
Protective CD8+ T Cell Response Induced by Modified Vaccinia Virus Ankara Delivering Ebola Virus Nucleoprotein

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plasmid Construction
2.2. Generation of Recombinant MVA Viruses
2.3. Characterization of Recombinant MVA Genomes
2.4. Detection of Recombinant EBOV Proteins
2.5. Immunization and EBOV Infection in Mice
2.6. Analysis of Antibody Response
2.7. Analysis of T Cell Response
2.8. Determination of EBOV Loads and Infectious Virus in Mouse Organs and Sera
2.9. Clinical Serum Chemistry
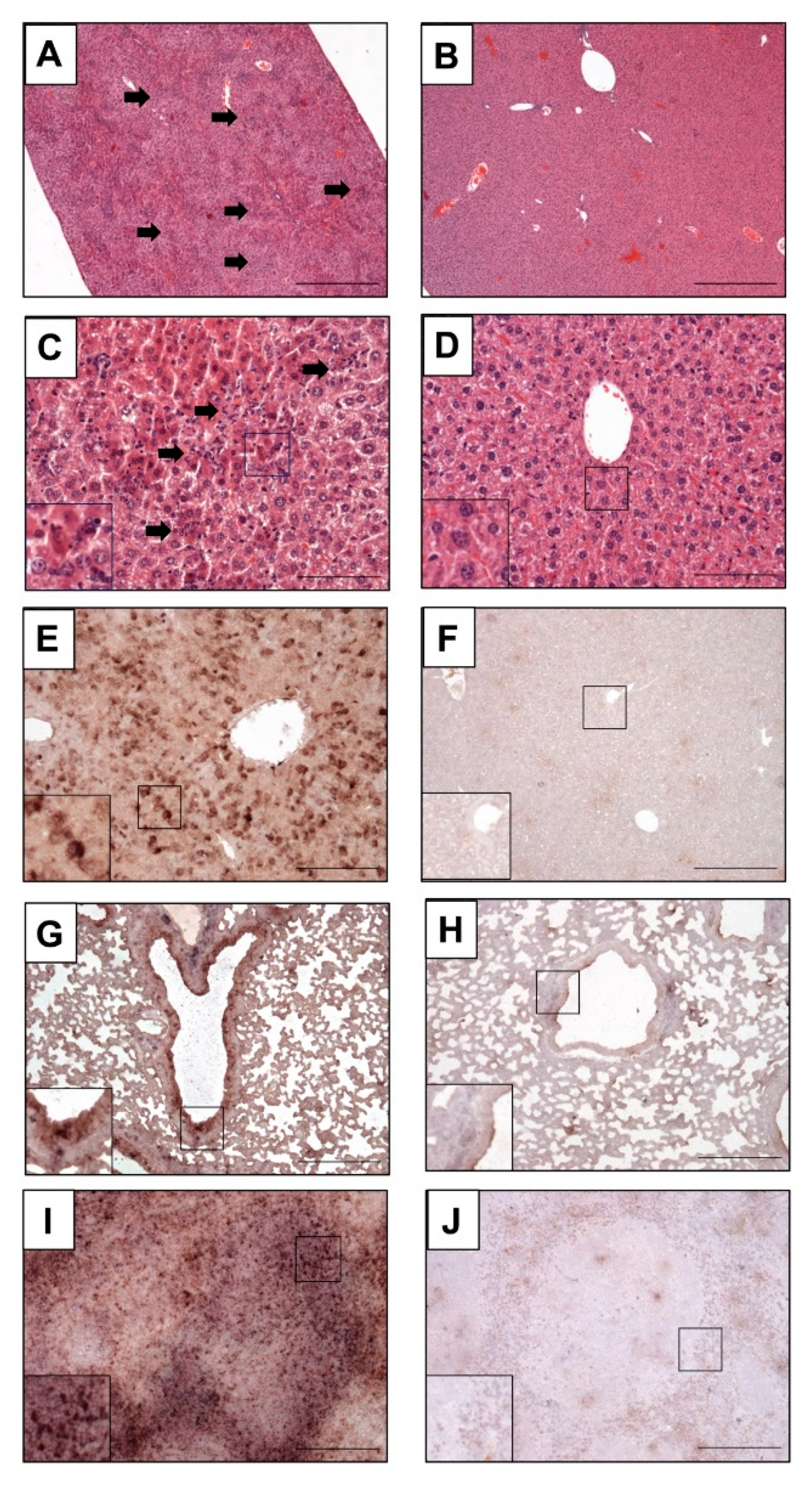
2.10. Histopathological Examination and In Situ Hybridization of Mouse Organs
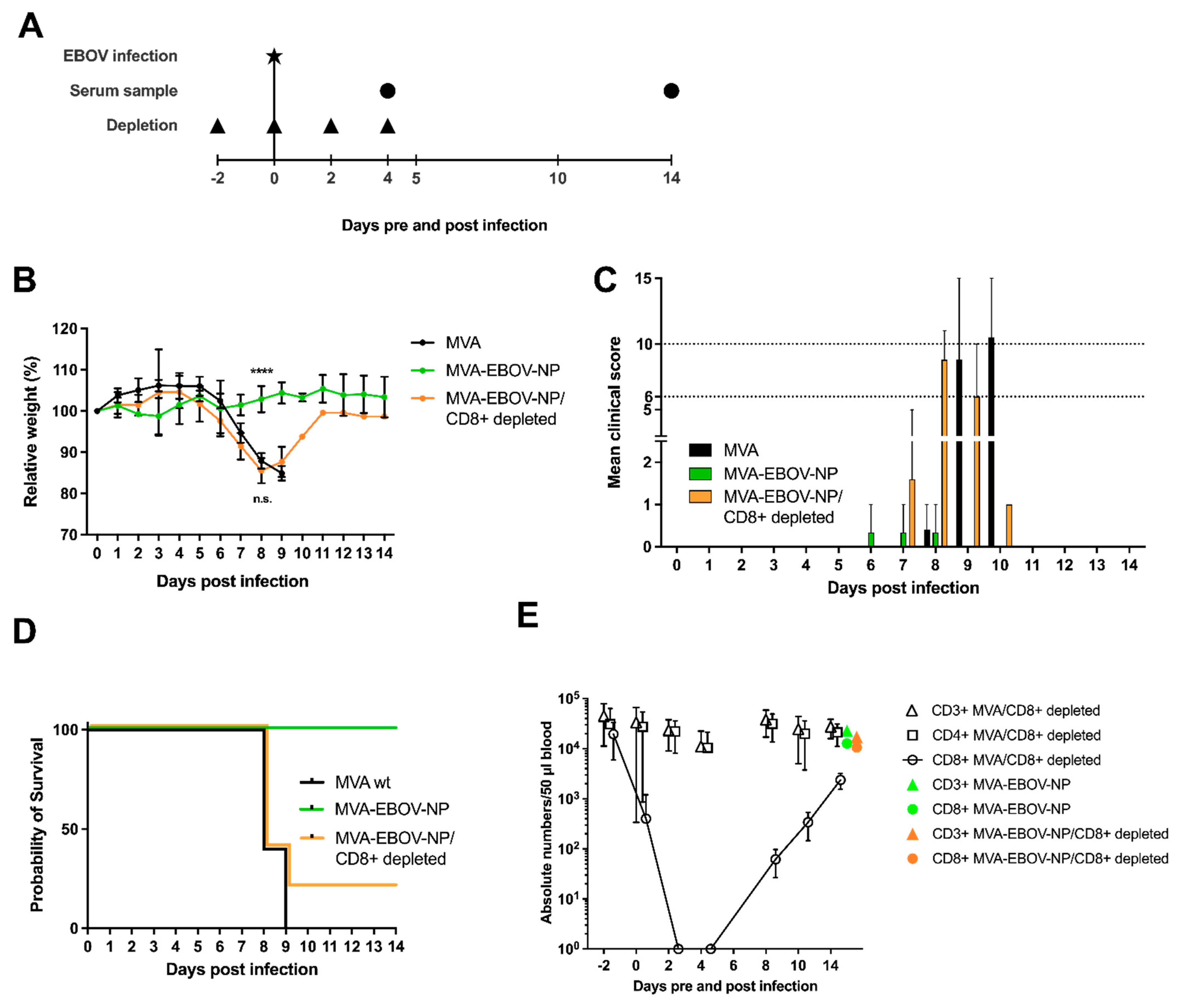
2.11. Depletion of CD8+ T Cells and Flow Cytometric Analysis
2.12. Statistical Analysis
3. Results
3.1. Construction and Characterization of Recombinant MVA Expressing EBOV NP or GP
3.2. EBOV-Specific Antibodies Are Induced by MVA-EBOV-NP and -GP
3.3. NP-Specific CD8+ T Cell Responses in MVA-EBOV-NP-Vaccinated Mice
3.4. MVA-EBOV-NP and MVA-EBOV-GP-Vaccinated Mice Are Protected against Lethal EBOV Challenge
3.5. Depletion of CD8+ T Cells in MVA-EBOV-NP-Vaccinated Mice Leads to Severe Disease Manifestation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Feldmann, H.; Geisbert, T.W. Ebola haemorrhagic fever. Lancet 2011, 377, 849–862. [Google Scholar] [CrossRef] [Green Version]
- Rougeron, V.; Feldmann, H.; Grard, G.; Becker, S.; Leroy, E.M. Ebola and Marburg haemorrhagic fever. J. Clin. Virol. 2015, 64, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Leroy, E.M.; Kumulungui, B.; Pourrut, X.; Rouquet, P.; Hassanin, A.; Yaba, P.; Delicat, A.; Paweska, J.T.; Gonzalez, J.P.; Swanepoel, R. Fruit bats as reservoirs of Ebola virus. Nature 2005, 438, 575–576. [Google Scholar] [CrossRef] [PubMed]
- Leroy, E.M.; Epelboin, A.; Mondonge, V.; Pourrut, X.; Gonzalez, J.P.; Muyembe-Tamfum, J.J.; Formenty, P. Human Ebola outbreak resulting from direct exposure to fruit bats in Luebo, Democratic Republic of Congo, 2007. Vector Borne Zoonotic Dis. 2009, 9, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Formenty, P.; Boesch, C.; Wyers, M.; Steiner, C.; Donati, F.; Dind, F.; Walker, F.; Le Guenno, B. Ebola virus outbreak among wild chimpanzees living in a rain forest of Côte d’Ivoire. J. Infect. Dis. 1999, 179 (Suppl. 1), S120–S126. [Google Scholar] [CrossRef] [Green Version]
- Leroy, E.M.; Rouquet, P.; Formenty, P.; Souquière, S.; Kilbourne, A.; Froment, J.M.; Bermejo, M.; Smit, S.; Karesh, W.; Swanepoel, R.; et al. Multiple Ebola virus transmission events and rapid decline of central African wildlife. Science 2004, 303, 387–390. [Google Scholar] [CrossRef] [Green Version]
- Bermejo, M.; Rodríguez-Teijeiro, J.D.; Illera, G.; Barroso, A.; Vilà, C.; Walsh, P.D. Ebola outbreak killed 5000 gorillas. Science 2006, 314, 1564. [Google Scholar] [CrossRef] [Green Version]
- Osterholm, M.T.; Moore, K.A.; Kelley, N.S.; Brosseau, L.M.; Wong, G.; Murphy, F.A.; Peters, C.J.; LeDuc, J.W.; Russell, P.K.; Van Herp, M.; et al. Transmission of Ebola viruses: What we know and what we do not know. MBio 2015, 6, e00137. [Google Scholar] [CrossRef] [Green Version]
- WHO. Statement on the 1st meeting of the IHR Emergency Committee on the 2014 Ebola outbreak in West Africa. 2014. Available online: https://www.who.int/news/item/08-08-2014-statement-on-the-1st-meeting-of-the-ihr-emergency-committee-on-the-2014-ebola-outbreak-in-west-africa (accessed on 24 March 2022).
- Mire, C.E.; Geisbert, T.W.; Feldmann, H.; Marzi, A. Ebola virus vaccines—Reality or fiction? Expert Rev. Vaccines 2016, 15, 1421–1430. [Google Scholar] [CrossRef] [Green Version]
- Henao-Restrepo, A.M.; Longini, I.M.; Egger, M.; Dean, N.E.; Edmunds, W.J.; Camacho, A.; Carroll, M.W.; Doumbia, M.; Draguez, B.; Duraffour, S.; et al. Efficacy and effectiveness of an rVSV-vectored vaccine expressing Ebola surface glycoprotein: Interim results from the Guinea ring vaccination cluster-randomised trial. Lancet 2015, 386, 857–866. [Google Scholar] [CrossRef]
- Huttner, A.; Dayer, J.A.; Yerly, S.; Combescure, C.; Auderset, F.; Desmeules, J.; Eickmann, M.; Finckh, A.; Goncalves, A.R.; Hooper, J.W.; et al. The effect of dose on the safety and immunogenicity of the VSV Ebola candidate vaccine: A randomised double-blind, placebo-controlled phase 1/2 trial. Lancet Infect. Dis. 2015, 15, 1156–1166. [Google Scholar] [CrossRef]
- Agnandji, S.T.; Huttner, A.; Zinser, M.E.; Njuguna, P.; Dahlke, C.; Fernandes, J.F.; Yerly, S.; Dayer, J.A.; Kraehling, V.; Kasonta, R.; et al. Phase 1 Trials of rVSV Ebola Vaccine in Africa and Europe. N. Engl. J. Med. 2016, 374, 1647–1660. [Google Scholar] [CrossRef]
- Milligan, I.D.; Gibani, M.M.; Sewell, R.; Clutterbuck, E.A.; Campbell, D.; Plested, E.; Nuthall, E.; Voysey, M.; Silva-Reyes, L.; McElrath, M.J.; et al. Safety and Immunogenicity of Novel Adenovirus Type 26- and Modified Vaccinia Ankara-Vectored Ebola Vaccines: A Randomized Clinical Trial. JAMA 2016, 315, 1610–1623. [Google Scholar] [CrossRef]
- Callendret, B.; Vellinga, J.; Wunderlich, K.; Rodriguez, A.; Steigerwald, R.; Dirmeier, U.; Cheminay, C.; Volkmann, A.; Brasel, T.; Carrion, R.; et al. A prophylactic multivalent vaccine against different filovirus species is immunogenic and provides protection from lethal infections with Ebolavirus and Marburgvirus species in non-human primates. PLoS ONE 2018, 13, e0192312. [Google Scholar] [CrossRef] [Green Version]
- Henao-Restrepo, A.M.; Camacho, A.; Longini, I.M.; Watson, C.H.; Edmunds, W.J.; Egger, M.; Carroll, M.W.; Dean, N.E.; Diatta, I.; Doumbia, M.; et al. Efficacy and effectiveness of an rVSV-vectored vaccine in preventing Ebola virus disease: Final results from the Guinea ring vaccination, open-label, cluster-randomised trial (Ebola Ça Suffit!). Lancet 2017, 389, 505–518. [Google Scholar] [CrossRef] [Green Version]
- Afolabi, M.O.; Ishola, D.; Manno, D.; Keshinro, B.; Bockstal, V.; Rogers, B.; Owusu-Kyei, K.; Serry-Bangura, A.; Swaray, I.; Lowe, B.; et al. Safety and immunogenicity of the two-dose heterologous Ad26.ZEBOV and MVA-BN-Filo Ebola vaccine regimen in children in Sierra Leone: A randomised, double-blind, controlled trial. Lancet Infect. Dis. 2022, 22, 110–122. [Google Scholar] [CrossRef]
- Ishola, D.; Manno, D.; Afolabi, M.O.; Keshinro, B.; Bockstal, V.; Rogers, B.; Owusu-Kyei, K.; Serry-Bangura, A.; Swaray, I.; Lowe, B.; et al. Safety and long-term immunogenicity of the two-dose heterologous Ad26.ZEBOV and MVA-BN-Filo Ebola vaccine regimen in adults in Sierra Leone: A combined open-label, non-randomised stage 1, and a randomised, double-blind, controlled stage 2 trial. Lancet Infect. Dis. 2022, 22, 97–109. [Google Scholar] [CrossRef]
- Pollard, A.J.; Launay, O.; Lelievre, J.D.; Lacabaratz, C.; Grande, S.; Goldstein, N.; Robinson, C.; Gaddah, A.; Bockstal, V.; Wiedemann, A.; et al. Safety and immunogenicity of a two-dose heterologous Ad26.ZEBOV and MVA-BN-Filo Ebola vaccine regimen in adults in Europe (EBOVAC2): A randomised, observer-blind, participant-blind, placebo-controlled, phase 2 trial. Lancet Infect. Dis. 2021, 21, 493–506. [Google Scholar] [CrossRef]
- Volz, A.; Sutter, G. Modified Vaccinia Virus Ankara: History, Value in Basic Research, and Current Perspectives for Vaccine Development. Adv. Virus Res. 2017, 97, 187–243. [Google Scholar] [CrossRef]
- Koch, T.; Dahlke, C.; Fathi, A.; Kupke, A.; Krähling, V.; Okba, N.M.A.; Halwe, S.; Rohde, C.; Eickmann, M.; Volz, A.; et al. Safety and immunogenicity of a modified vaccinia virus Ankara vector vaccine candidate for Middle East respiratory syndrome: An open-label, phase 1 trial. Lancet Infect. Dis. 2020, 20, 827–838. [Google Scholar] [CrossRef]
- Meyer, H.; Sutter, G.; Mayr, A. Mapping of deletions in the genome of the highly attenuated vaccinia virus MVA and their influence on virulence. J. Gen. Virol. 1991, 72 Pt 5, 1031–1038. [Google Scholar] [CrossRef]
- Antoine, G.; Scheiflinger, F.; Dorner, F.; Falkner, F.G. The complete genomic sequence of the modified vaccinia Ankara strain: Comparison with other orthopoxviruses. Virology 1998, 244, 365–396. [Google Scholar] [CrossRef]
- Drexler, I.; Heller, K.; Wahren, B.; Erfle, V.; Sutter, G. Highly attenuated modified vaccinia virus Ankara replicates in baby hamster kidney cells, a potential host for virus propagation, but not in various human transformed and primary cells. J. Gen. Virol. 1998, 79 Pt 2, 347–352. [Google Scholar] [CrossRef]
- Stittelaar, K.J.; Kuiken, T.; de Swart, R.L.; van Amerongen, G.; Vos, H.W.; Niesters, H.G.; van Schalkwijk, P.; van der Kwast, T.; Wyatt, L.S.; Moss, B.; et al. Safety of modified vaccinia virus Ankara (MVA) in immune-suppressed macaques. Vaccine 2001, 19, 3700–3709. [Google Scholar] [CrossRef]
- Langenmayer, M.C.; Lülf-Averhoff, A.T.; Adam-Neumair, S.; Fux, R.; Sutter, G.; Volz, A. Distribution and absence of generalized lesions in mice following single dose intramuscular inoculation of the vaccine candidate MVA-MERS-S. Biologicals 2018, 54, 58–62. [Google Scholar] [CrossRef]
- Kreijtz, J.H.; Goeijenbier, M.; Moesker, F.M.; van den Dries, L.; Goeijenbier, S.; De Gruyter, H.L.; Lehmann, M.H.; Mutsert, G.; van de Vijver, D.A.; Volz, A.; et al. Safety and immunogenicity of a modified-vaccinia-virus-Ankara-based influenza A H5N1 vaccine: A randomised, double-blind phase 1/2a clinical trial. Lancet Infect. Dis. 2014, 14, 1196–1207. [Google Scholar] [CrossRef]
- La Rosa, C.; Longmate, J.; Martinez, J.; Zhou, Q.; Kaltcheva, T.I.; Tsai, W.; Drake, J.; Carroll, M.; Wussow, F.; Chiuppesi, F.; et al. MVA vaccine encoding CMV antigens safely induces durable expansion of CMV-specific T cells in healthy adults. Blood 2017, 129, 114–125. [Google Scholar] [CrossRef]
- Overton, E.T.; Stapleton, J.; Frank, I.; Hassler, S.; Goepfert, P.A.; Barker, D.; Wagner, E.; von Krempelhuber, A.; Virgin, G.; Meyer, T.P.; et al. Safety and Immunogenicity of Modified Vaccinia Ankara-Bavarian Nordic Smallpox Vaccine in Vaccinia-Naive and Experienced Human Immunodeficiency Virus-Infected Individuals: An Open-Label, Controlled Clinical Phase II Trial. Open Forum Infect. Dis. 2015, 2, ofv040. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.; Heath, S.L.; Sweeton, B.; Williams, K.; Cunningham, P.; Keele, B.F.; Sen, S.; Palmer, B.E.; Chomont, N.; Xu, Y.; et al. DNA/MVA Vaccination of HIV-1 Infected Participants with Viral Suppression on Antiretroviral Therapy, followed by Treatment Interruption: Elicitation of Immune Responses without Control of Re-Emergent Virus. PLoS ONE 2016, 11, e0163164. [Google Scholar] [CrossRef]
- Lülf, A.T.; Freudenstein, A.; Marr, L.; Sutter, G.; Volz, A. Non-plaque-forming virions of Modified Vaccinia virus Ankara express viral genes. Virology 2016, 499, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Altenburg, A.F.; van de Sandt, C.E.; Li, B.W.S.; MacLoughlin, R.J.; Fouchier, R.A.M.; van Amerongen, G.; Volz, A.; Hendriks, R.W.; de Swart, R.L.; Sutter, G.; et al. Modified Vaccinia Virus Ankara Preferentially Targets Antigen Presenting Cells In Vitro, Ex Vivo and In Vivo. Sci. Rep. 2017, 7, 8580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waibler, Z.; Anzaghe, M.; Ludwig, H.; Akira, S.; Weiss, S.; Sutter, G.; Kalinke, U. Modified vaccinia virus Ankara induces Toll-like receptor-independent type I interferon responses. J. Virol. 2007, 81, 12102–12110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palgen, J.L.; Tchitchek, N.; Huot, N.; Elhmouzi-Younes, J.; Lefebvre, C.; Rosenbaum, P.; Dereuddre-Bosquet, N.; Martinon, F.; Hocini, H.; Cosma, A.; et al. NK cell immune responses differ after prime and boost vaccination. J. Leukoc. Biol. 2019, 105, 1055–1073. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, M.H.; Kastenmuller, W.; Kandemir, J.D.; Brandt, F.; Suezer, Y.; Sutter, G. Modified vaccinia virus ankara triggers chemotaxis of monocytes and early respiratory immigration of leukocytes by induction of CCL2 expression. J. Virol. 2009, 83, 2540–2552. [Google Scholar] [CrossRef] [Green Version]
- Price, P.J.; Torres-Dominguez, L.E.; Brandmuller, C.; Sutter, G.; Lehmann, M.H. Modified Vaccinia virus Ankara: Innate immune activation and induction of cellular signalling. Vaccine 2013, 31, 4231–4234. [Google Scholar] [CrossRef]
- Domi, A.; Feldmann, F.; Basu, R.; McCurley, N.; Shifflett, K.; Emanuel, J.; Hellerstein, M.S.; Guirakhoo, F.; Orlandi, C.; Flinko, R.; et al. A Single Dose of Modified Vaccinia Ankara expressing Ebola Virus Like Particles Protects Nonhuman Primates from Lethal Ebola Virus Challenge. Sci. Rep. 2018, 8, 864. [Google Scholar] [CrossRef] [Green Version]
- Cross, R.W.; Mire, C.E.; Feldmann, H.; Geisbert, T.W. Post-exposure treatments for Ebola and Marburg virus infections. Nat. Rev. Drug Discov. 2018, 17, 413–434. [Google Scholar] [CrossRef]
- Oestereich, L.; Ludtke, A.; Wurr, S.; Rieger, T.; Munoz-Fontela, C.; Gunther, S. Successful treatment of advanced Ebola virus infection with T-705 (favipiravir) in a small animal model. Antivir. Res. 2014, 105, 17–21. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, P.; Marzi, A. Ebola and Marburg virus vaccines. Virus Genes 2017, 53, 501–515. [Google Scholar] [CrossRef]
- Geisbert, T.W.; Daddario-Dicaprio, K.M.; Geisbert, J.B.; Reed, D.S.; Feldmann, F.; Grolla, A.; Stroher, U.; Fritz, E.A.; Hensley, L.E.; Jones, S.M.; et al. Vesicular stomatitis virus-based vaccines protect nonhuman primates against aerosol challenge with Ebola and Marburg viruses. Vaccine 2008, 26, 6894–6900. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, N.J.; Sanchez, A.; Rollin, P.E.; Yang, Z.Y.; Nabel, G.J. Development of a preventive vaccine for Ebola virus infection in primates. Nature 2000, 408, 605–609. [Google Scholar] [CrossRef]
- Sullivan, N.J.; Geisbert, T.W.; Geisbert, J.B.; Xu, L.; Yang, Z.Y.; Roederer, M.; Koup, R.A.; Jahrling, P.B.; Nabel, G.J. Accelerated vaccination for Ebola virus haemorrhagic fever in non-human primates. Nature 2003, 424, 681–684. [Google Scholar] [CrossRef]
- Wilson, J.A.; Hart, M.K. Protection from Ebola virus mediated by cytotoxic T lymphocytes specific for the viral nucleoprotein. J. Virol. 2001, 75, 2660–2664. [Google Scholar] [CrossRef] [Green Version]
- Simmons, G.; Lee, A.; Rennekamp, A.J.; Fan, X.; Bates, P.; Shen, H. Identification of murine T-cell epitopes in Ebola virus nucleoprotein. Virology 2004, 318, 224–230. [Google Scholar] [CrossRef] [Green Version]
- Tsuda, Y.; Caposio, P.; Parkins, C.J.; Botto, S.; Messaoudi, I.; Cicin-Sain, L.; Feldmann, H.; Jarvis, M.A. A replicating cytomegalovirus-based vaccine encoding a single Ebola virus nucleoprotein CTL epitope confers protection against Ebola virus. PLoS Negl. Trop. Dis. 2011, 5, e1275. [Google Scholar] [CrossRef] [Green Version]
- Wan, W.; Kolesnikova, L.; Clarke, M.; Koehler, A.; Noda, T.; Becker, S.; Briggs, J.A.G. Structure and assembly of the Ebola virus nucleocapsid. Nature 2017, 551, 394–397. [Google Scholar] [CrossRef] [Green Version]
- Noda, T.; Hagiwara, K.; Sagara, H.; Kawaoka, Y. Characterization of the Ebola virus nucleoprotein-RNA complex. J. Gen. Virol. 2010, 91, 1478–1483. [Google Scholar] [CrossRef]
- Nanbo, A.; Watanabe, S.; Halfmann, P.; Kawaoka, Y. The spatio-temporal distribution dynamics of Ebola virus proteins and RNA in infected cells. Sci. Rep. 2013, 3, 1206. [Google Scholar] [CrossRef]
- Kremer, M.; Suezer, Y.; Volz, A.; Frenz, T.; Majzoub, M.; Hanschmann, K.M.; Lehmann, M.H.; Kalinke, U.; Sutter, G. Critical role of perforin-dependent CD8+ T cell immunity for rapid protective vaccination in a murine model for human smallpox. PLoS Pathog. 2012, 8, e1002557. [Google Scholar] [CrossRef]
- Volz, A.; Langenmayer, M.; Jany, S.; Kalinke, U.; Sutter, G. Rapid expansion of CD8+ T cells in wild-type and type I interferon receptor-deficient mice correlates with protection after low-dose emergency immunization with modified vaccinia virus Ankara. J. Virol. 2014, 88, 10946–10957. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Cai, C.; Grifoni, A.; Müller, T.R.; Niessl, J.; Olofsson, A.; Humbert, M.; Hansson, L.; Österborg, A.; Bergman, P.; et al. Ancestral SARS-CoV-2-specific T cells cross-recognize the Omicron variant. Nat. Med. 2022, 28, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, L.S.; Earl, P.L.; Xiao, W.; Americo, J.L.; Cotter, C.A.; Vogt, J.; Moss, B. Elucidating and minimizing the loss by recombinant vaccinia virus of human immunodeficiency virus gene expression resulting from spontaneous mutations and positive selection. J. Virol. 2009, 83, 7176–7184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kremer, M.; Volz, A.; Kreijtz, J.H.; Fux, R.; Lehmann, M.H.; Sutter, G. Easy and efficient protocols for working with recombinant vaccinia virus MVA. Methods Mol. Biol. 2012, 890, 59–92. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, L.S.; Shors, S.T.; Murphy, B.R.; Moss, B. Development of a replication-deficient recombinant vaccinia virus vaccine effective against parainfluenza virus 3 infection in an animal model. Vaccine 1996, 14, 1451–1458. [Google Scholar] [CrossRef]
- Boukamp, P.; Petrussevska, R.T.; Breitkreutz, D.; Hornung, J.; Markham, A.; Fusenig, N.E. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J. Cell Biol. 1988, 106, 761–771. [Google Scholar] [CrossRef] [Green Version]
- Lucht, A.; Grunow, R.; Otterbein, C.; Möller, P.; Feldmann, H.; Becker, S. Production of monoclonal antibodies and development of an antigen capture ELISA directed against the envelope glycoprotein GP of Ebola virus. Med. Microbiol. Immunol. 2004, 193, 181–187. [Google Scholar] [CrossRef]
- Pauly, D.; Chacana, P.A.; Calzado, E.G.; Brembs, B.; Schade, R. IgY technology: Extraction of chicken antibodies from egg yolk by polyethylene glycol (PEG) precipitation. J. Vis. Exp. 2011, 51, e3084. [Google Scholar] [CrossRef]
- Müller, U.; Steinhoff, U.; Reis, L.F.; Hemmi, S.; Pavlovic, J.; Zinkernagel, R.M.; Aguet, M. Functional role of type I and type II interferons in antiviral defense. Science 1994, 264, 1918–1921. [Google Scholar] [CrossRef]
- Bray, M. The role of the Type I interferon response in the resistance of mice to filovirus infection. J. Gen. Virol. 2001, 82, 1365–1373. [Google Scholar] [CrossRef]
- Krähling, V.; Becker, D.; Rohde, C.; Eickmann, M.; Eroğlu, Y.; Herwig, A.; Kerber, R.; Kowalski, K.; Vergara-Alert, J.; Becker, S.; et al. Development of an antibody capture ELISA using inactivated Ebola Zaire Makona virus. Med. Microbiol. Immunol. 2016, 205, 173–183. [Google Scholar] [CrossRef]
- Gibb, T.R.; Norwood, D.A.; Woollen, N.; Henchal, E.A. Development and evaluation of a fluorogenic 5’ nuclease assay to detect and differentiate between Ebola virus subtypes Zaire and Sudan. J. Clin. Microbiol. 2001, 39, 4125–4130. [Google Scholar] [CrossRef] [Green Version]
- Werner-Keišs, N. Untersuchungen zur Expression von Strukturproteinen des Virus der Bornaschen Krankheit an Intrazerebral Infizierten Lewis-Ratten. Dissertation, Stiftung Tierärztliche Hochschule Hannover. 2006. Available online: https://elib.tiho-hannover.de/receive/etd_mods_00002011 (accessed on 24 March 2022).
- Werner-Keišs, N.; Garten, W.; Richt, J.A.; Porombka, D.; Algermissen, D.; Herzog, S.; Baumgartner, W.; Herden, C. Restricted expression of Borna disease virus glycoprotein in brains of experimentally infected Lewis rats. Neuropathol. Appl. Neurobiol. 2008, 34, 590–602. [Google Scholar] [CrossRef]
- Zurbriggen, A.; Muller, C.; Vandevelde, M. In situ hybridization of virulent canine distemper virus in brain tissue, using digoxigenin-labeled probes. Am. J. Vet. Res. 1993, 54, 1457–1461. [Google Scholar]
- Sutter, G.; Moss, B. Nonreplicating vaccinia vector efficiently expresses recombinant genes. Proc. Natl. Acad. Sci. USA 1992, 89, 10847–10851. [Google Scholar] [CrossRef] [Green Version]
- Elliott, L.H.; Kiley, M.P.; McCormick, J.B. Descriptive analysis of Ebola virus proteins. Virology 1985, 147, 169–176. [Google Scholar] [CrossRef]
- Prehaud, C.; Hellebrand, E.; Coudrier, D.; Volchkov, V.E.; Volchkova, V.A.; Feldmann, H.; Le Guenno, B.; Bouloy, M. Recombinant Ebola virus nucleoprotein and glycoprotein (Gabon 94 strain) provide new tools for the detection of human infections. J. Gen. Virol. 1998, 79 Pt 11, 2565–2572. [Google Scholar] [CrossRef] [Green Version]
- Volchkov, V.E.; Becker, S.; Volchkova, V.A.; Ternovoj, V.A.; Kotov, A.N.; Netesov, S.V.; Klenk, H.D. GP mRNA of Ebola virus is edited by the Ebola virus polymerase and by T7 and vaccinia virus polymerases. Virology 1995, 214, 421–430. [Google Scholar] [CrossRef] [Green Version]
- Volchkov, V.E.; Feldmann, H.; Volchkova, V.A.; Klenk, H.D. Processing of the Ebola virus glycoprotein by the proprotein convertase furin. Proc. Natl. Acad. Sci. USA 1998, 95, 5762–5767. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, A.; Yang, Z.Y.; Xu, L.; Nabel, G.J.; Crews, T.; Peters, C.J. Biochemical analysis of the secreted and virion glycoproteins of Ebola virus. J. Virol. 1998, 72, 6442–6447. [Google Scholar] [CrossRef] [Green Version]
- Meyer, M.; Garron, T.; Lubaki, N.M.; Mire, C.E.; Fenton, K.A.; Klages, C.; Olinger, G.G.; Geisbert, T.W.; Collins, P.L.; Bukreyev, A. Aerosolized Ebola vaccine protects primates and elicits lung-resident T cell responses. J. Clin. Investig. 2015, 125, 3241–3255. [Google Scholar] [CrossRef] [Green Version]
- Schweneker, M.; Laimbacher, A.S.; Zimmer, G.; Wagner, S.; Schraner, E.M.; Wolferstätter, M.; Klingenberg, M.; Dirmeier, U.; Steigerwald, R.; Lauterbach, H.; et al. Recombinant Modified Vaccinia Virus Ankara Generating Ebola Virus-Like Particles. J. Virol. 2017, 91, e00343-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lázaro-Frías, A.; Gómez-Medina, S.; Sánchez-Sampedro, L.; Ljungberg, K.; Ustav, M.; Liljeström, P.; Muñoz-Fontela, C.; Esteban, M.; García-Arriaza, J. Distinct Immunogenicity and Efficacy of Poxvirus-Based Vaccine Candidates against Ebola Virus Expressing GP and VP40 Proteins. J. Virol. 2018, 92, e00363-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pushko, P.; Bray, M.; Ludwig, G.V.; Parker, M.; Schmaljohn, A.; Sanchez, A.; Jahrling, P.B.; Smith, J.F. Recombinant RNA replicons derived from attenuated Venezuelan equine encephalitis virus protect guinea pigs and mice from Ebola hemorrhagic fever virus. Vaccine 2000, 19, 142–153. [Google Scholar] [CrossRef]
- Sullivan, N.J.; Geisbert, T.W.; Geisbert, J.B.; Shedlock, D.J.; Xu, L.; Lamoreaux, L.; Custers, J.H.; Popernack, P.M.; Yang, Z.Y.; Pau, M.G.; et al. Immune protection of nonhuman primates against Ebola virus with single low-dose adenovirus vectors encoding modified GPs. PLoS Med. 2006, 3, e177. [Google Scholar] [CrossRef]
- Padilla-Quirarte, H.O.; Lopez-Guerrero, D.V.; Gutierrez-Xicotencatl, L.; Esquivel-Guadarrama, F. Protective Antibodies Against Influenza Proteins. Front. Immunol. 2019, 10, 1677. [Google Scholar] [CrossRef] [Green Version]
- Rai, D.; Martin, M.D.; Badovinac, V.P. The longevity of memory CD8 T cell responses after repetitive antigen stimulations. J. Immunol. 2014, 192, 5652–5659. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kupke, A.; Volz, A.; Dietzel, E.; Freudenstein, A.; Schmidt, J.; Shams-Eldin, H.; Jany, S.; Sauerhering, L.; Krähling, V.; Gellhorn Serra, M.; et al. Protective CD8+ T Cell Response Induced by Modified Vaccinia Virus Ankara Delivering Ebola Virus Nucleoprotein. Vaccines 2022, 10, 533. https://doi.org/10.3390/vaccines10040533
Kupke A, Volz A, Dietzel E, Freudenstein A, Schmidt J, Shams-Eldin H, Jany S, Sauerhering L, Krähling V, Gellhorn Serra M, et al. Protective CD8+ T Cell Response Induced by Modified Vaccinia Virus Ankara Delivering Ebola Virus Nucleoprotein. Vaccines. 2022; 10(4):533. https://doi.org/10.3390/vaccines10040533
Chicago/Turabian StyleKupke, Alexandra, Asisa Volz, Erik Dietzel, Astrid Freudenstein, Jörg Schmidt, Hosam Shams-Eldin, Sylvia Jany, Lucie Sauerhering, Verena Krähling, Michelle Gellhorn Serra, and et al. 2022. "Protective CD8+ T Cell Response Induced by Modified Vaccinia Virus Ankara Delivering Ebola Virus Nucleoprotein" Vaccines 10, no. 4: 533. https://doi.org/10.3390/vaccines10040533
APA StyleKupke, A., Volz, A., Dietzel, E., Freudenstein, A., Schmidt, J., Shams-Eldin, H., Jany, S., Sauerhering, L., Krähling, V., Gellhorn Serra, M., Herden, C., Eickmann, M., Becker, S., & Sutter, G. (2022). Protective CD8+ T Cell Response Induced by Modified Vaccinia Virus Ankara Delivering Ebola Virus Nucleoprotein. Vaccines, 10(4), 533. https://doi.org/10.3390/vaccines10040533