
Application of mRNA Technology in Cancer Therapeutics

Abstract
:1. Introduction
2. Cancer Immunology and Immunotherapy
3. The Evolving Role of mRNA Technology in Cancer Immunotherapy
4. The Immunogenicity and Molecular Biology of mRNA-Based Immunotherapy
4.1. mRNA Vaccine Structure
4.2. mRNA Delivery Platforms
4.2.1. Synthetic Systems
- Lipid-based Delivery Systems
- 2.
- Polymer-based Delivery Systems
- 3.
- Peptide-based Delivery
4.2.2. Biological Systems
- Ex Vivo Transfected Cellular Systems
- Dendritic Cells
- b.
- CAR-T Cells
- 2.
- Viral Constructs
5. Clinical Applications
5.1. Personalized mRNA Vaccines
5.1.1. Naked mRNA Vaccines
5.1.2. LNP mRNA
5.1.3. Dendritic Cell-Based Vaccines
5.1.4. Viral-Based Self-Replicating mRNA Vaccines
5.2. mRNA-Engineered Cellular-Based Immunotherapy and Gene Editing
6. Boosting Immune Response
6.1. Modulation of the Tumor Microenvironment
6.2. Potential Combinations
7. Conclusions and Future Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cooley, W.B. The treatment of malignant tumors by repeated inoculations of erysipelas: With a report of ten original cases. Am. J. Med. Sci. 1893, 105, 487–511. [Google Scholar] [CrossRef]
- Small, E.J.; Schellhammer, P.F.; Higano, C.S.; Redfern, C.H.; Nemunaitis, J.J.; Valone, F.H.; Verjee, S.S.; Jones, L.A.; Hershberg, R.M. Placebo-controlled phase III trial of immunologic therapy with sipuleucel-T (APC8015) in patients with metastatic, asymptomatic hormone refractory prostate cancer. J. Clin. Oncol. 2006, 24, 3089–3094. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi, N.; Akbari, M.; Ghanaatian, M.; Roozbahani, M.P.; Adelian, S.; Borjian, B.M.; Yazdani, E.; Ahmadi, A.; Hamblin, M.R. Development of neoantigens: From identification in cancer cells to application in cancer vaccines. Expert. Rev. Vaccines 2021, 19, 941–955. [Google Scholar] [CrossRef]
- Fritsch, E.F.; Burkhardt, U.; Hacohen, N.; Wu, C.J. Personal Neoantigen Cancer Vaccines: A Road Not Fully Paved. Cancer Immunol. Res. 2020, 8, 1465–1469. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef]
- Sahin, U.; Karikó, K.; Türeci, Ö. mRNA-based therapeutics—Developing a new class of drugs. Nat. Rev. Drug Discov. 2014, 13, 759–780. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.P.; Simon, P.; Löwer, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrörs, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef]
- Xu, S.; Yang, K.; Li, R.; Zhang, L. mRNA Vaccine Era—Mechanisms, Drug Platform and Clinical Prospection. Int. J. Mol. Sci. 2020, 21, 6582. [Google Scholar] [CrossRef]
- Upadhyay, S.; Sharma, N.; Gupta, K.B.; Dhiman, M. Role of immune system in tumor progression and carcinogenesis. J. Cell Biochem. 2018, 119, 5028–5042. [Google Scholar] [CrossRef]
- Anari, F.; Ramamurthy, C.; Zibelman, M. Impact of tumor microenvironment composition on therapeutic responses and clinical outcomes in cancer. Future Oncol. 2018, 14, 1409–1421. [Google Scholar] [CrossRef]
- Khalaf, K.; Hana, D.; Chou, J.T.; Singh, C.; Mackiewicz, A.; Kaczmarek, M. Aspects of the Tumor Microenvironment Involved in Immune Resistance and Drug Resistance. Front. Immunol. 2021, 12, 656364. [Google Scholar] [CrossRef]
- Li, J.; Byrne, K.T.; Yan, F.; Yamazoe, T.; Chen, Z.; Baslan, T.; Richman, L.P.; Lin, J.H.; Sun, Y.H.; Rech, A.J.; et al. Tumor Cell-Intrinsic Factors Underlie Heterogeneity of Immune Cell Infiltration and Response to Immunotherapy. Immunity 2018, 49, 178–193. [Google Scholar] [CrossRef]
- Gerard, C.L.; Delyon, J.; Wicky, A.; Homicsko, K.; Cuendet, M.A.; Michielin, O. Turning tumors from cold to inflamed to improve immunotherapy response. Cancer Treat. Rev. 2021, 101, 102227. [Google Scholar] [CrossRef] [PubMed]
- Ito, F.; Chang, A.E. Cancer immunotherapy: Current status and future directions. Surg. Oncol. Clin. N. Am. 2013, 22, 765–783. [Google Scholar] [CrossRef] [PubMed]
- Ayana, R.; Kumar, A.R.; Devan, A.R.; Nair, B.; Vinod, B.S.; Nath, L.R. Harnessing the immune system against cancer: Current immunotherapy approaches and therapeutic targets. Mol. Biol. Rep. 2021, 48, 8075–8095. [Google Scholar]
- Stephan, K.; Matthias, I.; Sebastian, K. Advances in cancer immunotherapy 2019—Latest trends. Exp. Clin. Cancer Res. 2019, 38, 268. [Google Scholar]
- Marshall, H.T.; Djamgoz, M.B.A. Immuno-Oncology: Emerging targets and combination therapies. Front. Oncol. 2018, 8, 315. [Google Scholar] [CrossRef]
- Khalil, D.N.; Smith, E.L.; Brentjens, R.J.; Wolchok, J.D. The future of cancer treatment: Immunomodulation, CARs and combination immunotherapy. Nat. Rev. Clin. Oncol. 2016, 13, 273–290. [Google Scholar] [CrossRef]
- Wang, Z.; Cao, Y.J. Adoptive Cell Therapy Targeting Neoantigens: A Frontier for Cancer Research. Front. Immunol. 2020, 11, 176. [Google Scholar] [CrossRef] [PubMed]
- Bishani, A.; Chernolovskaya, E.L. Activation of innate immunity by therapeutic nucleic acids. Int. J. Mol. Sci. 2021, 22, 13360. [Google Scholar] [CrossRef]
- Miao, L.; Zhang, Y.; Huang, L. mRNA vaccine for cancer immunotherapy. Mol. Cancer 2021, 20, 41. [Google Scholar] [CrossRef]
- Beck, J.D.; Reidenbach, D.; Salomon, N.; Sahin, U.; Türeci, Ö.; Vormehr, M.; Kranz, L.M. mRNA therapeutics in cancer immunotherapy. Mol. Cancer 2021, 20, 69. [Google Scholar] [CrossRef] [PubMed]
- Pastor, F.; Berraondo, P.; Etxeberria, I.; Frederick, J.; Sahin, U.; Gilboa, E.; Melero, I. An RNA toolbox for cancer immunotherapy. Nat. Rev. Drug Discov. 2018, 17, 751–767. [Google Scholar] [CrossRef] [PubMed]
- Vormehr, M. Substantial improvement of cancer immunotherapy by an RNA encoded extended half-life Interleukin-2 variant. Abstract P626. In Proceedings of the 34th Annual Meeting & Pre-Conference Programs (SITC 2019), National Harbor, MD, USA, 6–10 November 2019. [Google Scholar]
- Waldmann, T.A. The shared and contrasting roles of IL2 and IL15 in the life and death of normal and neoplastic lymphocytes: Implications for cancer therapy. Cancer Immunol. Res. 2015, 3, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Etxeberria, I.; Bolaños, E.; Quetglas, J.I.; Gros, A.; Villanueva, A.; Palomero, J.; Sánchez-Paulete, A.R.; Piulats, J.M.; Matias-Guiu, X.; Olivera, I.; et al. Intratumor adoptive transfer of IL-12 mRNA transiently engineered antitumor CD8+ T cells. Cancer Cell 2019, 36, 613–629. [Google Scholar] [CrossRef]
- Hewitt, S.L.; Bailey, D.; Zielinski, J.; Apte, A.; Musenge, F.; Karp, R.; Burke, S.; Garcon, F.; Mishra, A.; Gurumurthy, S.; et al. Intratumoral IL12 mRNA therapy promotes TH1 transformation of the tumor microenvironment. Clin. Cancer Res. 2020, 26, 6284–6298. [Google Scholar] [CrossRef]
- Lai, I.; Swaminathan, S.; Baylot, V.; Mosley, A.; Dhanasekaran, R.; Gabay, M.; Felsher, D.W. Lipid nanoparticles that deliver IL-12 messenger RNA suppress tumorigenesis in MYC oncogene-driven hepatocellular carcinoma. J. Immunother. Cancer 2018, 6, 125. [Google Scholar] [CrossRef]
- Thran, M.; Mukherjee, J.; Ponisch, M.; Fiedler, K.; Thess, A.; Mui, B.L.; Hope, M.J.; Tam, Y.K.; Horscroft, N.; Heidenreich, R.; et al. mRNA mediates passive vaccination against infectious agents, toxins, and tumors. EMBO Mol. Med. 2017, 9, 1434–1447. [Google Scholar] [CrossRef]
- Rybakova, Y.; Kowalski, P.S.; Huang, Y.; Gonzalez, J.T.; Heartlein, M.W.; DeRosa, F.; Delcassian, D.; Anderson, D.G. mRNA delivery for therapeutic anti-HER2 antibody expression in vivo. Mol. Ther. 2019, 27, 1415–1423. [Google Scholar] [CrossRef]
- Stadler, C.R.; Bähr-Mahmud, H.; Celik, L.; Hebich, B.; Roth, A.S.; Roth, R.P.; Karikó, K.; Türeci, Ö.; Sahin, U. Elimination of large tumors in mice by mRNA-encoded bispecific antibodies. Nat. Med. 2017, 23, 815–817. [Google Scholar] [CrossRef]
- Mu, X.; Hur, S. Immunogenicity of In vitro transcribed RNA. Acc. Chem. Res. 2021, 54, 4012–4023. [Google Scholar] [CrossRef] [PubMed]
- Linares-Fernandez, S.; Lacroix, C.; Exposito, J.Y.; Verrier, B. Tailoring MRNA Vaccine to Balance Innate/Adaptive Immune Response. Trends Mol. Med. 2020, 26, 311–323. [Google Scholar] [CrossRef] [PubMed]
- De Beuckelaer, A.; Pollard, C.; Van Lint, S.; Roose, K.; Van Hoecke, L.; Naessens, T.; Udhayakumar, V.K.; Smet, M.; Sanders, N.; Lienenklaus, S.; et al. Type I Interferons Interfere with the Capacity of MRNA Lipoplex Vaccines to Elicit Cytolytic T Cell Responses. Mol. Ther. 2016, 24, 2012–2020. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Xia, P.; Li, S.; Zhang, T.; Wang, T.T.; Zhu, J. RNA sensors of the innate immune system and their detection of pathogens. IUBMB Life 2017, 69, 297–304. [Google Scholar] [CrossRef]
- Mian, M.F.; Ahmed, A.N.; Rad, M.; Babaian, A.; Bowdish, D.; Ashkar, A.A. Length of dsRNA (poly I:C) drives distinct innate immune responses, depending on the cell type. J. Leukoc. Biol. 2013, 94, 1025–1036. [Google Scholar] [CrossRef]
- Ramanathan, A.; Robb, G.B.; Chan, S.H. mRNA Capping: Biological Functions and Applications. Nucleic Acids Res. 2016, 44, 7511–7526. [Google Scholar] [CrossRef]
- Devarkar, S.C.; Wang, C.; Miller, M.T.; Ramanathan, A.; Jiang, F.; Khan, A.G.; Patel, S.S.; Marcotrigiano, J. Structural Basis for M7g Recognition and 2′-O-Methyl Discrimination in Capped RNAs by the Innate Immune Receptor RIG-I. Proc. Natl. Acad. Sci. USA 2016, 113, 596–601. [Google Scholar] [CrossRef]
- Mauro, V.P.; Chappell, S.A. A Critical Analysis of Codon Optimization in Human Therapeutics. Trends Mol. Med. 2014, 20, 604–613. [Google Scholar] [CrossRef]
- Gallie, D.R. The Cap and Poly (A) Tail Function Synergistically to Regulate mRNA Translational Efficiency. Genes Dev. 1991, 5, 2108–2116. [Google Scholar] [CrossRef]
- Jackson, N.A.; Kester, K.E.; Casimiro, D.; Gurunathan, S.; DeRosa, F. The Promise of mRNA Vaccines: A Biotech and Industrial Perspective. NPJ Vaccines 2020, 5, 11. [Google Scholar] [CrossRef]
- Bloom, K.; van den Berg, F.; Arbuthnot, P. Self-Amplifying RNA Vaccines for Infectious Diseases. Gene Ther. 2021, 28, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Lundstrom, K. Replicon RNA Viral Vectors as Vaccines. Vaccines 2016, 4, 39–61. [Google Scholar] [CrossRef]
- Singh, A.; Koutsoumpli, G.; van de Wall, S.; Daemen, T. An alphavirus-based therapeutic cancer vaccine: From design to clinical trial. Cancer Immunol. Immunother. 2019, 68, 849–859. [Google Scholar] [CrossRef]
- Kowalski, P.S.; Rudra, A.; Miao, L.; Anderson, D.G. Delivering the Messenger: Advances in Technologies for Therapeutic mRNA Delivery. Mol. Ther. 2019, 27, 710–728. [Google Scholar] [CrossRef] [PubMed]
- Fobian, S.F.; Cheng, Z.; Ten Hagen, T.L.M. Smart Lipid-Based Nanosystems for Therapeutic Immune Induction against Cancers: Perspectives and Outlooks. Pharmaceutics 2021, 14, 26. [Google Scholar] [CrossRef]
- Semple, S.C.; Akinc, A.; Chen, J.; Sandhu, A.P.; Mui, B.L.; Cho, C.K.; Sah, D.W.; Stebbing, D.; Crosley, E.J.; Yaworski, E.; et al. Rational design of cationic lipids for siRNA delivery. Nat. Biotechnol. 2010, 28, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Love, K.T.; Mahon, K.P.; Levins, C.G.; Whitehead, K.A.; Querbes, W.; Dorkin, J.R.; Qin, J.; Cantley, W.; Qin, L.L.; Racie, T.; et al. Lipid-like materials for low-dose, in vivo gene silencing. Proc. Natl. Acad. Sci. USA 2010, 107, 1864–1869. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, K.A.; Sahay, G.; Li, G.Z.; Love, K.T.; Alabi, C.A.; Ma, M.; Zurenko, C.; Querbes, W.; Langer, R.S.; Anderson, D.G. Synergistic silencing: Combinations of lipid-like materials for efficacious siRNA delivery. Mol. Ther. 2011, 19, 1688–1694. [Google Scholar] [CrossRef]
- Sabnis, S.; Kumarasinghe, E.S.; Salerno, T.; Mihai, C.; Ketova, T.; Senn, J.J.; Lynn, A.; Bulychev, A.; McFadyen, I.; Chan, J.; et al. A Novel Amino Lipid Series for mRNA Delivery: Improved Endosomal Escape and Sustained Pharmacology and Safety in Non-human Primates. Mol. Ther. 2018, 26, 1509–1519. [Google Scholar] [CrossRef]
- Tanaka, H.; Miyama, R.; Sakurai, Y.; Tamagawa, S.; Nakai, Y.; Tange, K.; Yoshioka, H.; Akita, H. Improvement of mRNA Delivery Efficiency to a T Cell Line by Modulating PEG-Lipid Content and Phospholipid Components of Lipid Nanoparticles. Pharmaceutics 2021, 13, 2097. [Google Scholar] [CrossRef]
- Yanez Arteta, M.; Kjellman, T.; Bartesaghi, S.; Wallin, S.; Wu, X.; Kvist, A.J.; Dabkowska, A.; Székely, N.; Radulescu, A.; Bergenholtz, J.; et al. Successful reprogramming of cellular protein production through mRNA delivered by functionalized lipid nanoparticles. Proc. Natl. Acad. Sci. USA 2018, 115, E3351–E3360. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Naradikian, M.S.; Parkhouse, K.; Cain, D.W.; Jones, L.; Moody, M.A.; Verkerke, H.P.; Myles, A.; Willis, E.; et al. Nucleoside-modified mRNA vaccines induce potent T follicular helper and germinal center B cell responses. J. Exp. Med. 2018, 215, 1571–1588. [Google Scholar] [CrossRef] [PubMed]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.J.; Moreira, E.D., Jr.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Pérez, M.G.; Polack, F.P.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine through 6 Months. N. Engl. J. Med. 2021, 385, 1761–1773. [Google Scholar] [CrossRef]
- Oberli, M.A.; Reichmuth, A.M.; Dorkin, J.R.; Mitchell, M.J.; Fenton, O.S.; Jaklenec, A.; Anderson, D.G.; Langer, R.; Blankschtein, D. Lipid Nanoparticle Assisted mRNA Delivery for Potent Cancer Immunotherapy. Nano Lett. 2017, 17, 1326–1335. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.N.; Li, M.; Luo, Y.L.; Chen, Q.; Wang, L.; Zhang, H.B.; Shen, S.; Gu, Z.; Wang, J. Cationic lipid-assisted nanoparticles for delivery of mRNA cancer vaccine. Biomater. Sci. 2018, 6, 3009–3018. [Google Scholar] [CrossRef]
- Zhang, Y.; Xie, F.; Yin, Y.; Zhang, Q.; Jin, H.; Wu, Y.; Pang, L.; Li, J.; Gao, J. Immunotherapy of Tumor RNA-Loaded Lipid Nanoparticles Against Hepatocellular Carcinoma. Int. J. Nanomed. 2021, 16, 1553–1564. [Google Scholar] [CrossRef] [PubMed]
- Kranz, L.M.; Diken, M.; Haas, H.; Kreiter, S.; Loquai, C.; Reuter, K.C.; Meng, M.; Fritz, D.; Vascotto, F.; Hefesha, H.; et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 2016, 534, 396–401. [Google Scholar] [CrossRef]
- Dahlman, J.E.; Barnes, C.; Khan, O.; Thiriot, A.; Jhunjunwala, S.; Shaw, T.E.; Xing, Y.; Sager, H.B.; Sahay, G.; Speciner, L.; et al. In vivo endothelial siRNA delivery using polymeric nanoparticles with low molecular weight. Nat. Nanotechnol. 2014, 9, 648–655. [Google Scholar] [CrossRef]
- Dong, Y.; Dorkin, J.R.; Wang, W.; Chang, P.H.; Webber, M.J.; Tang, B.C.; Yang, J.; Abutbul-Ionita, I.; Danino, D.; DeRosa, F.; et al. Poly(glycoamidoamine) Brushes Formulated Nanomaterials for Systemic siRNA and mRNA Delivery In Vivo. Nano Lett. 2016, 16, 842–848. [Google Scholar] [CrossRef]
- Kaczmarek, J.C.; Kauffman, K.J.; Fenton, O.S.; Sadtler, K.; Patel, A.K.; Heartlein, M.W.; DeRosa, F.; Anderson, D.G. Optimization of a Degradable Polymer-Lipid Nanoparticle for Potent Systemic Delivery of mRNA to the Lung Endothelium and Immune Cells. Nano Lett. 2018, 18, 6449–6454. [Google Scholar] [CrossRef] [PubMed]
- Chahal, J.S.; Khan, O.F.; Cooper, C.L.; McPartlan, J.S.; Tsosie, J.K.; Tilley, L.D.; Sidik, S.M.; Lourido, S.; Langer, R.; Bavari, S.; et al. Dendrimer-RNA nanoparticles generate protective immunity against lethal Ebola, H1N1 influenza, and Toxoplasma gondii challenges with a single dose. Proc. Natl. Acad. Sci. USA 2016, 113, E4133–E4142. [Google Scholar] [CrossRef] [PubMed]
- Chahal, J.S.; Fang, T.; Woodham, A.W.; Khan, O.F.; Ling, J.; Anderson, D.G.; Ploegh, H.L. An RNA nanoparticle vaccine against Zika virus elicits antibody and CD8+ T cell responses in a mouse model. Sci. Rep. 2017, 7, 252. [Google Scholar] [CrossRef] [PubMed]
- Uchida, S.; Kinoh, H.; Ishii, T.; Matsui, A.; Tockary, T.A.; Takeda, K.M.; Uchida, H.; Osada, K.; Itaka, K.; Kataoka, K. Systemic delivery of messenger RNA for the treatment of pancreatic cancer using polyplex nanomicelles with a cholesterol moiety. Biomaterials 2016, 82, 221–228. [Google Scholar] [CrossRef]
- Chen, Q.; Qi, R.; Chen, X.; Yang, X.; Wu, S.; Xiao, H.; Dong, W. A Targeted and Stable Polymeric Nanoformulation Enhances Systemic Delivery of mRNA to Tumors. Mol. Ther. 2017, 25, 92–101. [Google Scholar] [CrossRef]
- Islam, M.A.; Xu, Y.; Tao, W.; Ubellacker, J.M.; Lim, M.; Aum, D.; Lee, G.Y.; Zhou, K.; Zope, H.; Yu, M.; et al. Restoration of tumour-growth suppression in vivo via systemic nanoparticle-mediated delivery of PTEN mRNA. Nat. Biomed. Eng. 2018, 2, 850–864. [Google Scholar] [CrossRef]
- Hoerr, I.; Obst, R.; Rammensee, H.G.; Jung, G. In vivo application of RNA leads to induction of specific cytotoxic T lymphocytes and antibodies. Eur. J. Immunol. 2000, 30, 1–7. [Google Scholar] [CrossRef]
- Hoyer, J.; Neundorf, I. Peptide vectors for the nonviral delivery of nucleic acids. Acc. Chem. Res. 2012, 45, 1048–1056. [Google Scholar] [CrossRef] [PubMed]
- Nakase, I.; Akita, H.; Kogure, K.; Gräslund, A.; Langel, U.; Harashima, H.; Futaki, S. Efficient intracellular delivery of nucleic acid pharmaceuticals using cell-penetrating peptides. Acc. Chem. Res. 2012, 45, 1132–1139. [Google Scholar] [CrossRef]
- Reissmann, S. Cell penetration: Scope and limitations by the application of cell-penetrating peptides. J. Pept. Sci. 2014, 20, 760–784. [Google Scholar] [CrossRef]
- Li, H.; Tsui, T.Y.; Ma, W. Intracellular Delivery of Molecular Cargo Using Cell-Penetrating Peptides and the Combination Strategies. Int. J. Mol. Sci. 2015, 16, 19518–19536. [Google Scholar] [CrossRef] [PubMed]
- Kallen, K.J.; Heidenreich, R.; Schnee, M.; Petsch, B.; Schlake, T.; Thess, A.; Baumhof, P.; Scheel, B.; Koch, S.D.; Fotin-Mleczek, M. A novel, disruptive vaccination technology: Self-adjuvanted RNActive(®) vaccines. Hum. Vaccines Immunother. 2013, 9, 2263–2276. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Man, R.C.H.; Liao, Q.; Kung, K.L.K.; Chow, M.Y.T.; Lam, J.K.W. Effective mRNA pulmonary delivery by dry powder formulation of PEGylated synthetic KL4 peptide. J. Control. Release 2019, 314, 102–115. [Google Scholar] [CrossRef]
- Lou, B.; De Koker, S.; Lau, C.Y.J.; Hennink, W.E.; Mastrobattista, E. mRNA Polyplexes with Post-Conjugated GALA Peptides Efficiently Target, Transfect, and Activate Antigen Presenting Cells. Bioconjug. Chem. 2019, 30, 461–475. [Google Scholar] [CrossRef]
- Hajj, K.; Whitehead, K. Tools for translation: Non-viral materials for therapeutic mRNA delivery. Nat. Rev. Mater. 2017, 2, 17056. [Google Scholar] [CrossRef]
- Mastelic-Gavillet, B.; Balint, K.; Boudousquie, C.; Gannon, P.O.; Kandalaft, L.E. Personalized dendritic cell vaccines—Recent breakthroughs and encouraging clinical results. Front. Immunol. 2019, 10, 766. [Google Scholar] [CrossRef]
- Saxena, M.; Balan, S.; Roudko, V.; Bhardwaj, N. Towards superior dendritic-cell vaccines for cancer therapy. Nat. Biomed. Eng. 2018, 2, 341–346. [Google Scholar] [CrossRef]
- Perez, C.R.; De Palma, M. Engineering dendritic cell vaccines to improve cancer immunotherapy. Nat. Commun. 2019, 10, 5408–5417. [Google Scholar] [CrossRef]
- Nair, S.K.; Heiser, A.; Boczkowski, D.; Majumdar, A.; Naoe, M.; Lebkowski, J.S.; Vieweg, J.; Gilboa, E. Induction of cytotoxic T cell responses and tumor immunity against unrelated tumors using telomerase reverse transcriptase RNA transfected dendritic cells. Nat. Med. 2000, 6, 1011–1017. [Google Scholar] [CrossRef]
- Perche, F.; Benvegnu, T.; Berchel, M.; Lebegue, L.; Pichon, C.; Jaffrès, P.A.; Midoux, P. Enhancement of dendritic cells transfection in vivo and of vaccination against B16F10 melanoma with mannosylated histidylated lipopolyplexes loaded with tumor antigen messenger RNA. Nanomedicine 2011, 7, 445–453. [Google Scholar] [CrossRef]
- Eshhar, Z.; Waks, T.; Bendavid, A.; Schindler, D.G. Functional expression of chimeric receptor genes in human T cells. J. Immunol. Methods 2001, 248, 67–76. [Google Scholar] [CrossRef]
- Kalos, M.; June, C.H. Adoptive T cell transfer for cancer immunotherapy in the era of synthetic biology. Immunity 2013, 39, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Irving, M.; Lanitis, E.; Migliorini, D.; Ivics, Z.; Guedan, S. Choosing the Right Tool for Genetic Engineering: Clinical Lessons from Chimeric Antigen Receptor-T Cells. Hum. Gene Ther. 2021, 32, 1044–1058. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhou, G.; Zhang, L.; Zhao, Q. Building Potent Chimeric Antigen Receptor T Cells with CRISPR Genome Editing. Front. Immunol. 2019, 10, 456. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Chen, J.; Zhao, X.; Li, Y.; Harmon, J.; Huang, C.; Chen, J.; Xu, Q. In Vitro Engineering Chimeric Antigen Receptor Macrophages and T Cells by Lipid Nanoparticle-Mediated mRNA Delivery. ACS Biomater. Sci. Eng. 2022, 8, 722–733. [Google Scholar] [CrossRef]
- Hegde, M.; Corder, A.; Chow, K.K.; Mukherjee, M.; Ashoori, A.; Kew, Y.; Zhang, Y.J.; Baskin, D.S.; Merchant, F.A.; Brawley, V.S.; et al. Combinational targeting offsets antigen escape and enhances effector functions of adoptively transferred T cells in glioblastoma. Mol. Ther. 2013, 21, 2087–2101. [Google Scholar] [CrossRef]
- Barrett, D.M.; Zhao, Y.; Liu, X.; Jiang, S.; Carpenito, C.; Kalos, M.; Carroll, R.G.; June, C.H.; Grupp, S.A. Treatment of advanced leukemia in mice with mRNA engineered T cells. Hum. Gene Ther. 2011, 22, 1575–1586. [Google Scholar] [CrossRef]
- Barrett, D.M.; Liu, X.; Jiang, S.; June, C.H.; Grupp, S.A.; Zhao, Y. Regimen-specific effects of RNA-modified chimeric antigen receptor T cells in mice with advanced leukemia. Hum. Gene Ther. 2013, 24, 717–727. [Google Scholar] [CrossRef]
- Rajan, S.T.; Gugliandolo, A.; Bramanti, P.; Mazzon, E. In Vitro-Transcribed mRNA Chimeric Antigen Receptor T Cell (IVT mRNA CAR T) Therapy in Hematologic and Solid Tumor Management: A Preclinical Update. Int. J. Mol. Sci. 2020, 21, 6514. [Google Scholar] [CrossRef]
- Ljungberg, K.; Liljeström, P. Self-replicating alphavirus RNA vaccines. Expert Rev. Vaccines 2015, 14, 177–194. [Google Scholar] [CrossRef]
- Smerdou, C.; Liljeström, P. Two-helper RNA system for production of recombinant Semliki forest virus particles. J. Virol. 1999, 73, 1092–1098. [Google Scholar] [CrossRef]
- Beissert, T.; Perkovic, M.; Vogel, A.; Erbar, S.; Walzer, K.C.; Hempel, T.; Brill, S.; Haefner, E.; Becker, R.; Türeci, Ö.; et al. A Trans-amplifying RNA Vaccine Strategy for Induction of Potent Protective Immunity. Mol. Ther. 2020, 28, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Biddlecome, A.; Habte, H.H.; McGrath, K.M.; Sambanthamoorthy, S.; Wurm, M.; Sykora, M.M.; Knobler, C.M.; Lorenz, I.C.; Lasaro, M.; Elbers, K.; et al. Delivery of self-amplifying RNA vaccines in in vitro reconstituted virus-like particles. PLoS ONE 2019, 14, e0215031. [Google Scholar] [CrossRef] [PubMed]
- Nelson, E.L.; Prieto, D.; Alexander, T.G.; Pushko, P.; Lofts, L.A.; Rayner, J.O.; Kamrud, K.I.; Fralish, B.; Smith, J.F. Venezuelan equine encephalitis replicon immunization overcomes intrinsic tolerance and elicits effective anti-tumor immunity to the ‘self’ tumor-associated antigen, neu in a rat mammary tumor model. Breast Cancer Res. Treat. 2003, 82, 169–183. [Google Scholar] [CrossRef]
- Riabov, V.; Tretyakova, I.; Alexander, R.B.; Pushko, P.; Klyushnenkova, E.N. Anti-tumor effect of the alphavirus-based virus-like particle vector expressing prostate-specific antigen in a HLA-DR transgenic mouse model of prostate cancer. Vaccine 2015, 33, 5386–5395. [Google Scholar] [CrossRef]
- Zhang, X.; Mao, G.; van den Pol, A.N. Chikungunya-vesicular stomatitis chimeric virus targets and eliminates brain tumors. Virology 2018, 522, 244–259. [Google Scholar] [CrossRef]
- Velders, M.P.; McElhiney, S.; Cassetti, M.C.; Eiben, G.L.; Higgins, T.; Kovacs, G.R.; Elmishad, A.G.; Kast, W.M.; Smith, L.R. Eradication of established tumors by vaccination with Venezuelan equine encephalitis virus replicon particles delivering human papillomavirus 16 E7 RNA. Cancer Res. 2001, 61, 7861–7867. [Google Scholar] [PubMed]
- Sebastian, M.; Schröder, A.; Scheel, B.; Hong, H.S.; Muth, A.; von Boehmer, L.; Zippelius, A.; Mayer, F.; Reck, M.; Atanackovic, D.; et al. A phase I/IIa study of the mRNA-based cancer immunotherapy CV9201 in patients with stage IIIB/IV non-small cell lung cancer. Cancer Immunol. Immunother. 2019, 68, 799–812. [Google Scholar] [CrossRef]
- Kübler, H.; Scheel, B.; Gnad-Vogt, U.; Miller, K.; Schultze-Seemann, W.; Vom Dorp, F.; Parmiani, G.; Hampel, C.; Wedel, S.; Trojan, L.; et al. Self-adjuvanted mRNA vaccination in advanced prostate cancer patients: A first-in-man phase I/IIa study. J. Immunother. Cancer 2015, 3, 26. [Google Scholar] [CrossRef]
- Weide, B.; Pascolo, S.; Scheel, B.; Derhovanessian, E.; Pflugfelder, A.; Eigentler, T.K.; Pawelec, G.; Hoerr, I.; Rammensee, H.G.; Garbe, C. Direct injection of protamine-protected mRNA: Results of a phase 1/2 vaccination trial in metastatic melanoma patients. J. Immunother. 2009, 32, 498–507. [Google Scholar] [CrossRef]
- Rittig, S.M.; Haentschel, M.; Weimer, K.J.; Heine, A.; Muller, M.R.; Brugger, W.; Horger, M.S.; Maksimovic, O.; Stenzl, A.; Hoerr, I.; et al. Intradermal vaccinations with RNA coding for TAA generate CD8+ and CD4+ immune responses and induce clinical benefit in vaccinated patients. Mol. Ther. 2011, 19, 990–999. [Google Scholar] [CrossRef]
- Rittig, S.M.; Haentschel, M.; Weimer, K.J.; Heine, A.; Müller, M.R.; Brugger, W.; Horger, M.S.; Maksimovic, O.; Stenzl, A.; Hoerr, I.; et al. Long-term survival correlates with immunological responses in renal cell carcinoma patients treated with mRNA-based immunotherapy. Oncoimmunology 2015, 5, e1108511. [Google Scholar] [CrossRef] [PubMed]
- Papachristofilou, A.; Hipp, M.M.; Klinkhardt, U.; Früh, M.; Sebastian, M.; Weiss, C.; Pless, M.; Cathomas, R.; Hilbe, W.; Pall, G.; et al. Phase Ib evaluation of a self-adjuvanted protamine formulated mRNA-based active cancer immunotherapy, BI1361849 (CV9202), combined with local radiation treatment in patients with stage IV non-small cell lung cancer. J. Immunother. Cancer 2019, 7, 38. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Oehm, P.; Derhovanessian, E.; Jabulowsky, R.A.; Vormehr, M.; Gold, M.; Maurus, D.; Schwarck-Kokarakis, D.; Kuhn, A.N.; Omokoko, T.; et al. An RNA vaccine drives immunity in checkpoint-inhibitor-treated melanoma. Nature 2020, 585, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Heiser, A.; Coleman, D.; Dannull, J.; Yancey, D.; Maurice, M.A.; Lallas, C.D.; Dahm, P.; Niedzwiecki, D.; Gilboa, E.; Vieweg, J. Autologous dendritic cells transfected with prostate-specific antigen RNA stimulate CTL responses against metastatic prostate tumors. J. Clin. Investig. 2002, 109, 409–417. [Google Scholar] [CrossRef]
- Su, Z.; Dannull, J.; Heiser, A.; Yancey, D.; Pruitt, S.; Madden, J.; Coleman, D.; Niedzwiecki, D.; Gilboa, E.; Vieweg, J. Immunological and clinical responses in metastatic renal cancer patients vaccinated with tumor RNA-transfected dendritic cells. Cancer Res. 2003, 63, 2127–2133. [Google Scholar]
- Morse, M.A.; Nair, S.K.; Mosca, P.J.; Hobeika, A.C.; Clay, T.M.; Deng, Y.; Boczkowski, D.; Proia, A.; Neidzwiecki, D.; Clavien, P.A.; et al. Immunotherapy with autologous, human dendritic cells transfected with carcinoembryonic antigen mRNA. Cancer Investig. 2003, 21, 341–349. [Google Scholar] [CrossRef]
- Morse, M.A.; Nair, S.K.; Boczkowski, D.; Tyler, D.; Hurwitz, H.I.; Proia, A.; Clay, T.M.; Schlom, J.; Gilboa, E.; Lyerly, H.K. The feasibility and safety of immunotherapy with dendritic cells loaded with CEA mRNA following neoadjuvant chemoradiotherapy and resection of pancreatic cancer. Int. J. Gastrointest. Cancer 2002, 32, 1–6. [Google Scholar] [CrossRef]
- Wilgenhof, S.; Van Nuffel, A.M.T.; Benteyn, D.; Corthals, J.; Aerts, C.; Heirman, C.; Van Riet, I.; Bonehill, A.; Thielemans, K.; Neyns, B. A phase IB study on intravenous synthetic mRNA electroporated dendritic cell immunotherapy in pretreated advanced melanoma patients. Ann. Oncol. 2013, 24, 2686–2693. [Google Scholar] [CrossRef] [PubMed]
- Jansen, Y.; Kruse, V.; Corthals, J.; Schats, K.; van Dam, P.J.; Seremet, T.; Heirman, C.; Brochez, L.; Kockx, M.; Thielemans, K.; et al. A randomized controlled phase II clinical trial on mRNA electroporated autologous monocyte-derived dendritic cells (TriMixDC-MEL) as adjuvant treatment for stage III/IV melanoma patients who are disease-free following the resection of macrometastases. Cancer Immunol. Immunother. 2020, 69, 2589–2598. [Google Scholar] [CrossRef]
- Wilgenhof, S.; Corthals, J.; Heirman, C.; van Baren, N.; Lucas, S.; Kvistborg, P.; Thielemans, K.; Neyns, B. Phase II Study of Autologous Monocyte-Derived mRNA Electroporated Dendritic Cells (TriMixDC-MEL) Plus Ipilimumab in Patients with Pretreated Advanced Melanoma. J. Clin. Oncol. 2016, 34, 1330–1338. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.T.; Nie, Y.; Sun, S.N.; Lin, T.; Han, R.J.; Jiang, J.; Li, Z.; Li, J.Q.; Xiao, Y.P.; Fan, Y.Y.; et al. Tumor-associated antigen-based personalized dendritic cell vaccine in solid tumor patients. Cancer Immunol. Immunother. 2020, 69, 1375–1387. [Google Scholar] [CrossRef] [PubMed]
- Lundstrom, K. Self-Replicating RNA Viruses for Vaccine Development against Infectious Diseases and Cancer. Vaccines 2021, 9, 1187. [Google Scholar] [CrossRef] [PubMed]
- Komdeur, F.L.; Singh, A.; van de Wall, S.; Meulenberg, J.J.M.; Boerma, A.; Hoogeboom, B.N.; Paijens, S.T.; Oyarce, C.; de Bruyn, M.; Schuuring, E.; et al. First-in-Human Phase I Clinical Trial of an SFV-Based RNA Replicon Cancer Vaccine against HPV-Induced Cancers. Mol. Ther. 2021, 29, 611–625. [Google Scholar] [CrossRef] [PubMed]
- Morse, M.A.; Hobeika, A.C.; Osada, T.; Berglund, P.; Hubby, B.; Negri, S.; Niedzwiecki, D.; Devi, G.R.; Burnett, B.K.; Clay, T.M.; et al. An alphavirus vector overcomes the presence of neutralizing antibodies and elevated numbers of Tregs to induce immune responses in humans with advanced cancer. J. Clin. Investig. 2010, 120, 3234–3241. [Google Scholar] [CrossRef] [PubMed]
- Crosby, E.J.; Hobeika, A.C.; Niedzwiecki, D.; Rushing, C.; Hsu, D.; Berglund, P.; Smith, J.; Osada, T.; Gwin, W.R., III; Hartman, Z.C.; et al. Long-term survival of patients with stage III colon cancer treated with VRP-CEA(6D), an alphavirus vector that increases the CD8+ effector memory T cell to Treg ratio. J. Immunother. Cancer 2020, 8, e001662. [Google Scholar] [CrossRef]
- Crosby, E.J.; Gwin, W.; Blackwell, K.; Marcom, P.K.; Chang, S.; Maecker, H.T.; Broadwater, G.; Hyslop, T.; Kim, S.; Rogatko, A.; et al. Vaccine-Induced Memory CD8+T Cells Provide Clinical Benefit in HER2 Expressing Breast Cancer: A Mouse to Human Translational Study. Clin. Cancer Res. 2019, 25, 2725–2736. [Google Scholar] [CrossRef]
- Aliahmad, P.; Miyake-Stoner, S.J.; Geall, A.J.; Wang, N.S. Next generation self-replicating RNA vectors for vaccines and immunotherapies. Cancer Gene Ther. 2022, 22, 1–9. [Google Scholar] [CrossRef]
- Dimitri, A.; Herbst, F.; Fraietta, J.A. Engineering the next-generation of CAR T-cells with CRISPR-Cas9 gene editing. Mol. Cancer 2022, 21, 78. [Google Scholar] [CrossRef]
- Giuffrida, L.; Sek, K.; Henderson, M.A.; Lai, J.; Chen, A.X.Y.; Meyran, D.; Todd, K.; Petley, E.V.; Mardiana, S.; Mølck, C.; et al. CRISPR/Cas9 mediated deletion of the adenosine A2A receptor enhances CAR T cell efficacy. Nat. Commun. 2021, 12, 3236. [Google Scholar] [CrossRef]
- Zhang, P.; Lim, S.B.; Jiang, K.; Chew, T.W.; Low, B.C.; Lim, C.T. Distinct mRNAs in Cancer Extracellular Vesicles Activate Angiogenesis and Alter Transcriptome of Vascular Endothelial Cells. Cancers 2021, 13, 2009. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, C.; Luo, Y.; Zhang, G.; Wu, P.; Sun, N.; He, J. RNA N6-methyladenosine modification in the lethal teamwork of cancer stem cells and the tumor immune microenvironment: Current landscape and therapeutic potential. Clin. Transl. Med. 2021, 11, e525. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, S.L.; Bai, A.; Bailey, D.; Ichikawa, K.; Zielinski, J.; Karp, R.; Apte, A.; Arnold, K.; Zacharek, S.J.; Iliou, M.S.; et al. Durable anticancer immunity from intratumoral administration of IL-23, IL-36γ, and OX40L mRNAs. Sci. Transl. Med. 2019, 11, eaat9143. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.R.; Todd, M.B.; Jimeno, A.; Wang, D.; LoRusso, P.; Do, K.T.; Stemmer, S.M.; Maurice-Dror, C.; Geva, R.; Zacharek, S.; et al. A phase I study of mRNA-2752, a lipid nanoparticle encapsulating mRNAs encoding human OX40L, IL-23, and IL-36γ, for intratumoral (iTu) injection alone and in combination with durvalumab. J. Clin. Oncol. 2020, 38 (Suppl. 15), 3092. [Google Scholar] [CrossRef]
- Van Lint, S.; Renmans, D.; Broos, K.; Goethals, L.; Maenhout, S.; Benteyn, D.; Goyvaerts, C.; du Four, S.; van der Jeught, K.; Bialkowski, L.; et al. Intratumoral delivery of TriMix mRNA results in T-cell activation by cross-presenting dendritic cells. Cancer Immunol Res. 2016, 4, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Jimeno, A. A phase 1/2, open-label, multicenter, dose escalation and efficacy study of mRNA-2416, a lipid nanoparticle encapsulated mRNA encoding human OX40L, for intratumoral injection alone or in combination with durvalumab for patients with advanced malignancies. Abstract CT032. In Proceedings of the American Association for Cancer Research Annual Meeting, Philadelphia, PA, USA, 27–28 April 2020; 22–24 June 2020. [Google Scholar]
- Haabeth, O.A.W.; Blake, T.R.; McKinlay, C.J.; Tveita, A.A.; Sallets, A.; Waymouth, R.M.; Wender, P.A.; Levy, R. Local Delivery of Ox40l, Cd80, and Cd86mRNA Kindles Global Anticancer Immunity. Cancer Res. 2019, 79, 1624–1634. [Google Scholar] [CrossRef] [PubMed]
- Spiess, C.; Zhai, Q.; Carter, P.J. Alternative molecular formats and therapeutic applications for bispecific antibodies. Mol. Immunol. 2015, 67 Pt A, 95–106. [Google Scholar] [CrossRef]
- Pruitt, S.K.; Boczkowski, D.; de Rosa, N.; Haley, N.R.; Morse, M.A.; Tyler, D.S.; Dannull, J.; Nair, S. Enhancement of anti-tumor immunity through local modulation of CTLA-4 and GITR by dendritic cells. Eur. J. Immunol. 2011, 41, 3553–3563. [Google Scholar] [CrossRef]
- Nguyen, K.B.; Spranger, S. Modulation of the immune microenvironment by tumor-intrinsic oncogenic signaling. J. Cell Biol. 2020, 219, e201908224. [Google Scholar] [CrossRef]
- Haanen, J.B.; Mackensen, A.; Koenecke, C.; Alsdorf, W.; Desuki, A.; Wagner-Drouet, E.; Heudobler, D.; Borchmann, P.; Wiegert, E.; Schulz, C.; et al. BNT211: A Phase I trial to evaluate safety and efficacy of CLDN6 CAR-T cells and CARVac-mediated in vivo expansion in patients with CLDN6-positive advanced solid tumors. Abstract CT002. In Proceedings of the American Association for Cancer Research Annual Meeting, New Orleans, LA, USA, 8–13 April 2022. [Google Scholar]
- Barbier, A.J.; Jiang, A.Y.; Zhang, P.; Wooster, R.; Anderson, D.G. The clinical progress of mRNA vaccines and immunotherapies. Nat. Biotechnol. 2022, 40, 840–854. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Andresen, J.L.; Manan, R.S.; Langer, R. Nucleic acid delivery for therapeutic applications. Adv. Drug Deliv. Rev. 2021, 178, 113834. [Google Scholar] [CrossRef] [PubMed]
- Mendes, B.B.; Conniot, J.; Avital, A.; Yao, D.; Jiang, X.; Zhou, X.; Sharf-Pauker, N.; Xiao, Y.; Adir, O.; Liang, H.; et al. Nanodelivery of nucleic acids. Nat. Rev. Methods Primers 2022, 2, 24. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Wei, T.; Farbiak, L.; Johnson, L.T.; Dilliard, S.A.; Siegwart, D.J. Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR-Cas gene editing. Nat. Nanotechnol. 2020, 15, 313–320. [Google Scholar] [CrossRef] [PubMed]
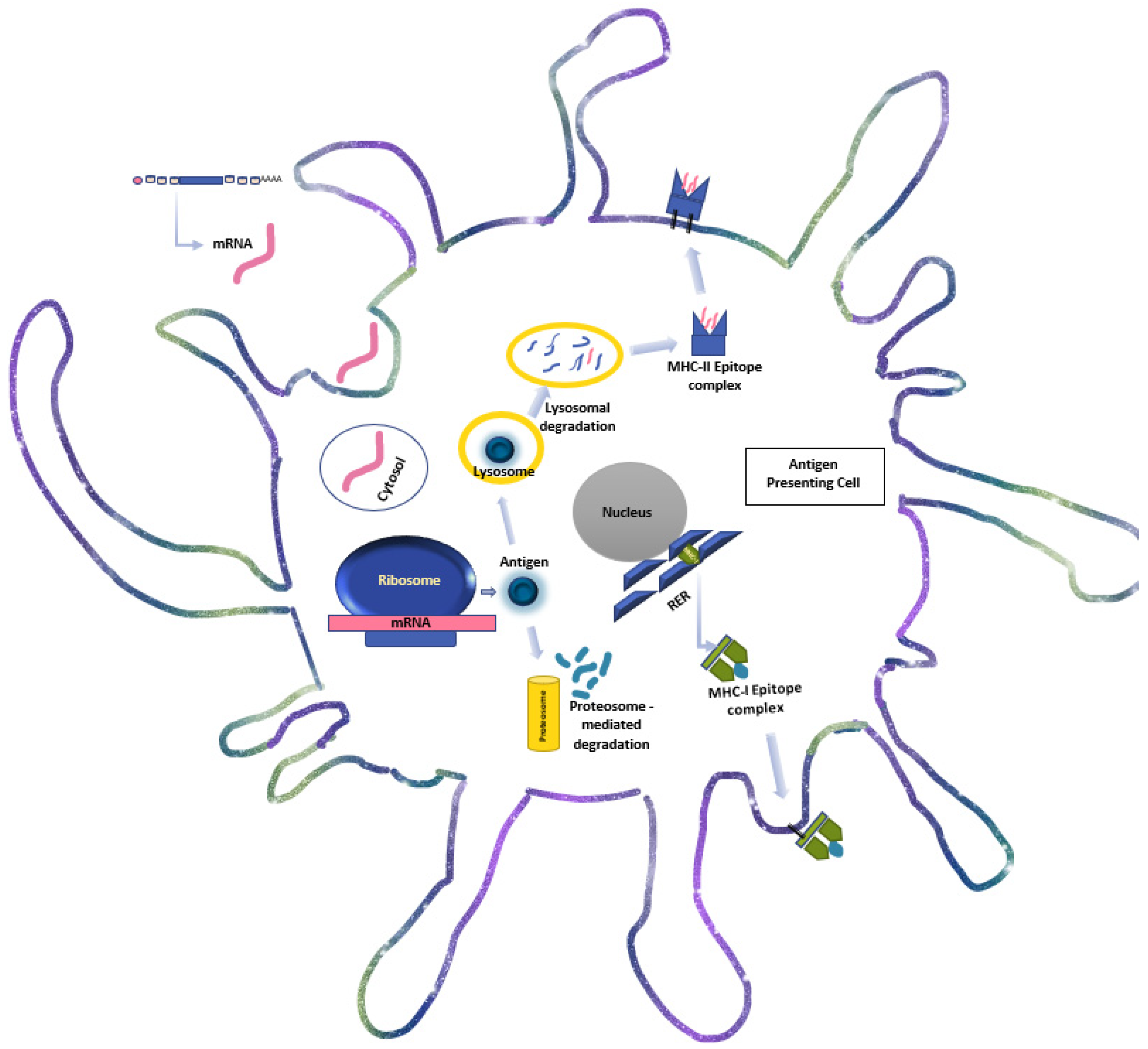


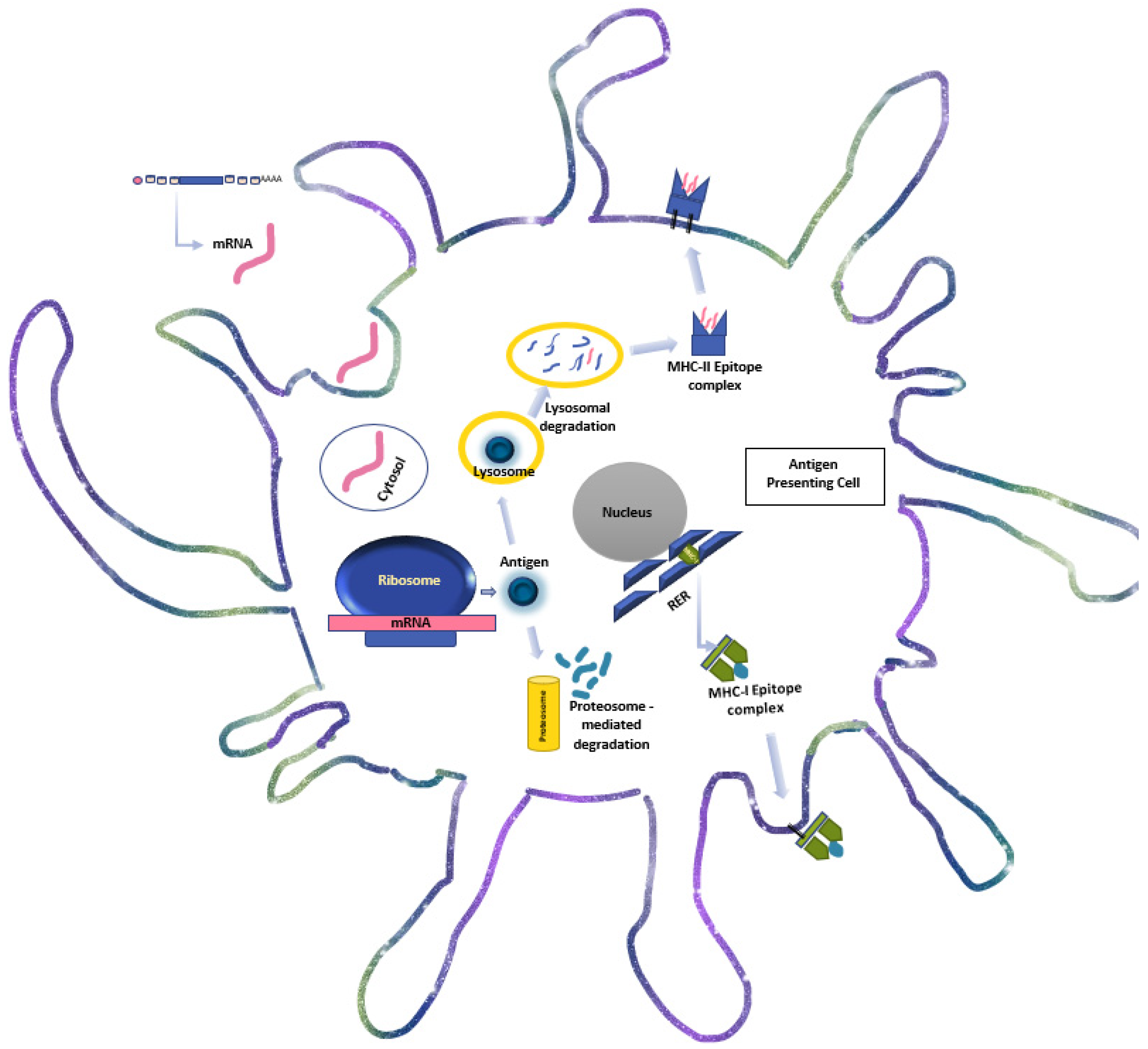{kind=link}
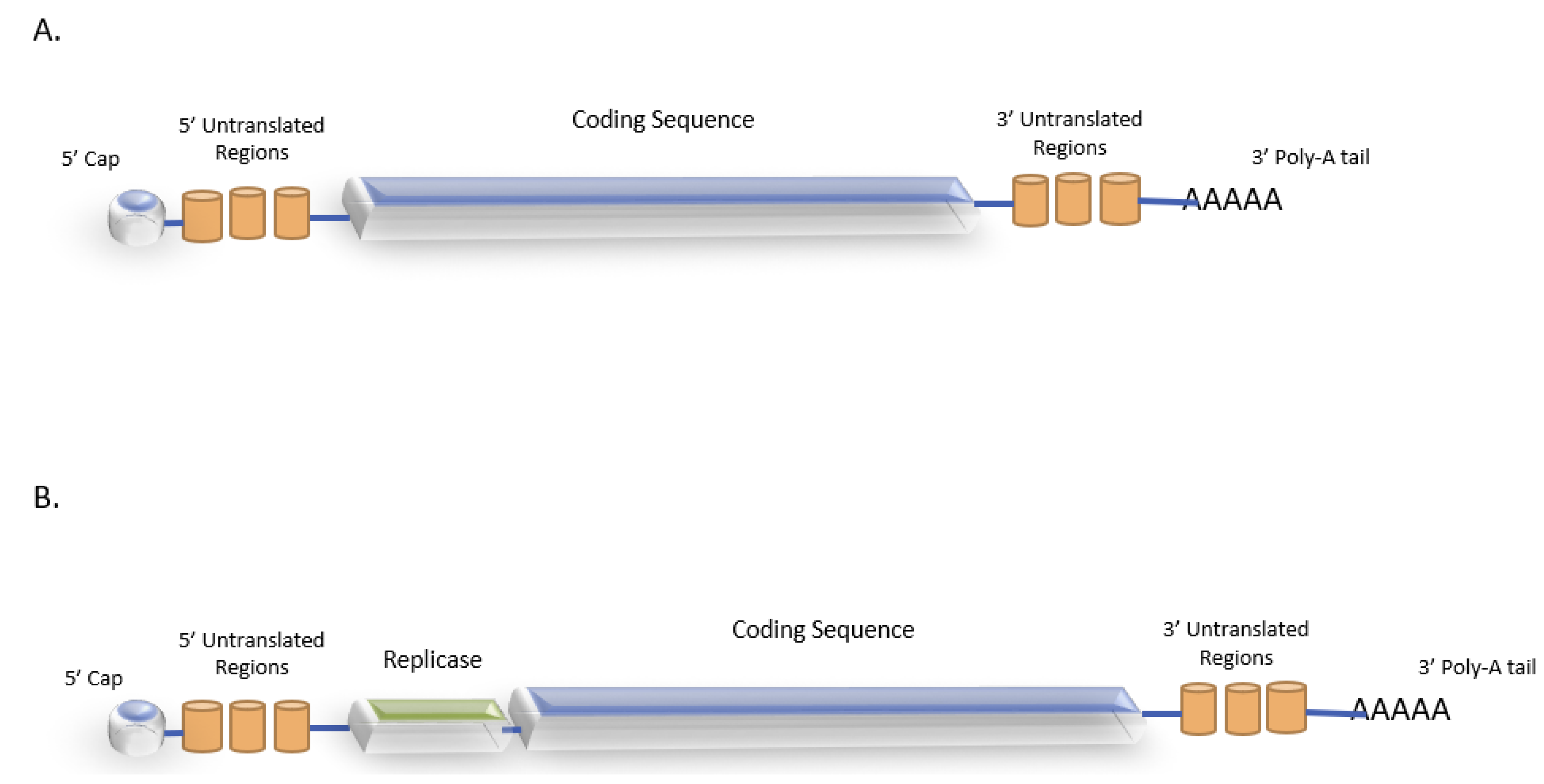{kind=link}
| Trial ID | Trial Design | Target Patient Population (n) | Cancer Type | Investigational Treatment | Primary Outcomes | Trial Responsible Party/Collaborators |
|---|---|---|---|---|---|---|
| Cancer vaccines: | ||||||
| NCT05192460 | Phase I; Dose escalation and expansion with mRNA vaccine (PGV002) | Adult patients (n: 36) | Advanced gastric cancer, esophageal cancer, and liver cancer | Dose expansion: vaccine + PD-1/L1 inhibitor | Safety Tolerability Feasibility | Affiliated Hospital of the Chinese Academy of Military Medical Sciences, China |
| NCT05202561 | Phase I; Open label; 2 arms; Arm I: mRNA cancer vaccine | Adult patients with HLA-A11:01 or C08:02 subtype (n: 10) | Refractory advanced solid tumors with KRAS mut | Arm II: vaccine + PD-1 inhibitor (Navuilumab) | Safety Tolerability Feasibility | Bengbu Medical College, China |
| NCT04534205 AHEAD-MERIT | Phase II; Open label nonrandomized 2 arm with run-in dose evaluation; mRNA vaccine + PD-1 inh vs. PD-1 inh monotherapy | Adult patients (n: 285) | Unresectable recurrent or metastatic HPV16+ HNSCC expressing PD-L1 with CPS ≥ 1 | BNT113 (HPV 16 B6/7 mRNA vaccine) + Pembrolizumab | Run-in: Safety Phase II: OS and ORR; QoL | BionTech SE |
| NCT03313778 KEYNOTE 603 | Phase I, Open label, dose escalation mRNA-4157 vaccine monotherapy (Part A); combined with PD-1 inhibitor (Part B, C, D); | Adult patients (n: 142) | Part A: clinically disease-free after early cancer diagnosis Part B, C: unresectable (locally advanced or metastatic) solid malignancies NSCLC, SCLC, HPV (-) HNSCC; Bladder urothelial; melanoma; MSI-H; high TMB Part D: resected melanoma | Part B, C, D: mRNA-4157 vaccine (lipid encapsulated mRNA vaccine encoding 20 tumor neoantigens) + Pembrolizumab | Safety | Moderna TX, Inc. |
| NCT01686334 WIDEA | Phase II randomized; Open label mRNA dendritic vaccine vs. surveillance | Adult patients (n: 130) | AML with minimal residual disease following front-line chemotherapy (morphological CR or CRi) | Autologous dendritic cells loaded by mRNA electroporation with the Wilms’ tumor antigen (WT1) | OS | Antwerp University Hospital; Belgium |
| NCT04526899 | Phase II randomized; Open label BNT111 and Cemiplimab Combination vs. single agents | Adult patients (n: 180) | Anti-PD-1-refractory/Relapsed, Unresectable Stage III or IV Melanoma; ≥1–5 prior lines treatment including nivolumab/pembrolizumab or BRAFinh | BNT111 and Cemiplimab Combination vs. BNT111 (mRNA vaccine encoding 4 melanoma tumor antigents- NY-ESO-1, MAGE-A3, tyrosinase, and TPTE) vs. Cemiplimab | ORR | BionTech SE |
| NCT04573140 PNOC020 | Phase I; dose escalation; Autologous LP-mRNA tumor vaccine | Pediatric and adult Patients (n: 28) | Newly diagnosed adult MGMT unmethylated glioblastoma and Pediatric High-Grade Gliomas (pHGG); <3 cm residual tumor following surgery and completed chemoradiation | Autologous total tumor mRNA and pp65 lysosomal associated membrane protein (LAMP) loaded lipid particles (liposomal vaccine) | Feasibility, Safety, Dose finding | University of Florida |
| NCT04911621 ADDICT-PedGLIO | Phase I–II mRNA loaded autologous mRNA dendritic cell vaccine | Pediatric Patients (Aged ≥ 12 months and < 18 years) (n: 10) | Adjuvant Dendritic Cell Immunotherapy complementing standard therapy in High-grade Glioma and Diffuse Intrinsic Pontine Glioma | WT1 mRNA-loaded autologous monocyte derived DC: Phase I newly diagnosed: combined with first line chemoradiation treatment Phase II prior therapy: Dendritic cell vaccination plus optional conventional antiglioma treatment | Feasibility, Safety | University Hospital, Antwerp, Belgium |
| NCT02465268 ATTAC-II | Phase II Randomized, Blinded, and Placebo-controlled; Autologous LP-mRNA dendritic cell vaccine with chemotherapy | Adult patients (n: 175) | Adjuvant CMV RNA-Pulsed Dendritic Cells with Tetanus–Diphtheria Toxoid Vaccine; Newly Diagnosed Glioblastoma with < 3 cm residual tumor following surgery and completed chemoradiation | mRNA DCs encoding the pp65 neoantigen and LAMP (lysosomal associated membrane protein) with GM-CSF vs. placebo and unpulsed PBMC combined with adjuvant TMZ | OS | Immunomic Therapeutics, Inc.; University of Florida; NCI |
| NCT03688178 DERIVe | Phase II Randomized, Blinded; Autologous LP-mRNA dendritic cell vaccine alone or combined with CD27 mab | Adult patients (n: 80) | Adjuvant CMV pp65-LAMP mRNA-pulsed autologous DCs ± Varlilumab; Newly Diagnosed Glioblastoma with < 1 cm residual tumor following surgery and completed chemoradiation | Adjuvant CMV RNA-Pulsed Dendritic Cells with pp65-lysosomal-associated membrane protein DCs ± anti CD27 mAb (Varlilumab) and Td preconditioning during adjuvant TMZ Group 1 and 2 (blinded) Group 3 (nonblinded) | OS Safety Change in Treg Depletion | Duke University Celldex Therapeutics |
| NCT05357898 | Phase I/II first in human, open labelEngineered vaccine alone and combined with chemotherapy | Adult patients (n: 60) | Recurrent, locally advanced, or metastatic HPV16+ solid tumors (head and neck, cervical, anal, vulvar, or penile cancer) | SQZ-eAPC-HPV vaccine (mRNA engineered APC-targeting multiple tumor antigens and encoding cytokines) as monotherapy and in combination with pembrolizumab | Safety, Dose-finding | SQZ Biotechnologies |
| NCT03548571 DEN-STEM | Phase II–III; Open, randomized study mRNA pulsed dendritic cell therapy vs. standard therapy | Adult patients (n: 60) | Newly diagnosed IDH wild-type, MGMT-methylated glioblastoma with <1 mm3 residual tumor following surgery and completed chemoradiation | Adjuvant autologous trivalent dendritic cells transfected with tm stem cells, survivin, and hTERT combined with TMZ compared to TMZ after surgery and RT | PFS | Oslo University Hospital |
| NCT04382898 PRO-MERIT | Phase I–II; Open label Dose expansion of W_pro1 vaccine alone and combined with PD-1 inhibitor | Adult patients (n: 130) | Metastatic castration-resistant prostate cancer (mCRPC) progressing after 2–3 prior lines of treatment; localized high risk prostate cancer (LPC) | W_pro1 liposomal mRNA vaccine encoding 5 tumor antigens Part 1, Part 2-1B (mCRPC): dose finding; Part 2-1A (mCRPC): vaccine + Cemiplimab Part 2-2 (LPC): vaccine; Part 2-3 (LPC): vaccine + Cemiplimab | Safety, ORR | BionTech SE |
| NCT03739931 | Phase I Open label, dose escalation study of mRNA-2752 alone and combined with PD-L1 inhibition | Adult patients (n: 264) | Advanced or metastatic solid tumor malignancies (TNBC, HNSCC, NSCLC, urothelial cancer, melanoma) or lymphoma progressing after standard 1 line of prior therapy | Arm A: mRNA 2752 alone Arm B: mRNA 2752 + Durvalumab | Safety, ORR | ModernaTX, Inc. AstraZeneca |
| NCT03788083 TMBA | Phase I Open label, intratumoral TriMix injection compared with placebo | Adult patients (n: 36) | Newly diagnosed stage 1–2 breast cancer; intratumoral administration before surgery | Dose escalation of TriMix (naked mRNA vaccine encoding CD70, CD40 ligand, and constitutively active TLR4 that activate dendritic cells) | Safety; Immune-modulatory Effect | Universitair Ziekenhuis, Brussels |
| Nonvaccine therapies | ||||||
| NCT04981691 (Amaretto) | Phase I, mRNA-engineered anti-Mesothelin CAR-T cells therapy | Adult patients (n: 12) | Unresectable or metastatic mesothelin expression-positive, advanced solid tumors | Dose-escalation of mRNA transduced mesothelin expressing CAR-T cells | Safety | Ruijin Hospital UTC Therapeutics Inc |
| NCT04683939 | Phase I/IIa dose escalation; Open label; BNT 141 alone and combined with chemotherapy | Adult patients (n: 96) | Unresectable or metastatic Claudin 18.2 (CLDN18.2)-positive GI, hepatobiliary or ovarian cancer | Part 1a: Dose-escalation monotherapy with BNT 141 (mRNA-encoded mAb targeting claudin 18.2) Part 1b: Dose escalation with Nab-Pac and gemcitabine | Safety, Dose finding | BionTech SE |
| NCT04995536 | Phase I CpG-STAT3 siRNA combined with RT | Adult patients (n: 18) | Recurrent/Refractory B-cell NHL; ≥2 prior lines treatment | Dose escalation of siRNA targeting TLR9 and STAT3 with local RT | Safety, Dose finding | City of Hope Medical Center NCI |
| NCT05392699 | Phase I ABOD2011 hsc IL-12 mRNA | Adult patients (n: 60) | Recurrent/Refractory solid tumors progressing after standard therapy | ABOD2011 (Humanized Single chain mRNA encoding IL-12) | Safety, Dose finding | Cancer Institute and Hospital, Chinese Academy of Medical Sciences |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eralp, Y. Application of mRNA Technology in Cancer Therapeutics. Vaccines 2022, 10, 1262. https://doi.org/10.3390/vaccines10081262
Eralp Y. Application of mRNA Technology in Cancer Therapeutics. Vaccines. 2022; 10(8):1262. https://doi.org/10.3390/vaccines10081262
Chicago/Turabian StyleEralp, Yesim. 2022. "Application of mRNA Technology in Cancer Therapeutics" Vaccines 10, no. 8: 1262. https://doi.org/10.3390/vaccines10081262
APA StyleEralp, Y. (2022). Application of mRNA Technology in Cancer Therapeutics. Vaccines, 10(8), 1262. https://doi.org/10.3390/vaccines10081262