklin, B.S.; Parroche, P.; Ataide, M.A.; Lauw, F.; Ropert, C.; de Oliveira, R.B.; Pereira, D.; Tada, M.S.; Nogueira, P.; da Silva, L.H.; et al. Malaria primes the innate immune response due to interferon-gamma induced enhancement of toll-like receptor expression and function. Proc. Natl. Acad. Sci. USA 2009, 106, 5789–5794. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Ding, Y.; Zhou, T.; Fu, X.; Xu, W. Plasmodium yoelii blood-stage primes macrophage-mediated innate immune response through modulation of toll-like receptor signalling. Malar. J. 2012, 11, 104. [Google Scholar] [CrossRef]
- Sun, T.; Chakrabarti, C. Schizonts, merozoites, and phagocytosis in falciparum malaria. Ann. Clin. Lab. Sci. 1985, 15, 465–469. [Google Scholar]
- Wickramasinghe, S.N.; Phillips, R.E.; Looareesuwan, S.; Warrell, D.A.; Hughes, M. The bone marrow in human cerebral malaria: Parasite sequestration within sinusoids. Br. J. Haematol. 1987, 66, 295–306. [Google Scholar] [CrossRef]
- Arora, G.; Hart, G.T.; Manzella-Lapeira, J.; Doritchamou, J.Y.; Narum, D.L.; Thomas, L.M.; Brzostowski, J.; Rajagopalan, S.; Doumbo, O.K.; Traore, B.; et al. NK cells inhibit Plasmodium falciparum growth in red blood cells via antibody-dependent cellular cytotoxicity. eLife 2018, 7, e36806. [Google Scholar] [CrossRef]
- Burrack, K.S.; Hart, G.T.; Hamilton, S.E. Contributions of natural killer cells to the immune response against Plasmodium. Malar. J. 2019, 18, 321. [Google Scholar] [CrossRef]
- Hart, G.T.; Tran, T.M.; Theorell, J.; Schlums, H.; Arora, G.; Rajagopalan, S.; Sangala, A.D.J.; Welsh, K.J.; Traore, B.; Pierce, S.K.; et al. Adaptive NK cells in people exposed to Plasmodium falciparum correlate with protection from malaria. J. Exp. Med. 2019, 216, 1280–1290. [Google Scholar] [CrossRef]
- Sherratt, S.; Patel, A.; Baker, D.A.; Riley, E.M.; Goodier, M.R. Differential IL-18 Dependence of Canonical and Adaptive NK Cells for Antibody Dependent Responses to P. falciparum. Front. Immunol. 2020, 11, 533. [Google Scholar] [CrossRef]
- Jagannathan, P.; Lutwama, F.; Boyle, M.J.; Nankya, F.; Farrington, L.A.; McIntyre, T.I.; Bowen, K.; Naluwu, K.; Nalubega, M.; Musinguzi, K. Vδ2+ T cell response to malaria correlates with protection from infection but is attenuated with repeated exposure. Sci. Rep. 2017, 7, 11487. [Google Scholar] [CrossRef]
- Schofield, L.; Novakovic, S.; Gerold, P.; Schwarz, R.T.; McConville, M.J.; Tachado, S.D. Glycosylphosphatidylinositol toxin of Plasmodium up-regulates intercellular adhesion molecule-1, vascular cell adhesion molecule-1, and E-selectin expression in vascular endothelial cells and increases leukocyte and parasite cytoadherence via tyrosine kinase-dependent signal transduction. J. Immunol. 1996, 156, 1886–1896. [Google Scholar]
- Gowda, D.C.; Wu, X. Parasite Recognition and Signaling Mechanisms in Innate Immune Responses to Malaria. Front. Immunol. 2018, 9, 3006. [Google Scholar] [CrossRef]
- Schofield, L.; Hackett, F. Signal transduction in host cells by a glycosylphosphatidylinositol toxin of malaria parasites. J. Exp. Med. 1993, 177, 145–153. [Google Scholar] [CrossRef]
- Schofield, L.; Hewitt, M.C.; Evans, K.; Siomos, M.A.; Seeberger, P.H. Synthetic GPI as a candidate anti-toxic vaccine in a model of malaria. Nature 2002, 418, 785–789. [Google Scholar] [CrossRef]
- Vijaykumar, M.; Naik, R.S.; Gowda, D.C. Plasmodium falciparum glycosylphosphatidylinositol-induced TNF-alpha secretion by macrophages is mediated without membrane insertion or endocytosis. J. Biol. Chem. 2001, 276, 6909–6912. [Google Scholar] [CrossRef]
- Krishnegowda, G.; Hajjar, A.M.; Zhu, J.; Douglass, E.J.; Uematsu, S.; Akira, S.; Woods, A.S.; Gowda, D.C. Induction of proinflammatory responses in macrophages by the glycosylphosphatidylinositols of Plasmodium falciparum: Cell signaling receptors, glycosylphosphatidylinositol (GPI) structural requirement, and regulation of GPI activity. J. Biol. Chem. 2005, 280, 8606–8616. [Google Scholar] [CrossRef]
- Franklin, B.S.; Rodrigues, S.O.; Antonelli, L.R.; Oliveira, R.V.; Goncalves, A.M.; Sales-Junior, P.A.; Valente, E.P.; Alvarez-Leite, J.I.; Ropert, C.; Golenbock, D.T.; et al. MyD88-dependent activation of dendritic cells and CD4(+) T lymphocytes mediates symptoms, but is not required for the immunological control of parasites during rodent malaria. Microbes Infect. 2007, 9, 881–890. [Google Scholar] [CrossRef]
- Gowda, N.M.; Wu, X.; Gowda, D.C. TLR9 and MyD88 are crucial for the development of protective immunity to malaria. J. Immunol. 2012, 188, 5073–5085. [Google Scholar] [CrossRef]
- Coban, C.; Ishii, K.J.; Uematsu, S.; Arisue, N.; Sato, S.; Yamamoto, M.; Kawai, T.; Takeuchi, O.; Hisaeda, H.; Horii, T.; et al. Pathological role of Toll-like receptor signaling in cerebral malaria. Int. Immunol. 2007, 19, 67–79. [Google Scholar] [CrossRef]
- Pham, T.T.; Lamb, T.J.; Deroost, K.; Opdenakker, G.; Van den Steen, P.E. Hemozoin in Malarial Complications: More Questions Than Answers. Trends Parasitol. 2021, 37, 226–239. [Google Scholar] [CrossRef]
- Pichyangkul, S.; Yongvanitchit, K.; Kum-arb, U.; Hemmi, H.; Akira, S.; Krieg, A.M.; Heppner, D.G.; Stewart, V.A.; Hasegawa, H.; Looareesuwan, S.; et al. Malaria blood stage parasites activate human plasmacytoid dendritic cells and murine dendritic cells through a Toll-like receptor 9-dependent pathway. J. Immunol. 2004, 172, 4926–4933. [Google Scholar] [CrossRef]
- Lamphier, M.S.; Sirois, C.M.; Verma, A.; Golenbock, D.T.; Latz, E. TLR9 and the recognition of self and non-self nucleic acids. Ann. N. Y. Acad. Sci. 2006, 1082, 31–43. [Google Scholar] [CrossRef]
- Wu, X.; Gowda, N.M.; Kumar, S.; Gowda, D.C. Protein-DNA complex is the exclusive malaria parasite component that activates dendritic cells and triggers innate immune responses. J. Immunol. 2010, 184, 4338–4348. [Google Scholar] [CrossRef]
- Parroche, P.; Lauw, F.N.; Goutagny, N.; Latz, E.; Monks, B.G.; Visintin, A.; Halmen, K.A.; Lamphier, M.; Olivier, M.; Bartholomeu, D.C.; et al. Malaria hemozoin is immunologically inert but radically enhances innate responses by presenting malaria DNA to Toll-like receptor 9. Proc. Natl. Acad. Sci. USA 2007, 104, 1919–1924. [Google Scholar] [CrossRef]
- Millington, O.R.; Di Lorenzo, C.; Phillips, R.S.; Garside, P.; Brewer, J.M. Suppression of adaptive immunity to heterologous antigens during Plasmodium infection through hemozoin-induced failure of dendritic cell function. J. Biol. 2006, 5, 5. [Google Scholar] [CrossRef]
- Rivera-Correa, J.; Guthmiller, J.J.; Vijay, R.; Fernandez-Arias, C.; Pardo-Ruge, M.A.; Gonzalez, S.; Butler, N.S.; Rodriguez, A. Plasmodium DNA-mediated TLR9 activation of T-bet(+) B cells contributes to autoimmune anaemia during malaria. Nat. Commun. 2017, 8, 1282. [Google Scholar] [CrossRef]
- Jaschke, A.; Coulibaly, B.; Remarque, E.J.; Bujard, H.; Epp, C. Merozoite Surface Protein 1 from Plasmodium falciparum Is a Major Target of Opsonizing Antibodies in Individuals with Acquired Immunity against Malaria. Clin. Vaccine Immunol. 2017, 24, e00155-17. [Google Scholar] [CrossRef]
- Antonelli, L.R.; Leoratti, F.M.; Costa, P.A.; Rocha, B.C.; Diniz, S.Q.; Tada, M.S.; Pereira, D.B.; Teixeira-Carvalho, A.; Golenbock, D.T.; Goncalves, R.; et al. The CD14+CD16+ inflammatory monocyte subset displays increased mitochondrial activity and effector function during acute Plasmodium vivax malaria. PLoS Pathog. 2014, 10, e1004393. [Google Scholar] [CrossRef]
- McGilvray, I.D.; Serghides, L.; Kapus, A.; Rotstein, O.D.; Kain, K.C. Nonopsonic monocyte/macrophage phagocytosis of Plasmodium falciparum-parasitized erythrocytes: A role for CD36 in malarial clearance. Blood 2000, 96, 3231–3240. [Google Scholar] [CrossRef]
- Rodrigues, D.A.S.; Prestes, E.B.; Gama, A.M.S.; Silva, L.S.; Pinheiro, A.A.S.; Ribeiro, J.M.C.; Campos, R.M.P.; Pimentel-Coelho, P.M.; De Souza, H.S.; Dicko, A.; et al. CXCR4 and MIF are required for neutrophil extracellular trap release triggered by Plasmodium-infected erythrocytes. PLoS Pathog. 2020, 16, e1008230. [Google Scholar] [CrossRef] [PubMed]
- Korbel, D.S.; Newman, K.C.; Almeida, C.R.; Davis, D.M.; Riley, E.M. Heterogeneous human NK cell responses to Plasmodium falciparum-infected erythrocytes. J. Immunol. 2005, 175, 7466–7473. [Google Scholar] [CrossRef] [PubMed]
- Artavanis-Tsakonas, K.; Riley, E.M. Innate immune response to malaria: Rapid induction of IFN-gamma from human NK cells by live Plasmodium falciparum-infected erythrocytes. J. Immunol. 2002, 169, 2956–2963. [Google Scholar] [CrossRef] [PubMed]
- Newman, K.C.; Korbel, D.S.; Hafalla, J.C.; Riley, E.M. Cross-talk with myeloid accessory cells regulates human natural killer cell interferon-gamma responses to malaria. PLoS Pathog. 2006, 2, e118. [Google Scholar] [CrossRef] [PubMed]
- McCall, M.B.; Roestenberg, M.; Ploemen, I.; Teirlinck, A.; Hopman, J.; de Mast, Q.; Dolo, A.; Doumbo, O.K.; Luty, A.; van der Ven, A.J.; et al. Memory-like IFN-gamma response by NK cells following malaria infection reveals the crucial role of T cells in NK cell activation by P. falciparum. Eur. J. Immunol. 2010, 40, 3472–3477. [Google Scholar] [CrossRef]
- Stegmann, K.A.; De Souza, J.B.; Riley, E.M. IL-18-induced expression of high-affinity IL-2R on murine NK cells is essential for NK-cell IFN-γ production during murine Plasmodium yoelii infection. Eur. J. Immunol. 2015, 45, 3431–3440. [Google Scholar] [CrossRef]
- Jagannathan, P.; Kim, C.C.; Greenhouse, B.; Nankya, F.; Bowen, K.; Eccles-James, I.; Muhindo, M.K.; Arinaitwe, E.; Tappero, J.W.; Kamya, M.R. Loss and dysfunction of Vδ2+ γδ T cells are associated with clinical tolerance to malaria. Sci. Transl. Med. 2014, 6, 251ra117. [Google Scholar] [CrossRef]
- Farrington, L.A.; Jagannathan, P.; McIntyre, T.I.; Vance, H.M.; Bowen, K.; Boyle, M.J.; Nankya, F.; Wamala, S.; Auma, A.; Nalubega, M. Frequent malaria drives progressive Vδ2 T-cell loss, dysfunction, and CD16 up-regulation during early childhood. J. Infect. Dis. 2016, 213, 1483–1490. [Google Scholar] [CrossRef]
- Inoue, S.I.; Niikura, M.; Asahi, H.; Kawakami, Y.; Kobayashi, F. γδ T cells modulate humoral immunity against Plasmodium berghei infection. Immunology 2018, 155, 519–532. [Google Scholar] [CrossRef]
- Farrington, L.A.; Callaway, P.C.; Vance, H.M.; Baskevitch, K.; Lutz, E.; Warrier, L.; McIntyre, T.I.; Budker, R.; Jagannathan, P.; Nankya, F. Opsonized antigen activates Vδ2+ T cells via CD16/FCγRIIIa in individuals with chronic malaria exposure. PLoS Pathog. 2020, 16, e1008997. [Google Scholar] [CrossRef]
- Junqueira, C.; Polidoro, R.B.; Castro, G.; Absalon, S.; Liang, Z.; Sen Santara, S.; Crespo, A.; Pereira, D.B.; Gazzinelli, R.T.; Dvorin, J.D.; et al. Gammadelta T cells suppress Plasmodium falciparum blood-stage infection by direct killing and phagocytosis. Nat. Immunol. 2021, 22, 347–357. [Google Scholar] [CrossRef]
- Mohan, K.; Moulin, P.; Stevenson, M.M. Natural killer cell cytokine production, not cytotoxicity, contributes to resistance against blood-stage Plasmodium chabaudi AS infection. J. Immunol. 1997, 159, 4990–4998. [Google Scholar]
- Ing, R.; Stevenson, M.M. Dendritic cell and NK cell reciprocal cross talk promotes gamma interferon-dependent immunity to blood-stage Plasmodium chabaudi AS infection in mice. Infect. Immun. 2009, 77, 770–782. [Google Scholar] [CrossRef]
- Ng, S.S.; Souza-Fonseca-Guimaraes, F.; Rivera, F.L.; Amante, F.H.; Kumar, R.; Gao, Y.; Sheel, M.; Beattie, L.; Montes de Oca, M.; Guillerey, C.; et al. Rapid loss of group 1 innate lymphoid cells during blood stage Plasmodium infection. Clin. Transl. Immunol. 2018, 7, e1003. [Google Scholar] [CrossRef]
- Teirlinck, A.C.; McCall, M.B.; Roestenberg, M.; Scholzen, A.; Woestenenk, R.; de Mast, Q.; van der Ven, A.J.; Hermsen, C.C.; Luty, A.J.; Sauerwein, R.W. Longevity and composition of cellular immune responses following experimental Plasmodium falciparum malaria infection in humans. PLoS Pathog. 2011, 7, e1002389. [Google Scholar] [CrossRef]
- Walther, M.; Tongren, J.E.; Andrews, L.; Korbel, D.; King, E.; Fletcher, H.; Andersen, R.F.; Bejon, P.; Thompson, F.; Dunachie, S.J.; et al. Upregulation of TGF-beta, FOXP3, and CD4+CD25+ regulatory T cells correlates with more rapid parasite growth in human malaria infection. Immunity 2005, 23, 287–296. [Google Scholar] [CrossRef]
- Portugal, S.; Moebius, J.; Skinner, J.; Doumbo, S.; Doumtabe, D.; Kone, Y.; Dia, S.; Kanakabandi, K.; Sturdevant, D.E.; Virtaneva, K. Exposure-dependent control of malaria-induced inflammation in children. PLoS Pathog. 2014, 10, e1004079. [Google Scholar] [CrossRef]
- Portugal, S.; Tran, T.M.; Ongoiba, A.; Bathily, A.; Li, S.; Doumbo, S.; Skinner, J.; Doumtabe, D.; Kone, Y.; Sangala, J. Treatment of chronic asymptomatic Plasmodium falciparum infection does not increase the risk of clinical malaria upon reinfection. Clin. Infect. Dis. 2017, 64, 645–653. [Google Scholar] [CrossRef]
- Su, Z.; Stevenson, M.M. Central role of endogenous gamma interferon in protective immunity against blood-stage Plasmodium chabaudi AS infection. Infect. Immun. 2000, 68, 4399–4406. [Google Scholar] [CrossRef]
- King, T.; Lamb, T. Interferon-γ: The Jekyll and Hyde of malaria. PLoS Pathog. 2015, 11, e1005118. [Google Scholar] [CrossRef]
- Walther, M.; Jeffries, D.; Finney, O.C.; Njie, M.; Ebonyi, A.; Deininger, S.; Lawrence, E.; Ngwa-Amambua, A.; Jayasooriya, S.; Cheeseman, I.H.; et al. Distinct roles for FOXP3 and FOXP3 CD4 T cells in regulating cellular immunity to uncomplicated and severe Plasmodium falciparum malaria. PLoS Pathog. 2009, 5, e1000364. [Google Scholar] [CrossRef]
- Wenisch, C.; Parschalk, B.; Burgmann, H.; Looareesuwan, S.; Graninger, W. Decreased serum levels of TGF-beta in patients with acute Plasmodium falciparum malaria. J. Clin. Immunol. 1995, 15, 69–73. [Google Scholar] [CrossRef]
- Perkins, D.J.; Weinberg, J.B.; Kremsner, P.G. Reduced interleukin-12 and transforming growth factor-beta1 in severe childhood malaria: Relationship of cytokine balance with disease severity. J. Infect. Dis. 2000, 182, 988–992. [Google Scholar] [CrossRef]
- Chaiyaroj, S.C.; Rutta, A.S.; Muenthaisong, K.; Watkins, P.; Na Ubol, M.; Looareesuwan, S. Reduced levels of transforming growth factor-beta1, interleukin-12 and increased migration inhibitory factor are associated with severe malaria. Acta Trop. 2004, 89, 319–327. [Google Scholar] [CrossRef]
- Imai, T.; Shen, J.; Chou, B.; Duan, X.; Tu, L.; Tetsutani, K.; Moriya, C.; Ishida, H.; Hamano, S.; Shimokawa, C.; et al. Involvement of CD8+ T cells in protective immunity against murine blood-stage infection with Plasmodium yoelii 17XL strain. Eur. J. Immunol. 2010, 40, 1053–1061. [Google Scholar] [CrossRef]
- Imai, T.; Ishida, H.; Suzue, K.; Taniguchi, T.; Okada, H.; Shimokawa, C.; Hisaeda, H. Cytotoxic activities of CD8(+) T cells collaborate with macrophages to protect against blood-stage murine malaria. eLife 2015, 4, e04232. [Google Scholar] [CrossRef]
- Horne-Debets, J.M.; Faleiro, R.; Karunarathne, D.S.; Liu, X.Q.; Lineburg, K.E.; Poh, C.M.; Grotenbreg, G.M.; Hill, G.R.; MacDonald, K.P.; Good, M.F.; et al. PD-1 dependent exhaustion of CD8+ T cells drives chronic malaria. Cell Rep. 2013, 5, 1204–1213. [Google Scholar] [CrossRef]
- Horne-Debets, J.M.; Karunarathne, D.S.; Faleiro, R.J.; Poh, C.M.; Renia, L.; Wykes, M.N. Mice lacking Programmed cell death-1 show a role for CD8(+) T cells in long-term immunity against blood-stage malaria. Sci. Rep. 2016, 6, 26210. [Google Scholar] [CrossRef]
- Podoba, J.E.; Stevenson, M.M. CD4+ and CD8+ T lymphocytes both contribute to acquired immunity to blood-stage Plasmodium chabaudi AS. Infect. Immun. 1991, 59, 51–58. [Google Scholar] [CrossRef]
- Van der Heyde, H.C.; Manning, D.D.; Roopenian, D.C.; Weidanz, W.P. Resolution of blood-stage malarial infections in CD8+ cell-deficient beta 2-m0/0 mice. J. Immunol. 1993, 151, 3187–3191. [Google Scholar]
- Vinetz, J.M.; Kumar, S.; Good, M.F.; Fowlkes, B.J.; Berzofsky, J.A.; Miller, L.H. Adoptive transfer of CD8+ T cells from immune animals does not transfer immunity to blood stage Plasmodium yoelii malaria. J. Immunol. 1990, 144, 1069–1074. [Google Scholar] [PubMed]
- Kobayashi, F.; Morii, T.; Matsui, T.; Fujino, T.; Watanabe, Y.; Weidanz, W.P.; Tsuji, M. Production of interleukin 10 during malaria caused by lethal and nonlethal variants of Plasmodium yoelii yoelii. Parasitol. Res. 1996, 82, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Do Rosário, A.P.F.; Lamb, T.; Spence, P.; Stephens, R.; Lang, A.; Roers, A.; Muller, W.; O’Garra, A.; Langhorne, J. IL-27 promotes IL-10 production by effector Th1 CD4+ T cells: A critical mechanism for protection from severe immunopathology during malaria infection. J. Immunol. 2012, 188, 1178–1190. [Google Scholar] [CrossRef] [PubMed]
- Jagannathan, P.; Eccles-James, I.; Bowen, K.; Nankya, F.; Auma, A.; Wamala, S.; Ebusu, C.; Muhindo, M.K.; Arinaitwe, E.; Briggs, J.; et al. IFNgamma/IL-10 co-producing cells dominate the CD4 response to malaria in highly exposed children. PLoS Pathog. 2014, 10, e1003864. [Google Scholar] [CrossRef] [PubMed]
- Sukhbaatar, O.; Kimura, D.; Miyakoda, M.; Nakamae, S.; Kimura, K.; Hara, H.; Yoshida, H.; Inoue, S.-I.; Yui, K. Activation and IL-10 production of specific CD4+ T cells are regulated by IL-27 during chronic infection with Plasmodium chabaudi. Parasitol. Int. 2020, 74, 101994. [Google Scholar] [CrossRef] [PubMed]
- Villegas-Mendez, A.; Inkson, C.A.; Shaw, T.N.; Strangward, P.; Couper, K.N. Long-lived CD4+ IFN-γ+ T cells rather than short-lived CD4+ IFN-γ+ IL-10+ T cells initiate rapid IL-10 production to suppress anamnestic T cell responses during secondary malaria infection. J. Immunol. 2016, 197, 3152–3164. [Google Scholar] [CrossRef]
- Chen, Q.; Schlichtherle, M.; Wahlgren, M. Molecular aspects of severe malaria. Clin. Microbiol. Rev. 2000, 13, 439–450. [Google Scholar] [CrossRef]
- Dondorp, A.M.; Kager, P.A.; Vreeken, J.; White, N.J. Abnormal blood flow and red blood cell deformability in severe malaria. Parasitol. Today 2000, 16, 228–232. [Google Scholar] [CrossRef]
- Shelby, J.P.; White, J.; Ganesan, K.; Rathod, P.K.; Chiu, D.T. A microfluidic model for single-cell capillary obstruction by Plasmodium falciparum-infected erythrocytes. Proc. Natl. Acad. Sci. 2003, 100, 14618–14622. [Google Scholar] [CrossRef]
- Barber, B.E.; Russell, B.; Grigg, M.J.; Zhang, R.; William, T.; Amir, A.; Lau, Y.L.; Chatfield, M.D.; Dondorp, A.M.; Anstey, N.M. Reduced red blood cell deformability in Plasmodium knowlesi malaria. Blood Adv. 2018, 2, 433–443. [Google Scholar] [CrossRef]
- Mwakingwe, A.; Ting, L.M.; Hochman, S.; Chen, J.; Sinnis, P.; Kim, K. Noninvasive real-time monitoring of liver-stage development of bioluminescent Plasmodium parasites. J. Infect. Dis. 2009, 200, 1470–1478. [Google Scholar] [CrossRef]
- Franke-Fayard, B.; Waters, A.P.; Janse, C.J. Real-time in vivo imaging of transgenic bioluminescent blood stages of rodent malaria parasites in mice. Nat. Protoc. 2006, 1, 476–485. [Google Scholar] [CrossRef]
- Nallandhighal, S.; Park, G.S.; Ho, Y.-Y.; Opoka, R.O.; John, C.C.; Tran, T.M. Whole-Blood Transcriptional Signatures Composed of Erythropoietic and NRF2-Regulated Genes Differ Between Cerebral Malaria and Severe Malarial Anemia. J. Infect. Dis. 2019, 219, 154–164. [Google Scholar] [CrossRef]
- White, N.J. Anaemia and malaria. Malar. J. 2018, 17, 371. [Google Scholar] [CrossRef]
- Jakeman, G.; Saul, A.; Hogarth, W.; Collins, W. Anaemia of acute malaria infections in non-immune patients primarily results from destruction of uninfected erythrocytes. Parasitology 1999, 119, 127–133. [Google Scholar] [CrossRef]
- Abdalla, S.; Weatherall, D.; Wickramasinghe, S.; Hughes, M. The anaemia of P. falciparum malaria. Br. J. Haematol. 1980, 46, 171–183. [Google Scholar] [CrossRef]
- Aguilar, R.; Moraleda, C.; Achtman, A.H.; Mayor, A.; Quinto, L.; Cistero, P.; Nhabomba, A.; Macete, E.; Schofield, L.; Alonso, P.L.; et al. Severity of anaemia is associated with bone marrow haemozoin in children exposed to Plasmodium falciparum. Br. J. Haematol. 2014, 164, 877–887. [Google Scholar] [CrossRef]
- Chang, K.H.; Tam, M.; Stevenson, M.M. Inappropriately low reticulocytosis in severe malarial anemia correlates with suppression in the development of late erythroid precursors. Blood 2004, 103, 3727–3735. [Google Scholar] [CrossRef]
- Mohan, K.; Stevenson, M.M. Dyserythropoiesis and severe anaemia associated with malaria correlate with deficient interleukin-12 production. Br. J. Haematol. 1998, 103, 942–949. [Google Scholar] [CrossRef]
- McDevitt, M.A.; Xie, J.; Ganapathy-Kanniappan, S.; Griffith, J.; Liu, A.; McDonald, C.; Thuma, P.; Gordeuk, V.R.; Metz, C.N.; Mitchell, R.; et al. A critical role for the host mediator macrophage migration inhibitory factor in the pathogenesis of malarial anemia. J. Exp. Med. 2006, 203, 1185–1196. [Google Scholar] [CrossRef]
- Thawani, N.; Tam, M.; Stevenson, M.M. STAT6-mediated suppression of erythropoiesis in an experimental model of malarial anemia. Haematologica 2009, 94, 195–204. [Google Scholar] [CrossRef]
- O’Dea, K.P.; Pasvol, G. Optimal tumor necrosis factor induction by Plasmodium falciparum requires the highly localized release of parasite products. Infect. Immun. 2003, 71, 3155–3164. [Google Scholar] [CrossRef]
- Rudin, W.; Quesniaux, V.; Favre, N.; Bordmann, G. Malaria toxins from P. chabaudi chabaudi AS and P. berghei ANKA cause dyserythropoiesis in C57BL/6 mice. Parasitology 1997, 115 Pt 5, 467–474. [Google Scholar] [CrossRef]
- Bordmann, G.; Favre, N.; Rudin, W. Malaria toxins: Effects on murine spleen and bone marrow cell proliferation and cytokine production in vitro. Parasitology 1997, 115 Pt 5, 475–483. [Google Scholar] [CrossRef]
- Thawani, N.; Tam, M.; Bellemare, M.J.; Bohle, D.S.; Olivier, M.; de Souza, J.B.; Stevenson, M.M. Plasmodium products contribute to severe malarial anemia by inhibiting erythropoietin-induced proliferation of erythroid precursors. J. Infect. Dis. 2014, 209, 140–149. [Google Scholar] [CrossRef]
- Chang, K.H.; Stevenson, M.M. Effect of anemia and renal cytokine production on erythropoietin production during blood-stage malaria. Kidney Int. 2004, 65, 1640–1646. [Google Scholar] [CrossRef]
- Spottiswoode, N.; Armitage, A.E.; Williams, A.R.; Fyfe, A.J.; Biswas, S.; Hodgson, S.H.; Llewellyn, D.; Choudhary, P.; Draper, S.J.; Duffy, P.E.; et al. Role of Activins in Hepcidin Regulation during Malaria. Infect. Immun. 2017, 85, e00191-17. [Google Scholar] [CrossRef]
- Maretty, L.; Sharp, R.E.; Andersson, M.; Kurtzhals, J.A. Intravenous ferric carboxymaltose accelerates erythropoietic recovery from experimental malarial anemia. J. Infect. Dis. 2012, 205, 1173–1177. [Google Scholar] [CrossRef]
- Harris, J.V.; Bohr, T.M.; Stracener, C.; Landmesser, M.E.; Torres, V.; Mbugua, A.; Moratz, C.; Stoute, J.A. Sequential Plasmodium chabaudi and Plasmodium berghei infections provide a novel model of severe malarial anemia. Infect. Immun. 2012, 80, 2997–3007. [Google Scholar] [CrossRef]
- Mourao, L.C.; Cardoso-Oliveira, G.P.; Braga, E.M. Autoantibodies and Malaria: Where We Stand? Insights into Pathogenesis and Protection. Front. Cell. Infect. Microbiol. 2020, 10, 262. [Google Scholar] [CrossRef]
- Van den Akker, E.; Satchwell, T.J.; Williamson, R.C.; Toye, A.M. Band 3 multiprotein complexes in the red cell membrane; of mice and men. Blood Cells Mol. Dis. 2010, 45, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Wesseling, M.C.; Wagner-Britz, L.; Huppert, H.; Hanf, B.; Hertz, L.; Nguyen, D.B.; Bernhardt, I. Phosphatidylserine Exposure in Human Red Blood Cells Depending on Cell Age. Cell. Physiol. Biochem. 2016, 38, 1376–1390. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Zhang, C.; Zhao, Q.; Li, D. Spectrin: Structure, function and disease. Sci. China Life Sci. 2013, 56, 1076–1085. [Google Scholar] [CrossRef] [PubMed]
- Mourao, L.C.; Baptista, R.P.; de Almeida, Z.B.; Grynberg, P.; Pucci, M.M.; Castro-Gomes, T.; Fontes, C.J.F.; Rathore, S.; Sharma, Y.D.; da Silva-Pereira, R.A.; et al. Anti-band 3 and anti-spectrin antibodies are increased in Plasmodium vivax infection and are associated with anemia. Sci. Rep. 2018, 8, 8762. [Google Scholar] [CrossRef]
- Hogh, B.; Petersen, E.; Crandall, I.; Gottschau, A.; Sherman, I.W. Immune responses to band 3 neoantigens on Plasmodium falciparum-infected erythrocytes in subjects living in an area of intense malaria transmission are associated with low parasite density and high hematocrit value. Infect. Immun. 1994, 62, 4362–4366. [Google Scholar] [CrossRef]
- Fernandez-Arias, C.; Rivera-Correa, J.; Gallego-Delgado, J.; Rudlaff, R.; Fernandez, C.; Roussel, C.; Gotz, A.; Gonzalez, S.; Mohanty, A.; Mohanty, S.; et al. Anti-Self Phosphatidylserine Antibodies Recognize Uninfected Erythrocytes Promoting Malarial Anemia. Cell Host Microbe 2016, 19, 194–203. [Google Scholar] [CrossRef]
- Ternynck, T.; Falanga, P.B.; Unterkirscher, C.; Gregoire, J.; da Silva, L.P.; Avrameas, S. Induction of high levels of IgG autoantibodies in mice infected with Plasmodium chabaudi. Int. Immunol. 1991, 3, 29–37. [Google Scholar] [CrossRef]
- Romero, H.; Warburton, C.; Sanchez-Dardon, J.; Scorza, T. Osteoclasts Are Required for Hematopoietic Stem and Progenitor Cell Mobilization but Not for Stress Erythropoiesis in Plasmodium chabaudi adami Murine Malaria. Mediat. Inflamm. 2016, 2016, 3909614. [Google Scholar] [CrossRef]
- Yap, G.S.; Stevenson, M.M. Plasmodium chabaudi AS: Erythropoietic responses during infection in resistant and susceptible mice. Exp. Parasitol. 1992, 75, 340–352. [Google Scholar] [CrossRef]
- Asami, M.; Owhashi, M.; Abe, T.; Nawa, Y. A comparative study of the kinetic changes of hemopoietic stem cells in mice infected with lethal and non-lethal malaria. Int. J. Parasitol. 1992, 22, 43–47. [Google Scholar] [CrossRef]
- Bruneel, F. Human cerebral malaria: 2019 mini review. Rev. Neurol. 2019, 175, 445–450. [Google Scholar] [CrossRef]
- Sahu, P.K.; Satpathi, S.; Behera, P.K.; Mishra, S.K.; Mohanty, S.; Wassmer, S.C. Pathogenesis of cerebral malaria: New diagnostic tools, biomarkers, and therapeutic approaches. Front. Cell. Infect. Microbiol. 2015, 5, 75. [Google Scholar] [CrossRef]
- Ghazanfari, N.; Mueller, S.N.; Heath, W.R. Cerebral Malaria in Mouse and Man. Front. Immunol. 2018, 9, 2016. [Google Scholar] [CrossRef]
- Belnoue, E.; Potter, S.M.; Rosa, D.S.; Mauduit, M.; Gruner, A.C.; Kayibanda, M.; Mitchell, A.J.; Hunt, N.H.; Renia, L. Control of pathogenic CD8+ T cell migration to the brain by IFN-gamma during experimental cerebral malaria. Parasite Immunol. 2008, 30, 544–553. [Google Scholar] [CrossRef]
- Strangward, P.; Haley, M.J.; Shaw, T.N.; Schwartz, J.-M.; Greig, R.; Mironov, A.; de Souza, J.-B.; Cruickshank, S.M.; Craig, A.G.; Milner, D.A., Jr. A quantitative brain map of experimental cerebral malaria pathology. PLoS Pathog. 2017, 13, e1006267. [Google Scholar] [CrossRef]
- Howland, S.W.; Poh, C.M.; Gun, S.Y.; Claser, C.; Malleret, B.; Shastri, N.; Ginhoux, F.; Grotenbreg, G.M.; Renia, L. Brain microvessel cross-presentation is a hallmark of experimental cerebral malaria. EMBO Mol. Med. 2013, 5, 984–999. [Google Scholar] [CrossRef]
- Howland, S.W.; Poh, C.M.; Renia, L. Activated Brain Endothelial Cells Cross-Present Malaria Antigen. PLoS Pathog. 2015, 11, e1004963. [Google Scholar] [CrossRef]
- Newbold, C.; Craig, A.; Kyes, S.; Rowe, A.; Fernandez-Reyes, D.; Fagan, T. Cytoadherence, pathogenesis and the infected red cell surface in Plasmodium falciparum. Int. J. Parasitol. 1999, 29, 927–937. [Google Scholar] [CrossRef]
- Turner, G.D.; Morrison, H.; Jones, M.; Davis, T.M.; Looareesuwan, S.; Buley, I.D.; Gatter, K.C.; Newbold, C.I.; Pukritayakamee, S.; Nagachinta, B.; et al. An immunohistochemical study of the pathology of fatal malaria. Evidence for widespread endothelial activation and a potential role for intercellular adhesion molecule-1 in cerebral sequestration. Am. J. Pathol. 1994, 145, 1057–1069. [Google Scholar]
- Fonager, J.; Pasini, E.M.; Braks, J.A.; Klop, O.; Ramesar, J.; Remarque, E.J.; Vroegrijk, I.O.; van Duinen, S.G.; Thomas, A.W.; Khan, S.M.; et al. Reduced CD36-dependent tissue sequestration of Plasmodium-infected erythrocytes is detrimental to malaria parasite growth in vivo. J. Exp. Med. 2012, 209, 93–107. [Google Scholar] [CrossRef]
- Tuikue Ndam, N.; Moussiliou, A.; Lavstsen, T.; Kamaliddin, C.; Jensen, A.T.; Mama, A.; Tahar, R.; Wang, C.W.; Jespersen, J.S.; Alao, J.M. Parasites causing cerebral falciparum malaria bind multiple endothelial receptors and express EPCR and ICAM-1-binding PfEMP1. J. Infect. Dis. 2017, 215, 1918–1925. [Google Scholar] [CrossRef]
- Nacer, A.; Movila, A.; Baer, K.; Mikolajczak, S.A.; Kappe, S.H.; Frevert, U. Neuroimmunological blood brain barrier opening in experimental cerebral malaria. PLoS Pathog. 2012, 8, e1002982. [Google Scholar] [CrossRef]
- Frevert, U.; Nacer, A.; Cabrera, M.; Movila, A.; Leberl, M. Imaging Plasmodium immunobiology in the liver, brain, and lung. Parasitol. Int. 2014, 63, 171–186. [Google Scholar] [CrossRef]
- Rénia, L.; Howland, S.W. Targeting the olfactory bulb during experimental cerebral malaria. Trends Parasitol. 2014, 30, 375–376. [Google Scholar] [CrossRef]
- Franke-Fayard, B.; Djokovic, D.; Dooren, M.; Ramesar, J.; Waters, A.; Falade, M.; Kranendonk, M.; Martinelli, A.; Cravo, P.; Janse, C. Simple and sensitive antimalarial drug screening in vitro and in vivo using transgenic luciferase expressing Plasmodium berghei parasites. Int. J. Parasitol. 2008, 38, 1651–1662. [Google Scholar] [CrossRef]
- Ploemen, I.H.; Prudêncio, M.; Douradinha, B.G.; Ramesar, J.; Fonager, J.; van Gemert, G.-J.; Luty, A.J.; Hermsen, C.C.; Sauerwein, R.W.; Baptista, F.G. Visualisation and quantitative analysis of the rodent malaria liver stage by real time imaging. PLoS ONE 2009, 4, e7881. [Google Scholar] [CrossRef]
- Franke-Fayard, B.; Fonager, J.; Braks, A.; Khan, S.M.; Janse, C.J. Sequestration and tissue accumulation of human malaria parasites: Can we learn anything from rodent models of malaria? PLoS Pathog. 2010, 6, e1001032. [Google Scholar] [CrossRef]
- Claser, C.; Malleret, B.; Gun, S.Y.; Wong, A.Y.W.; Chang, Z.W.; Teo, P.; See, P.C.E.; Howland, S.W.; Ginhoux, F.; Rénia, L. CD8+ T cells and IFN-γ mediate the time-dependent accumulation of infected red blood cells in deep organs during experimental cerebral malaria. PLoS ONE 2011, 6, e18720. [Google Scholar] [CrossRef]
- Kwiatkowski, D.; Molyneux, M.E.; Stephens, S.; Curtis, N.; Klein, N.; Pointaire, P.; Smit, M.; Allan, R.; Brewster, D.R.; Grau, G.E.; et al. Anti-TNF therapy inhibits fever in cerebral malaria. Q. J. Med. 1993, 86, 91–98. [Google Scholar]
- Togbe, D.; de Sousa, P.L.; Fauconnier, M.; Boissay, V.; Fick, L.; Scheu, S.; Pfeffer, K.; Menard, R.; Grau, G.E.; Doan, B.T.; et al. Both functional LTbeta receptor and TNF receptor 2 are required for the development of experimental cerebral malaria. PLoS ONE 2008, 3, e2608. [Google Scholar] [CrossRef]
- Engwerda, C.R.; Mynott, T.L.; Sawhney, S.; De Souza, J.B.; Bickle, Q.D.; Kaye, P.M. Locally up-regulated lymphotoxin alpha, not systemic tumor necrosis factor alpha, is the principle mediator of murine cerebral malaria. J. Exp. Med. 2002, 195, 1371–1377. [Google Scholar] [CrossRef] [PubMed]
- Feintuch, C.M.; Tare, A.; Cusumano, L.R.; Benayoun, J.; Ryu, S.; Sixpence, A.; Seydel, K.; Laufer, M.; Taylor, T.; Suh, Y. Type I interferon receptor variants in gene regulatory regions are associated with susceptibility to cerebral malaria in Malawi. Am. J. Trop. Med. Hyg. 2018, 98, 1692–1698. [Google Scholar] [CrossRef] [PubMed]
- Aucan, C.; Walley, A.; Hennig, B.; Fitness, J.; Frodsham, A.; Zhang, L.; Kwiatkowski, D.; Hill, A. Interferon-alpha receptor-1 (IFNAR1) variants are associated with protection against cerebral malaria in the Gambia. Genes Immun. 2003, 4, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Ball, E.A.; Sambo, M.R.; Martins, M.; Trovoada, M.J.; Benchimol, C.; Costa, J.; Gonçalves, L.A.; Coutinho, A.; Penha-Gonçalves, C. IFNAR1 controls progression to cerebral malaria in children and CD8+ T cell brain pathology in Plasmodium berghei-infected mice. J. Immunol. 2013, 190, 5118–5127. [Google Scholar] [CrossRef] [PubMed]
- Krupka, M.; Seydel, K.; Feintuch, C.M.; Yee, K.; Kim, R.; Lin, C.-Y.; Calder, R.B.; Petersen, C.; Taylor, T.; Daily, J. Mild Plasmodium falciparum malaria following an episode of severe malaria is associated with induction of the interferon pathway in Malawian children. Infect. Immun. 2012, 80, 1150–1155. [Google Scholar] [CrossRef]
- Sharma, S.; DeOliveira, R.B.; Kalantari, P.; Parroche, P.; Goutagny, N.; Jiang, Z.; Chan, J.; Bartholomeu, D.C.; Lauw, F.; Hall, J.P.; et al. Innate immune recognition of an AT-rich stem-loop DNA motif in the Plasmodium falciparum genome. Immunity 2011, 35, 194–207. [Google Scholar] [CrossRef]
- Koch, O.; Awomoyi, A.; Usen, S.; Jallow, M.; Richardson, A.; Hull, J.; Pinder, M.; Newport, M.; Kwiatkowski, D. IFNGR1 gene promoter polymorphisms and susceptibility to cerebral malaria. J. Infect. Dis. 2002, 185, 1684–1687. [Google Scholar] [CrossRef][Green Version]
- Cabantous, S.; Poudiougou, B.; Traore, A.; Keita, M.; Cisse, M.B.; Doumbo, O.; Dessein, A.J.; Marquet, S. Evidence that interferon-gamma plays a protective role during cerebral malaria. J. Infect. Dis. 2005, 192, 854–860. [Google Scholar] [CrossRef]
- Villegas-Mendez, A.; Strangward, P.; Shaw, T.N.; Rajkovic, I.; Tosevski, V.; Forman, R.; Muller, W.; Couper, K.N. Gamma Interferon Mediates Experimental Cerebral Malaria by Signaling within Both the Hematopoietic and Nonhematopoietic Compartments. Infect. Immun. 2017, 85, e01035-16. [Google Scholar] [CrossRef]
- Villegas-Mendez, A.; Greig, R.; Shaw, T.N.; de Souza, J.B.; Gwyer Findlay, E.; Stumhofer, J.S.; Hafalla, J.C.; Blount, D.G.; Hunter, C.A.; Riley, E.M.; et al. IFN-gamma-producing CD4+ T cells promote experimental cerebral malaria by modulating CD8+ T cell accumulation within the brain. J. Immunol. 2012, 189, 968–979. [Google Scholar] [CrossRef]
- Van den Steen, P.E.; Deroost, K.; Aelst, I.V.; Geurts, N.; Martens, E.; Struyf, S.; Nie, C.Q.; Hansen, D.S.; Matthys, P.; Damme, J.V. CXCR3 determines strain susceptibility to murine cerebral malaria by mediating T lymphocyte migration toward IFN-γ-induced chemokines. Eur. J. Immunol. 2008, 38, 1082–1095. [Google Scholar] [CrossRef]
- Hansen, D.S.; Bernard, N.J.; Nie, C.Q.; Schofield, L. NK cells stimulate recruitment of CXCR3+ T cells to the brain during Plasmodium berghei-mediated cerebral malaria. J. Immunol. 2007, 178, 5779–5788. [Google Scholar] [CrossRef]
- Belnoue, E.; Kayibanda, M.; Deschemin, J.-C.; Viguier, M.; Mack, M.; Kuziel, W.A.; Rénia, L. CCR5 deficiency decreases susceptibility to experimental cerebral malaria. Blood 2003, 101, 4253–4259. [Google Scholar] [CrossRef]
- Dorovini-Zis, K.; Schmidt, K.; Huynh, H.; Fu, W.; Whitten, R.O.; Milner, D.; Kamiza, S.; Molyneux, M.; Taylor, T.E. The neuropathology of fatal cerebral malaria in malawian children. Am. J. Pathol. 2011, 178, 2146–2158. [Google Scholar] [CrossRef]
- Barrera, V.; Haley, M.J.; Strangward, P.; Attree, E.; Kamiza, S.; Seydel, K.; Taylor, T.; Milner, D.; Craig, A.; Couper, K.N. Comparison of CD8+ T cell accumulation in the brain during human and murine cerebral malaria. Front. Immunol. 2019, 10, 1747. [Google Scholar] [CrossRef]
- Riggle, B.A.; Manglani, M.; Maric, D.; Johnson, K.R.; Lee, M.-H.; Neto, O.L.A.; Taylor, T.E.; Seydel, K.B.; Nath, A.; Miller, L.H. CD8+ T cells target cerebrovasculature in children with cerebral malaria. J. Clin. Investing. 2020, 130, 1128–1138. [Google Scholar] [CrossRef]
- Haque, A.; Best, S.E.; Unosson, K.; Amante, F.H.; de Labastida, F.; Anstey, N.M.; Karupiah, G.; Smyth, M.J.; Heath, W.R.; Engwerda, C.R. Granzyme B expression by CD8+ T cells is required for the development of experimental cerebral malaria. J. Immunol. 2011, 186, 6148–6156. [Google Scholar] [CrossRef]
- Huggins, M.A.; Johnson, H.L.; Jin, F.; N’ Songo, A.; Hanson, L.M.; LaFrance, S.J.; Butler, N.S.; Harty, J.T.; Johnson, A.J. Perforin Expression by CD8 T Cells Is Sufficient To Cause Fatal Brain Edema during Experimental Cerebral Malaria. Infect. Immun. 2017, 85, e00985-16. [Google Scholar] [CrossRef]
- Punsawad, C.; Maneerat, Y.; Chaisri, U.; Nantavisai, K.; Viriyavejakul, P. Nuclear factor kappa B modulates apoptosis in the brain endothelial cells and intravascular leukocytes of fatal cerebral malaria. Malar. J. 2013, 12, 260. [Google Scholar] [CrossRef]
- Pino, P.; Vouldoukis, I.; Kolb, J.P.; Mahmoudi, N.; Desportes-Livage, I.; Bricaire, F.; Danis, M.; Dugas, B.; Mazier, D. Plasmodium falciparum-infected erythrocyte adhesion induces caspase activation and apoptosis in human endothelial cells. J. Infect. Dis. 2003, 187, 1283–1290. [Google Scholar] [CrossRef]
- Tsunoda, I.; Kuang, L.-Q.; Fujinami, R.S. Induction of autoreactive CD8+ cytotoxic T cells during Theiler’s murine encephalomyelitis virus infection: Implications for autoimmunity. J. Virol. 2002, 76, 12834–12844. [Google Scholar] [CrossRef]
- Cong, X.; Kong, W. Endothelial tight junctions and their regulatory signaling pathways in vascular homeostasis and disease. Cell. Signal. 2020, 66, 109485. [Google Scholar] [CrossRef]
- Brown, H.; Hien, T.; Day, N.; Mai, N.; Chuong, L.; Chau, T.; Loc, P.; Phu, N.; Bethell, D.; Farrar, J. Evidence of blood-brain barrier dysfunction in human cerebral malaria. Neuropathol. Appl. Neurobiol. 1999, 25, 331–340. [Google Scholar] [CrossRef]
- Suidan, G.L.; McDole, J.R.; Chen, Y.; Pirko, I.; Johnson, A.J. Induction of blood brain barrier tight junction protein alterations by CD8 T cells. PLoS ONE 2008, 3, e3037. [Google Scholar] [CrossRef]
- Tunon-Ortiz, A.; Lamb, T.J. Blood brain barrier disruption in cerebral malaria: Beyond endothelial cell activation. PLoS Pathog. 2019, 15, e1007786. [Google Scholar] [CrossRef]
- Shrivastava, S.K.; Dalko, E.; Delcroix-Genete, D.; Herbert, F.; Cazenave, P.A.; Pied, S. Uptake of parasite-derived vesicles by astrocytes and microglial phagocytosis of infected erythrocytes may drive neuroinflammation in cerebral malaria. Glia 2017, 65, 75–92. [Google Scholar] [CrossRef]
- Promeneur, D.; Lunde, L.K.; Amiry-Moghaddam, M.; Agre, P. Protective role of brain water channel AQP4 in murine cerebral malaria. Proc. Natl. Acad. Sci. USA 2013, 110, 1035–1040. [Google Scholar] [CrossRef]
- Capuccini, B.; Lin, J.; Talavera-Lopez, C.; Khan, S.M.; Sodenkamp, J.; Spaccapelo, R.; Langhorne, J. Transcriptomic profiling of microglia reveals signatures of cell activation and immune response, during experimental cerebral malaria. Sci. Rep. 2016, 6, 39258. [Google Scholar] [CrossRef]
- Medana, I.M.; Idro, R.; Newton, C.R. Axonal and astrocyte injury markers in the cerebrospinal fluid of Kenyan children with severe malaria. J. Neurol. Sci. 2007, 258, 93–98. [Google Scholar] [CrossRef]
- Schluesener, H.J.; Kremsner, P.G.; Meyermann, R. Widespread expression of MRP8 and MRP14 in human cerebral malaria by microglial cells. Acta Neuropathol. 1998, 96, 575–580. [Google Scholar] [CrossRef]
- Poh, C.M.; Howland, S.W.; Grotenbreg, G.M.; Renia, L. Damage to the blood-brain barrier during experimental cerebral malaria results from synergistic effects of CD8+ T cells with different specificities. Infect. Immun. 2014, 82, 4854–4864. [Google Scholar] [CrossRef] [PubMed]
- Besnard, A.-G.; Guabiraba, R.; Niedbala, W.; Palomo, J.; Reverchon, F.; Shaw, T.N.; Couper, K.N.; Ryffel, B.; Liew, F.Y. IL-33-mediated protection against experimental cerebral malaria is linked to induction of type 2 innate lymphoid cells, M2 macrophages and regulatory T cells. PLoS Pathog. 2015, 11, e1004607. [Google Scholar] [CrossRef] [PubMed]
- Burrack, K.S.; Huggins, M.A.; Taras, E.; Dougherty, P.; Henzler, C.M.; Yang, R.; Alter, S.; Jeng, E.K.; Wong, H.C.; Felices, M.; et al. Interleukin-15 Complex Treatment Protects Mice from Cerebral Malaria by Inducing Interleukin-10-Producing Natural Killer Cells. Immunity 2018, 48, 760–772.e4. [Google Scholar] [CrossRef] [PubMed]
- Charoenpan, P.; Indraprasit, S.; Kiatboonsri, S.; Suvachittanont, O.; Tanomsup, S. Pulmonary edema in severe falciparum malaria: Hemodynamic study and clinicophysiologic correlation. Chest 1990, 97, 1190–1197. [Google Scholar] [CrossRef] [PubMed]
- Feldman, R.M.; Singer, C. Noncardiogenic pulmonary edema and pulmonary fibrosis in falciparum malaria. Clin. Infect. Dis. 1987, 9, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Ando, M.; Miyazaki, E.; Abe, T.; Ehara, C.; Goto, A.; Masuda, T.; Nishio, S.; Fujisaki, H.; Yamasue, M.; Ishii, T. Angiopoietin-2 expression in patients with an acute exacerbation of idiopathic interstitial pneumonias. Respir. Med. 2016, 117, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.-T.; Punsawad, C.; Glaharn, S.; De Meyer, S.F.; Viriyavejakul, P.; Van den Steen, P.E. Release of endothelial activation markers in lungs of patients with malaria-associated acute respiratory distress syndrome. Malar. J. 2019, 18, 395. [Google Scholar] [CrossRef] [PubMed]
- Avril, M.; Bernabeu, M.; Benjamin, M.; Brazier, A.J.; Smith, J.D. Interaction between endothelial protein C receptor and intercellular adhesion molecule 1 to mediate binding of Plasmodium falciparum-infected erythrocytes to endothelial cells. MBio 2016, 7, e00615-16. [Google Scholar] [CrossRef]
- Lovegrove, F.E.; Gharib, S.A.; Peña-Castillo, L.; Patel, S.N.; Ruzinski, J.T.; Hughes, T.R.; Liles, W.C.; Kain, K.C. Parasite burden and CD36-mediated sequestration are determinants of acute lung injury in an experimental malaria model. PLoS Pathog. 2008, 4, e1000068. [Google Scholar] [CrossRef]
- Togbe, D.; Schofield, L.; Grau, G.E.; Schnyder, B.; Boissay, V.; Charron, S.; Rose, S.; Beutler, B.; Quesniaux, V.F.; Ryffel, B. Murine cerebral malaria development is independent of toll-like receptor signaling. Am. J. Pathol. 2007, 170, 1640–1648. [Google Scholar] [CrossRef]
- Kraisin, S.; Verhenne, S.; Pham, T.T.; Martinod, K.; Tersteeg, C.; Vandeputte, N.; Deckmyn, H.; Vanhoorelbeke, K.; Van den Steen, P.E.; De Meyer, S.F. Von Willebrand factor in experimental malaria-associated acute respiratory distress syndrome. J. Thromb. Haemost. 2019, 17, 1372–1383. [Google Scholar] [CrossRef]
- Zielinska, K.A.; de Cauwer, L.; Knoops, S.; Van der Molen, K.; Sneyers, A.; Thommis, J.; De Souza, J.B.; Opdenakker, G.; De Bosscher, K.; Van den Steen, P.E. Plasmodium berghei NK65 in Combination with IFN-gamma Induces Endothelial Glucocorticoid Resistance via Sustained Activation of p38 and JNK. Front. Immunol. 2017, 8, 1199. [Google Scholar] [CrossRef]
- Claser, C.; Nguee, S.Y.T.; Balachander, A.; Wu Howland, S.; Becht, E.; Gunasegaran, B.; Hartimath, S.V.; Lee, A.W.; Theng Theng Ho, J.; Bing Ong, C.; et al. Lung endothelial cell antigen cross-presentation to CD8(+)T cells drives malaria-associated lung injury. Nat. Commun. 2019, 10, 4241. [Google Scholar] [CrossRef]
- Sercundes, M.K.; Ortolan, L.S.; Debone, D.; Soeiro-Pereira, P.V.; Gomes, E.; Aitken, E.H.; Condino-Neto, A.; Russo, M.; Neto, A.C.; Alvarez, J.M.; et al. Targeting Neutrophils to Prevent Malaria-Associated Acute Lung Injury/Acute Respiratory Distress Syndrome in Mice. PLoS Pathog. 2016, 12, e1006054. [Google Scholar] [CrossRef]
- Lagasse, H.A.; Anidi, I.U.; Craig, J.M.; Limjunyawong, N.; Poupore, A.K.; Mitzner, W.; Scott, A.L. Recruited monocytes modulate malaria-induced lung injury through CD36-mediated clearance of sequestered infected erythrocytes. J. Leukoc. Biol. 2016, 99, 659–671. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olatunde, A.C.; Cornwall, D.H.; Roedel, M.; Lamb, T.J. Mouse Models for Unravelling Immunology of Blood Stage Malaria. Vaccines 2022, 10, 1525. https://doi.org/10.3390/vaccines10091525
Olatunde AC, Cornwall DH, Roedel M, Lamb TJ. Mouse Models for Unravelling Immunology of Blood Stage Malaria. Vaccines. 2022; 10(9):1525. https://doi.org/10.3390/vaccines10091525
Chicago/Turabian StyleOlatunde, Adesola C., Douglas H. Cornwall, Marshall Roedel, and Tracey J. Lamb. 2022. "Mouse Models for Unravelling Immunology of Blood Stage Malaria" Vaccines 10, no. 9: 1525. https://doi.org/10.3390/vaccines10091525
APA StyleOlatunde, A. C., Cornwall, D. H., Roedel, M., & Lamb, T. J. (2022). Mouse Models for Unravelling Immunology of Blood Stage Malaria. Vaccines, 10(9), 1525. https://doi.org/10.3390/vaccines10091525