
Success of Current COVID-19 Vaccine Strategies vs. the Epitope Topology of SARS-CoV-2 Spike Protein-Receptor Binding Domain (RBD): A Computational Study of RBD Topology to Guide Future Vaccine Design

 , ,
, ,  and
and {kind=link}
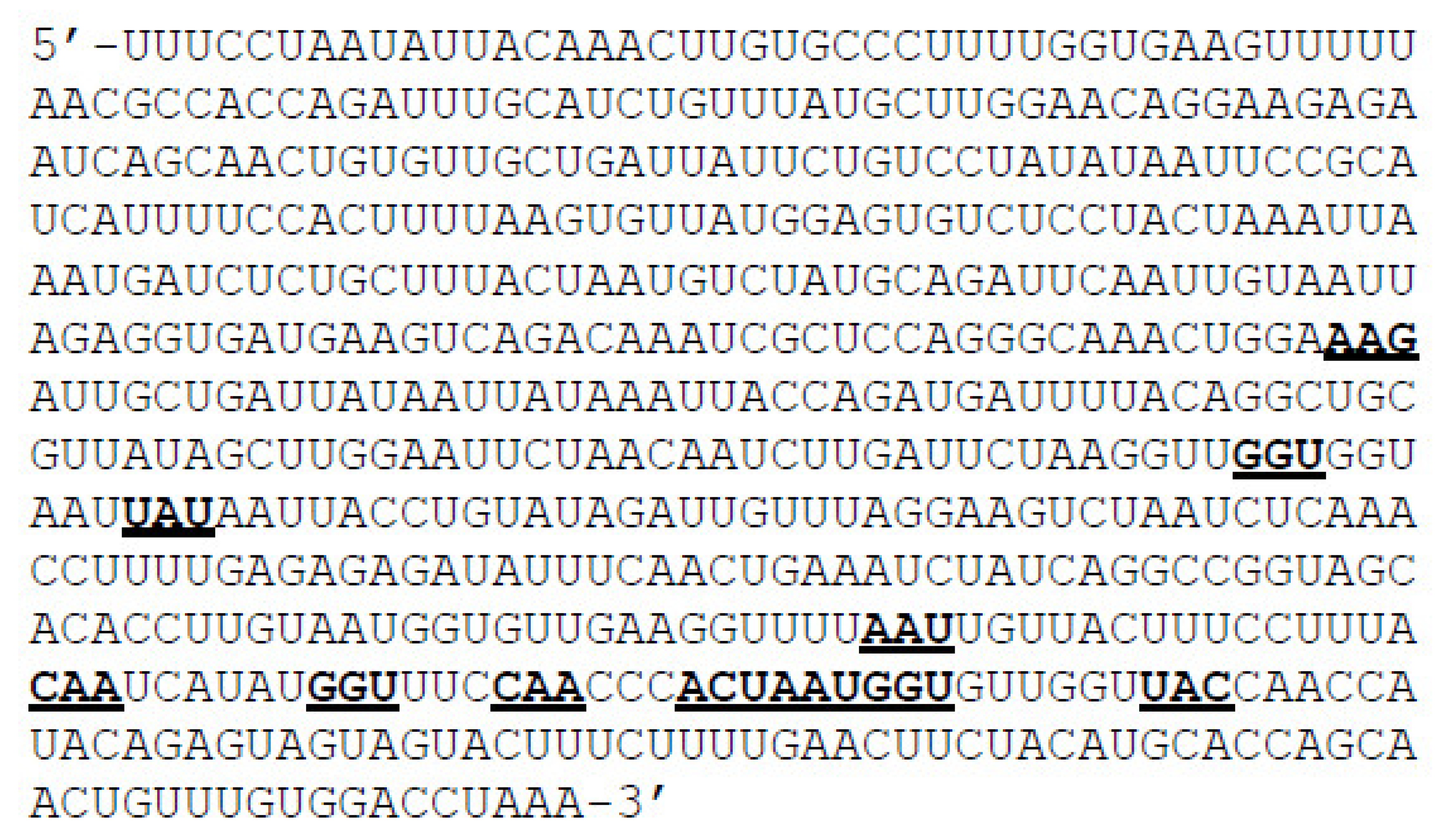{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Preparation of Mutant RNA and Protein Sequences
2.2. RNA Folding and Energy Calculations
2.3. Generation of Mutant Protein Models
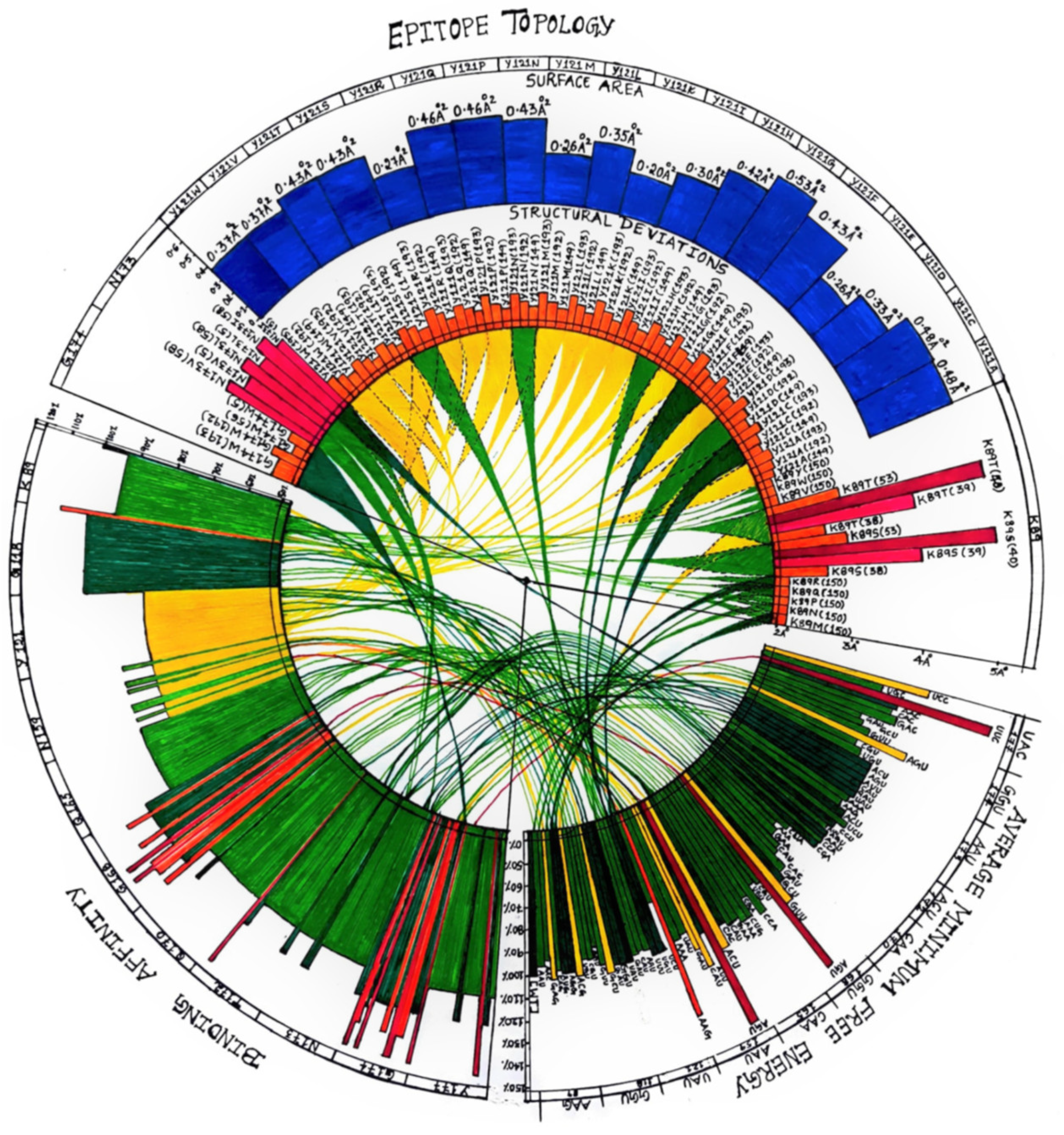
2.4. Structural Analysis
2.5. Binding Affinity
2.6. Viral Fitness
3. Results and Discussion
3.1. Mutant mRNA Sequences Show Higher Stability than Wild Type
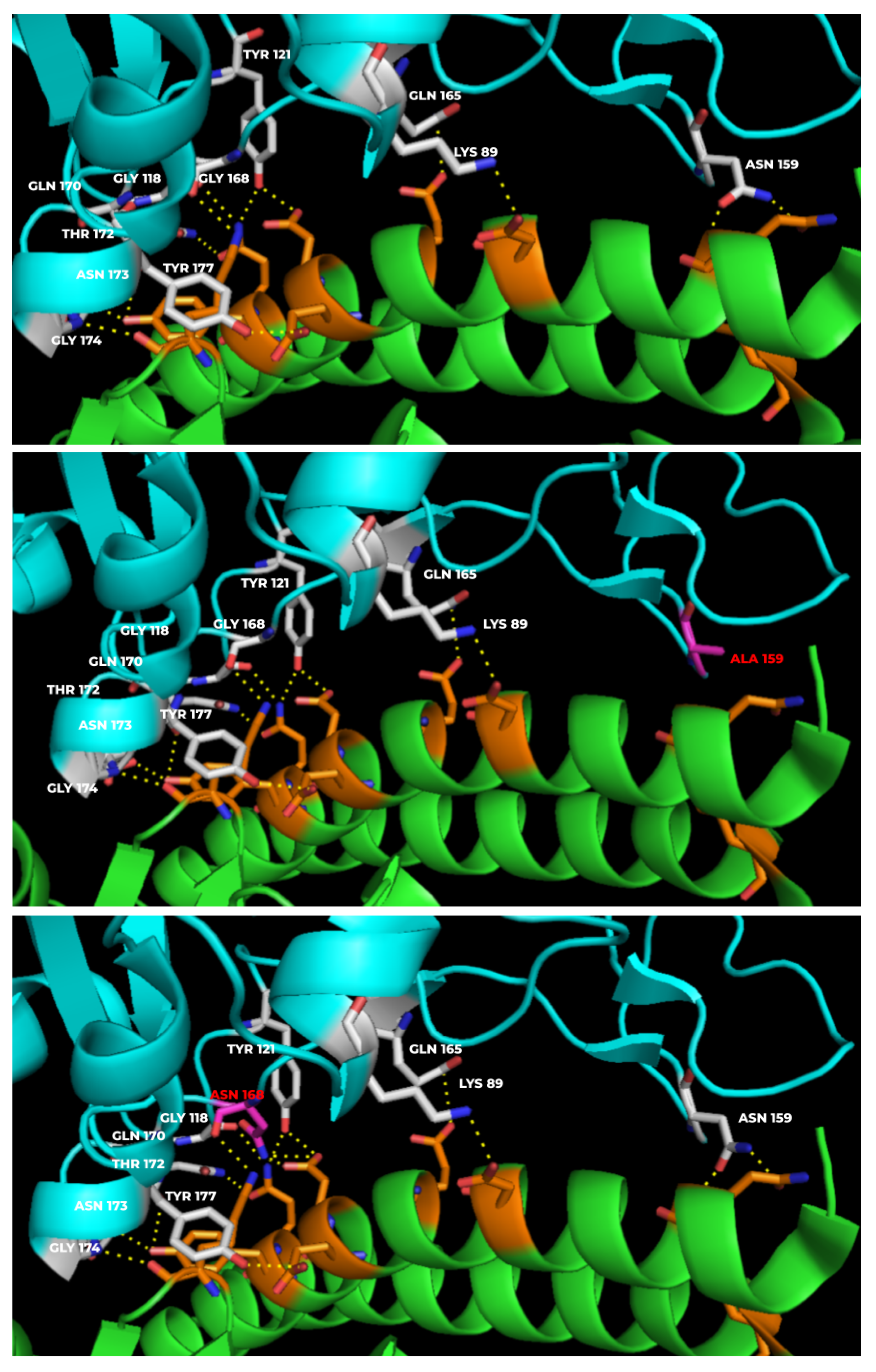
3.2. Mutants Show Altered RBD-hACE-2r Interface Hydrogen Bonds
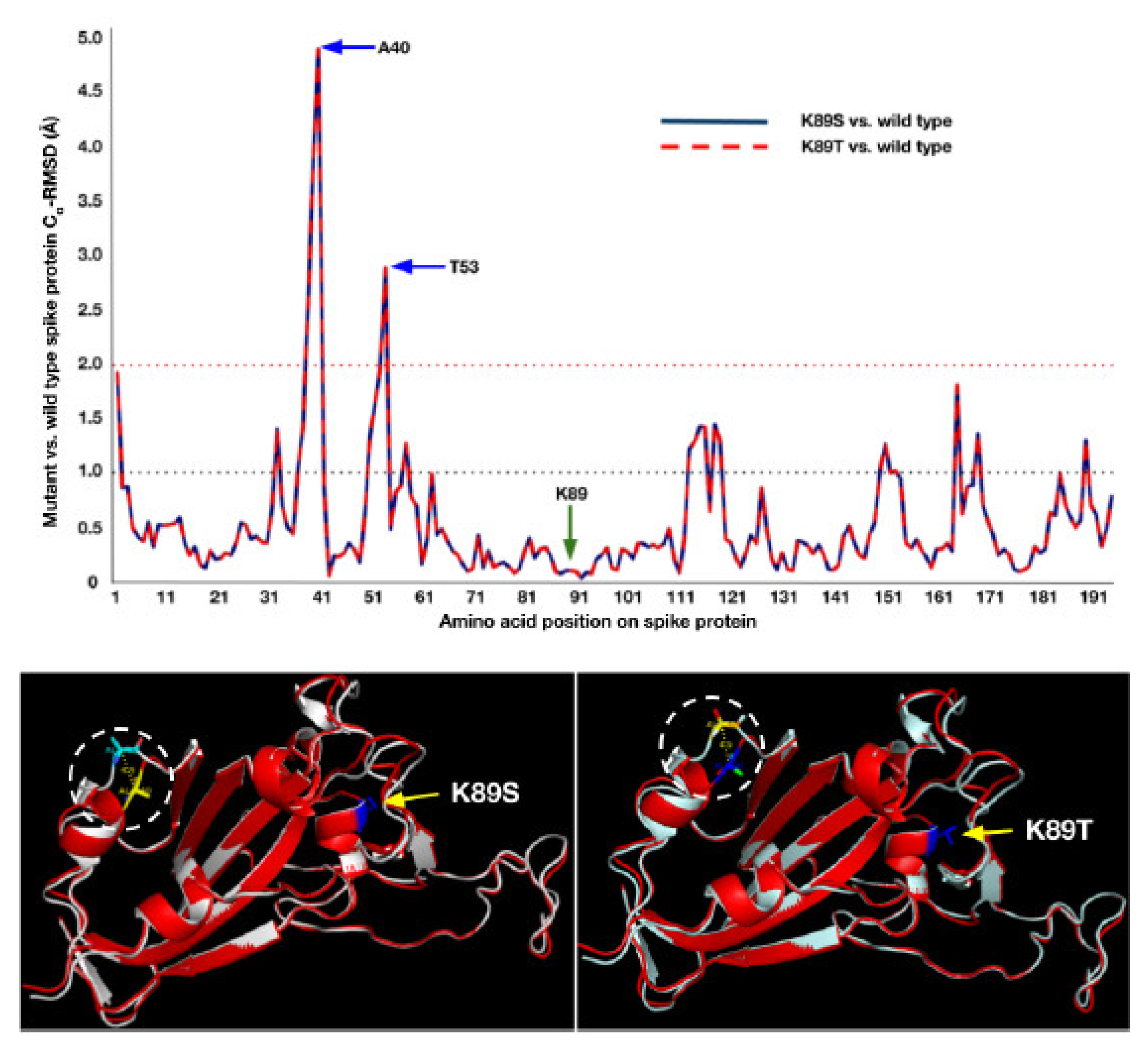
3.3. Mutant Models Show Structural Deviations Onsite and Offsite
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zheng, J. SARS-CoV-2: An Emerging Coronavirus that Causes a Global Threat. Int. J. Biol. Sci. 2020, 16, 1678–1685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Available online: https://en.wikipedia.org/wiki/COVID-19_pandemic (accessed on 1 January 2022).
- Pachetti, M.; Marini, B.; Benedetti, F.; Giudici, F.; Mauro, E.; Storici, P.; Masciovecchio, C.; Angeletti, S.; Ciccozzi, M.; Gallo, R.C.; et al. Emerging SARS-CoV-2 mutation hot spots include a novel RNA-dependent-RNA polymerase variant. J. Transl. Med. 2020, 18, 179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- V’Kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Baloch, Z.; Ikram, A.; Hakim, M.S.; Awan, F.M. The Impact of Mutations on the Pathogenic and Antigenic Activity of SARS-CoV-2 during the First Wave of the COVID-19 Pandemic: A Comprehensive Immunoinformatics Analysis. Vaccines 2021, 9, 1410. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Cui, H.; Gao, Z.; Liu, M.; Lu, S.; Mkandawire, W.; Narykov, O.; Sun, M.; Korkin, D. Structural Genomics of SARS-CoV-2 Indicates Evolutionary Conserved Functional Regions of Viral Proteins. Viruses 2020, 12, 360. [Google Scholar] [CrossRef] [Green Version]
- Xia, X. Domains and Functions of Spike Protein in SARS-Cov-2 in the Context of Vaccine Design. Viruses 2021, 13, 109. [Google Scholar] [CrossRef]
- Available online: https://www.ncbi.nlm.nih.gov/nuccore/NC_045512.2?report=genbank&from=21563&to=25384 (accessed on 1 January 2022).
- Hoffmann, M.; Kleine-Weber, H.; Pöhlmann, S. A Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells. Mol. Cell 2020, 78, 779–784.e5. [Google Scholar] [CrossRef]
- Dai, L.; Gao, G.F. Viral targets for vaccines against COVID-19. Nat. Rev. Immunol. 2021, 21, 73–82. [Google Scholar] [CrossRef]
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef] [Green Version]
- Vzorov, A.N.; Samokhvalov, E.I.; Chebanenko, V.V.; Scheblyakov, D.V.; Gintsburg, A.L. Modification of the Spike Protein for Vaccines against Enveloped RNA Viruses. Mol. Biol. 2021, 55, 538–547. [Google Scholar] [CrossRef]
- Yang, Z.-Y.; Kong, W.-P.; Huang, Y.; Roberts, A.; Murphy, B.R.; Subbarao, K.; Nabel, G.J. A DNA vaccine induces SARS coronavirus neutralization and protective immunity in mice. Nature 2004, 428, 561–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Gao, X. Immunological responses against SARS-coronavirus infection in humans. Cell Mol. Immunol. 2004, 1, 119–122. [Google Scholar] [PubMed]
- Available online: https://en.wikipedia.org/wiki/Epitope (accessed on 1 January 2022).
- Bakhtawar, N.; Usman, M.; Khan, M.M.U. Convalescent Plasma Therapy and Its Effects On COVID-19 Patient Outcomes: A Systematic Review of Current Literature. Cureus 2020, 12, e9535. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; He, Y. Challenges of Convalescent Plasma Therapy on COVID-19. J. Clin. Virol. 2020, 127, 104358. [Google Scholar] [CrossRef] [PubMed]
- MacLennan, S.; Barbara, J.A. Risks and side effects of therapy with plasma and plasma fractions. Best Pract. Res. Clin. Haematol. 2006, 19, 169–189. [Google Scholar] [CrossRef]
- He, J.; Huang, F.; Zhang, J.; Chen, Q.; Zheng, Z.; Zhou, Q.; Chen, D.; Li, J.; Chen, J. Vaccine design based on 16 epitopes of SARS-CoV-2 spike protein. J. Med. Virol. 2021, 93, 2115–2131. [Google Scholar] [CrossRef]
- Barnes, C.O.; West, A.P., Jr.; Huey-Tubman, K.E.; Hoffmann, M.A.G.; Sharaf, N.G.; Hoffman, P.R.; Koranda, N.; Gristick, H.B.; Gaebler, C.; Muecksch, F.; et al. Structures of Human Antibodies Bound to SARS-CoV-2 Spike Reveal Common Epitopes and Recurrent Features of Antibodies. Cell 2020, 182, 828–842.e16. [Google Scholar] [CrossRef]
- Wikipedia Contributors. Vaccine. Available online: https://en.wikipedia.org/wiki/Vaccine (accessed on 3 December 2021).
- Frazer, I.H. Development and implementation of papillomavirus prophylactic vaccines. J. Immunol. 2014, 192, 4007–4011. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://www.hhs.gov/immunization/basics/types/index.html (accessed on 1 January 2022).
- Wikipedia Contributors. Inactivated vaccine. Available online: https://en.wikipedia.org/wiki/Inactivated_vaccine (accessed on 4 December 2021).
- Petrovsky, N.; Aguilar, J.C. Vaccine adjuvants: Current state and future trends. Immunol. Cell Biol. 2004, 82, 488–496. [Google Scholar] [CrossRef]
- Types of Vaccines. U.S. Department of Health and Human Services. 23 July 2013. Available online: https://www.vaccines.gov/ (accessed on 9 June 2013).
- Wodi, A.P.; Morelli, V. Chapter 1: Principles of Vaccination. In Epidemiology and Prevention of Vaccine-Preventable Diseases, 14th ed.; Hall, E., Wodi, A.P., Hamborsky, J., Morelli, V., Schilllie, S., Eds.; Public Health Foundation, Centers for Disease Control and Prevention: Washington, DC, USA, 2021. [Google Scholar]
- Available online: https://www.bharatbiotech.com/covaxin.html (accessed on 1 January 2022).
- Ella, R.; Vadrevu, K.M.; Jogdand, H.; Prasad, S.; Reddy, S.; Sarangi, V.; Ganneru, B.; Sapkal, G.; Yadav, P.; Abraham, P.; et al. Safety and immunogenicity of an inactivated SARS-CoV-2 vaccine, BBV152: A double-blind, randomised, phase 1 trial. Lancet Infect. Dis. 2021, 21, 637–646, Erratum in 2021, 21, e81. [Google Scholar] [CrossRef]
- Ella, R.; Reddy, S.; Blackwelder, W.; Potdar, V.; Yadav, P.; Sarangi, V.; Aileni, V.K.; Kanungo, S.; Rai, S.; Reddy, P.; et al. COVAXIN Study Group. Efficacy, safety, and lot-to-lot immunogenicity of an inactivated SARS-CoV-2 vaccine (BBV152): Interim results of a randomised, double-blind, controlled, phase 3 trial. Lancet 2021, 398, 2173–2184. [Google Scholar] [CrossRef]
- Plotkin, S.; Plotkin, S. The development of vaccines: How the past led to the future. Nat. Rev. Microbiol. 2011, 9, 889–893. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://medlineplus.gov/genetics/understanding/therapy/mrnavaccines/ (accessed on 1 January 2022).
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tartof, S.Y.; Slezak, J.M.; Fischer, H.; Hong, V.; Ackerson, B.K.; Ranasinghe, O.N.; Frankland, T.B.; Ogun, O.A.; Zamparo, J.M.; Gray, S.; et al. Effectiveness of mRNA BNT162b2 COVID-19 vaccine up to 6 months in a large integrated health system in the USA: A retrospective cohort study. Lancet 2021, 398, 1407–1416. [Google Scholar] [CrossRef]
- El Sahly, H.M.; Baden, L.R.; Essink, B.; Doblecki-Lewis, S.; Martin, J.M.; Anderson, E.J.; Campbell, T.B.; Clark, J.; Jackson, L.A.; Fichtenbaum, C.J.; et al. COVE Study Group. Efficacy of the mRNA-1273 SARS-CoV-2 Vaccine at Completion of Blinded Phase. N. Engl. J. Med. 2021, 385, 1774–1785. [Google Scholar] [CrossRef]
- Gilbert, P.B.; Montefiori, D.C.; McDermott, A.; Fong, Y.; Benkeser, D.; Deng, W.; Zhou, H.; Houchens, C.R.; Martins, K.; Jayashankar, L.; et al. Immune Correlates Analysis of the mRNA-1273 COVID-19 Vaccine Efficacy Trial. medRxiv 2021. [Google Scholar] [CrossRef]
- Khalaj-Hedayati, A.; Chua, C.L.L.; Smooker, P.; Lee, K.W. Nanoparticles in influenza subunit vaccine development: Immunogenicity enhancement. Influ. Other Respir Viruses 2020, 14, 92–101. [Google Scholar] [CrossRef]
- López-Sanguos, C.; Rivero Calle, I.; Rodriguez Tenreiro, C.; Raguindin, P.F.; Martinón-Torres, F. Safety and immunogenicity of pneumococcal conjugate vaccines in preterm infants. Expert Opin. Drug Saf. 2019, 18, 253–259. [Google Scholar] [CrossRef]
- Sun, B.; Yu, S.; Zhao, D.; Guo, S.; Wang, X.; Zhao, K. Polysaccharides as vaccine adjuvants. Vaccine 2018, 36, 5226–5234. [Google Scholar] [CrossRef]
- Yadav, T.; Srivastava, N.; Mishra, G.; Dhama, K.; Kumar, S.; Puri, B.; Saxena, S.K. Recombinant vaccines for COVID-19. Hum. Vaccin Immunother. 2020, 16, 2905–2912. [Google Scholar] [CrossRef]
- Bastola, R.; Noh, G.; Keum, T.; Bashyal, S.; Seo, J.E.; Choi, J.; Oh, Y.; Cho, Y.; Lee, S. Vaccine adjuvants: Smart components to boost the immune system. Arch. Pharm. Res. 2017, 40, 1238–1248. [Google Scholar] [CrossRef] [PubMed]
- Syed, Y.Y. DTaP-IPV-HepB-Hib Vaccine (Hexyon®): An Updated Review of its Use in Primary and Booster Vaccination. Paediatr. Drugs 2019, 21, 397–408, Erratum in Paediatr. Drugs 2019, 21, 501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haber, P.; Moro, P.L.; Ng, C.; Dores, G.M.; Perez-Vilar, S.; Marquez, P.L.; Cano, M. Safety review of tetanus toxoid, reduced diphtheria toxoid, acellular pertussis vaccines (Tdap) in adults aged ≥65 years, Vaccine Adverse Event reporting System (VAERS), United States, September 2010–December 2018. Vaccine 2020, 38, 1476–1480. [Google Scholar] [CrossRef] [PubMed]
- Ewer, K.J.; Lambe, T.; Rollier, C.S.; Spencer, A.J.; Hill, A.V.; Dorrell, L. Viral vectors as vaccine platforms: From immunogenicity to impact. Curr. Opin. Immunol. 2016, 41, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Ura, T.; Yamashita, A.; Mizuki, N.; Okuda, K.; Shimada, M. New vaccine production platforms used in developing SARS-CoV-2 vaccine candidates. Vaccine 2021, 39, 197–201. [Google Scholar] [CrossRef]
- Voysey, M.; Clemens, S.A.C.; Madhi, S.A.; Weckx, L.Y.; Folegatti, P.M.; Aley, P.K.; Angus, B.; Baillie, V.L.; Barnabas, S.L.; Bhorat, Q.E.; et al. Oxford COVID Vaccine Trial Group. Safety and efficacy of the ChAdOx1 nCoV-19 vaccine (AZD1222) against SARS-CoV-2: An interim analysis of four randomised controlled trials in Brazil, South Africa, and the UK. Lancet 2021, 397, 99–111, Erratum in Lancet 2021, 397, 98. [Google Scholar] [CrossRef]
- Ramasamy, M.N.; Minassian, A.M.; Ewer, K.J.; Flaxman, A.L.; Folegatti, P.M.; Owens, D.R.; Voysey, M.; Aley, P.K.; Angus, B.; Babbage, G.; et al. Oxford COVID Vaccine Trial Group. Safety and immunogenicity of ChAdOx1 nCoV-19 vaccine administered in a prime-boost regimen in young and old adults (COV002): A single-blind, randomised, controlled, phase 2/3 trial. Lancet 2021, 396, 1979–1993, Erratum in Lancet 2021, 396, 1978; Erratum in 2021, 397, 1350. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [Green Version]
- Cao, Z.; Liu, L.; Du, L.; Zhang, C.; Jiang, S.; Li, T.; He, Y. Potent and persistent antibody responses against the receptor-binding domain of SARS-CoV spike protein in recovered patients. Virol J. 2010, 7, 299. [Google Scholar] [CrossRef] [Green Version]
- Vissapragada MAddala SSodasani, K.; Yedidi, R.S. Major structural deviations in the receptor binding domain of SARS-CoV-2 spike protein may pose threat to the existing vaccines. TCABSE J. 2021, 1, 12–14. [Google Scholar]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Addala, S.; Vissapragada, M.; Aggunna, M.; Mukala, N.; Lanka, M.; Gampa, S.; Sodasani, M.; Chintalapati, J.; Kamidi, A.; Veeranna, R.P.; et al. Success of Current COVID-19 Vaccine Strategies vs. the Epitope Topology of SARS-CoV-2 Spike Protein-Receptor Binding Domain (RBD): A Computational Study of RBD Topology to Guide Future Vaccine Design. Vaccines 2022, 10, 841. https://doi.org/10.3390/vaccines10060841
Addala S, Vissapragada M, Aggunna M, Mukala N, Lanka M, Gampa S, Sodasani M, Chintalapati J, Kamidi A, Veeranna RP, et al. Success of Current COVID-19 Vaccine Strategies vs. the Epitope Topology of SARS-CoV-2 Spike Protein-Receptor Binding Domain (RBD): A Computational Study of RBD Topology to Guide Future Vaccine Design. Vaccines. 2022; 10(6):841. https://doi.org/10.3390/vaccines10060841
Chicago/Turabian StyleAddala, Santhinissi, Madhuri Vissapragada, Madhumita Aggunna, Niharikha Mukala, Manisha Lanka, Shyamkumar Gampa, Manikanta Sodasani, Jahnavi Chintalapati, Akhila Kamidi, Ravindra P. Veeranna, and et al. 2022. "Success of Current COVID-19 Vaccine Strategies vs. the Epitope Topology of SARS-CoV-2 Spike Protein-Receptor Binding Domain (RBD): A Computational Study of RBD Topology to Guide Future Vaccine Design" Vaccines 10, no. 6: 841. https://doi.org/10.3390/vaccines10060841
APA StyleAddala, S., Vissapragada, M., Aggunna, M., Mukala, N., Lanka, M., Gampa, S., Sodasani, M., Chintalapati, J., Kamidi, A., Veeranna, R. P., & Yedidi, R. S. (2022). Success of Current COVID-19 Vaccine Strategies vs. the Epitope Topology of SARS-CoV-2 Spike Protein-Receptor Binding Domain (RBD): A Computational Study of RBD Topology to Guide Future Vaccine Design. Vaccines, 10(6), 841. https://doi.org/10.3390/vaccines10060841