
A Rapid and Consistent Method to Identify Four SARS-CoV-2 Variants during the First Half of 2021 by RT-PCR

 ,
,  ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Detection of SARS-CoV-2 Virus by Real-Time Polymerase Chain Reaction (RT-PCR)
2.3. RNA Extraction and cDNA Synthesis
2.4. TaqMan RT-PCR Assay
2.5. SNP Genotyping Panel Design for SARS-CoV-2 Variants
2.6. Criteria for SARS-CoV-2 Variant Definitions
2.7. Library Preparation and Sequencing
2.8. SARS-CoV-2 Variant and Mutation Characterization
2.9. External Validation Dataset
2.10. Statistical Analysis
3. Results
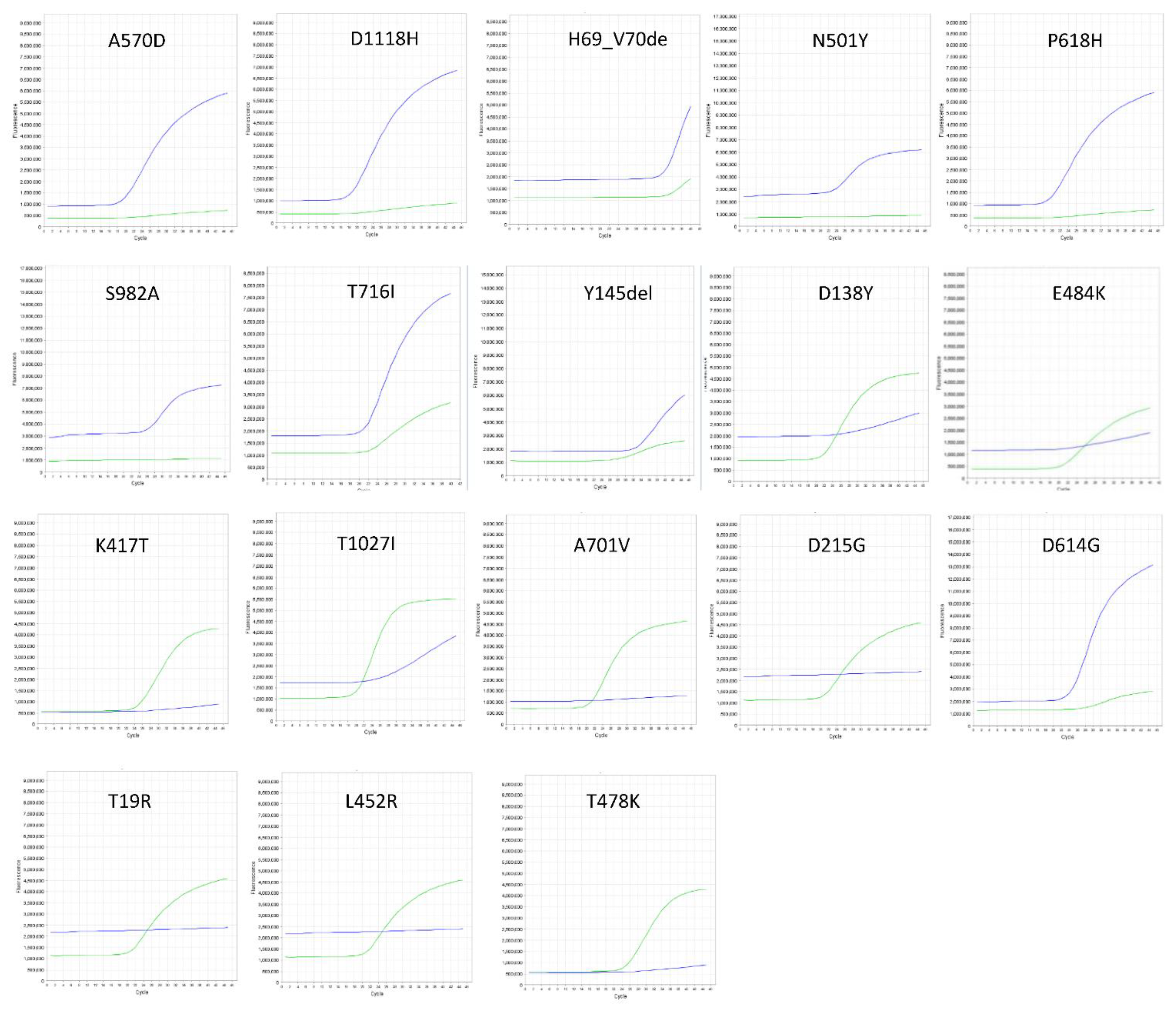
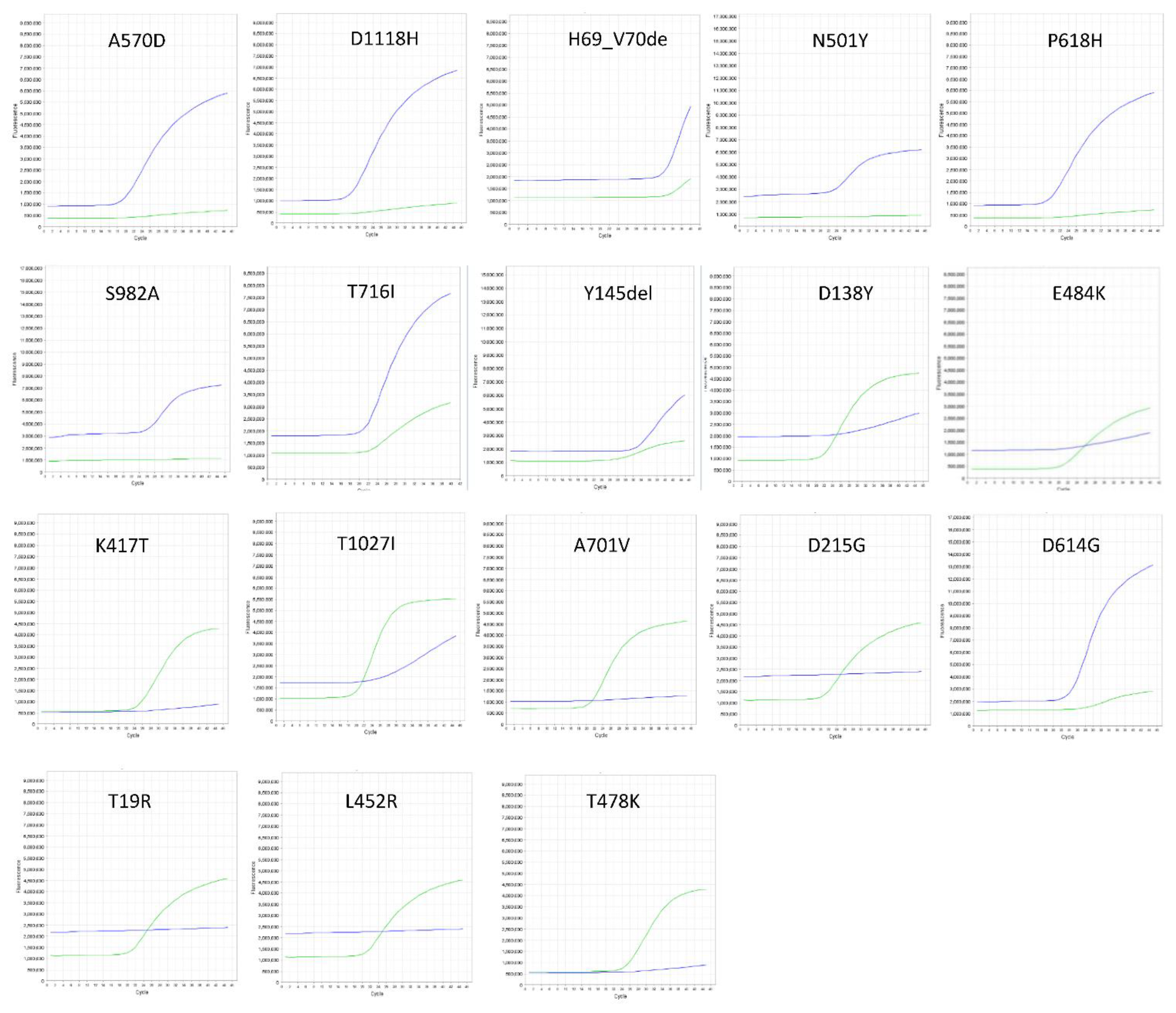
3.1. Genotyping Clinical SARS-CoV-2 Samples
3.2. Comparison between External Dataset and SNP Genotyping Panel
3.3. Frequency and Informativity of the SNP Marker for SARS-CoV-2 Variant Characterization by RT-PCR Probe
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lu, H.; Stratton, C.W.; Tang, Y.W. Outbreak of pneumonia of unknown etiology in Wuhan, China: The mystery and the miracle. J. Med. Virol. 2020, 92, 401–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Volz, E.M.S.; Chand, M.; Barrett, J.C.; Johnson, R.; Hopkins, S.; Gandy, A.; Rambaut, A.; Ferguson, N.M. Transmission of SARS-CoV-2 Lineage B.1.1.7 in England: Insights from linking epidemiological and genetic data. medRxiv 2021. [Google Scholar] [CrossRef]
- du Plessis, L.; McCrone, J.T.; Zarebski, A.E.; Hill, V.; Ruis, C.; Gutierrez, B.; Raghwani, J.; Ashworth, J.; Colquhoun, R.; Connor, T.R.; et al. Establishment and lineage dynamics of the SARS-CoV-2 epidemic in the UK. Science 2021, 371, 708–712. [Google Scholar] [CrossRef] [PubMed]
- Investigation of Novel SARS-CoV-2 Variant Variant of Concern 202012/01 Technical Briefing 5. 2021. Available online: https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/959426/Variant_of_Concern_VOC_202012_01_Technical_Briefing_5.pdf (accessed on 27 November 2021).
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N.; et al. Detection of a SARS-CoV-2 variant of concern in South Africa. Nature 2021, 592, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, Q.; Inchakalody, V.P.; Merhi, M.; Mestiri, S.; Taib, N.; Moustafa Abo El-Ella, D.; Bedhiafi, T.; Raza, A.; Al-Zaidan, L.; Mohsen, M.O.; et al. Emerging COVID-19 variants and their impact on SARS-CoV-2 diagnosis, therapeutics and vaccines. Ann. Med. 2022, 54, 524–540. [Google Scholar] [CrossRef] [PubMed]
- Joshi, M.; Kumar, M.; Srivastava, V.; Kumar, D.; Rathore, D.; Pandit, R.; Chaitanya, G. First detection of SARS-CoV-2 Delta variant (B.1.617.2) in the wastewater of (Ahmedabad), India Capsule: Delta variant found a month earlier in the wastewater of Ahmedabad than the first confirMed. Clinical Patient Report. medRxiv 2021. [Google Scholar] [CrossRef]
- Weisblum, Y.; Schmidt, F.; Zhang, F.; DaSilva, J.; Poston, D.; Lorenzi, J.C.; Muecksch, F.; Rutkowska, M.; HoffmAnn, H.H.; Michailidis, E.; et al. Escape from neutralizing antibodies by SARS-CoV-2 spike protein variants. Elife 2020, 9, e61312. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization Classification of Omicron (B.1.1.529): SARS-CoV-2 Variant of Concern. 2021. Available online: https://www.who.int/news/item/26-11-2021-classification-of-omicron-(B.1.1.529)-sars-cov-2-variant-of-concern (accessed on 27 November 2021).
- Wang, L.; Cheng, G. Sequence analysis of the emerging SARS-CoV-2 variant Omicron in South Africa. J. Med. Virol. 2022, 94, 1728–1733. [Google Scholar] [CrossRef]
- Vogels, C.B.F.; Breban, M.I.; Ott, I.M.; Alpert, T.; Petrone, M.E.; Watkins, A.E.; Kalinich, C.C.; Earnest, R.; Rothman, J.E.; Goes de Jesus, J.; et al. Multiplex qPCR discriminates variants of concern to enhance global surveillance of SARS-CoV-2. PLoS Biol. 2021, 19, e3001236. [Google Scholar] [CrossRef] [PubMed]
- Korukluoglu, G.; Kolukirik, M.; Bayrakdar, F.; Ozgumus, G.G.; Altas, A.B.; Cosgun, Y.; Ketre Kolukirik, C.Z. 40 minutes RT-qPCR assay for screening Spike N501Y and HV69-70del mutations. bioRxiv 2021. [Google Scholar]
- Vogels, C.B.F.; Breban, M.I.; Alpert, T.; Petrone, M.E.; Watkins, A.E.; Ott, I.M.; de Jesus, J.G.; Claro, I.M.; Ferreira, G.M.; Crispim, M.A.E.; et al. PCR assay to enhance global surveillance for SARS-CoV-2 variants of concern. medRxiv 2021. [Google Scholar] [CrossRef]
- Khan, A.; Zia, T.; Suleman, M.; Khan, T.; Ali, S.S.; Abbasi, A.A.; Mohammad, A.; Wei, D.Q. Higher infectivity of the SARS-CoV-2 new variants is associated with K417N/T, E484K, and N501Y mutants: An insight from structural data. J. Cell Physiol. 2021, 236, 7045–7057. [Google Scholar] [CrossRef] [PubMed]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827.e19. [Google Scholar] [CrossRef] [PubMed]
- WHO Update on Omicron. Available online: https://www.who.int/news/item/28-11-2021-update-on-omicron (accessed on 28 November 2021).
- GISAID Tracking of Variants. 2021. Available online: https://www.gisaid.org/hcov19-variants/ (accessed on 27 November 2021).
- Garson, J.A.; Badru, S.; Parker, E.; Tedder, R.S.; McClure, M.O. Highly sensitive and specific detection of the SARS-CoV-2 Delta variant by double-mismatch allele-specific real time reverse transcription PCR. J. Clin. Virol. 2022, 146, 105049. [Google Scholar] [CrossRef] [PubMed]
- Bezerra, M.F.; Machado, L.C.; De Carvalho, V.; Docena, C.; Brandao-Filho, S.P.; Ayres, C.F.J.; Paiva, M.H.S.; Wallau, G.L. A Sanger-based approach for scaling up screening of SARS-CoV-2 variants of interest and concern. Infect. Genet. Evol. 2021, 92, 104910. [Google Scholar] [CrossRef] [PubMed]
- Aoki, A.; Mori, Y.; Okamoto, Y.; Jinno, H. Development of a genotyping platform for SARS-CoV-2 variants using high-resolution melting analysis. J. Infect. Chemother. 2021, 27, 1336–1341. [Google Scholar] [CrossRef] [PubMed]
- Aoki, A.; Adachi, H.; Mori, Y.; Ito, M.; Sato, K.; Okuda, K.; Sakakibara, T.; Okamoto, Y.; Jinno, H. A rapid screening assay for L452R and T478K spike mutations in SARS-CoV-2 Delta variant using high-resolution melting analysis. J. Toxicol Sci. 2021, 46, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Butler-Laporte, G.; Lawandi, A.; Schiller, I.; Yao, M.; Dendukuri, N.; McDonald, E.G.; Lee, T.C. Comparison of Saliva and Nasopharyngeal Swab Nucleic Acid Amplification Testing for Detection of SARS-CoV-2: A Systematic Review and Meta-analysis. JAMA Intern. Med. 2021, 181, 353–360. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Stage | Temperature | Time |
|---|---|---|
| UNG incubation | 50 °C | 2 min |
| Polymarase activation | 95 °C | 10 min |
| PCR (40 cycles) | 95 °C | 15 s |
| 60 °C | 1 min |
| Sars-Cov-2 VOC | ||||
|---|---|---|---|---|
| Mutation | Alpha | Beta | Gamma | Delta |
| A570D | Present | - | - | - |
| D1118H | Present | - | - | - |
| H69_V70del | Present | - | - | - |
| N501Y | Present | Present | Present | - |
| P681H | Present | - | - | - |
| S982A | Present | - | - | - |
| T716I | Present | - | - | - |
| Y145del | Present | - | - | - |
| D138Y | - | - | Present | - |
| E484K | - | Present | Present | - |
| K417T | - | - | Present | - |
| T1027I | - | - | Present | - |
| A701V | - | Present | - | - |
| D215G | - | Present | - | - |
| D614G | Present | Present | Present | Present |
| L452R | - | - | - | Present |
| T19R | - | - | - | Present |
| T478K | - | - | - | Present |
| Lineage | N | Concordance, RT-PCR vs. NGS |
|---|---|---|
| Alpha (B.1.1.7) | 61/61 | 100% (95% CI 94.1–100%) |
| Beta (B.1.351) | 5/5 | 100%(95% CI 47.8–100%) |
| Gamma (P.1) | 12/12 | 100% (95% CI 73.5–100%) |
| Delta (B.1.617.2) | 32/32 | 100% (95% CI 84.6–100%) |
| B.1 with D614G | 22/22 | 100% (95% CI 84.6–100%) |
| Frequency (%) | Informativity * (%) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Other Variants | Alpha | Gamma | Beta | Delta | Other Variants | Alpha | Gamma | Beta | Delta | |
| A570D | 0 | 98% | 0 | 0 | 0 | 0 | 98% | 0 | 0 | 0 |
| D1118H | 0 | 100% | 0 | 0 | 0 | 0 | 100% | 0 | 0 | 0 |
| H69_V70del | 0 | 97% | 0 | 0 | 0 | 0 | 97% | 0 | 0 | 0 |
| N501Y | 0 | 94% | 92% | 100% | 0 | 0 | 0 | 0 | 0 | 0 |
| P681H | 5% | 100% | 0 | 0 | 10% | 0 | 85% | 0 | 0 | 0 |
| S982A | 0 | 99% | 0 | 0 | 0 | 0 | 99% | 0 | 0 | 0 |
| T716I | 0 | 99% | 0 | 0 | 0 | 0 | 99% | 0 | 0 | 0 |
| Y145del | 5% | 94% | 0 | 0 | 0 | 0 | 89% | 0 | 0 | 0 |
| D138Y | 0 | 0 | 92% | 0 | 0 | 0 | 0 | 92% | 0 | 0 |
| E484K | 5% | 0 | 100% | 100% | 0 | 0 | 0 | 0 | 0 | 0 |
| K417T | 0 | 0 | 92% | 0 | 0 | 0 | 0 | 92% | 0 | 0 |
| T1027I | 5% | 0 | 92% | 0 | 0 | 0 | 0 | 87% | 0 | 0 |
| A701V | 0 | 1% | 0 | 100% | 0 | 0 | 0 | 0 | 99% | 0 |
| D215G | 0 | 0 | 0 | 100% | 0 | 0 | 0 | 0 | 100% | 0 |
| D614G | 100% | 99% | 92% | 100% | 97% | 0 | 0 | 0 | 0 | 0 |
| L452R | 0 | 0 | 0 | 0 | 100% | 0 | 0 | 0 | 0 | 100% |
| T19R | 0 | 0 | 0 | 0 | 91% | 0 | 0 | 0 | 0 | 91% |
| T478K | 0 | 0 | 0 | 0 | 97% | 0 | 0 | 0 | 0 | 97% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fabiani, M.; Margiotti, K.; Sabatino, M.; Viola, A.; Mesoraca, A.; Giorlandino, C. A Rapid and Consistent Method to Identify Four SARS-CoV-2 Variants during the First Half of 2021 by RT-PCR. Vaccines 2022, 10, 483. https://doi.org/10.3390/vaccines10030483
Fabiani M, Margiotti K, Sabatino M, Viola A, Mesoraca A, Giorlandino C. A Rapid and Consistent Method to Identify Four SARS-CoV-2 Variants during the First Half of 2021 by RT-PCR. Vaccines. 2022; 10(3):483. https://doi.org/10.3390/vaccines10030483
Chicago/Turabian StyleFabiani, Marco, Katia Margiotti, Manuela Sabatino, Antonella Viola, Alvaro Mesoraca, and Claudio Giorlandino. 2022. "A Rapid and Consistent Method to Identify Four SARS-CoV-2 Variants during the First Half of 2021 by RT-PCR" Vaccines 10, no. 3: 483. https://doi.org/10.3390/vaccines10030483
APA StyleFabiani, M., Margiotti, K., Sabatino, M., Viola, A., Mesoraca, A., & Giorlandino, C. (2022). A Rapid and Consistent Method to Identify Four SARS-CoV-2 Variants during the First Half of 2021 by RT-PCR. Vaccines, 10(3), 483. https://doi.org/10.3390/vaccines10030483