
Genome Similarities between Human-Derived and Mink-Derived SARS-CoV-2 Make Mink a Potential Reservoir of the Virus


Abstract
:1. Introduction
2. Methods
2.1. Mink-Derived SARS-CoV-2 Genome Sequences
2.2. Mutational Analysis of the Mink-Derived SARS-CoV-2 Genome Sequences
3. Results
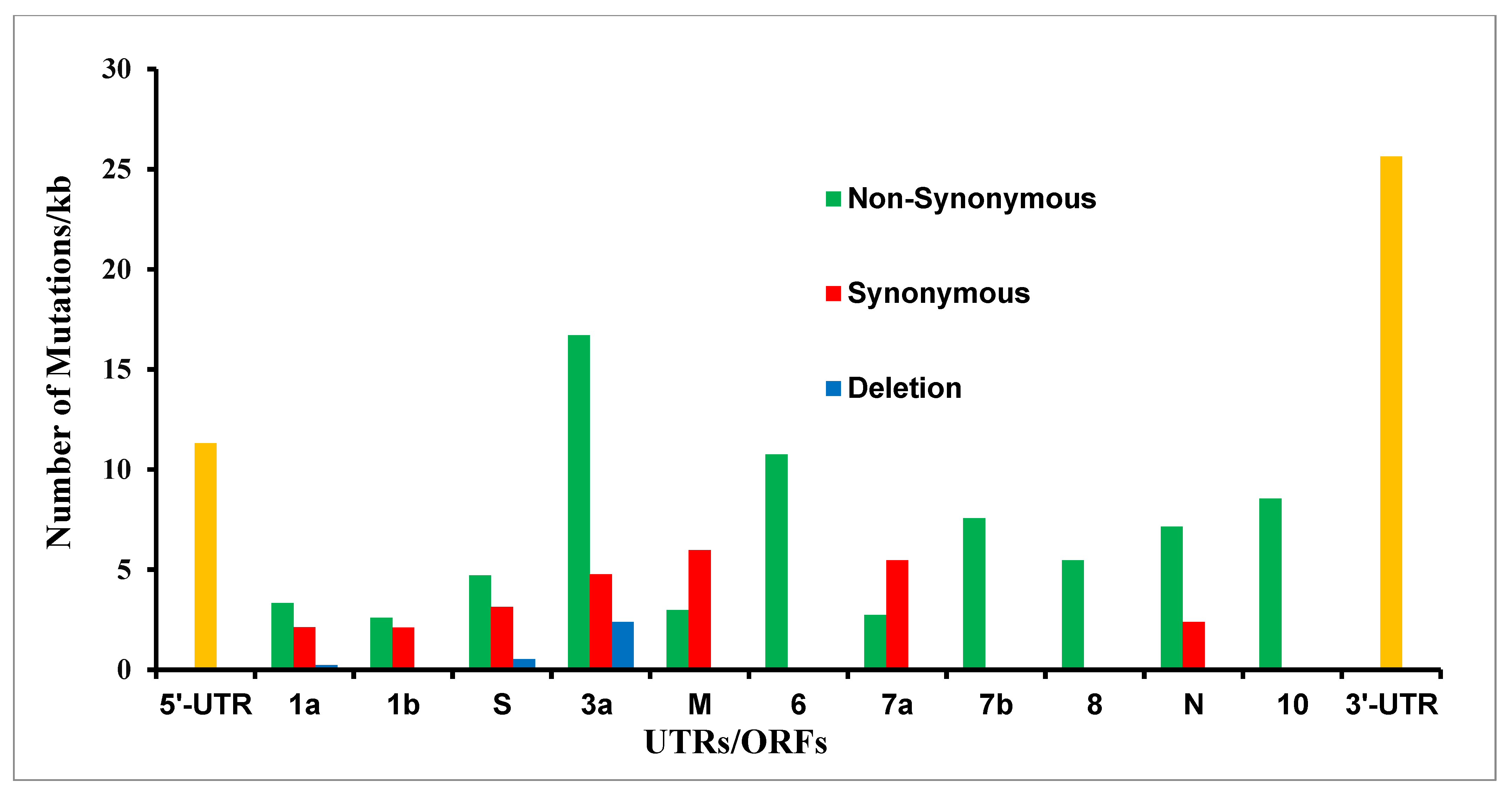
3.1. Pipeline for the Mutation Analysis
3.2. SARS-CoV-2 Hosts Range
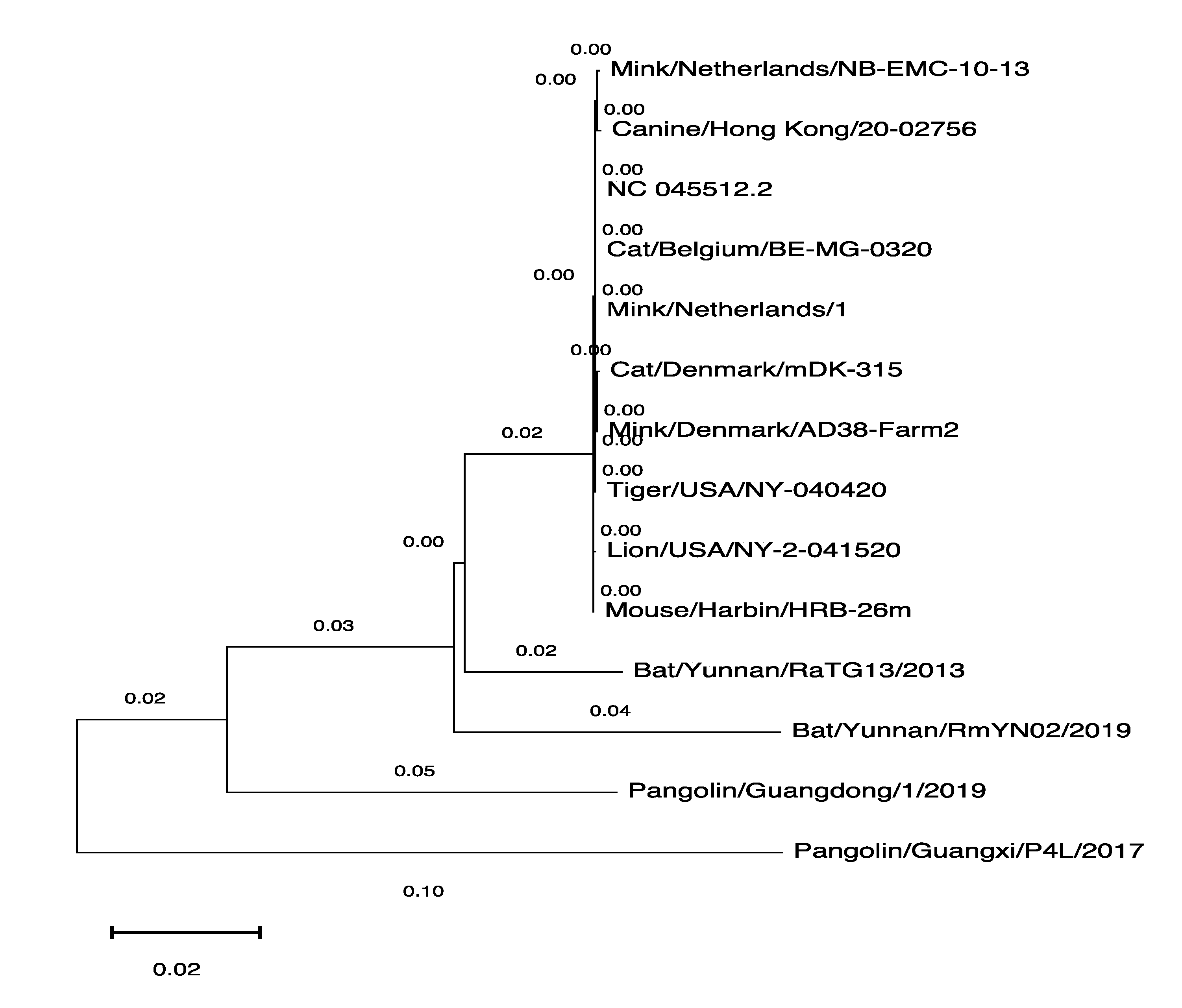
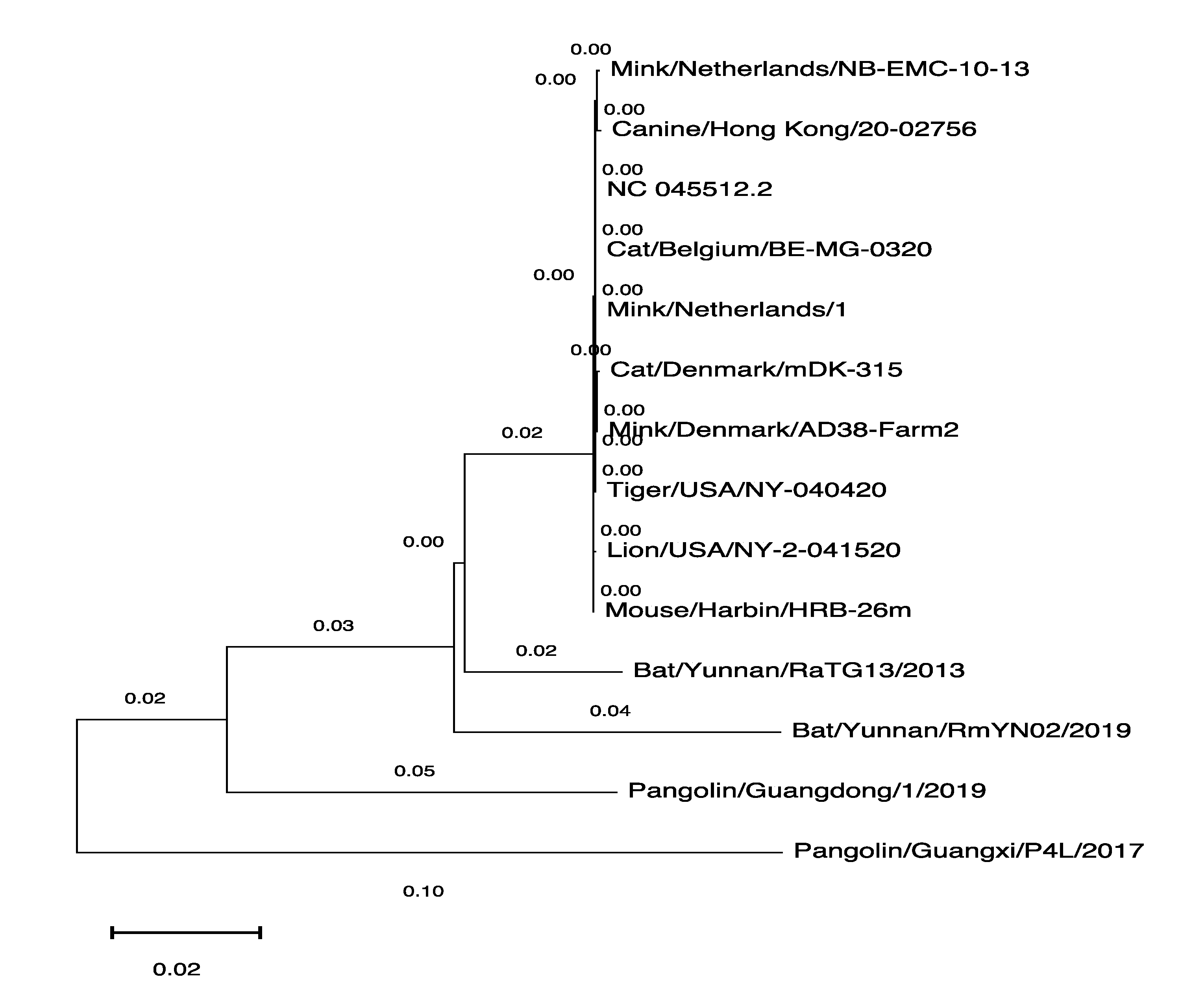
3.3. The Phylogenetic Tree of SARS-CoV-2 Genomes Derived from Various Hosts
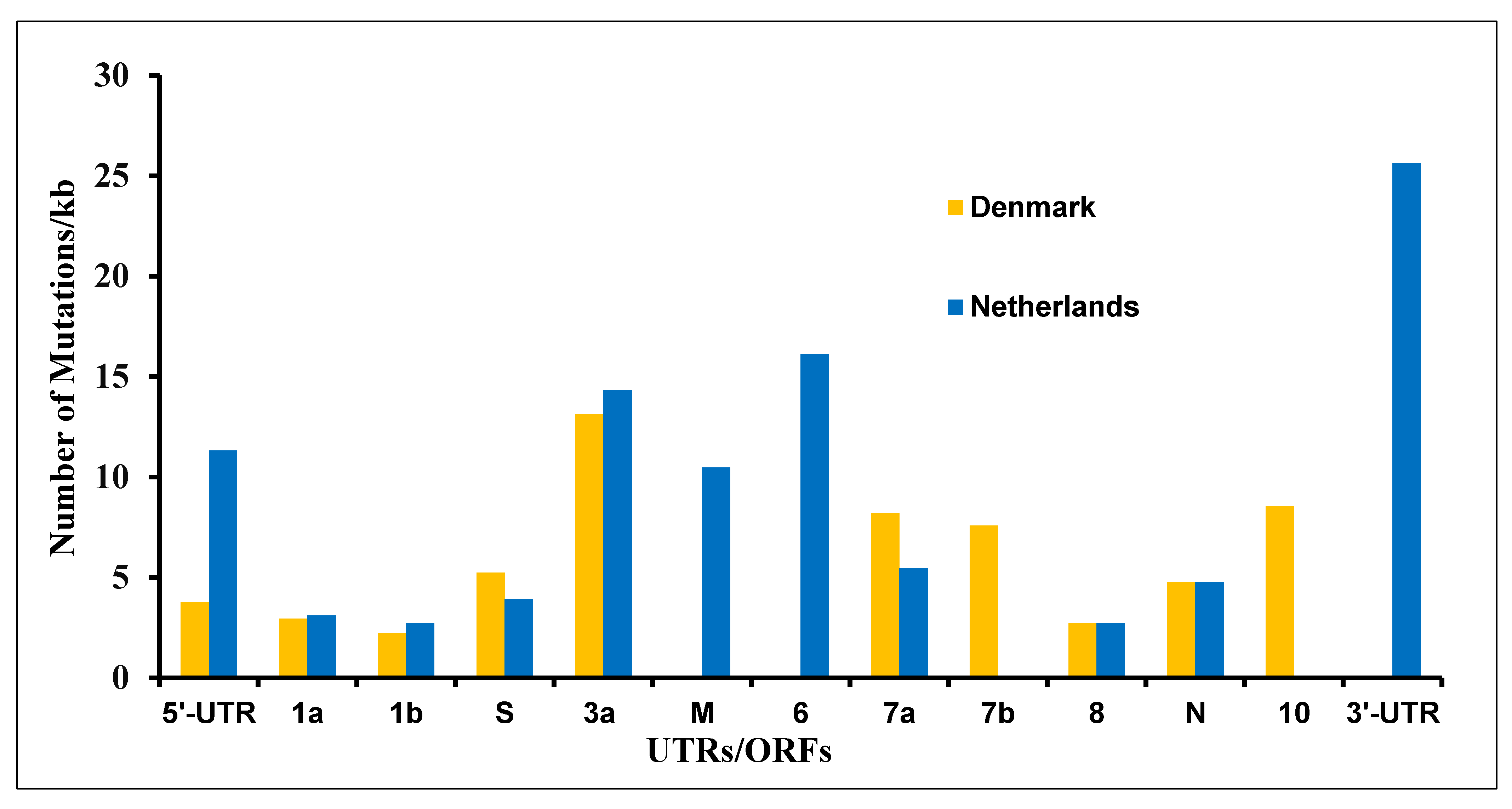
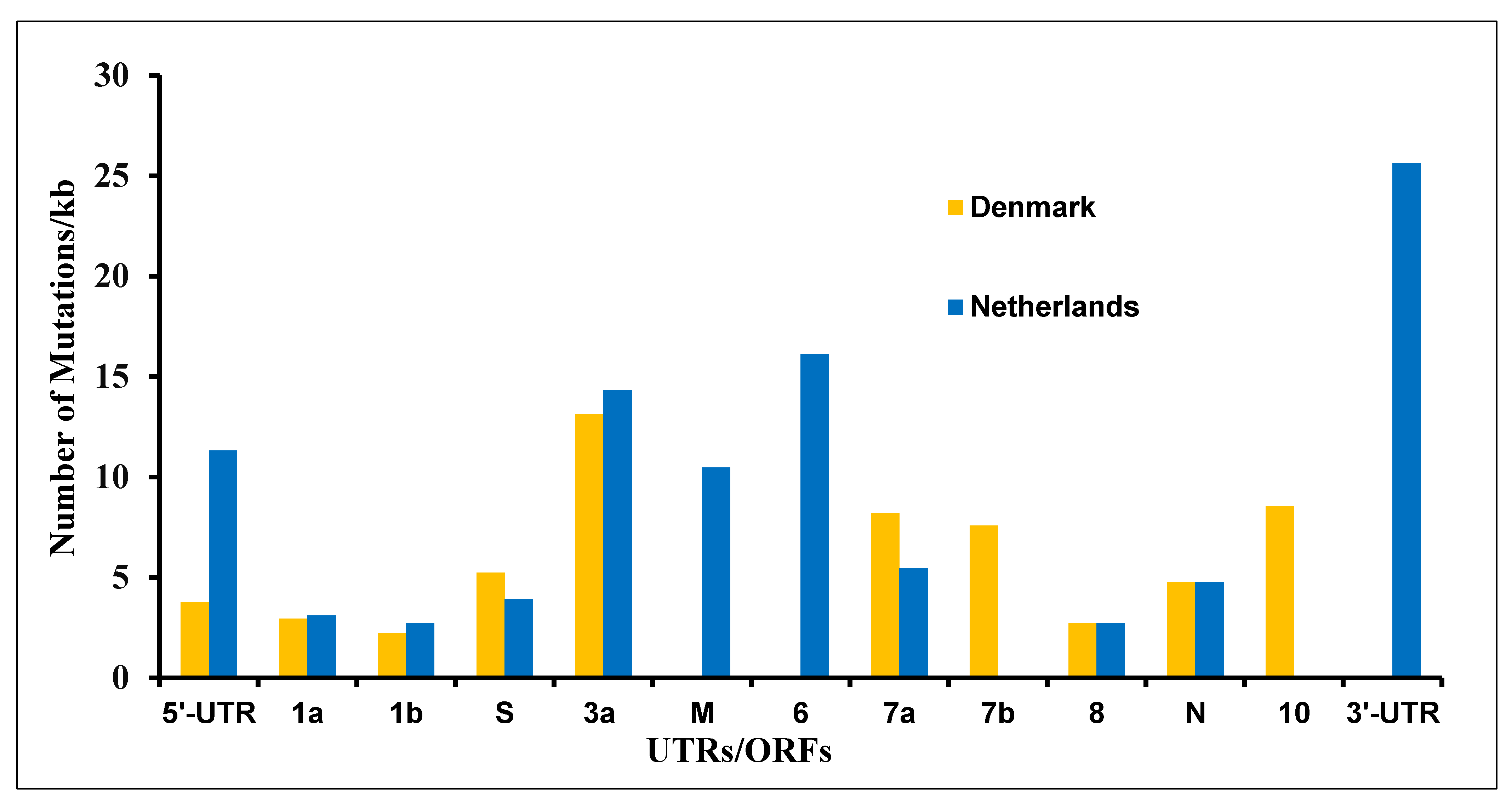
3.4. The Mutations in SARS-CoV-2 Genome Derived from the Netherlands and Denmark Mink Farms
3.5. The ΔH69/V70 and Several Other Mutations Were Prevalent in Mink-Derived SARS-CoV-2 Genome
3.6. Mutations at Amino Acid Level
3.7. The Frame-Shift and Nonsense Mutations
4. Discussions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| GISAID | Global initiative on sharing all influenza data |
| NCBI | National Center for Biotechnology Information |
| NSP6 | Non-structural protein 6 |
| ORF-8 | Open Reading Frame-8 |
| UTRs | Untranslated regions |
References
- Mori, M.; Capasso, C.; Carta, F.; a Donald, W.; Supuran, C.T. A deadly spillover: SARS-CoV-2 outbreak. Expert Opin. Ther. Pat. 2020, 30, 481–485. [Google Scholar] [CrossRef]
- Kemp, S.A.; Datir, R.P.; Collier, D.A.; Ferreira, I.A.T.M.; Carabelli, A.; Harvey, W.; Robertson, D.L.; Gupta, R.K. Recurrent emergence and transmission of a SARS-CoV-2 Spike deletion ΔH69/V70. bioRxiv 2021, 14, 422555. [Google Scholar] [CrossRef]
- New Coronavirus Variant: What Do We know? Available online: https://www.bbc.com/news/health-55388846 (accessed on 20 December 2020).
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.Y.; et al. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell 2020, 181, 894–904. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef]
- Oreshkova, N.; Molenaar, R.J.; Vreman, S.; Harders, F.; Munnink, B.B.O.; Hakze-van der Honing, R.W.; Gerhards, N.; Tolsma, P.; Bouwstra, R.; Sikkema, R.S.; et al. SARS-CoV-2 infection in farmed minks, the Netherlands, April and May 2020. Eurosurveillance 2020, 25, 2001005. [Google Scholar] [CrossRef] [PubMed]
- Munnink, B.B.O.; Sikkema, R.S.; Nieuwenhuijse, D.F.; Molenaar, R.J.; Munger, E.; Molenkamp, R.; van der Spek, A.; Tolsma, P.; Rietveld, A.; Brouwer, M.; et al. Transmission of SARS-CoV-2 on mink farms between humans and mink and back to humans. Science 2021, 371, 172–177. [Google Scholar] [CrossRef]
- Enserink, M. Coronavirus rips through Dutch mink farms, triggering culls. Science 2020, 368, 1169. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, E.C.; Reid, T.J. Animals and SARS-CoV-2: Species susceptibility and viral transmission in experimental and natural conditions, and the potential implications for community transmission. Transbound. Emerg. Dis. 2020, 68, 1850–1867. [Google Scholar] [CrossRef] [PubMed]
- Toby, S. Mink Infected Two Humans with Coronavirus: Dutch Government; Reuters: London, UK, 2020; Available online: https://www.reuters.com/article/us-h (accessed on 19 October 2020).
- Animal Guardians. Available online: http://animalguardians.us/issues/fur-industry (accessed on 7 February 2021).
- Chan, J.F.; Kok, K.H.; Zhu, Z.; Chu, H.; To, K.K.; Yuan, S.; Yuen, K.Y. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 2020, 9, 221–236. [Google Scholar] [CrossRef]
- Peck, K.M.; Lauring, A.S. Complexities of viral mutation rates. J. Virol. 2018, 92, e01031-17. [Google Scholar] [CrossRef]
- Pachetti, M.; Marini, B.; Benedetti, F.; Giudici, F.; Mauro, E.; Storici, P.; Masciovecchio, C.; Angeletti, S.; Ciccozzi, M.; Gallo, R.; et al. Emerging SARS-CoV-2 mutation hot spots include a novel RNA-dependent-RNA polymerase variant. J. Transl. Med. 2020, 18, 179. [Google Scholar] [CrossRef]
- Phan, T. Genetic diversity and evolution of SARS-CoV-2. Infect. Genet. Evol. 2020, 81, 104260. [Google Scholar] [CrossRef]
- Tang, X.; Wu, C.; Li, X.; Song, Y.; Yao, X.; Wu, X.; Duan, Y.; Zhang, H.; Wang, Y.; Qian, Z.; et al. On the origin and continuing evolution of SARS-CoV-2. Natl. Sci. Rev. 2020, 7, 1012–1023. [Google Scholar] [CrossRef]
- Wang, C.; Liu, Z.; Chen, Z.; Huang, X.; Xu, M.; He, T.; Zhang, Z. The establishment of reference sequence for SARS-CoV-2 and variation analysis. J. Med. Virol. 2020, 92, 667–674. [Google Scholar] [CrossRef]
- GISAID. Available online: https://www.gisaid.org (accessed on 4 November 2020).
- Khalid, M.; Murphy, D.; Shoai, M.; Al-ebini, Y. Host’s Specific SARS-CoV-2 Mutations: Insertion of the Phenylalanine in the NSP6 Linked to the United Kingdom and Premature Termination of the ORF-8 Associated with the European and the United States of America Derived Samples. bioRxiv 2021, 29, 424530. [Google Scholar] [CrossRef]
- MultAlin. Available online: http://multalin.toulouse.inra.fr/multalin/ (accessed on 25 November 2020).
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. Author Correction: A new coronavirus associated with human respiratory disease in China. Nature 2020, 580, E7. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827. [Google Scholar] [CrossRef]
- Jo, W.K.; de Oliveira-Filho, E.F.; Rasche, A.; Greenwood, A.D.; Osterrieder, K.; Drexler, J.F. Potential zoonotic sources of SARS-CoV-2 infections. Transbound. Emerg. Dis. 2021, 68, 1824–1834. [Google Scholar] [CrossRef]
- Fenollar, F.; Mediannikov, O.; Maurin, M.; Devaux, C.; Colson, P.; Levasseur, A.; Fournier, P.E.; Raoult, D. Mink, SARS-CoV-2, and the Human-Animal Interface. Front. Microbiol. 2021, 12, 663815. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, A.; Chaudhari, M.; Mahera, S.; Saiyed, Z.; Nathani, N.M.; Shukla, S.; Patel, D.; Patel, C.; Joshi, M.; Joshi, C.G. In-Silico analysis reveals lower transcription efficiency of C241T variant of SARS-CoV-2 with host replication factors MADP1 and hnRNP-1. Inform. Med. Unlocked 2021, 25, 100670. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, J.; Zhu, W.; Tonnu, N.; Singer, O.; Hunter, T.; Ryan, A.L.; Pao, G.M. The D614G mutation in the SARS-CoV2 Spike protein increases infectivity in an ACE2 receptor dependent manner. bioRxiv 2020, 21, 214932. [Google Scholar] [CrossRef]
- Maurya, R.; Mishra, P.; Swaminathan, A.; Ravi, V.; Saifi, S.; Kanakan, A.; Mehta, P.; Devi, P.; Praveen, S.; Budhiraja, S.; et al. SARS-CoV-2 Mutations and COVID-19 Clinical Outcome: Mutation Global Frequency Dynamics and Structural Modulation Hold the Key. Front. Cell. Infect. Microbiol. 2022, 12, 868414. [Google Scholar] [CrossRef]
- Raheja, H.; Das, S.; Banerjee, A.; Dikshaya, P.; Deepika, C.; Mukhopadhyay, D.; Ramachandra, S.G.; Das, S. RG203KR Mutations in SARS-CoV-2 Nucleocapsid: Assessing the Impact Using a Virus-Like Particle Model System. Microbiol. Spectr. [CrossRef]
- Meng, B.; Kemp, S.A.; Papa, G.; Datir, R.; Ferreira, I.A.; Marelli, S.; Harvey, W.T.; Lytras, S.; Mohamed, A.; Gallo, G.; et al. Recurrent emergence of SARS-CoV-2 spike deletion H69/V70 and its role in the Alpha variant B.1.1.7. Cell Rep. 2021, 35, 109292. [Google Scholar] [CrossRef]
- Zinzula, L. Lost in deletion: The enigmatic ORF8 protein of SARS-CoV-2. Biochem. Biophys. Res. Commun. 2020, 538, 116–124. [Google Scholar] [CrossRef]
- Pereira, F. Evolutionary dynamics of the SARS-CoV-2 ORF8 accessory gene. Infect. Genet. Evol. 2020, 85, 104525. [Google Scholar] [CrossRef]
- Muth, D.; Corman, V.M.; Roth, H.; Binger, T.; Dijkman, R.; Gottula, L.T.; Gloza-Rausch, F.; Balboni, A.; Battilani, M.; Rihtarič, D.; et al. Attenuation of replication by a 29 nucleotide deletion in SARS-coronavirus acquired during the early stages of human-to-human transmission. Sci. Rep. 2018, 8, 15177. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No. | Name | Sequences |
|---|---|---|
| 1 | Human (Homo sapiens) | 3,240,911 |
| 2 | Mink (Neovison vison) | 994 |
| 3 | Cat (Felis catus) | 70 |
| 4 | Lion (Panthera leo) | 37 |
| 5 | Dog (Canis lupus familiaries | 28 |
| 6 | Pangolin (Manis javanica) | 19 |
| 7 | Tiger (Panthera tigris jacksoni) | 13 |
| 8 | Otter (Aonyx cinereus) | 5 |
| 9 | Mouse (Mus musculus) | 4 |
| 10 | Bat (Rhinolophus malayanus) | 4 |
| 11 | Bat (Rhinolophus shameli) | 2 |
| 12 | Bat (Rhinolophus affinis) | 1 |
| 13 | Monkey (Chlorocebus sabaeus) | 1 |
| 14 | Pangolin (Manis pentadactyla) | 1 |
| S. No. | Mutation | Amino Acid | ORFs | Denmark % | Netherlands % |
|---|---|---|---|---|---|
| 1 | C241T | N/A | 5′-UTR | 100.0 | 90.7 |
| 2 | TTA516--- | M84 deletion | 1a | 83.5 | 0.0 |
| 3 | C1380T | A372V | 1a | 0.0 | 50.9 |
| 4 | T3037C | Silent | 1a | 100.0 | 77.6 |
| 5 | C5144T | Silent | 1a | 84.2 | 0.0 |
| 6 | ATA6510--- | S2082 deletion | 1a | 84.2 | 16.0 |
| 7 | C11776T | Silent | 1a | 84.2 | 0.0 |
| 8 | G14274A | Silent | 1b | 0.0 | 51.4 |
| 9 | C14408T | P314L | 1b | 100.0 | 89.3 |
| 10 | C15656T | T730I | 1b | 93.2 | 5.7 |
| 11 | ACATGT21766------ | H69/V70 deletion | S | 84.2 | 0.0 |
| 12 | A22920T | Y453F | S | 90.2 | 19.8 |
| 13 | A23403G | D614G | S | 100.0 | 89.3 |
| 14 | A24862G | Silent | S | 0.0 | 51.4 |
| 15 | C25936T | H182Y | 3a | 93.2 | 43.9 |
| 16 | G28854T | S194L | N | 84.2 | 0.0 |
| 17 | GGG28881AAC | RG203KR | N | 97.7 | 0.0 |
| 18 | T29726- | - | 10/3′-UTR | 83.5 | 0.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khalid, M.; Alshishani, A.; Al-ebini, Y. Genome Similarities between Human-Derived and Mink-Derived SARS-CoV-2 Make Mink a Potential Reservoir of the Virus. Vaccines 2022, 10, 1352. https://doi.org/10.3390/vaccines10081352
Khalid M, Alshishani A, Al-ebini Y. Genome Similarities between Human-Derived and Mink-Derived SARS-CoV-2 Make Mink a Potential Reservoir of the Virus. Vaccines. 2022; 10(8):1352. https://doi.org/10.3390/vaccines10081352
Chicago/Turabian StyleKhalid, Mohammad, Anas Alshishani, and Yousef Al-ebini. 2022. "Genome Similarities between Human-Derived and Mink-Derived SARS-CoV-2 Make Mink a Potential Reservoir of the Virus" Vaccines 10, no. 8: 1352. https://doi.org/10.3390/vaccines10081352
APA StyleKhalid, M., Alshishani, A., & Al-ebini, Y. (2022). Genome Similarities between Human-Derived and Mink-Derived SARS-CoV-2 Make Mink a Potential Reservoir of the Virus. Vaccines, 10(8), 1352. https://doi.org/10.3390/vaccines10081352