Development of a Universal Epitope-Based Influenza Vaccine and Evaluation of Its Effectiveness in Mice

 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Epitope Selection
2.2. Cell Lines and Viruses
2.3. Construction of Plasmids
2.4. Obtaining Recombinant MVA
2.5. Western Blot
2.6. Experimental Animals
2.7. Mouse Immunization and Study Design
2.8. HAI Test
2.9. The Neutralization Test
2.10. Determination of the Protective Activity of Vaccine Variants
2.11. Determination of the Viral Titer in the Lungs of Mice
2.12. Data Analysis
3. Results
3.1. Constructing the Variants of the Epitope Vaccine
3.2. Constructing Reference Constructs Variants for Comparison
3.3. Obtaining Recombinant MVA and Checking Antigen Expression
3.4. Study of the Antibody Response to Different Strains of Influenza Virus Types A and B
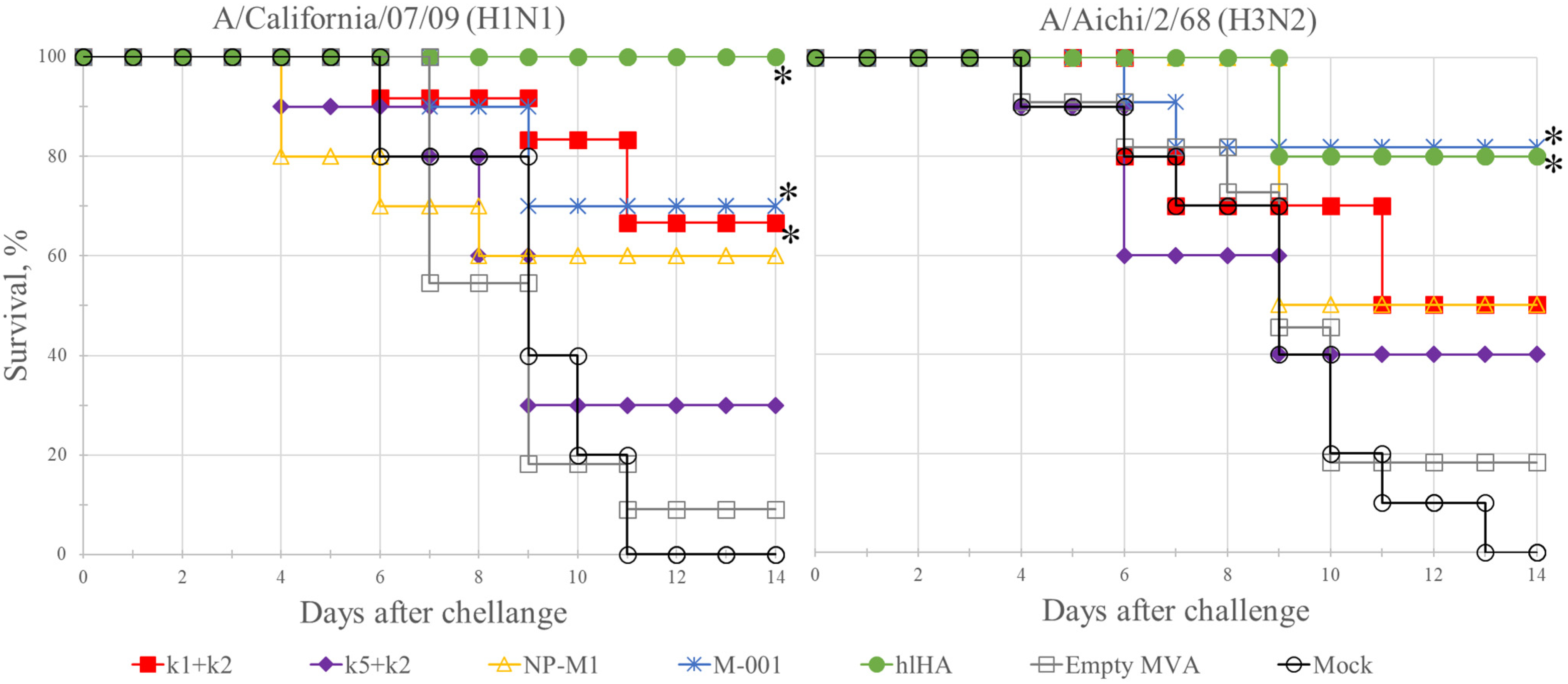
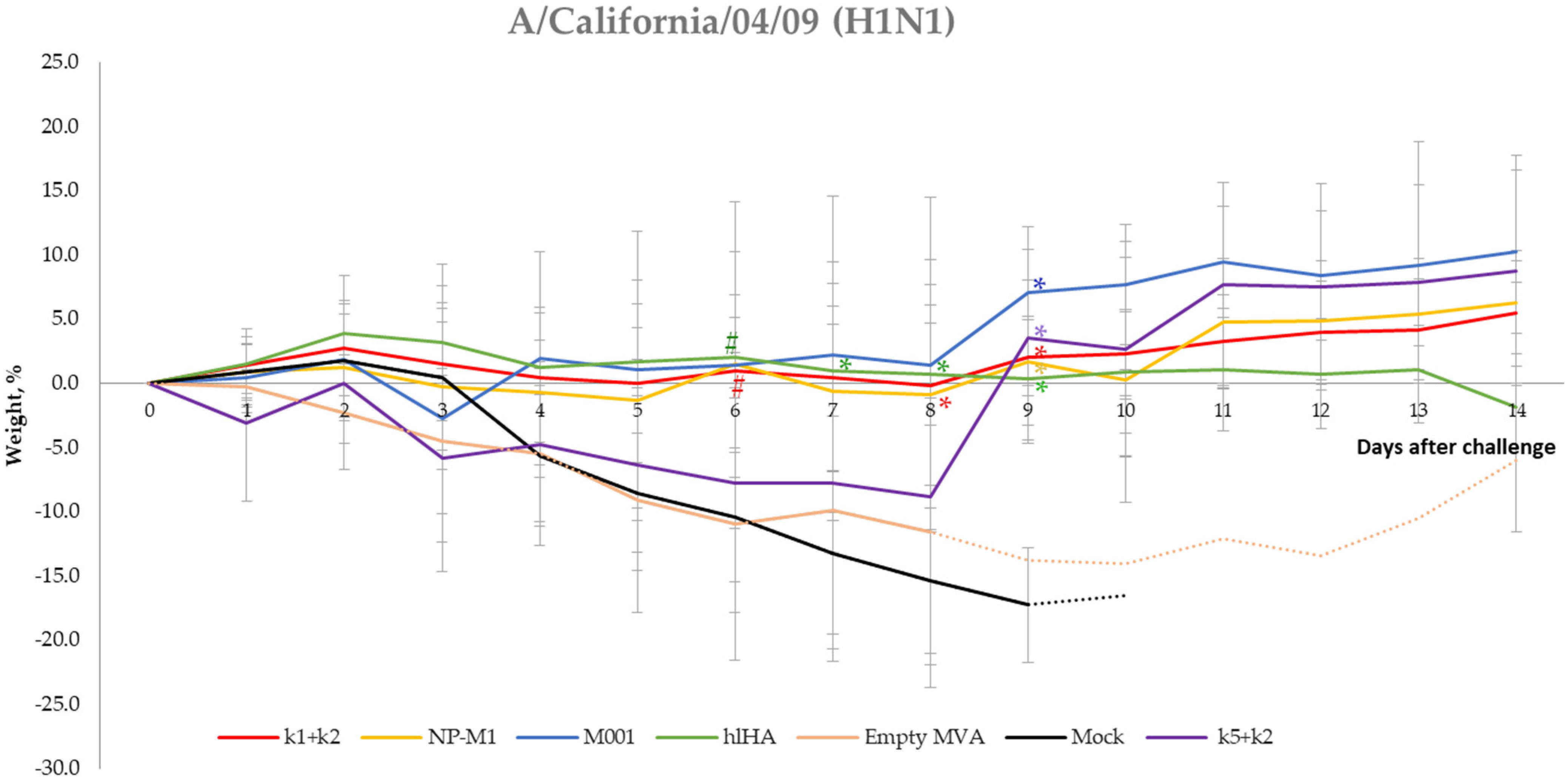
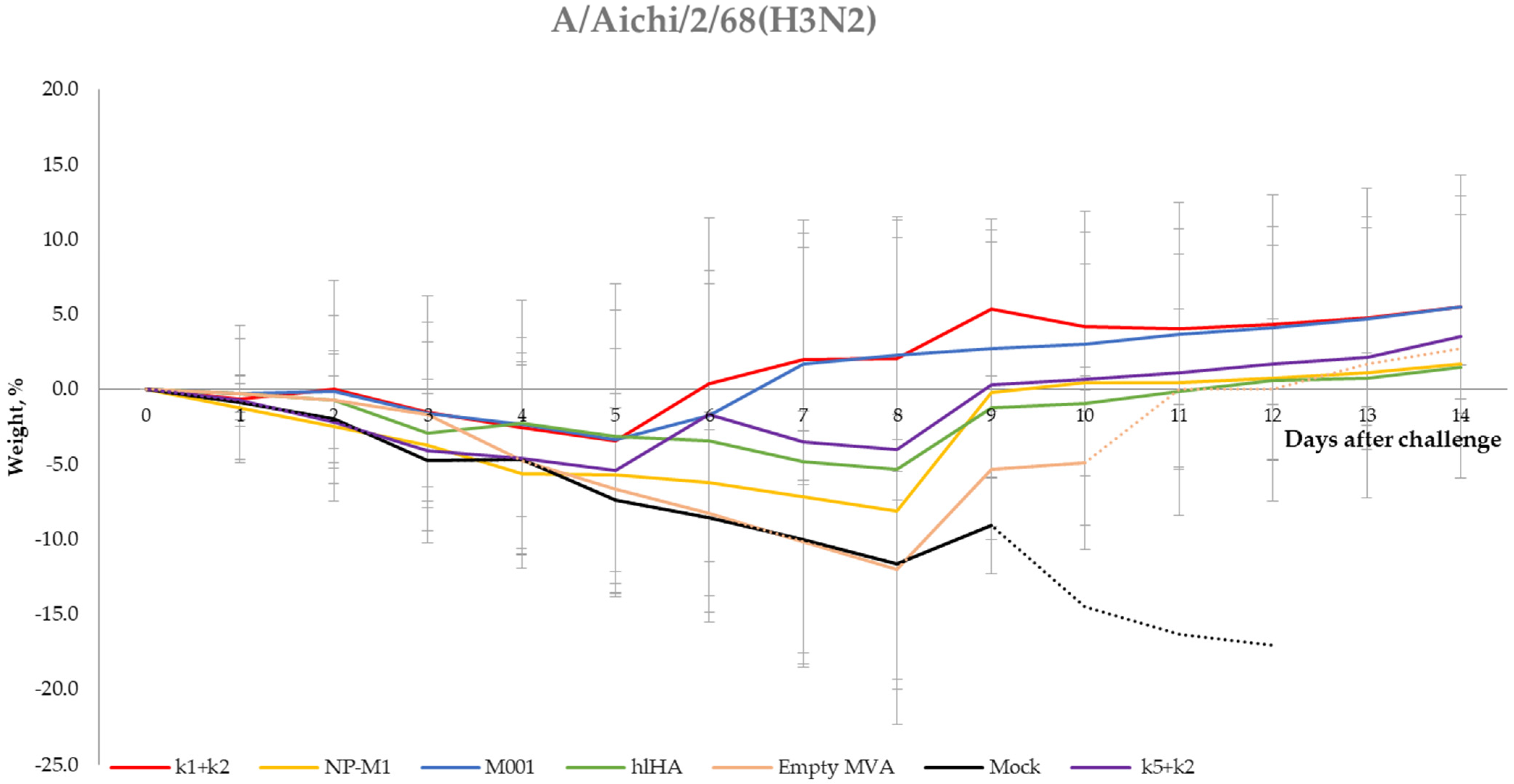
3.5. Protection of Mice against Challenge Infection
3.6. Viral Titer in the Lungs of Mice after Infection with H1N1 and H3N2 Influenza Virus Subtypes
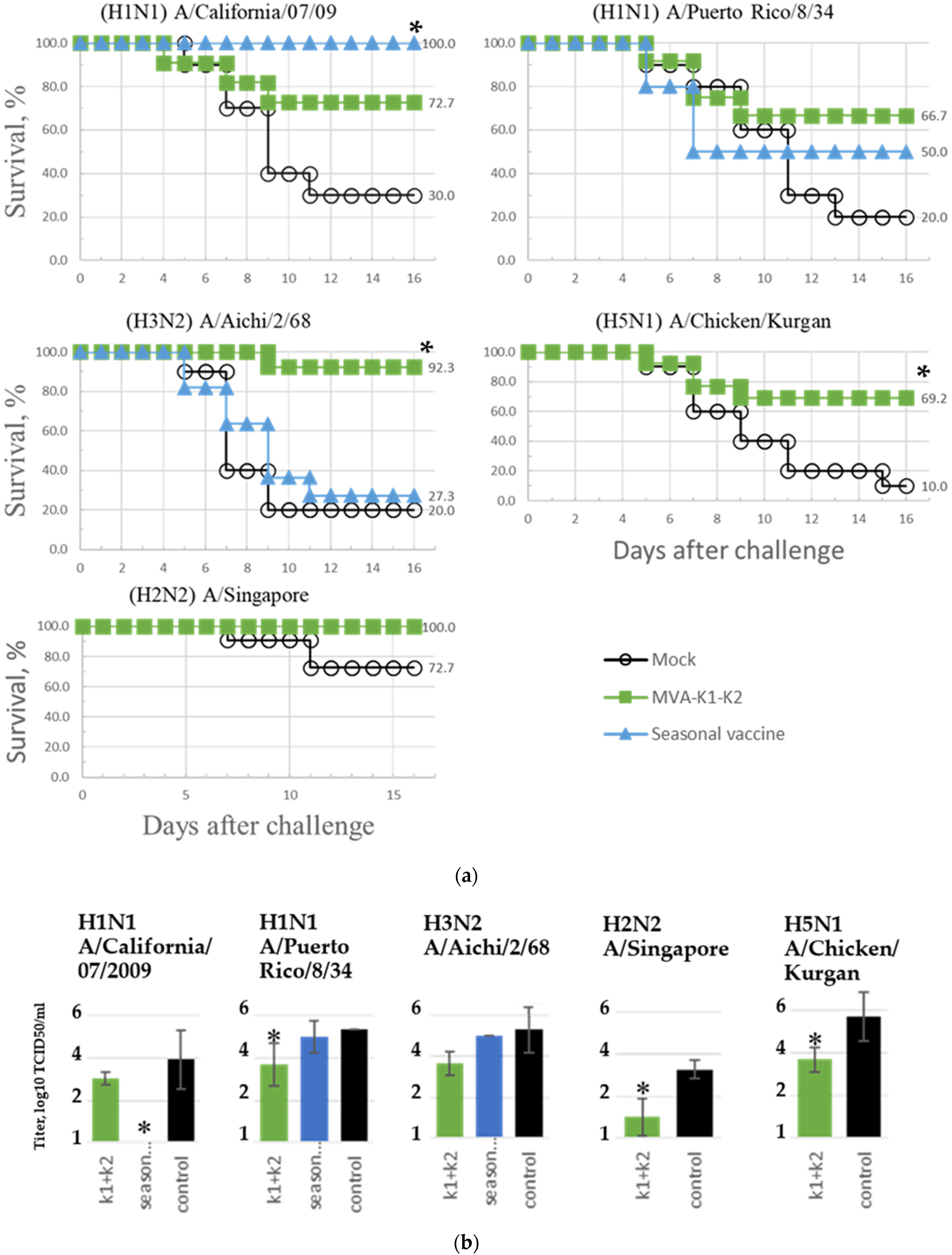
3.7. Assessment of the Breadth of Immunogenicity and Protection of the rMVA-k1-k2 Vaccine Variant
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | Strain | Source |
|---|---|---|
| 1 | A/California/07/2009 (pdm H1N1 2009) adapted to mice | WHO |
| 2 | A/Puerto Rico/8/34 (H1N1) | Smorodintsev Research Institute of Influenza |
| 3 | A/Chicken/Kurgan (H5N1) | Smorodintsev Research Institute of Influenza |
| 4 | A/Singapore (H2N2) | Smorodintsev Research Institute of Influenza |
| 5 | A/Aichi/2/68 (H3N2) adapted to mice | The National Research Center for Epidemiology and Microbiology named after the Honorary Academician N.F. Gamaleya of the Ministry of Health of the Russian Federation |
| # | Strain | Source |
|---|---|---|
| 1 | A/California/07/2009 (pdm H1N1 2009), adapted to mice | WHO |
| 2 | A/Puerto Rico/8/34 (H1N1) | Smorodintsev Research Institute of Influenza |
| 3 | A/Chicken/Kurgan (H5N1) | Smorodintsev Research Institute of Influenza |
| 5 | A/Aichi/2/68 (H3N2), adapted to mice | The National Research Center for Epidemiology and Microbiology named after the Honorary Academician N.F. Gamaleya of the Ministry of Health of the Russian Federation |
| 6 | A/Texas/50/2012 (H3N2) | CDC (Atlanta, GA, USA) |
| 7 | B/Brisbane/60/2008 | CDC (Atlanta, GA, USA) |
| 8 | B/Colorado/06/2017 | CDC (Atlanta, GA, USA) |
| 9 | B/Wisconsin/1/2010 | CDC (Atlanta, GA, USA) |


References
- Iuliano, A.D.; Rogusk, K.M.; Chang, H.H.; Muscatello, D.J.; Palekar, R.; Tempia, S.; Cohen, C.; Gran, J.M.; Schanzer, D.; Cowling, B.J.; et al. Estimates of global seasonal influenza-associated respiratory mortality: A modelling study. Lancet 2018, 39, 1285–1300. [Google Scholar] [CrossRef]
- Flahault, A.; Vergu, E.; Boëlle, P.Y. Potential for a global dynamic of Influenza A (H1N1). BMC Infect. Dis. 2009, 9, 129. [Google Scholar] [CrossRef] [PubMed]
- Krammer, F. Emerging influenza viruses and the prospect of a universal influenza virus vaccine. Biotechnol. J. 2015, 10, 690–701. [Google Scholar] [CrossRef]
- DiazGranados, C.A.; Denis, M.; Plotkin, S. Seasonal influenza vaccine efficacy and its determinants in children and non-elderly adults: A systematic review with meta-analyses of controlled trials. Vaccine 2012, 31, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention, DC Seasonal Flu Vaccine Effectiveness Studies. Available online: https://www.cdc.gov/flu/vaccines-work/effectiveness-studies.htm (accessed on 2 November 2021).
- Young, B.; Sadarangani, S.; Jiang, L.; Wilder-Smith, A.; Chen, M.I. Duration of Influenza Vaccine Effectiveness: A Systematic Review, Meta-analysis, and Meta-regression of Test-Negative Design Case-Control Studies. J. Infect. Dis. 2018, 217, 731–741. [Google Scholar] [CrossRef]
- Wei, C.J.; Crank, M.C.; Shiver, J.; Graham, B.S.; Mascola, J.R.; Nabel, G.J. Next-generation influenza vaccines: Opportunities and challenges. Nat. Rev. Drug Discov. 2020, 19, 239–252. [Google Scholar] [CrossRef]
- Corder, B.N.; Bullard, B.L.; Poland, G.A.; Weaver, E.A. A Decade in Review: A Systematic Review of Universal Influenza Vaccines in Clinical Trials during the 2010 Decade. Viruses 2020, 12, 1186. [Google Scholar] [CrossRef]
- Liu, H.; Frijlink, H.W.; Huckriede, A.; van Doorn, E.; Schmidt, E.; Leroy, O.; Rimmelzwaan, G.; McCullough, K.; Whelan, M.; Hak, E. Influenza Vaccine Research funded by the European Commission FP7-Health-2013-Innovation-1 project. Vaccine 2016, 34, 5845–5854. [Google Scholar] [CrossRef]
- Wu, C.; Zanker, D.; Valkenburg, S.; Tan, B.; Kedzierska, K.; Zou, Q.M.; Doherty, P.C.; Chen, W. Systematic identification of immunodominant CD8+ T-cell responses to influenza A virus in HLA-A2 individuals. Proc. Natl. Acad. Sci. USA 2011, 108, 9178–9183. [Google Scholar] [CrossRef]
- Impagliazzo, A.; Milder, F.; Kuipers, H.; Wagner, M.V.; Zhu, X.; Hoffman, R.M.; van Meersbergen, R.; Huizingh, J.; Wan-ningen, P.; Verspuij, J.; et al. Stable trimeric influenza hemagglutinin stem as a broadly protective immunogen. Science 2015, 349, 1301–1306. [Google Scholar] [CrossRef]
- Berthoud, T.K.; Hamill, M.; Lillie, P.J.; Hwenda, L.; Collins, K.A.; Ewer, K.J.; Milicic, A.; Poyntz, H.C.; Lambe, T.; Fletcher, H.A.; et al. Potent CD8+ T-cell immunogenicity in humans of a novel heterosubtypic influenza A vaccine, MVA-NP+M1. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2011, 52, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Lillie, P.J.; Berthoud, T.K.; Powell, T.J.; Lambe, T.; Mullarkey, C.; Spencer, A.J.; Hamill, M.; Peng, Y.; Blais, M.E.; Duncan, C.J.; et al. Preliminary assessment of the efficacy of a T-cell-based influenza vaccine, MVA-NP+M1, in humans. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2012, 55, 19–25. [Google Scholar] [CrossRef] [PubMed]
- van Doorn, E.; Liu, H.; Ben-Yedidia, T.; Hassin, S.; Visontai, I.; Norley, S.; Frijlink, H.W.; Hak, E. Evaluating the immuno-genicity and safety of a BiondVax-developed universal influenza vaccine (Multimeric-001) either as a standalone vaccine or as a primer to H5N1 influenza vaccine: Phase IIb study protocol. Medicine 2017, 96, e6339. [Google Scholar] [CrossRef] [PubMed]
- Immune Epitope Database. Available online: http://www.iedb.org (accessed on 2 November 2021).
- Zhang, Y.; Aevermann, B.; Anderson, T.; Burke, D.; Dauphin, G.; Gu, Z.; He, S.; Kumar, S.; Larsen, C.; Lee, A.; et al. Influenza Research Database: An integrated bioinformatics resource for influenza virus research. Nucleic Acids Res. 2017, 45, D466–D474. [Google Scholar] [CrossRef]
- Katoh, K.; Rozewicki, J.; Yamada, K. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef]
- GitHub/Lioj/Bioinformatics/MSA_stat.py at Master. Available online: https://github.com/lioj/bioinformatics/blob/master/py/MSA_stat.py (accessed on 2 November 2021).
- Shenkin, P.S.; Erman, B.; Mastrandrea, L.D. Information-Theoretical Entropy as a Measure of Sequence Variability. Proteins Struct. Funct. Bioinform. 1991, 11, 297–313. [Google Scholar] [CrossRef]
- Mintaev, R.R.; Alexeevski, A.V.; Kordyukova, L.V. Co-evolution analysis to predict protein-protein interactions within in-fluenza virus envelope. J. Bioinform. Comput. Biol. 2014, 12, 144100843. [Google Scholar] [CrossRef]
- PyMOL Program. Available online: http://pymol.org (accessed on 2 November 2021).
- PDB Database. Available online: http://www.rcsb.org (accessed on 2 November 2021).
- Gonzalez-Galarza, F.F.; McCabe, A.; Santos, E.J.; Jones, J.; Takeshita, L.Y.; Ortega-Rivera, N.D.; Del Cid-Pavon, G.M.; Rams-bottom, K.; Ghattaoraya, G.S.; Alfirevic, A.; et al. Allele frequency net database (AFND) 2020 update: Gold-standard data classification, open access genotype data and new query tools. Nucleic Acid Res. 2020, 48, 783–788. [Google Scholar] [CrossRef]
- Kremer, M.; Volz, A.; Kreijtz, J.; Fux, R.; Lehmann, M.; Sutter, G. Easy and efficient protocols for working with recombinant vaccinia virus MVA. In Vaccinia Virus and Poxvirology; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2012; Volume 890, pp. 59–92. [Google Scholar]
- Reed, L.; Muenchm, H. A simple method of estimating fifty per cent endpoints. Am. J. Epidemiol. 1938, 27, 493–497. [Google Scholar] [CrossRef]
- Orlova, O.V.; Glazkova, D.V.; Tsyganova, G.M.; Antoshkina, I.V.; Mintaev, R.R.; Tikhonov, A.S.; Bogoslovskaya, E.V.; Shipulin, G.A. Application of real-time PCR to significantly reduce the time to obtain recombinant MVA virus. J. Virol. Methods 2021, 289, 114056. [Google Scholar] [CrossRef] [PubMed]
- You, M.; Yang, Y.; Zhong, C.; Chen, F.; Wang, X.; Jia, T.; Chen, Y.; Zhou, B.; Mi, Q.; Zhao, Q.; et al. Efficient mAb production in CHO cells with optimized signal peptide, codon, and UTR. Appl. Microbiol. Biotechnol. 2018, 102, 5953–5964. [Google Scholar] [CrossRef] [PubMed]
- Güler-Gane, G.; Kidd, S.; Sridharan, S.; Vaughan, T.J.; Wilkinson, T.C.; Tigue, N.J. Overcoming the Refractory Expression of Secreted Recombinant Proteins in Mammalian Cells through Modification of the Signal Peptide and Adjacent Amino Acids. PLoS ONE 2016, 11, e0155340. [Google Scholar] [CrossRef] [PubMed]
- Mironov, A. Guidelines for Conducting Preclinical Studies of Drugs (Immunobiological Drugs); Part Two; Aquarium: Tula, Russia, 2012; p. 212. [Google Scholar]
- WHO Manual on Animal Influenza Diagnosis and Surveillance. Available online: https://www.who.int/csr/resources/publications/influenza/whocdscsrncs20025rev.pdf (accessed on 2 November 2021).
- Manual for the Laboratory Diagnosis and Virological Surveillance of Influenza, WHO Global Influenza Surveillance Network. Available online: https://apps.who.int/iris/bitstream/handle/10665/44518/9789241548090_eng.pdf?sequence=1 (accessed on 2 November 2021).
- Cottey, R.; Rowe, C.A.; Bender, B.S. Influenza virus. Curr. Protoc. Immunol. 2001, 42, 19.11.1–19.11.32. [Google Scholar] [CrossRef]
- Gauger, P.C.; Vincent, A.L. Serum virus neutralization assay for detection and quantitation of serum-neutralizing antibodies to influenza A virus in swine. In Methods in Molecular Biology, 3rd ed.; Spackman, E., Ed.; Humana: New York, NY, USA, 2014; Volume 1161, p. 24. [Google Scholar]
- Knossow, M.; Skehel, J.J. Variation and infectivity neutralization in influenza. Immunology 2006, 119, 1–7. [Google Scholar] [CrossRef]
- Chianese-Bullock, K.A.; Russell, H.I.; Moller, C.; Gerhard, W.; Monaco, J.J.; Eisenlohr, L.C. Antigen processing of two H2-IEd-restricted epitopes is differentially influenced by the structural changes in a viral glycoprotein. J. Immunol. 1998, 161, 1599–1607. [Google Scholar]
- Guo, C.; Zhang, H.; Xie, X.; Liu, Y.; Sun, L.; Li, H.; Yu, P.; Hu, H.; Sun, J.; Li, Y.; et al. H1N1 influenza virus epitopes classified by monoclonal antibodies. Exp. Ther. Med. 2018, 16, 2001–2007. [Google Scholar] [CrossRef]
- Chong, L.C.; Khan, A.M. Identification of highly conserved, serotype-specific dengue virus sequences: Implications for vaccine design. BMC Genom. 2019, 20, 921. [Google Scholar] [CrossRef]
- Simeckova-Rosenberg, J.; Yun, Z.; Wyde, P.R.; Atassi, M.Z. Protection of mice against lethal viral infection by synthetic peptides corresponding to B- and T-cell recognition sites of influenza A hemagglutinin. Vaccine 1995, 13, 927–932. [Google Scholar] [CrossRef]
- Atassi, M.Z.; Webster, R.G. Localization, synthesis, and activity of an antigenic site on influenza virus hemagglutinin. Proc. Natl. Acad. Sci. USA 1983, 80, 840–844. [Google Scholar] [CrossRef] [PubMed]
- Atassi, M.Z.; Kurisaki, J. A novel approach for localization of the continuous protein antigenic sites by comprehensive synthetic surface scanning: Antibody and T-cell activity to several influenza hemagglutinin synthetic sites. Immunol. Commun. 1984, 13, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Price, J.V.; Jarrell, J.A.; Furman, D.; Kattah, N.H.; Newell, E.; Dekker, C.L.; Davis, M.M.; Utz, P.J. Characterization of influenza vaccine immunogenicity using influenza antigen microarrays. PLoS ONE 2013, 8, e64555. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, E.; Liang, X.; Ingallinella, P.; Finotto, M.; Chastain, M.A.; Fan, J.; Fu, T.M.; Song, H.C.; Horton, M.S.; Freed, D.C.; et al. Universal influenza B vaccine based on the maturational cleavage site of the hemagglutinin precursor. J. Virol. 2005, 79, 7380–7388. [Google Scholar] [CrossRef]
- Zeliszewski, D.; Golvano, J.J.; Gaudebout, P.; Dorval, I.; Borras-Cuesta, F.; Sterkers, G. Binding of ALA-substituted analogs of HA306-320 to DR1101, DR1301, and DR0402 molecules: Correlation of DR-peptide interactions with recognition by a single TCR. Hum. Immunol. 1996, 50, 61–69. [Google Scholar] [CrossRef]
- Moise, L.; Terry, F.; Ardito, M.; Tassone, R.; Latimer, H.; Boyle, C.; Martin, W.D.; De Groot, A.S. Universal H1N1 influenza vaccine development: Identification of consensus class II hemagglutinin and neuraminidase epitopes derived from strains circulating between 1980 and 2011. Hum. Vaccines Immunother. 2013, 9, 1598–1607. [Google Scholar] [CrossRef]
- Danke, N.A.; Kwok, W.W. HLA class II-restricted CD4+ T cell responses directed against influenza viral antigens postinfluenza vaccination. J. Immunol. 2003, 171, 3163–3169. [Google Scholar] [CrossRef]
- Quiñones-Parra, S.; Grant, E.; Loh, L.; Nguyen, T.H.; Campbell, K.A.; Tong, S.Y.; Miller, A.; Doherty, P.C.; Vijaykrishna, D.; Rossjohn, J.; et al. Preexisting CD8+ T-cell immunity to the H7N9 influenza A virus varies across ethnicities. Proc. Natl. Acad. Sci. USA 2014, 111, 1049–1054. [Google Scholar] [CrossRef]
- Berkhoff, E.G.; Geelhoed-Mieras, M.M.; Verschuren, E.J.; van Baalen, C.A.; Gruters, R.A.; Fouchier, R.A.; Osterhaus, A.D.; Rimmelzwaan, G.F. The loss of immunodominant epitopes affects interferon-gamma production and lytic activity of the human influenza virus-specific cytotoxic T lymphocyte response in vitro. Clin. Exp. Immunol. 2007, 148, 296–306. [Google Scholar] [CrossRef]
- Eickhoff, C.S.; Terry, F.E.; Peng, L.; Meza, K.A.; Sakala, I.G.; Van Aartsen, D.; Moise, L.; Martin, W.D.; Schriewer, J.; Buller, R.M.; et al. Highly conserved influenza T cell epitopes induce broadly protective immunity. Vaccine 2019, 37, 5371–5381. [Google Scholar] [CrossRef]
- Terajima, M.; Cruz, J.; Leporati, A.M.; Orphin, L.; Babon, J.A.; Co, M.D.; Pazoles, P.; Jameson, J.; Ennis, F.A. Influenza A virus matrix protein 1-specific human CD8+ T-cell response induced in trivalent inactivated vaccine recipients. J. Virol. 2008, 82, 9283–9287. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Robbins, P.A.; Garboczi, D.N.; Strominger, J.L. HLA-A*0201 complexes with two 10-Mer peptides differing at the P2 anchor residue have distinct refolding kinetics. J. Immunol. 1995, 154, 703–709. [Google Scholar] [PubMed]
- Ben-Yedidia, T.; Batya, M.; Singer, Y.; Hashmonaim, I.L. Multimeric Multiepitope Influenza Vaccine. U.S. Patent Application No. US009353159B2, 31 May 2016. [Google Scholar]
- Atsmon, J.; Caraco, Y.; Ziv-Sefer, S.; Shaikevich, D.; Abramov, E.; Volokhov, I.; Bruzil, S.; Haima, K.Y.; Gottlieb, T.; Ben-Yedidia, T. Priming by a novel universal influenza vaccine (Multimeric-001)-a gateway for improving immune response in the elderly population. Vaccine 2014, 32, 5816–5823. [Google Scholar] [CrossRef] [PubMed]
- Sarah, G.; Adrian, H.; Anne, M. MVA-Based Compositions and Methods for Inducing an Immune Response against Influenza. European Patent Application EP2044947A1, 8 April 2009. [Google Scholar]
- Bajic, G.; van der Poel, C.E.; Kuraoka, M.; Schmidt, A.G.; Carroll, M.C.; Kelsoe, G.; Harrison, S.C. Autoreactivity profiles of influenza hemagglutinin broadly neutralizing antibodies. Sci. Rep. 2019, 9, 3492. [Google Scholar] [CrossRef] [PubMed]
- Sicca, F.; Martinuzzi, D.; Montomoli, E.; Huckriede, A. Comparison of influenza-specific neutralizing antibody titers determined using different assay readouts and hemagglutination inhibition titers: Good correlation but poor agreement. Vaccine 2020, 38, 2527–2541. [Google Scholar] [CrossRef]
- Viboud, C.; Simonsen, L.; Fuentes, R.; Flores, J.; Miller, M.A.; Chowell, G. Global Mortality Impact of the 1957–1959 Influenza Pandemic. J. Infect. Dis. 2016, 213, 738–745. [Google Scholar] [CrossRef]
- Kilbourne, E.D. Influenza pandemics of the 20th century. Emerg. Infect. Dis. 2006, 12, 9–14. [Google Scholar] [CrossRef]
- Luke, C.J.; Subbarao, K. Vaccines for pandemic influenza. Emerg. Infect. Dis. 2006, 12, 66–72. [Google Scholar] [CrossRef]




| No. | Protein | Position | Sequence | Type/Subtype (Conservation) | Immune Response Type | References |
|---|---|---|---|---|---|---|
| 1 | HA | 124–136 | SVSSFERFEIFPK | A/H1 (100%) | B cell response | [36] |
| 2 | HA | 18–32 | DTLCIGYHANNSTDT | A/H1 (100%) | B cell response | [37,38] |
| 3 | HA | 345–355 | GLFGAIAGFIE | A/all (100%) and B (90%) | B cell response | [39,40] |
| 4 | HA | 411–427 | KEFSEVEGRIQDLEKYV | A/H3 (100%) | B cell response | [39,41] |
| 5 | HA | 247–256 | PNQTEDGGLP | B (51%) | B cell response | [42] |
| 6 | HA | 247–256 | PDQTEDGGLP | B (49%) | B cell response | [42] |
| 7 | HA | 299–307 | KGSLPLIGE | B (100%) | B cell response | [42] |
| 8 | HA | 354–372 | PAKLIKERGFFGAIAGFLE | B (100%) | B cell response | [43] |
| 9 | HA | 318–332 | GKCPKYVKSTKLRLATGLRN | A/H1 (74%) | Th response | [44] |
| 10 | HA | 343–359 | SRGLFGAIAGFIEGGWT | A/H1 (100%) | B cell + Th responses | [45] |
| 11 | NP | 205–229 | NFWRGENGRKTRSAYERMCNILKGK | A (92%) | Th + CTL responses | [46,47] |
| 12 | NP | 37–54 | GRFYIQMCTELKLSDYEG | A (100%) | CTL response | [48] |
| 13 | M1 | 180–197 | VLASTTAKAMEQMAGSSE | A (100%) | Th response | [49] |
| 14 | M1 | 58–66 | GILGFVFTL | A (100%) | CTL response | [50] |
| 15 | NP | 85–94 | KLGEFYNQM | B (100%) | CTL response | [51] |
| Name | Order of Epitopes 1 | Immune Response Type |
|---|---|---|
| k1 | 8-1-10-7-2-9-4-5′ 2 | B cell and Th responses |
| k2 | 11-12-13-14-15 | Th and CTL responses |
| k5 | 8-1-9-10-5′-7-4-2 | B cell and Th responses |
| # | Group Name | Number of Mice | First Immunization | Second Immunization |
|---|---|---|---|---|
| 1 | rMVA-k1-k2 | 28 | rMVA-k1 + rMVA-k2 | rMVA-k1 + rMVA-k2 |
| 2 | rMVA-k5-k2 | 26 | rMVA-k5 + rMVA-k2 | rMVA-k5 + rMVA-k2 |
| 3 | rMVA-NP+M1 | 26 | rMVA-NP+M1 | rMVA-NP+M1 |
| 4 | rMVA-M001 | 27 | rMVA-M001 | rMVA-M001 |
| 5 | rMVA-hlHA | 26 | rMVA-hlHA | rMVA-hlHA |
| 6 | Empty MVA | 28 | wt MVA | wt MVA |
| 7 | Mock | 26 | PBS | PBS |
| # | Strain | Groups Vaccinated with | Mock Group | |
|---|---|---|---|---|
| rMVA-k1-k2 | Seasonal Vaccine (2019–2020) | |||
| 1 | A/California/07/2009 (H1N1) | 30 | 120 | <10 |
| 2 | A/Puerto Rico/8/34 (H1N1) | 27 | <10 | <10 |
| 3 | A/Aichi/2/68 (H3N2) | 27 | <10 | <10 |
| 4 | A/Texas/50/2012 (H3N2) | 20 | - | <10 |
| 5 | A/Singapore (H2N2) | <10 | - | <10 |
| 6 | A/Chicken/Kurgan (H5N1) | 40 | <10 | <10 |
| 7 | B/Colorado/06/2017 | <10 | 80 | <10 |
| 8 | B/Phuket/3073/2013 | <10 | <10 | <10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mintaev, R.R.; Glazkova, D.V.; Orlova, O.V.; Bogoslovskaya, E.V.; Shipulin, G.A. Development of a Universal Epitope-Based Influenza Vaccine and Evaluation of Its Effectiveness in Mice. Vaccines 2022, 10, 534. https://doi.org/10.3390/vaccines10040534
Mintaev RR, Glazkova DV, Orlova OV, Bogoslovskaya EV, Shipulin GA. Development of a Universal Epitope-Based Influenza Vaccine and Evaluation of Its Effectiveness in Mice. Vaccines. 2022; 10(4):534. https://doi.org/10.3390/vaccines10040534
Chicago/Turabian StyleMintaev, Ramil R., Dina V. Glazkova, Olga V. Orlova, Elena V. Bogoslovskaya, and German A. Shipulin. 2022. "Development of a Universal Epitope-Based Influenza Vaccine and Evaluation of Its Effectiveness in Mice" Vaccines 10, no. 4: 534. https://doi.org/10.3390/vaccines10040534
APA StyleMintaev, R. R., Glazkova, D. V., Orlova, O. V., Bogoslovskaya, E. V., & Shipulin, G. A. (2022). Development of a Universal Epitope-Based Influenza Vaccine and Evaluation of Its Effectiveness in Mice. Vaccines, 10(4), 534. https://doi.org/10.3390/vaccines10040534