
Immunoinformatics-Aided Analysis of RSV Fusion and Attachment Glycoproteins to Design a Potent Multi-Epitope Vaccine




Abstract
1. Introduction
2. Materials and Methods
2.1. Collection of the RSV Glycoproteins F and G Sequences
2.2. Prioritization of T-Cell Epitopes in the RSV F and G Glycoproteins
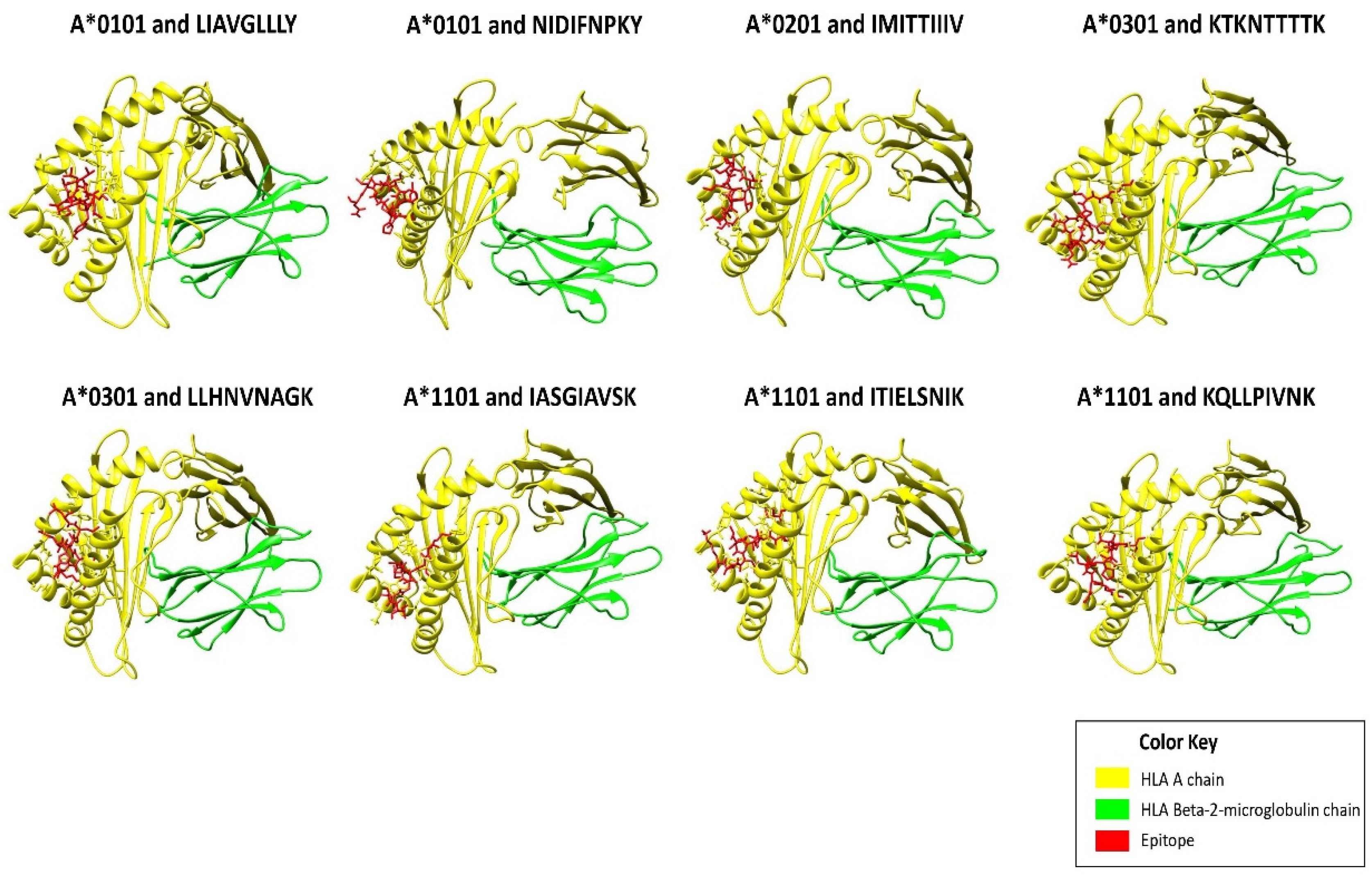
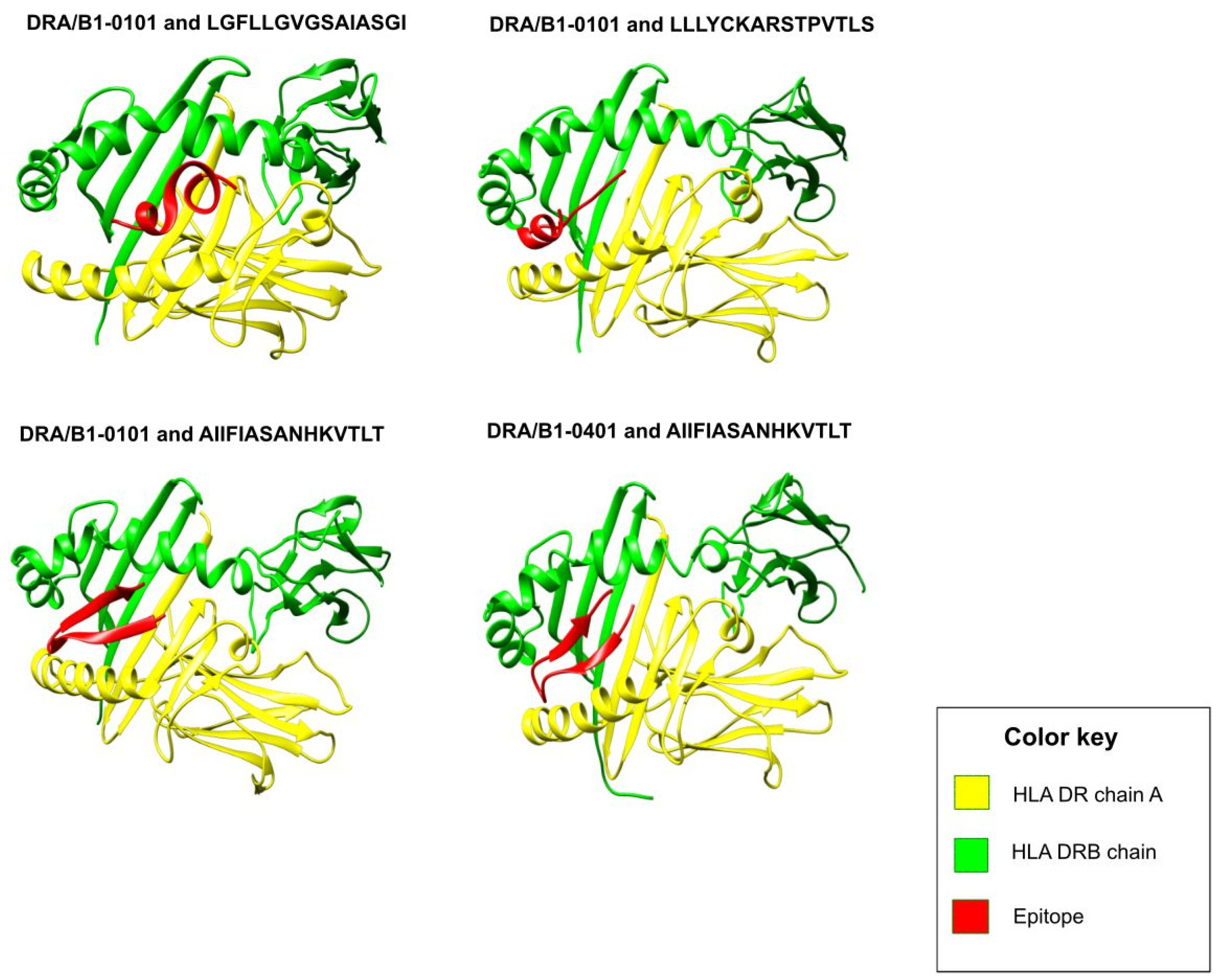
2.3. Molecular Docking of Epitopes with HLA Alleles
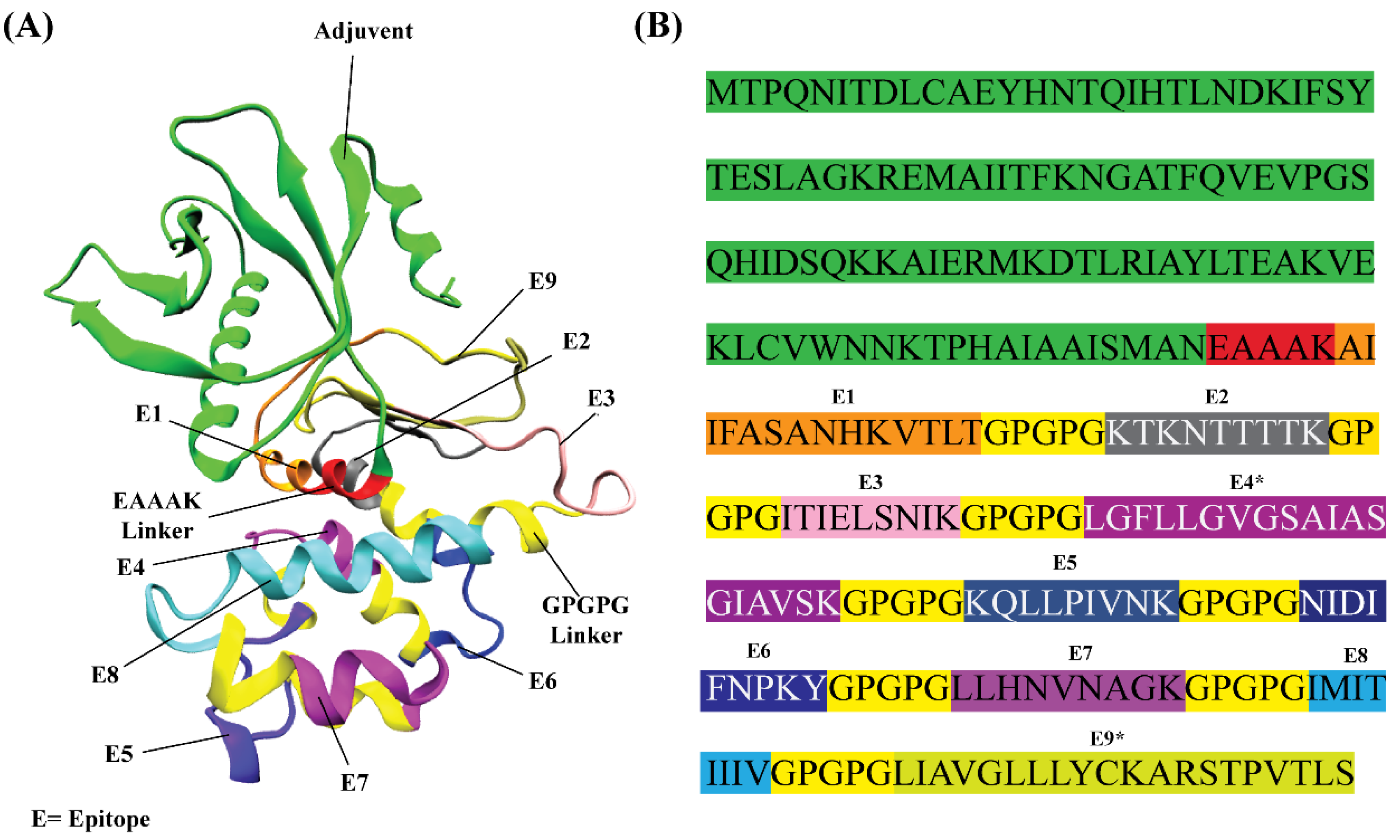
2.4. In Silico Design of RSV Multi-Epitope Vaccine
2.5. Structural Modeling of the RSV Multi-Epitope Vaccine
2.6. Molecular Docking Analysis of Vaccine with Toll-Like Receptor 4
2.7. Molecular Dynamics Analysis on Vaccine-TLR4 Complex
2.8. In Silico Immune Simulation Analysis
2.9. Cloning of Designed Vaccine
3. Results
3.1. Prioritization of T-Cell Epitopes in the RSV F and G Glycoproteins
3.2. In Silico Designing, Physiochemical and Immunological Properties Evaluation of Multi-Epitope Vaccine
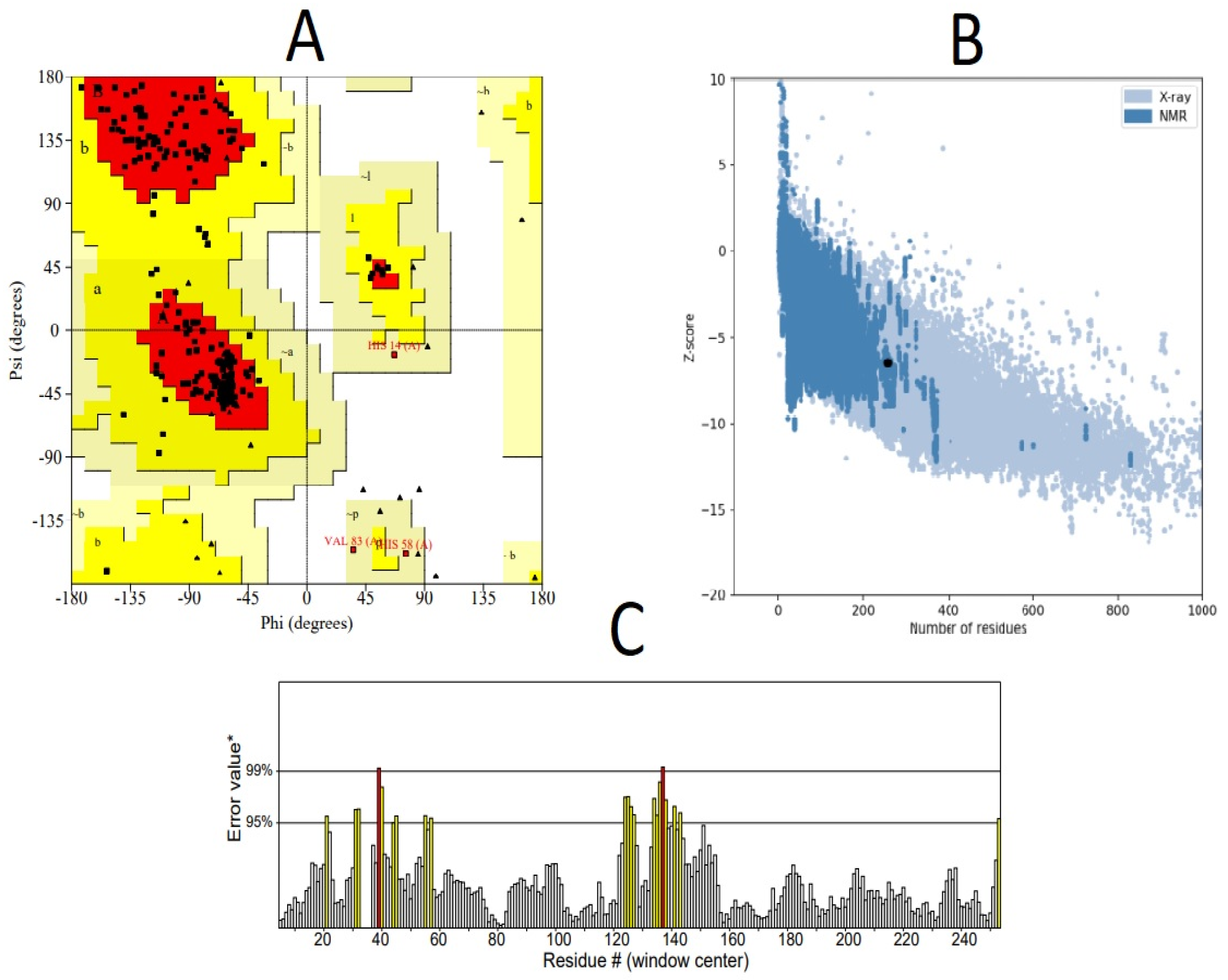
3.3. Structural Modeling of RSV Multi-Epitope Vaccine
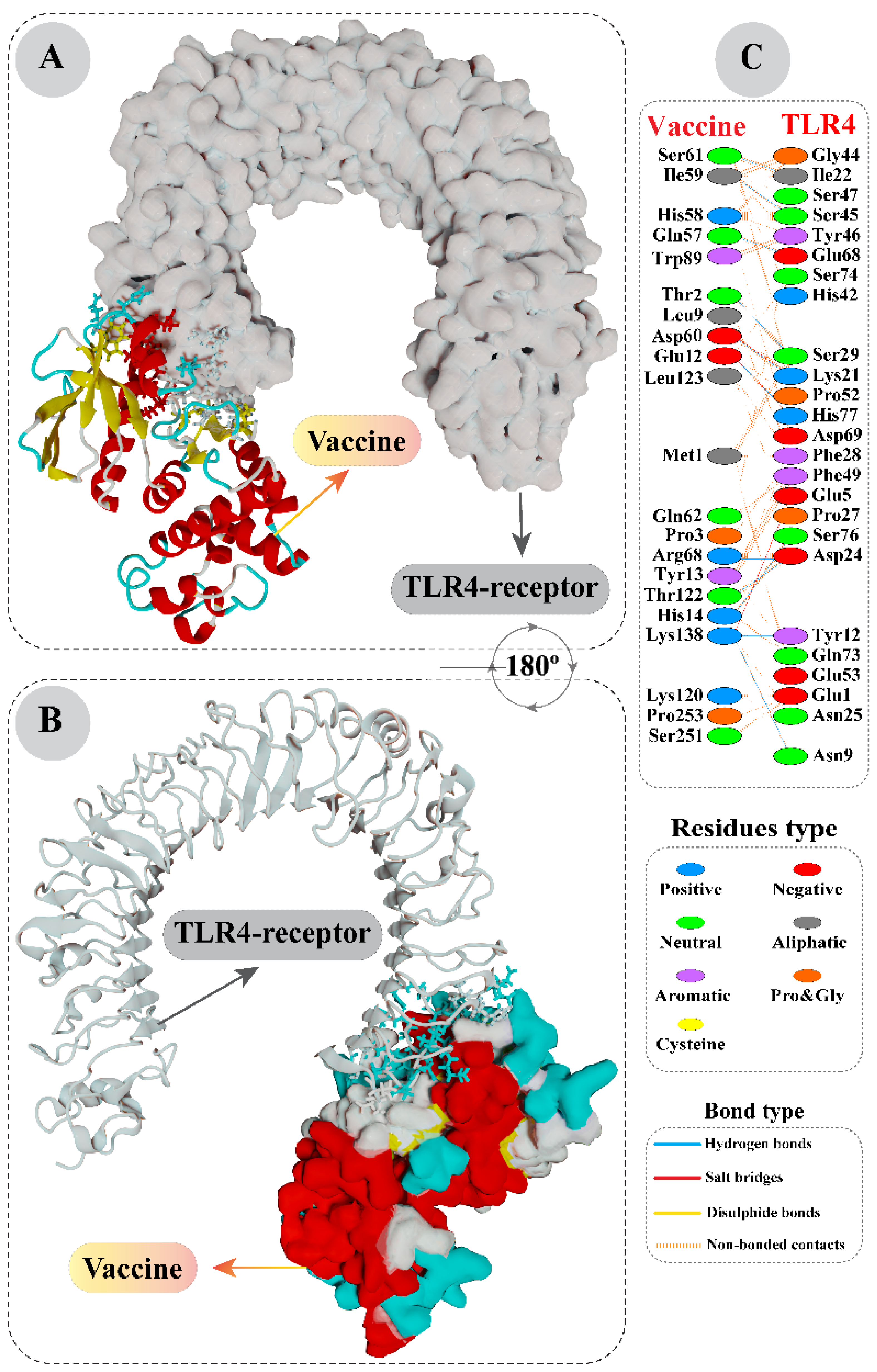
3.4. Interaction Analysis of Vaccine with Toll-Like Receptor 4 by Molecular Docking
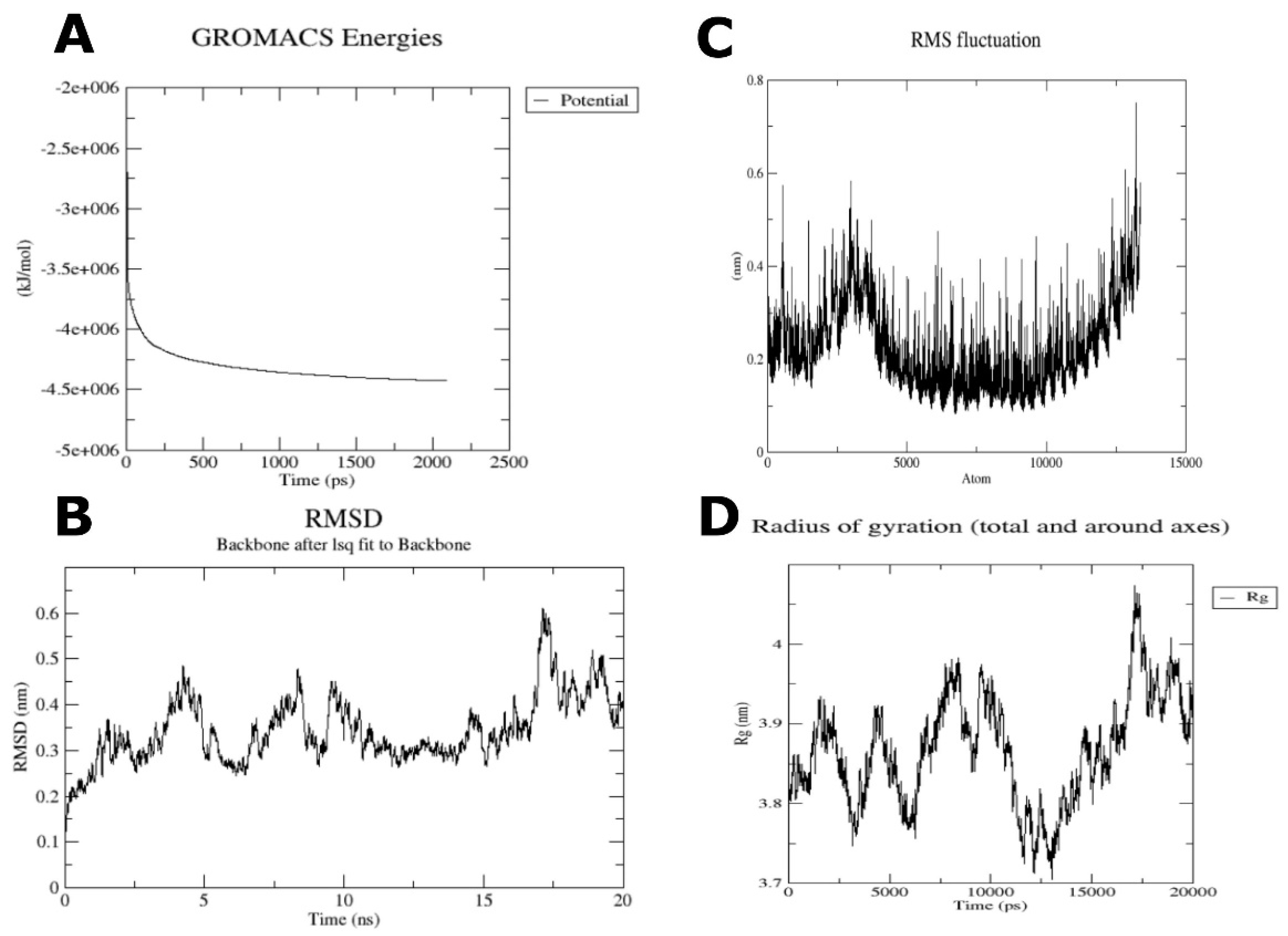
3.5. Molecular Dynamics Analysis on Vaccine-TLR4 Complex
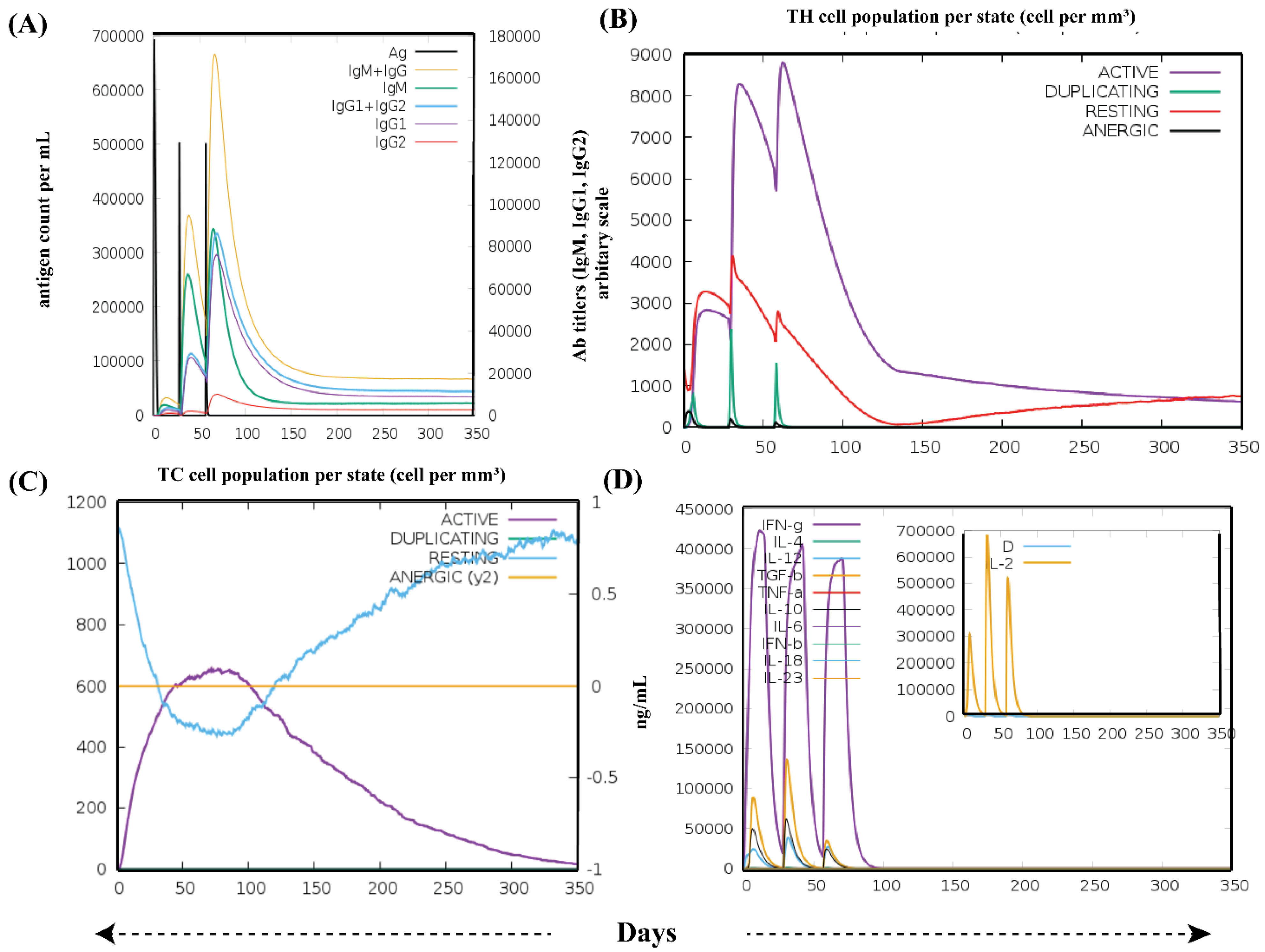
3.6. Immune Simulation Analysis
3.7. Optimized Cloning of the RSV Vaccine Candidate
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Elhalik, M.; El-Atawi, K.; Dash, S.K.; Faquih, A.; Satyan, A.D.; Gourshettiwar, N.; Khan, A.; Varughese, S.; Ramesh, A.; Khamis, E. Palivizumab Prophylaxis among Infants at Increased Risk of Hospitalization Due to Respiratory Syncytial Virus Infection in UAE: A Hospital-Based Study. Can. Respir. J. 2019, 2019, 2986286. [Google Scholar] [CrossRef]
- Griffiths, C.; Drews, S.J.; Marchant, D.J. Respiratory Syncytial Virus: Infection, Detection, and New Options for Prevention and Treatment. Clin. Microbiol. Rev. 2017, 30, 277–319. [Google Scholar] [CrossRef]
- Afonso, C.L.; Amarasinghe, G.K.; Bányai, K.; Bào, Y.; Basler, C.F.; Bavari, S.; Bejerman, N.; Blasdell, K.R.; Briand, F.-X.; Briese, T.; et al. Taxonomy of the Order Mononegavirales: Update 2016. Arch. Virol. 2016, 161, 2351–2360. [Google Scholar] [CrossRef]
- ul Qamar, M.; Shokat, Z.; Muneer, I.; Ashfaq, U.A.; Javed, H.; Anwar, F.; Bari, A.; Zahid, B.; Saari, N. Multiepitope-Based Subunit Vaccine Design and Evaluation against Respiratory Syncytial Virus Using Reverse Vaccinology Approach. Vaccines 2020, 8, 288. [Google Scholar] [CrossRef]
- Mufson, M.A.; Örvell, C.; Rafnar, B.; Norrby, E. Two Distinct Subtypes of Human Respiratory Syncytial Virus. J. Gen. Virol. 1985, 66, 2111–2124. [Google Scholar] [CrossRef]
- Thongpan, I.; Mauleekoonphairoj, J.; Vichiwattana, P.; Korkong, S.; Wasitthankasem, R.; Vongpunsawad, S.; Poovorawan, Y. Respiratory Syncytial Virus Genotypes NA1, ON1, and BA9 Are Prevalent in Thailand, 2012–2015. PeerJ 2017, 5, e3970. [Google Scholar] [CrossRef]
- Collins, P.L.; Fearns, R.; Graham, B.S. Respiratory Syncytial Virus: Virology, Reverse Genetics, and Pathogenesis of Disease. In Challenges and Opportunities for Respiratory Syncytial Virus Vaccines; Springer: Berlin/Heidelberg, Germany, 2013; pp. 3–38. [Google Scholar]
- Carvajal, J.J.; Avellaneda, A.M.; Salazar-Ardiles, C.N.; Maya, J.E.; Kalergis, A.M.; Lay, M.K.-L. Host Components Contributing to Respiratory Syncytial Virus Pathogenesis. Front. Immunol. 2019, 10, 2152. [Google Scholar] [CrossRef]
- Mastrangelo, P.; Hegele, R.G. RSV Fusion: Time for a New Model. Viruses 2013, 5, 873–885. [Google Scholar] [CrossRef]
- McLellan, J.S.; Ray, W.C.; Peeples, M.E. Structure and Function of Respiratory Syncytial Virus Surface Glycoproteins. In Challenges and Opportunities for Respiratory Syncytial Virus Vaccines; Springer: Berlin/Heidelberg, Germany, 2013; pp. 83–104. [Google Scholar]
- Naz, R.; Gul, A.; Javed, U.; Urooj, A.; Amin, S.; Fatima, Z. Etiology of Acute Viral Respiratory Infections Common in Pakistan: A Review. Rev. Med. Virol. 2019, 29, e2024. [Google Scholar] [CrossRef]
- Goins, W.P.; Talbot, H.K.; Talbot, T.R. Health Care—Acquired Viral Respiratory Diseases. Infect. Dis. Clin. 2011, 25, 227–244. [Google Scholar] [CrossRef][Green Version]
- Schmidt, M.R.; McGinnes, L.W.; Kenward, S.A.; Willems, K.N.; Woodland, R.T.; Morrison, T.G. Long-Term and Memory Immune Responses in Mice against Newcastle Disease Virus-like Particles Containing Respiratory Syncytial Virus Glycoprotein Ectodomains. J. Virol. 2012, 86, 11654–11662. [Google Scholar] [CrossRef]
- Schweitzer, J.W.; Justice, N.A. Respiratory Syncytial Virus Infection. 2017. Available online: https://www.ncbi.nlm.nih.gov/books/NBK459215/ (accessed on 3 July 2022).
- Falsey, A.R.; McCann, R.M.; Hall, W.J.; Criddle, M.M.; Formica, M.A.; Wycoff, D.; Kolassa, J.E. The “Common Cold” in Frail Older Persons: Impact of Rhinovirus and Coronavirus in a Senior Daycare Center. J. Am. Geriatr. Soc. 1997, 45, 706–711. [Google Scholar] [CrossRef]
- Saravolatz, L.D.; Empey, K.M.; Peebles Jr, R.S.; Kolls, J.K. Pharmacologic Advances in the Treatment and Prevention of Respiratory Syncytial Virus. Clin. Infect. Dis. 2010, 50, 1258–1267. [Google Scholar]
- IMpact-RSV Study Group. Palivizumab, a Humanized Respiratory Syncytial Virus Monoclonal Antibody, Reduces Hospitalization from Respiratory Syncytial Virus Infection in High-Risk Infants. Pediatrics 1998, 102, 531–537. [Google Scholar] [CrossRef]
- Ventre, K.; Randolph, A. Ribavirin for Respiratory Syncytial Virus Infection of the Lower Respiratory Tract in Infants and Young Children. Cochrane Database Syst. Rev. 2007, 1, CD000181. [Google Scholar]
- Hall, C.B.; McBride, J.T.; Walsh, E.E.; Bell, D.M.; Gala, C.L.; Hildreth, S.; Ten Eyck, L.G.; Hall, W.J. Aerosolized Ribavirin Treatment of Infants with Respiratory Syncytial Viral Infection: A Randomized Double-Blind Study. N. Engl. J. Med. 1983, 308, 1443–1447. [Google Scholar] [CrossRef]
- Sette, A.; Rappuoli, R. Reverse Vaccinology: Developing Vaccines in the Era of Genomics. Immunity 2010, 33, 530–541. [Google Scholar] [CrossRef]
- Dar, H.A.; Zaheer, T.; Shehroz, M.; Ullah, N.; Naz, K.; Muhammad, S.A.; Zhang, T.; Ali, A. Immunoinformatics-Aided Design and Evaluation of a Potential Multi-Epitope Vaccine against Klebsiella Pneumoniae. Vaccines 2019, 7, 88. [Google Scholar] [CrossRef]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. T Cells and MHC Proteins. In Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002. [Google Scholar]
- Ayub, G.; Waheed, Y.; Najmi, M.H. Prediction and Conservancy Analysis of Promiscuous T-Cell Binding Epitopes of Ebola Virus L Protein: An in Silico Approach. Asian Pacific J. Trop. Dis. 2016, 6, 169–173. [Google Scholar] [CrossRef]
- Oyarzun, P.; Ellis, J.J.; Gonzalez-Galarza, F.F.; Jones, A.R.; Middleton, D.; Boden, M.; Kobe, B. A Bioinformatics Tool for Epitope-Based Vaccine Design That Accounts for Human Ethnic Diversity: Application to Emerging Infectious Diseases. Vaccine 2015, 33, 1267–1273. [Google Scholar] [CrossRef]
- Sette, A.; Fikes, J. Epitope-Based Vaccines: An Update on Epitope Identification, Vaccine Design and Delivery. Curr. Opin. Immunol. 2003, 15, 461–470. [Google Scholar] [CrossRef]
- Patronov, A.; Doytchinova, I. T-Cell Epitope Vaccine Design by Immunoinformatics. Open Biol. 2013, 3, 120139. [Google Scholar] [CrossRef]
- Barh, D.; Barve, N.; Gupta, K.; Chandra, S.; Jain, N.; Tiwari, S.; Leon-Sicairos, N.; Canizalez-Roman, A.; dos Santos, A.R.; Hassan, S.S.; et al. Exoproteome and Secretome Derived Broad Spectrum Novel Drug and Vaccine Candidates in Vibrio Cholerae Targeted by Piper Betel Derived Compounds. PLoS ONE 2013, 8, e52773. [Google Scholar] [CrossRef]
- Compton, T.; Kurt-Jones, E.A.; Boehme, K.W.; Belko, J.; Latz, E.; Golenbock, D.T.; Finberg, R.W. Human Cytomegalovirus Activates Inflammatory Cytokine Responses via CD14 and Toll-like Receptor 2. J. Virol. 2003, 77, 4588–4596. [Google Scholar] [CrossRef]
- Vijay, K. Toll-like Receptors in Immunity and Inflammatory Diseases: Past, Present, and Future. Int. Immunopharmacol. 2018, 59, 391–412. [Google Scholar] [CrossRef]
- Kerepesi, L.A.; Leon, O.; Lustigman, S.; Abraham, D. Protective Immunity to the Larval Stages of Onchocerca Volvulus Is Dependent on Toll-like Receptor 4. Infect. Immun. 2005, 73, 8291–8297. [Google Scholar] [CrossRef]
- Andreatta, M.; Nielsen, M. Gapped Sequence Alignment Using Artificial Neural Networks: Application to the MHC Class I System. Bioinformatics 2015, 32, 511–517. [Google Scholar] [CrossRef]
- Nielsen, M.; Lundegaard, C.; Worning, P.; Lauemøller, S.L.; Lamberth, K.; Buus, S.; Brunak, S.; Lund, O. Reliable Prediction of T-Cell Epitopes Using Neural Networks with Novel Sequence Representations. Protein Sci. 2003, 12, 1007–1017. [Google Scholar] [CrossRef]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A Server for Prediction of Protective Antigens, Tumour Antigens and Subunit Vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef]
- Calis, J.J.A.; Maybeno, M.; Greenbaum, J.A.; Weiskopf, D.; De Silva, A.D.; Sette, A.; Keşmir, C.; Peters, B. Properties of MHC Class I Presented Peptides That Enhance Immunogenicity. PLoS Comput. Biol. 2013, 9, e1003266. [Google Scholar] [CrossRef]
- Guan, P.; Doytchinova, I.A.; Zygouri, C.; Flower, D.R. MHCPred: A Server for Quantitative Prediction of Peptide--MHC Binding. Nucleic Acids Res. 2003, 31, 3621–3624. [Google Scholar] [CrossRef]
- Jensen, K.K.; Andreatta, M.; Marcatili, P.; Buus, S.; Greenbaum, J.A.; Yan, Z.; Sette, A.; Peters, B.; Nielsen, M. Improved Methods for Predicting Peptide Binding Affinity to MHC Class II Molecules. Immunology 2018, 154, 394–406. [Google Scholar] [CrossRef]
- Chen, H.; Zhou, H.-X. Prediction of Solvent Accessibility and Sites of Deleterious Mutations from Protein Sequence. Nucleic Acids Res. 2005, 33, 3193–3199. [Google Scholar] [CrossRef]
- Lamiable, A.; Thévenet, P.; Rey, J.; Vavrusa, M.; Derreumaux, P.; Tufféry, P. PEP-FOLD3: Faster de Novo Structure Prediction for Linear Peptides in Solution and in Complex. Nucleic Acids Res. 2016, 44, W449–W454. [Google Scholar] [CrossRef]
- van Zundert, G.C.P.; Rodrigues, J.P.G.L.M.; Trellet, M.; Schmitz, C.; Kastritis, P.L.; Karaca, E.; Melquiond, A.S.J.; van Dijk, M.; de Vries, S.J.; Bonvin, A.M.J.J. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef]
- De Vries, S.J.; Van Dijk, M.; Bonvin, A.M.J.J. The HADDOCK Web Server for Data-Driven Biomolecular Docking. Nat. Protoc. 2010, 5, 883. [Google Scholar] [CrossRef]
- Gerstner, C.; Dubnovitsky, A.; Sandin, C.; Kozhukh, G.; Uchtenhagen, H.; James, E.A.; Rönnelid, J.; Ytterberg, A.J.; Pieper, J.; Reed, E.; et al. Functional and Structural Characterization of a Novel HLA-DRB1* 04: 01-Restricted $α$-Enolase T Cell Epitope in Rheumatoid Arthritis. Front. Immunol. 2016, 7, 494. [Google Scholar] [CrossRef]
- Quiñones-Parra, S.; Grant, E.; Loh, L.; Nguyen, T.H.O.; Campbell, K.-A.; Tong, S.Y.C.; Miller, A.; Doherty, P.C.; Vijaykrishna, D.; Rossjohn, J.; et al. Preexisting CD8+ T-Cell Immunity to the H7N9 Influenza A Virus Varies across Ethnicities. Proc. Natl. Acad. Sci. USA 2014, 111, 1049–1054. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, J.; Cheng, H.; Tan, S.; Qi, J.; Yan, J.; Gao, G.F. Structural Basis of Cross-Allele Presentation by HLA-A* 0301 and HLA-A* 1101 Revealed by Two HIV-Derived Peptide Complexes. Mol. Immunol. 2011, 49, 395–401. [Google Scholar] [CrossRef]
- Li, L.; Bouvier, M. Structures of HLA-A* 1101 Complexed with Immunodominant Nonamer and Decamer HIV-1 Epitopes Clearly Reveal the Presence of a Middle, Secondary Anchor Residue. J. Immunol. 2004, 172, 6175–6184. [Google Scholar] [CrossRef]
- Murthy, V.L.; Stern, L.J. The Class II MHC Protein HLA-DR1 in Complex with an Endogenous Peptide: Implications for the Structural Basis of the Specificity of Peptide Binding. Structure 1997, 5, 1385–1396. [Google Scholar] [CrossRef]
- Khan, A.R.; Baker, B.M.; Ghosh, P.; Biddison, W.E.; Wiley, D.C. The Structure and Stability of an HLA-A* 0201/Octameric Tax Peptide Complex with an Empty Conserved Peptide-N-Terminal Binding Site. J. Immunol. 2000, 164, 6398–6405. [Google Scholar] [CrossRef]
- Laskowski, R.A. PDBsum: Summaries and Analyses of PDB Structures. Nucleic Acids Res. 2001, 29, 221–222. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Gasteiger, E.; Gattiker, A.; Hoogland, C.; Ivanyi, I.; Appel, R.D.; Bairoch, A. ExPASy: The Proteomics Server for in-Depth Protein Knowledge and Analysis. Nucleic Acids Res. 2003, 31, 3784–3788. [Google Scholar] [CrossRef]
- Magnan, C.N.; Zeller, M.; Kayala, M.A.; Vigil, A.; Randall, A.; Felgner, P.L.; Baldi, P. High-Throughput Prediction of Protein Antigenicity Using Protein Microarray Data. Bioinformatics 2010, 26, 2936–2943. [Google Scholar] [CrossRef]
- Dimitrov, I.; Bangov, I.; Flower, D.R.; Doytchinova, I. AllerTOP v. 2—a Server for in Silico Prediction of Allergens. J. Mol. Model. 2014, 20, 2278. [Google Scholar] [CrossRef]
- Dimitrov, I.; Naneva, L.; Doytchinova, I.; Bangov, I. AllergenFP: Allergenicity Prediction by Descriptor Fingerprints. Bioinformatics 2014, 30, 846–851. [Google Scholar] [CrossRef]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 Web Portal for Protein Modeling, Prediction and Analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef]
- Zhang, Y. I-TASSER Server for Protein 3D Structure Prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef]
- Källberg, M.; Wang, H.; Wang, S.; Peng, J.; Wang, Z.; Lu, H.; Xu, J. Template-Based Protein Structure Modeling Using the RaptorX Web Server. Nat. Protoc. 2012, 7, 1511–1522. [Google Scholar] [CrossRef] [PubMed]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate Prediction of Protein Structures and Interactions Using a Three-Track Neural Network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Randall, A.Z.; Sweredoski, M.J.; Baldi, P. SCRATCH: A Protein Structure and Structural Feature Prediction Server. Nucleic Acids Res. 2005, 33, W72–W76. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A Program to Check the Stereochemical Quality of Protein Structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Colovos, C.; Yeates, T. ERRAT: An Empirical Atom-Based Method for Validating Protein Structures. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. ProSA-Web: Interactive Web Service for the Recognition of Errors in Three-Dimensional Structures of Proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef]
- Sippl, M.J. Recognition of Errors in Three-Dimensional Structures of Proteins. Proteins Struct. Funct. Bioinform. 1993, 17, 355–362. [Google Scholar] [CrossRef]
- Heo, L.; Park, H.; Seok, C. GalaxyRefine: Protein Structure Refinement Driven by Side-Chain Repacking. Nucleic Acids Res. 2013, 41, W384–W388. [Google Scholar] [CrossRef]
- Park, B.S.; Song, D.H.; Kim, H.M.; Choi, B.-S.; Lee, H.; Lee, J.-O. The Structural Basis of Lipopolysaccharide Recognition by the TLR4--MD-2 Complex. Nature 2009, 458, 1191–1195. [Google Scholar] [CrossRef]
- de Vries, S.J.; Bonvin, A.M.J.J. CPORT: A Consensus Interface Predictor and Its Performance in Prediction-Driven Docking with HADDOCK. PLoS ONE 2011, 6, e17695. [Google Scholar] [CrossRef]
- Xue, L.C.; Rodrigues, J.P.; Kastritis, P.L.; Bonvin, A.M.; Vangone, A. PRODIGY: A Web Server for Predicting the Binding Affinity of Protein--Protein Complexes. Bioinformatics 2016, 32, 3676–3678. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Kaminski, G.A.; Friesner, R.A.; Tirado-Rives, J.; Jorgensen, W.L. Evaluation and Reparametrization of the OPLS-AA Force Field for Proteins via Comparison with Accurate Quantum Chemical Calculations on Peptides. J. Phys. Chem. B 2001, 105, 6474–6487. [Google Scholar] [CrossRef]
- Turner, P. XMGRACE, Version 5.1. 19; Center for Coastal and Land-Margin Research, Oregon Graduate Institute of Science and Technology: Beaverton, OR, USA, 2005. [Google Scholar]
- Rapin, N.; Lund, O.; Bernaschi, M.; Castiglione, F. Computational Immunology Meets Bioinformatics: The Use of Prediction Tools for Molecular Binding in the Simulation of the Immune System. PLoS ONE 2010, 5, e9862. [Google Scholar] [CrossRef] [PubMed]
- Ismail, S.; Ahmad, S.; Azam, S.S. Immunoinformatics Characterization of SARS-CoV-2 Spike Glycoprotein for Prioritization of Epitope Based Multivalent Peptide Vaccine. J. Mol. Liq. 2020, 314, 113612. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, V.; Singh, M.P. Immuno-Informatics Approach to Design a Multi-Epitope Vaccine to Combat Cytomegalovirus Infection. Eur. J. Pharm. Sci. 2020, 147, 105279. [Google Scholar] [CrossRef]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI Search and Sequence Analysis Tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef]
- Grote, A.; Hiller, K.; Scheer, M.; Münch, R.; Nörtemann, B.; Hempel, D.C.; Jahn, D. JCat: A Novel Tool to Adapt Codon Usage of a Target Gene to Its Potential Expression Host. Nucleic Acids Res. 2005, 33, W526–W531. [Google Scholar] [CrossRef]
- Krishnarjuna, B.; Andrew, D.; MacRaild, C.A.; Morales, R.A.V.; Beeson, J.G.; Anders, R.F.; Richards, J.S.; Norton, R.S. Strain-Transcending Immune Response Generated by Chimeras of the Malaria Vaccine Candidate Merozoite Surface Protein 2. Sci. Rep. 2016, 6, 20613. [Google Scholar] [CrossRef]
- Trimaille, T.; Verrier, B. Micelle-Based Adjuvants for Subunit Vaccine Delivery. Vaccines 2015, 3, 803–813. [Google Scholar] [CrossRef]
- Zaheer, T.; Waseem, M.; Waqar, W.; Dar, H.A.; Shehroz, M.; Naz, K.; Ishaq, Z.; Ahmad, T.; Ullah, N.; Bakhtiar, S.M.; et al. Anti-COVID-19 Multi-Epitope Vaccine Designs Employing Global Viral Genome Sequences. PeerJ 2020, 8, e9541. [Google Scholar] [CrossRef]
- Khatoon, N.; Pandey, R.K.; Prajapati, V.K. Exploring Leishmania Secretory Proteins to Design B and T Cell Multi-Epitope Subunit Vaccine Using Immunoinformatics Approach. Sci. Rep. 2017, 7, 8285. [Google Scholar] [CrossRef] [PubMed]
- Jabbar, B.; Rafique, S.; Salo-Ahen, O.M.H.; Ali, A.; Munir, M.; Idrees, M.; Mirza, M.U.; Vanmeert, M.; Shah, S.Z.; Jabbar, I.; et al. Antigenic Peptide Prediction from E6 and E7 Oncoproteins of HPV Types 16 and 18 for Therapeutic Vaccine Design Using Immunoinformatics and MD Simulation Analysis. Front. Immunol. 2018, 9, 3000. [Google Scholar] [CrossRef] [PubMed]
- Almanzar, G.; Herndler-Brandstetter, D.; Chaparro, S.V.; Jenewein, B.; Keller, M.; Grubeck-Loebenstein, B. Immunodominant Peptides from Conserved Influenza Proteins--A Tool for More Efficient Vaccination in the Elderly? Wien. Med. Wochenschr. 2007, 157, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Oyarzún, P.; Kobe, B. Recombinant and Epitope-Based Vaccines on the Road to the Market and Implications for Vaccine Design and Production. Hum. Vaccines Immunother. 2016, 12, 763–767. [Google Scholar] [CrossRef]
- Dong, R.; Chu, Z.; Yu, F.; Zha, Y. Contriving Multi-Epitope Subunit of Vaccine for COVID-19: Immunoinformatics Approaches. Front. Immunol. 2020, 11, 1784. [Google Scholar] [CrossRef]
- Zhang, L. Multi-Epitope Vaccines: A Promising Strategy against Tumors and Viral Infections. Cell. Mol. Immunol. 2018, 15, 182–184. [Google Scholar] [CrossRef]
- Alam, A.; Khan, A.; Imam, N.; Siddiqui, M.F.; Waseem, M.; Malik, M.Z.; Ishrat, R. Design of an Epitope-Based Peptide Vaccine against the SARS-CoV-2: A Vaccine-Informatics Approach. Brief. Bioinform. 2021, 22, 1309–1323. [Google Scholar] [CrossRef]
- Mahmud, S.; Rafi, M.; Paul, G.K.; Promi, M.M.; Shimu, M.; Sultana, S.; Biswas, S.; Emran, T.B.; Dhama, K.; Alyami, S.A.; et al. Designing a Multi-Epitope Vaccine Candidate to Combat MERS-CoV by Employing an Immunoinformatics Approach. Sci. Rep. 2021, 11, 15431. [Google Scholar] [CrossRef]
- MacRaild, C.A.; Zachrdla, M.; Andrew, D.; Krishnarjuna, B.; Nováček, J.; Žídek, L.; Sklenář, V.; Richards, J.S.; Beeson, J.G.; Anders, R.F.; et al. Conformational Dynamics and Antigenicity in the Disordered Malaria Antigen Merozoite Surface Protein 2. PLoS ONE 2015, 10, e0119899. [Google Scholar] [CrossRef]
- Jain, S.; Baranwal, M. Conserved Peptide Vaccine Candidates Containing Multiple Ebola Nucleoprotein Epitopes Display Interactions with Diverse HLA Molecules. Med. Microbiol. Immunol. 2019, 208, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Fritsche, P.J. Vaccine Hypersensitivity-Update and Overview. Swiss Med. Wkly. 2010, 140, 238–246. [Google Scholar] [PubMed]
- Cohen, A.D.; Shoenfeld, Y. Vaccine-Induced Autoimmunity. J. Autoimmun. 1996, 9, 699. [Google Scholar] [CrossRef] [PubMed]
- Meyboom, R.H.B.; Fucik, H.; Edwards, I.R. Thrombocytopenia Reported in Association with Hepatitis B and A Vaccines. Lancet 1995, 345, 1638. [Google Scholar] [CrossRef]
- Topaloglu, H.; Berker, M.; Kansu, T.; Saatci, U.; Renda, Y. Optic Neuritis and Myelitis after Booster Tetanus Toxoid Vaccination. Lancet (Br. Ed.) 1992, 339, 178–179. [Google Scholar] [CrossRef]
- Akya, A.; Farasat, A.; Ghadiri, K.; Rostamian, M. Identification of HLA-I Restricted Epitopes in Six Vaccine Candidates of Leishmania Tropica Using Immunoinformatics and Molecular Dynamics Simulation Approaches. Infect. Genet. Evol. 2019, 75, 103953. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | Epitopes | HLA Alleles | Protein | VaxiJen Score | Imunogenicity Score | IC50 Value |
|---|---|---|---|---|---|---|
| 383–391 | NIDIFNPKY | HLA-A*0101 | Fusion glycoprotein | 0.7826 | 0.11367 | 49.43 |
| 541–549 | LIAVGLLLY | 0.8288 | 0.06096 | 30.13 | ||
| 525–533 | IMITTIIIV | HLA-A*0201 | Fusion glycoprotein | 0.5548 | 0.43542 | 77.62 |
| 512–520 | LLHNNAGK | HLA-A*0301 | Fusion glycoprotein | 0.5354 | 0.09092 | 60.53 |
| 132–140 | KTKNTTTTK | Attachment glycoprotein | 0.8053 | 0.05327 | 54.83 | |
| 57–64 | ITIELNIK | HLA-A*1101 | Fusion glycoprotein | 1.4507 | 0.04972 | 13.03 |
| 201–210 | KQLPIVNK | 0.8386 | 0.13078 | 27.04 | ||
| 148–156 | IASGAVSK | 0.8027 | 0.08501 | 36.48 |
| Position | Epitope | HLA Alleles | Protein | VaxJen Score | IC50 Value |
|---|---|---|---|---|---|
| 138–152 | LGFLLGVGSAIASGI | DRB1*0101 | Fusion glycoprotein | 0.6271 | 37.33 |
| 546–560 | LLLYCKARSTPVTLS | DRB1*0101 | 1.2472 | 2.75 | |
| AIIFIASANHKVTLT | DRB1*0101 | Attachment glycoprotein | 0.7845 | 40.18 | |
| 58–72 | AIIFIASANHKVTLT | DRB1*0401 | 264.24 |
| Model No. | Ramachandran Plot Analysis | ERRAT Score | |||
|---|---|---|---|---|---|
| Most Favored Region | Additionally Allowed Regions | Generously Allowed Regions | Outlier Residues | ||
| Model 1 | 89% | 9% | 1% | 1% | 89.9194 |
| Model 2 | 84.50% | 13.50% | 1% | 1% | 96.6942 |
| Model 3 | 87.50% | 11% | 0% | 1.50% | 91.5323 |
| Model 4 | 89.50% | 8% | 1% | 1.50% | 90.1639 |
| Model 5 | 89% | 9.50% | 1.50% | 0% | 91.4286 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dar, H.A.; Almajhdi, F.N.; Aziz, S.; Waheed, Y. Immunoinformatics-Aided Analysis of RSV Fusion and Attachment Glycoproteins to Design a Potent Multi-Epitope Vaccine. Vaccines 2022, 10, 1381. https://doi.org/10.3390/vaccines10091381
Dar HA, Almajhdi FN, Aziz S, Waheed Y. Immunoinformatics-Aided Analysis of RSV Fusion and Attachment Glycoproteins to Design a Potent Multi-Epitope Vaccine. Vaccines. 2022; 10(9):1381. https://doi.org/10.3390/vaccines10091381
Chicago/Turabian StyleDar, Hamza Arshad, Fahad Nasser Almajhdi, Shahkaar Aziz, and Yasir Waheed. 2022. "Immunoinformatics-Aided Analysis of RSV Fusion and Attachment Glycoproteins to Design a Potent Multi-Epitope Vaccine" Vaccines 10, no. 9: 1381. https://doi.org/10.3390/vaccines10091381
APA StyleDar, H. A., Almajhdi, F. N., Aziz, S., & Waheed, Y. (2022). Immunoinformatics-Aided Analysis of RSV Fusion and Attachment Glycoproteins to Design a Potent Multi-Epitope Vaccine. Vaccines, 10(9), 1381. https://doi.org/10.3390/vaccines10091381