
Genetic Modification of T Cells for the Immunotherapy of Cancer


Abstract
1. Introduction
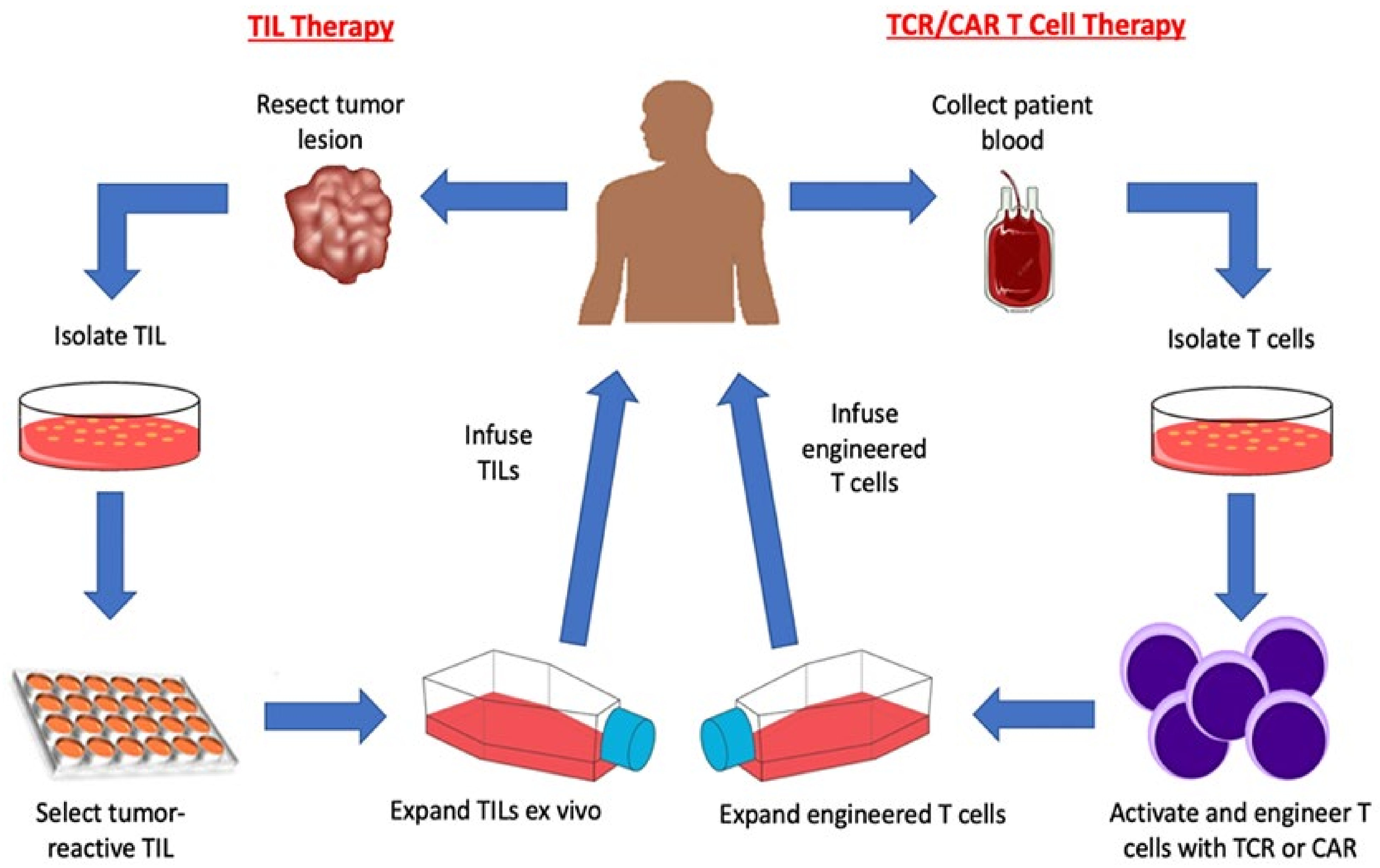
2. Adoptive T Cell Transfer
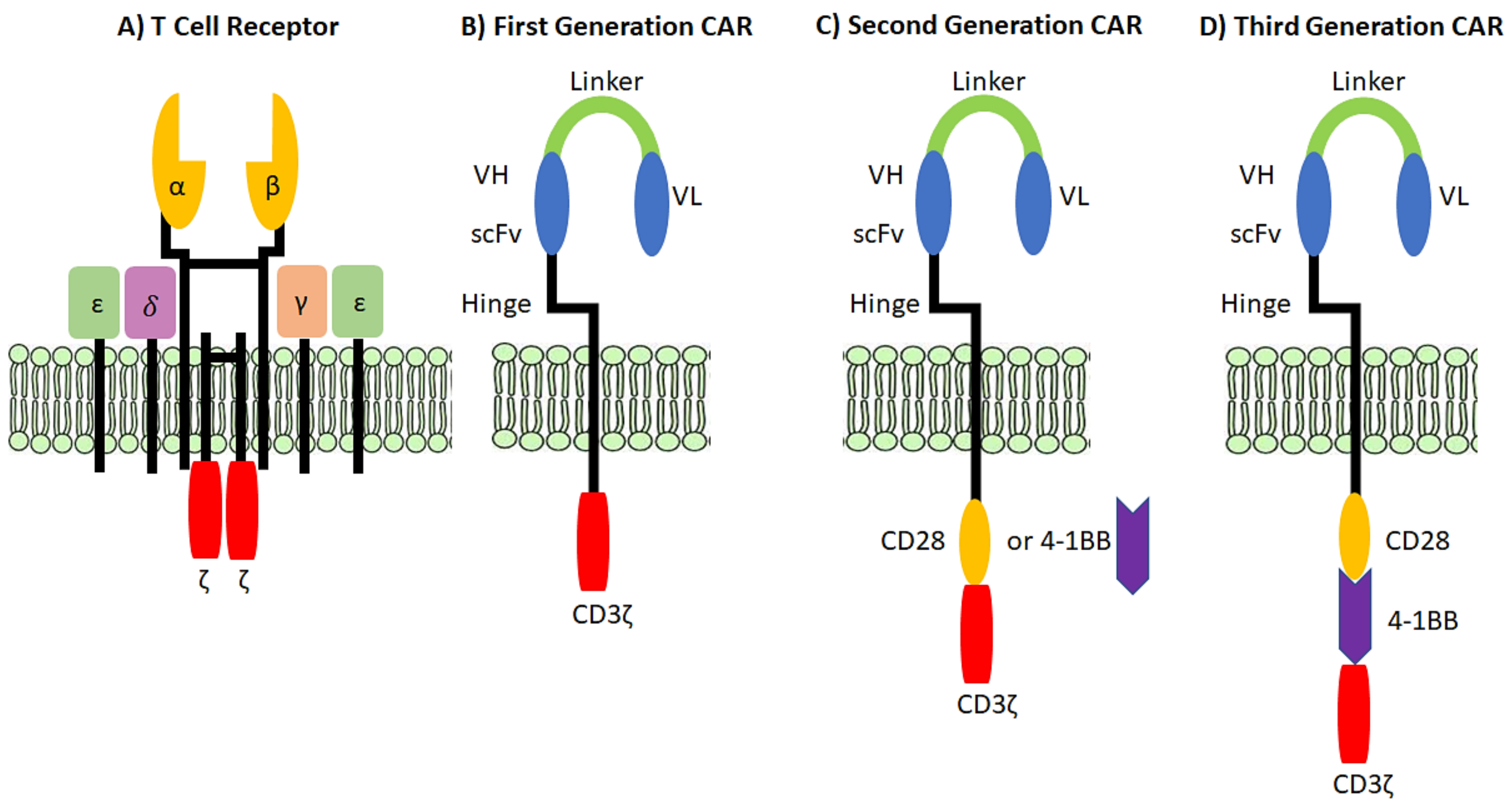
2.1. Antigen Recognition by T Cells
2.2. Tumor Infiltrating Lymphocytes
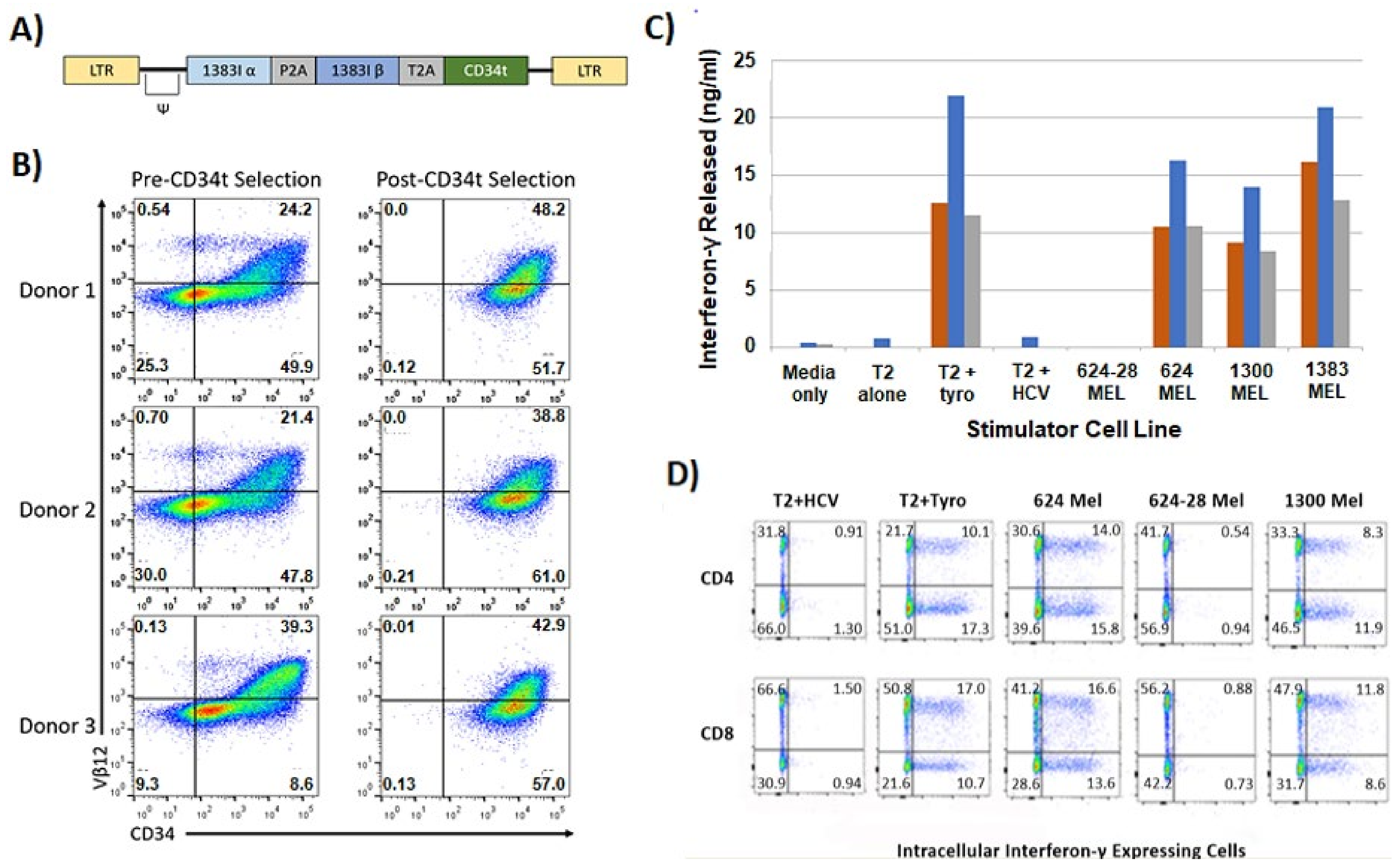
2.3. Genetically Modified T Cell Receptor Transduced T Cells
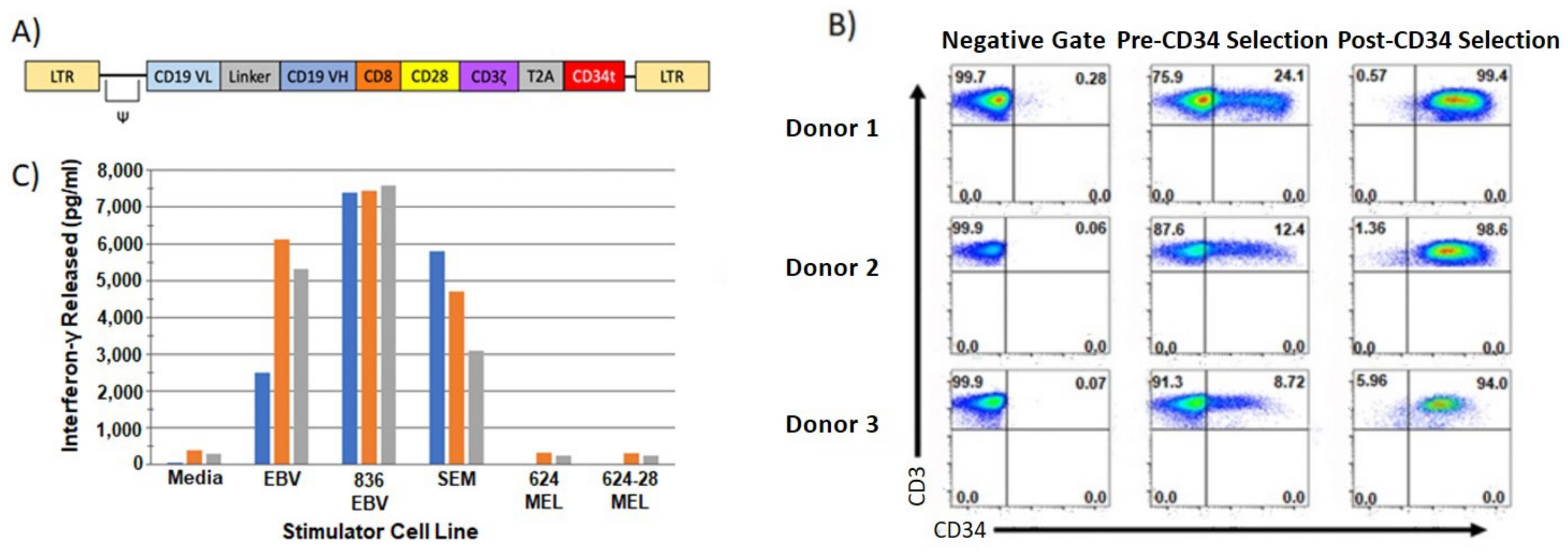
2.4. Chimeric Antigen Receptor (CAR) T cells
3. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yang, J.C.; Rosenberg, S.A. Adoptive T-Cell Therapy for Cancer. Adv. Immunol. 2016, 130, 279–294. [Google Scholar] [PubMed]
- Spear, T.T.; Nagato, K.; Nishimura, M.I. Strategies to genetically engineer T cells for cancer immunotherapy. Cancer Immunol. Immunother. 2016, 65, 631–649. [Google Scholar] [CrossRef] [PubMed]
- Allison, J.P.; McIntyre, B.W.; Bloch, D. Tumor-specific antigen of murine T-lymphoma defined with monoclonal antibody. J. Immunol. 1982, 129, 2293–2300. [Google Scholar] [PubMed]
- Yanagi, Y.; Yoshikai, Y.; Leggett, K.; Clark, S.P.; Aleksander, I.; Mak, T.W. A human T cell-specific cDNA clone encodes a protein having extensive homology to immunoglobulin chains. Nature 1984, 308, 145–149. [Google Scholar] [CrossRef]
- Hedrick, S.M.; Cohen, D.I.; Nielsen, E.A.; Davis, M.M. Isolation of cDNA clones encoding T cell-specific membrane-associated proteins. Nature 1984, 308, 149–153. [Google Scholar] [CrossRef]
- Call, M.E.; Pyrdol, J.; Wiedmann, M.; Wucherpfennig, K.W. The organizing principle in the formation of the T cell receptor-CD3 complex. Cell 2002, 111, 967–979. [Google Scholar] [CrossRef]
- Pageon, S.V.; Tabarin, T.; Yamamoto, Y.; Ma, Y.; Nicovich, P.R.; Bridgeman, J.S.; Cohnen, A.; Benzing, C.; Gao, Y.; Crowther, M.D.; et al. Functional role of T-cell receptor nanoclusters in signal initiation and antigen discrimination. Proc. Natl. Acad. Sci. USA 2016, 113, E5454–E5463. [Google Scholar] [CrossRef]
- Smith-Garvin, J.E.; Koretzky, G.A.; Jordan, M.S. T cell activation. Annu. Rev. Immunol. 2009, 27, 591–619. [Google Scholar] [CrossRef]
- Miceli, M.C.; Parnes, J.R. The roles of CD4 and CD8 in T cell activation. Semin. Immunol. 1991, 3, 133–141. [Google Scholar]
- Rangarajan, S.; Mariuzza, R.A. T cell receptor bias for MHC: Co-evolution or co-receptors? Cell. Mol. Life Sci. 2014, 71, 3059–3068. [Google Scholar] [CrossRef]
- Wang, J.H.; Reinherz, E.L. The structural basis of αβ T-lineage immune recognition: TCR docking topologies, mechanotransduction, and co-receptor function. Immunol. Rev. 2012, 250, 102–119. [Google Scholar] [CrossRef]
- Johnson, D.K.; Magoffin, W.; Myers, S.J.; Finnell, J.G.; Hancock, J.C.; Orton, T.S.; Persaud, S.P.; Christensen, K.A.; Weber, K.S. CD4 Inhibits Helper T Cell Activation at Lower Affinity Threshold for Full-Length T Cell Receptors Than Single Chain Signaling Constructs. Front. Immunol. 2020, 11, 561889. [Google Scholar]
- Rock, K.L.; Reits, E.; Neefjes, J. Present Yourself! By MHC Class I and MHC Class II Molecules. Trends Immunol. 2016, 37, 724–737. [Google Scholar] [CrossRef]
- Tendeiro Rego, R.; Morris, E.C.; Lowdell, M.W. T-cell receptor gene-modified cells: Past promises, present methodologies and future challenges. Cytotherapy 2019, 21, 341–357. [Google Scholar] [CrossRef]
- Sicard, A.; Boardman, D.A.; Levings, M.K. Taking regulatory T-cell therapy one step further. Curr. Opin. Organ Transpl. 2018, 23, 509–515. [Google Scholar] [CrossRef]
- Kishton, R.J.; Sukumar, M.; Restifo, N.P. Metabolic Regulation of T Cell Longevity and Function in Tumor Immunotherapy. Cell Metab. 2017, 26, 94–109. [Google Scholar] [CrossRef]
- Van den Berg, J.H.; Heemskerk, B.; van Rooij, N.; Gomez-Eerland, R.; Michels, S.; van Zon, M.; de Boer, R.; Bakker, N.A.M.; Jorritsma-Smit, A.; van Buuren, M.M.; et al. Tumor infiltrating lymphocytes (TIL) therapy in metastatic melanoma: Boosting of neoantigen-specific T cell reactivity and long-term follow-up. J. Immunother. Cancer 2020, 8, e000848. [Google Scholar] [CrossRef]
- Peng, S.; Zaretsky, J.M.; Ng, A.H.C.; Chour, W.; Bethune, M.T.; Choi, J.; Hsu, A.; Holman, E.; Ding, X.; Guo, K.; et al. Sensitive Detection and Analysis of Neoantigen-Specific T Cell Populations from Tumors and Blood. Cell Rep. 2019, 28, 2728–2738. [Google Scholar] [CrossRef]
- Leko, V.; McDuffie, L.A.; Zheng, Z.; Gartner, J.J.; Prickett, T.D.; Apolo, A.B.; Agarwal, P.K.; Rosenberg, S.A.; Lu, Y.C. Identification of Neoantigen-Reactive Tumor-Infiltrating Lymphocytes in Primary Bladder Cancer. J. Immunol. 2019, 202, 3458–3467. [Google Scholar] [CrossRef]
- Foley, K.C.; Nishimura, M.I.; Moore, T.V. Combination immunotherapies implementing adoptive T-cell transfer for advanced-stage melanoma. Melanoma Res. 2018, 28, 171. [Google Scholar] [CrossRef]
- Draghi, A.; Chamberlain, C.A.; Khan, S.; Papp, K.; Lauss, M.; Soraggi, S.; Radic, H.D.; Presti, M.; Harbst, K.; Gokuldass, A.; et al. Rapid Identification of the Tumor-Specific Reactive TIL Repertoire via Combined Detection of CD137, TNF, and IFNγ, Following Recognition of Autologous Tumor-Antigens. Front. Immunol. 2021, 12, 705422. [Google Scholar] [CrossRef]
- Kongkaew, T.; Thaiwong, R.; Tudsamran, S.; Sae-Jung, T.; Sengprasert, P.; Vasuratna, A.; Suppipat, K.; Reantragoon, R. TIL expansion with high dose IL-2 or low dose IL-2 with anti-CD3/anti-CD28 stimulation provides different quality of TIL-expanded T cell clones. J. Immunol. Methods 2022, 503, 113229. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Packard, B.S.; Aebersold, P.M.; Solomon, D.; Topalian, S.L.; Toy, S.T.; Simon, P.; Lotze, M.T.; Yang, J.C.; Seipp, C.A.; et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N. Engl. J. Med. 1988, 319, 1676–1680. [Google Scholar] [CrossRef]
- Santoiemma, P.P.; Powell, D.J., Jr. Tumor infiltrating lymphocytes in ovarian cancer. Cancer Biol. Ther. 2015, 16, 807–820. [Google Scholar] [CrossRef]
- Stanton, S.E.; Disis, M.L. Clinical significance of tumor-infiltrating lymphocytes in breast cancer. J. Immunother. Cancer 2016, 4, 59. [Google Scholar] [CrossRef]
- Andersen, R.; Donia, M.; Westergaard, M.C.; Pedersen, M.; Hansen, M.; Svane, I.M. Tumor infiltrating lymphocyte therapy for ovarian cancer and renal cell carcinoma. Hum. Vaccines Immunother. 2015, 11, 2790–2795. [Google Scholar] [CrossRef] [PubMed]
- Eshhar, Z.; Waks, T.; Gross, G.; Schindler, D.G. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc. Natl. Acad. Sci. USA 1993, 90, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Clay, T.M.; Custer, M.C.; Sachs, J.; Hwu, P.; Rosenberg, S.A.; Nishimura, M.I. Efficient transfer of a tumor antigen-reactive TCR to human peripheral blood lymphocytes confers anti-tumor reactivity. J. Immunol. 1999, 163, 507–513. [Google Scholar] [PubMed]
- Crowther, M.D.; Svane, I.M.; Met, Ö. T-Cell Gene Therapy in Cancer Immunotherapy: Why It Is No Longer Just CARs on The Road. Cells 2020, 9, 1588. [Google Scholar] [CrossRef] [PubMed]
- Bertoletti, A.; Tan, A.T.; Koh, S. T-cell therapy for chronic viral hepatitis. Cytotherapy 2017, 19, 1317–1324. [Google Scholar] [CrossRef]
- Lang, F.; Schrörs, B.; Löwer, M.; Türeci, Ö.; Sahin, U. Identification of neoantigens for individualized therapeutic cancer vaccines. Nat. Rev. Drug. Discov. 2022, 1–22. [Google Scholar] [CrossRef]
- Duval, L.; Schmidt, H.; Kaltoft, K.; Fode, K.; Jensen, J.J.; Sorensen, S.M.; Nishimura, M.I.; von der Maase, H. Adoptive transfer of allogeneic cytotoxic T lymphocytes equipped with a HLA-A2 restricted MART-1 T-cell receptor: A phase I trial in metastatic melanoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2006, 12, 1229–1236. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, L. The Emerging World of TCR-T Cell Trials Against Cancer: A Systematic Review. Technol. Cancer Res. Treat. 2019, 18, 1533033819831068. [Google Scholar] [CrossRef]
- Kageyama, S.; Ikeda, H.; Miyahara, Y.; Imai, N.; Ishihara, M.; Saito, K.; Sugino, S.; Ueda, S.; Ishikawa, T.; Kokura, S.; et al. Adoptive Transfer of MAGE-A4 T-cell Receptor Gene-Transduced Lymphocytes in Patients with Recurrent Esophageal Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 2268–2277. [Google Scholar] [CrossRef]
- Tawara, I.; Kageyama, S.; Miyahara, Y.; Fujiwara, H.; Nishida, T.; Akatsuka, Y.; Ikedaa, H.; Tanimoto, K.; Terakura, S.; Murata, M.; et al. Safety and persistence of WT1-specific T-cell receptor gene-transduced lymphocytes in patients with AML and MDS. Blood 2017, 130, 1985–1994. [Google Scholar] [CrossRef]
- Chapuis, A.G.; Egan, D.N.; Bar, M.; Schmitt, T.M.; McAfee, M.S.; Paulson, K.G.; Voillet, V.; Gottardo, R.; Ragnarsson, G.B.; Bleakley, M.; et al. T cell receptor gene therapy targeting WT1 prevents acute myeloid leukemia relapse post-transplant. Nat. Med. 2019, 25, 1064–1072. [Google Scholar] [CrossRef]
- Robbins, P.F.; Morgan, R.A.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; Dudley, M.E.; Wunderlich, J.R.; Nahvi, A.V.; Helman, L.J.; Mackall, C.L.; et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2011, 29, 917–924. [Google Scholar] [CrossRef]
- Xue, S.; Gillmore, R.; Gao, L.; Bendle, G.; Holler, A.; Downs, A.M.; Tsaillos, A.; Ramirez, F.; Ghani, Y.; Hart, D.; et al. Use of the allogeneic TCR repertoire to enhance anti-tumor immunity. J. Biol. Regul. Homeost. Agents 2004, 18, 131–133. [Google Scholar]
- Nishimura, M.I.; Avichezer, D.; Custer, M.C.; Lee, C.S.; Chen, C.; Parkhurst, M.R.; Diamond, R.A.; Robbins, P.F.; Schwartzentruber, D.J.; Rosenberg, S.A. MHC class I-restricted recognition of a melanoma antigen by a human CD4+ tumor infiltrating lymphocyte. Cancer Res. 1999, 59, 6230–6238. [Google Scholar]
- Roszkowski, J.J.; Lyons, G.E.; Kast, W.M.; Yee, C.; Van Besien, K.; Nishimura, M.I. Simultaneous generation of CD8+ and CD4+ melanoma-reactive T cells by retroviral-mediated transfer of a single T-cell receptor. Cancer Res. 2005, 65, 1570–1576. [Google Scholar] [CrossRef]
- Roszkowski, J.J.; Yu, D.C.; Rubinstein, M.P.; McKee, M.D.; Cole, D.J.; Nishimura, M.I. CD8-independent tumor cell recognition is a property of the T cell receptor and not the T cell. J. Immunol. 2003, 170, 2582–2589. [Google Scholar] [CrossRef]
- Moore, T.; Wagner, C.R.; Scurti, G.M.; Hutchens, K.A.; Godellas, C.; Clark, A.L.; Kolawole, E.M.; Hellman, L.M.; Singh, N.K.; Huyke, F.A.; et al. Clinical and immunologic evaluation of three metastatic melanoma patients treated with autologous melanoma-reactive TCR-transduced T cells. Cancer Immunol. Immunother. 2018, 67, 311–325. [Google Scholar] [CrossRef]
- Norell, H.; Zhang, Y.; McCracken, J.; Martins da Palma, T.; Lesher, A.; Liu, Y.; Roszkowski, J.J.; Temple, A.; Callender, G.G.; Clay, T.; et al. CD34-based enrichment of genetically engineered human T cells for clinical use results in dramatically enhanced tumor targeting. Cancer Immunol. Immunother. 2010, 59, 851–862. [Google Scholar] [CrossRef]
- Cole, D.J.; Weil, D.P.; Shilyansky, J.; Custer, M.; Kawakami, Y.; Rosenberg, S.A.; Nishimura, M.I. Characterization of the functional specificity of a cloned T-cell receptor heterodimer recognizing the MART-1 melanoma antigen. Cancer Res. 1995, 55, 748–752. [Google Scholar]
- Moore, T.V.; Lyons, G.E.; Brasic, N.; Roszkowski, J.J.; Voelkl, S.; Mackensen, A.; Kast, W.M.; Le Poole, I.C.; Nishimura, M.I. Relationship between CD8-dependent antigen recognition, T cell functional avidity, and tumor cell recognition. Cancer Immunol. Immunother. 2009, 58, 719–728. [Google Scholar] [CrossRef]
- Callender, G.G.; Rosen, H.R.; Roszkowski, J.J.; Lyons, G.E.; Li, M.; Moore, T.; Brasic, N.; McKee, M.D.; Nishimura, M.I. Identification of a hepatitis C virus-reactive T cell receptor that does not require CD8 for target cell recognition. Hepatology 2006, 43, 973–981. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Y.; Moxley, K.M.; Golden-Mason, L.; Hughes, M.G.; Liu, T.; Heemskerk, M.H.; Rosen, H.R.; Nishimura, M.I. Transduction of human T cells with a novel T-cell receptor confers anti-HCV reactivity. PLoS Pathog. 2010, 6, e1001018. [Google Scholar] [CrossRef] [PubMed]
- Spear, T.T.; Foley, K.C.; Garrett-Mayer, E.; Nishimura, M.I. TCR modifications that enhance chain pairing in gene-modified T cells can augment cross-reactivity and alleviate CD8 dependence. J. Leukoc. Biol. 2018, 103, 973–983. [Google Scholar] [CrossRef]
- Wilde, S.; Schendel, D.J. High-quality and high-avidity T cell clones specific for tumor-associated antigens and how to find them. Oncoimmunology 2012, 1, 1643–1644. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Parkhurst, M.R.; Joo, J.; Riley, J.P.; Yu, Z.; Li, Y.; Robbins, P.F.; Rosenberg, S.A. Characterization of genetically modified T-cell receptors that recognize the CEA:691–699 peptide in the context of HLA-A2.1 on human colorectal cancer cells. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Sandri, S.; Bobisse, S.; Moxley, K.; Lamolinara, A.; De Sanctis, F.; Boschi, F.; Sbarbati, A.; Fracasso, G.; Ferrarini, G.; Hendriks, R.W.; et al. Feasibility of Telomerase-Specific Adoptive T-cell Therapy for B-cell Chronic Lymphocytic Leukemia and Solid Malignancies. Cancer Res. 2016, 76, 2540–2551. [Google Scholar] [CrossRef]
- Sandri, S.; De Sanctis, F.; Lamolinara, A.; Boschi, F.; Poffe, O.; Trovato, R.; Flore, A.; Sartori, S.; Sbarbati, A.; Bondanza, A.; et al. Effective control of acute myeloid leukaemia and acute lymphoblastic leukaemia progression by telomerase specific adoptive T-cell therapy. Oncotarget 2017, 8, 86987–87001. [Google Scholar] [CrossRef]
- Stanislawski, T.; Voss, R.H.; Lotz, C.; Sadovnikova, E.; Willemsen, R.A.; Kuball, J.; Ruppert, T.; Bolhuis, R.L.; Melief, C.J.; Huber, C.; et al. Circumventing tolerance to a human MDM2-derived tumor antigen by TCR gene transfer. Nat. Immunol. 2001, 2, 962–970. [Google Scholar] [CrossRef]
- Kuball, J.; Schmitz, F.W.; Voss, R.H.; Ferreira, E.A.; Engel, R.; Guillaume, P.; Strand, S.; Romero, P.; Huber, C.; Sherman, L.A.; et al. Cooperation of human tumor-reactive CD4+ and CD8+ T cells after redirection of their specificity by a high-affinity p53A2.1-specific TCR. Immunity 2005, 22, 117–129. [Google Scholar] [CrossRef]
- Voss, R.H.; Kuball, J.; Engel, R.; Guillaume, P.; Romero, P.; Huber, C.; Theobald, M. Redirection of T cells by delivering a transgenic mouse-derived MDM2 tumor antigen-specific TCR and its humanized derivative is governed by the CD8 coreceptor and affects natural human TCR expression. Immunol. Res. 2006, 34, 67–87. [Google Scholar] [CrossRef]
- Houot, R.; Schultz, L.M.; Marabelle, A.; Kohrt, H. T-cell-based Immunotherapy: Adoptive Cell Transfer and Checkpoint Inhibition. Cancer Immunol. Res. 2015, 3, 1115–1122. [Google Scholar] [CrossRef]
- Varela-Rohena, A.; Molloy, P.E.; Dunn, S.M.; Li, Y.; Suhoski, M.M.; Carroll, R.G.; Milicic, A.; Mahon, T.; Sutton, D.H.; Laugel, B.; et al. Control of HIV-1 immune escape by CD8 T cells expressing enhanced T-cell receptor. Nat. Med. 2008, 14, 1390–1395. [Google Scholar] [CrossRef]
- Chlewicki, L.K.; Holler, P.D.; Monti, B.C.; Clutter, M.R.; Kranz, D.M. High-affinity, peptide-specific T cell receptors can be generated by mutations in CDR1, CDR2 or CDR3. J. Mol. Biol. 2005, 346, 223–239. [Google Scholar] [CrossRef]
- Harris, D.T.; Hager, M.V.; Smith, S.N.; Cai, Q.; Stone, J.D.; Kruger, P.; Lever, M.; Dushek, O.; Schmitt, T.M.; Greenberg, P.D.; et al. Comparison of T Cell Activities Mediated by Human TCRs and CARs That Use the Same Recognition Domains. J. Immunol. 2018, 200, 1088–1100. [Google Scholar] [CrossRef]
- Jones, L.L.; Brophy, S.E.; Bankovich, A.J.; Colf, L.A.; Hanick, N.A.; Garcia, K.C.; Kranz, D.M. Engineering and characterization of a stabilized alpha1/alpha2 module of the class I major histocompatibility complex product Ld. J. Biol. Chem. 2006, 281, 25734–25744. [Google Scholar] [CrossRef]
- Schmitt, T.M.; Aggen, D.H.; Stromnes, I.M.; Dossett, M.L.; Richman, S.A.; Kranz, D.M.; Greenberg, P.D. Enhanced-affinity murine T-cell receptors for tumor/self-antigens can be safe in gene therapy despite surpassing the threshold for thymic selection. Blood 2013, 122, 348–356. [Google Scholar] [CrossRef]
- Wilde, S.; Geiger, C.; Milosevic, S.; Mosetter, B.; Eichenlaub, S.; Schendel, D.J. Generation of allo-restricted peptide-specific T cells using RNA-pulsed dendritic cells: A three phase experimental procedure. Oncoimmunology 2012, 1, 129–140. [Google Scholar] [CrossRef]
- Wilde, S.; Sommermeyer, D.; Frankenberger, B.; Schiemann, M.; Milosevic, S.; Spranger, S.; Pohla, H.; Uckert, W.; Busch, D.H.; Schendel, D.J. Dendritic cells pulsed with RNA encoding allogeneic MHC and antigen induce T cells with superior antitumor activity and higher TCR functional avidity. Blood 2009, 114, 2131–2139. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.A.; Morgan, R.A.; Dudley, M.E.; Cassard, L.; Yang, J.C.; Hughes, M.S.; Kammula, U.S.; Royal, R.E.; Sherry, R.M.; Wunderlich, J.R.; et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood 2009, 114, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Parkhurst, M.R.; Yang, J.C.; Langan, R.C.; Dudley, M.E.; Nathan, D.A.; Feldman, S.A.; Davis, J.L.; Morgan, R.A.; Merino, M.J.; Sherry, R.M.; et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol. Ther. J. Am. Soc. Gene Ther. 2011, 19, 620–626. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Chinnasamy, N.; Abate-Daga, D.; Gros, A.; Robbins, P.F.; Zheng, Z.; Dudley, M.E.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J. Immunother. 2013, 36, 133–151. [Google Scholar] [CrossRef]
- Cameron, B.J.; Gerry, A.B.; Dukes, J.; Harper, J.V.; Kannan, V.; Bianchi, F.C.; Grand, F.; Brewer, J.E.; Gupta, M.; Plesa, G. Identification of a Titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci. Transl. Med. 2013, 5, 197ra103. [Google Scholar] [CrossRef]
- Robbins, P.F.; Kassim, S.H.; Tran, T.L.; Crystal, J.S.; Morgan, R.A.; Feldman, S.A.; Yang, J.C.; Dudley, M.E.; Wunderlich, J.R.; Sherry, R.M.; et al. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: Long-term follow-up and correlates with response. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 1019–1027. [Google Scholar] [CrossRef]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef]
- Deniger, D.C.; Pasetto, A.; Tran, E.; Parkhurst, M.R.; Cohen, C.J.; Robbins, P.F.; Cooper, J.; Rosenberg, S.A. Stable, Nonviral Expression of Mutated Tumor Neoantigen-specific T-cell Receptors Using the Sleeping Beauty Transposon/Transposase System. Mol. Ther. J. Am. Soc. Gene Ther. 2016, 24, 1078–1089. [Google Scholar] [CrossRef]
- McGranahan, N.; Furness, A.J.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef]
- Gross, L. The Specificity of Acquired Tumor Immunity. J. Immunol. 1945, 50, 91–99. [Google Scholar]
- Foley, E.J. Antigenic Properties of Methylcholanthrene-induced Tumors in Mice of the Strain of Origin. Cancer Res. 1953, 13, 835–837. [Google Scholar]
- Klein, G.; Sjogren, H.O.; Klein, E.; Hellstrom, K.E. Demonstration of resistance against methylcholanthrene-induced sarcomas in the primary autochthonous host. Cancer Res. 1960, 20, 1561–1572. [Google Scholar]
- Coulie, P.G.; Lehmann, F.; Lethé, B.; Herman, J.; Lurquin, C.; Andrawiss, M.; Boon, T. A mutated intron sequence codes for an antigenic peptide recognized by cytolytic T lymphocytes on a human melanoma. Proc. Natl. Acad. Sci. USA 1995, 92, 7976–7980. [Google Scholar] [CrossRef]
- Wölfel, T.; Hauer, M.; Schneider, J.; Serrano, M.; Wölfel, C.; Klehmann-Hieb, E.; De Plaen, E.; Hankeln, T.; Zum Büschenfelde, K.-H.M.; Beach, D. A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science 1995, 269, 1281–1284. [Google Scholar] [CrossRef]
- Robbins, P.F.; El-Gamil, M.; Li, Y.F.; Kawakami, Y.; Loftus, D.; Appella, E.; Rosenberg, S.A. A mutated beta-catenin gene encodes a melanoma-specific antigen recognized by tumor infiltrating lymphocytes. J. Exp. Med. 1996, 183, 1185–1192. [Google Scholar] [CrossRef]
- Pieper, R.; Christian, R.E.; Gonzales, M.I.; Nishimura, M.I.; Gupta, G.; Settlage, R.E.; Shabanowitz, J.; Rosenberg, S.A.; Hunt, D.F.; Topalian, S.L. Biochemical identification of a mutated human melanoma antigen recognized by CD4(+) T cells. J. Exp. Med. 1999, 189, 757–766. [Google Scholar] [CrossRef]
- Shilyansky, J.; Nishimura, M.I.; Yannelli, J.R.; Kawakami, Y.; Jacknin, L.S.; Charmley, P.; Rosenberg, S.A. T-cell receptor usage by melanoma-specific clonal and highly oligoclonal tumor-infiltrating lymphocyte lines. Proc. Natl. Acad. Sci. USA 1994, 91, 2829–2833. [Google Scholar] [CrossRef]
- Nishimura, M.I.; Custer, M.C.; Schwarz, S.L.; Parker, L.L.; Mixon, A.; Clay, T.M.; Yanelli, J.R.; Rosenberg, S.A. T cell-receptor V gene use by CD4+ melanoma-reactive clonal and oligoclonal T-cell lines. J. Immunother. 1998, 21, 352–362. [Google Scholar] [CrossRef]
- Topalian, S.L.; Rivoltini, L.; Mancini, M.; Ng, J.; Hartzman, R.J.; Rosenberg, S.A. Melanoma-specific CD4+ T lymphocytes recognize human melanoma antigens processed and presented by Epstein-Barr virus-transformed B cells. Int. J. Cancer 1994, 58, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Veatch, J.R.; Lee, S.M.; Fitzgibbon, M.; Chow, I.T.; Jesernig, B.; Schmitt, T.; Kong, Y.Y.; Kargl, J.; Houghton, A.M.; Thompson, J.A.; et al. Tumor-infiltrating BRAFV600E-specific CD4+ T cells correlated with complete clinical response in melanoma. J. Clin. Investig. 2018, 128, 1563–1568. [Google Scholar] [CrossRef] [PubMed]
- Veatch, J.R.; Jesernig, B.L.; Kargl, J.; Fitzgibbon, M.; Lee, S.M.; Baik, C.; Martins, R.; Houghton, A.M.; Riddell, S.R. Endogenous CD4(+) T Cells Recognize Neoantigens in Lung Cancer Patients, Including Recurrent Oncogenic KRAS and ERBB2 (Her2) Driver Mutations. Cancer Immunol. Res. 2019, 7, 910–922. [Google Scholar] [CrossRef]
- Van der Lee, D.I.; Reijmers, R.M.; Honders, M.W.; Hagedoorn, R.S.; de Jong, R.C.; Kester, M.G.; van de Steen, D.M.; de Ru, A.H.; Kweekel, C.; Bijen, H.M. Mutated nucleophosmin 1 as immunotherapy target in acute myeloid leukemia. J. Clin. Investig. 2019, 129, 774–785. [Google Scholar] [CrossRef] [PubMed]
- Parkhurst, M.; Gros, A.; Pasetto, A.; Prickett, T.; Crystal, J.S.; Robbins, P.; Rosenberg, S.A. Isolation of T-Cell Receptors Specifically Reactive with Mutated Tumor-Associated Antigens from Tumor-Infiltrating Lymphocytes Based on CD137 Expression. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 2491–2505. [Google Scholar] [CrossRef] [PubMed]
- Paria, B.C.; Levin, N.; Lowery, F.J.; Pasetto, A.; Deniger, D.C.; Parkhurst, M.R.; Yossef, R.; Kim, S.P.; Florentin, M.; Ngo, L.T.; et al. Rapid Identification and Evaluation of Neoantigen-reactive T-Cell Receptors From Single Cells. J. Immunother. 2021, 44, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.C.; Zheng, Z.; Robbins, P.F.; Tran, E.; Prickett, T.D.; Gartner, J.J.; Li, Y.F.; Ray, S.; Franco, Z.; Bliskovsky, V.; et al. An Efficient Single-Cell RNA-Seq Approach to Identify Neoantigen-Specific T Cell Receptors. Mol. Ther. J. Am. Soc. Gene Ther. 2018, 26, 379–389. [Google Scholar] [CrossRef]
- Liu, S.; Matsuzaki, J.; Wei, L.; Tsuji, T.; Battaglia, S.; Hu, Q.; Cortes, E.; Wong, L.; Yan, L.; Long, M.; et al. Efficient identification of neoantigen-specific T-cell responses in advanced human ovarian cancer. J. Immunother. Cancer 2019, 7, 156. [Google Scholar] [CrossRef]
- Inderberg, E.M.; Wälchli, S.; Myhre, M.R.; Trachsel, S.; Almåsbak, H.; Kvalheim, G.; Gaudernack, G. T cell therapy targeting a public neoantigen in microsatellite instable colon cancer reduces in vivo tumor growth. Oncoimmunology 2017, 6, e1302631. [Google Scholar] [CrossRef]
- Wang, Q.J.; Yu, Z.; Griffith, K.; Hanada, K.; Restifo, N.P.; Yang, J.C. Identification of T-cell Receptors Targeting KRAS-Mutated Human Tumors. Cancer Immunol. Res. 2016, 4, 204–214. [Google Scholar] [CrossRef]
- Dillard, P.; Casey, N.; Pollmann, S.; Vernhoff, P.; Gaudernack, G.; Kvalheim, G.; Wälchli, S.; Inderberg, E.M. Targeting KRAS mutations with HLA class II-restricted TCRs for the treatment of solid tumors. Oncoimmunology 2021, 10, 1936757. [Google Scholar] [CrossRef] [PubMed]
- Yossef, R.; Tran, E.; Deniger, D.C.; Gros, A.; Pasetto, A.; Parkhurst, M.R.; Gartner, J.J.; Prickett, T.D.; Cafri, G.; Robbins, P.F.; et al. Enhanced detection of neoantigen-reactive T cells targeting unique and shared oncogenes for personalized cancer immunotherapy. JCI Insight 2018, 3, e122467. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Gallagher, D.T.; Gowthaman, R.; Pierce, B.G.; Mariuzza, R.A. Structural basis for oligoclonal T cell recognition of a shared p53 cancer neoantigen. Nat. Commun. 2020, 11, 2908. [Google Scholar] [CrossRef]
- Jazirehi, A.R. Molecular Analysis of Elements of Melanoma Insensitivity to TCR-Engineered Adoptive Cell Therapy. Int. J. Mol. Sci. 2021, 22, 11726. [Google Scholar] [CrossRef] [PubMed]
- Seliger, B. Novel insights into the molecular mechanisms of HLA class I abnormalities. Cancer Immunol. Immunother. 2012, 61, 249–254. [Google Scholar] [CrossRef]
- Eshhar, Z.; Waks, T.; Gross, G. The emergence of T-bodies/CAR T cells. Cancer J. 2014, 20, 123–126. [Google Scholar] [CrossRef]
- Sermer, D.; Brentjens, R. CAR T-cell therapy: Full speed ahead. Hematol. Oncol. 2019, 37, 95–100. [Google Scholar] [CrossRef]
- Almåsbak, H.; Aarvak, T.; Vemuri, M.C. CAR T Cell Therapy: A Game Changer in Cancer Treatment. J. Immunol. Res. 2016, 2016, 5474602. [Google Scholar] [CrossRef]
- Feins, S.; Kong, W.; Williams, E.F.; Milone, M.C.; Fraietta, J.A. An introduction to chimeric antigen receptor (CAR) T-cell immunotherapy for human cancer. Am. J. Hematol. 2019, 94, S3–S9. [Google Scholar] [CrossRef]
- Branella, G.M.; Spencer, H.T. Natural Receptor- and Ligand-Based Chimeric Antigen Receptors: Strategies Using Natural Ligands and Receptors for Targeted Cell Killing. Cells 2021, 11, 21. [Google Scholar] [CrossRef]
- Sadelain, M.; Brentjens, R.; Riviere, I. The promise and potential pitfalls of chimeric antigen receptors. Curr. Opin. Immunol. 2009, 21, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Veillette, A.; Bookman, M.A.; Horak, E.M.; Bolen, J.B. The CD4 and CD8 T cell surface antigens are associated with the internal membrane tyrosine-protein kinase p56lck. Cell 1988, 55, 301–308. [Google Scholar] [CrossRef]
- Sadelain, M.; Brentjens, R.; Riviere, I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013, 3, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Van der Stegen, S.J.; Hamieh, M.; Sadelain, M. The pharmacology of second-generation chimeric antigen receptors. Nat. Rev. Drug Discov. 2015, 14, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Huang, J.; Ma, W.; Yang, W.; Hu, B. The Antitumor Activity of CAR-T-PD1 Cells Enhanced by HPV16mE7-Pulsed and SOCS1-Silenced DCs in Cervical Cancer Models. Cancer Manag. Res. 2021, 13, 6045–6053. [Google Scholar] [CrossRef]
- Weinkove, R.; George, P.; Dasyam, N.; McLellan, A.D. Selecting costimulatory domains for chimeric antigen receptors: Functional and clinical considerations. Clin. Transl. Immunol. 2019, 8, e1049. [Google Scholar] [CrossRef]
- Duong, C.P.; Westwood, J.A.; Yong, C.S.; Murphy, A.; Devaud, C.; John, L.B.; Darcy, P.K.; Kershaw, M.H. Engineering T cell function using chimeric antigen receptors identified using a DNA library approach. PLoS ONE 2013, 8, e63037. [Google Scholar] [CrossRef]
- Wenthe, J.; Naseri, S.; Labani-Motlagh, A.; Enblad, G.; Wikström, K.I.; Eriksson, E.; Loskog, A.; Lövgren, T. Boosting CAR T-cell responses in lymphoma by simultaneous targeting of CD40/4–1BB using oncolytic viral gene therapy. Cancer Immunol. Immunother. 2021, 70, 2851–2865. [Google Scholar] [CrossRef]
- Brentjens, R.J.; Santos, E.; Nikhamin, Y.; Yeh, R.; Matsushita, M.; La Perle, K.; Quintás-Cardama, A.; Larson, S.M.; Sadelain, M. Genetically targeted T cells eradicate systemic acute lymphoblastic leukemia xenografts. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2007, 13, 5426–5435. [Google Scholar] [CrossRef]
- Milone, M.C.; Fish, J.D.; Carpenito, C.; Carroll, R.G.; Binder, G.K.; Teachey, D.; Samantha, M.; Lakhal, M.; Gloss, B.; Danet-Desnoyers, G.; et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol. Ther. J. Am. Soc. Gene Ther. 2009, 17, 1453–1464. [Google Scholar] [CrossRef]
- Kochenderfer, J.N.; Feldman, S.A.; Zhao, Y.; Xu, H.; Black, M.A.; Morgan, R.A.; Wilson, W.; Rosenberg, S.A. Construction and preclinical evaluation of an anti-CD19 chimeric antigen receptor. J. Immunother. 2009, 32, 689–702. [Google Scholar] [CrossRef]
- Almåsbak, H.; Walseng, E.; Kristian, A.; Myhre, M.R.; Suso, E.M.; Munthe, L.A.; Andersen, J.; Wang, M.Y.; Kvalheim, G.; Gaudernack, G.; et al. Inclusion of an IgG1-Fc spacer abrogates efficacy of CD19 CAR T cells in a xenograft mouse model. Gene Ther. 2015, 22, 391–403. [Google Scholar] [CrossRef]
- Magnani, C.F.; Turazzi, N.; Benedicenti, F.; Calabria, A.; Tenderini, E.; Tettamanti, S.; Attianese, G.M.; Cooper, L.J.; Aiuti, A.; Montini, E.; et al. Immunotherapy of acute leukemia by chimeric antigen receptor-modified lymphocytes using an improved Sleeping Beauty transposon platform. Oncotarget 2016, 7, 51581–51597. [Google Scholar] [CrossRef]
- Simonetta, F.; Alam, I.S.; Lohmeyer, J.K.; Sahaf, B.; Good, Z.; Chen, W.; Xiao, Z.; Hirai, T.; Scheller, L.; Engels, P.; et al. Molecular Imaging of Chimeric Antigen Receptor T Cells by ICOS-ImmunoPET. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020, 136, 5–6. [Google Scholar] [CrossRef]
- Frigault, M.J.; Lee, J.; Basil, M.C.; Carpenito, C.; Motohashi, S.; Scholler, J.; Kawalekar, O.U.; Guedan, S.; McGettigan, S.E.; Posey, A.D.; et al. Identification of chimeric antigen receptors that mediate constitutive or inducible proliferation of T cells. Cancer Immunol. Res. 2015, 3, 356–367. [Google Scholar] [CrossRef]
- Toulouie, S.; Johanning, G.; Shi, Y. Chimeric antigen receptor T-cell immunotherapy in breast cancer: Development and challenges. J. Cancer 2021, 12, 1212–1219. [Google Scholar] [CrossRef]
- Guedan, S.; Posey, A.D., Jr.; Shaw, C.; Wing, A.; Da, T.; Patel, P.R.; McGettigan, S.E.; Casado-Medrano, V.; Kawalekar, O.U.; Uribe-Herranz, M.; et al. Enhancing CAR T cell persistence through ICOS and 4–1BB costimulation. JCI Insight 2018, 3, e96976. [Google Scholar] [CrossRef]
- Goff, S.L.; Morgan, R.A.; Yang, J.C.; Sherry, R.M.; Robbins, P.F.; Restifo, N.P.; Feldman, S.A.; Lu, Y.C.; Lu, L.; Zheng, Z.; et al. Pilot Trial of Adoptive Transfer of Chimeric Antigen Receptor-transduced T Cells Targeting EGFRvIII in Patients with Glioblastoma. J. Immunother. 2019, 42, 126–135. [Google Scholar] [CrossRef]
- Morgan, R.A.; Johnson, L.A.; Davis, J.L.; Zheng, Z.; Woolard, K.D.; Reap, E.A.; Feldman, S.A.; Chinnasamy, N.; Kuan, C.T.; Song, H.; et al. Recognition of glioma stem cells by genetically modified T cells targeting EGFRvIII and development of adoptive cell therapy for glioma. Hum. Gene Ther. 2012, 23, 1043–1053. [Google Scholar] [CrossRef]
- Zhong, X.S.; Matsushita, M.; Plotkin, J.; Riviere, I.; Sadelain, M. Chimeric antigen receptors combining 4–1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Mol. Ther. J. Am. Soc. Gene Ther. 2010, 18, 413–420. [Google Scholar] [CrossRef]
- Long, A.H.; Haso, W.M.; Shern, J.F.; Wanhainen, K.M.; Murgai, M.; Ingaramo, M.; Smith, J.P.; Walker, A.J.; Kohler, M.E.; Venkateshwara, V.R.; et al. 4–1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med. 2015, 21, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Savoldo, B.; Ramos, C.A.; Liu, E.; Mims, M.P.; Keating, M.J.; Carrum, G.; Kamble, R.T.; Bollard, C.M.; Gee, A.P.; Mei, Z.; et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J. Clin. Investig. 2011, 121, 1822–1826. [Google Scholar] [CrossRef] [PubMed]
- Wutti-In, Y.; Sujjitjoon, J.; Sawasdee, N.; Panya, A.; Kongkla, K.; Yuti, P.; Yongpitakwattana, P.; Thepmalee, C.; Junking, M.; Chieochansin, T.; et al. Development of a Novel Anti-CD19 CAR Containing a Fully Human scFv and Three Costimulatory Domains. Front. Oncol. 2021, 11, 802876. [Google Scholar] [CrossRef] [PubMed]
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef]
- Kochenderfer, J.N.; Dudley, M.E.; Feldman, S.A.; Wilson, W.H.; Spaner, D.E.; Maric, I.; Stetler-Stevenson, M.; Phan, G.Q.; Hughes, M.S.; Sherry, R.M.; et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood 2012, 119, 2709–2720. [Google Scholar] [CrossRef]
- Kochenderfer, J.N.; Wilson, W.H.; Janik, J.E.; Dudley, M.E.; Stetler-Stevenson, M.; Feldman, S.A.; Maric, I.; Raffeld, M.; Nathan, D.A.; Lanier, B.J.; et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood 2010, 116, 4099–4102. [Google Scholar] [CrossRef]
- Shah, N.N.; Highfill, S.L.; Shalabi, H.; Yates, B.; Jin, J.; Wolters, P.L.; Ombrello, A.; Steinberg, S.M.; Martin, S.; Delbrook, C.; et al. CD4/CD8 T-Cell Selection Affects Chimeric Antigen Receptor (CAR) T-Cell Potency and Toxicity: Updated Results From a Phase I Anti-CD22 CAR T-Cell Trial. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020, 38, 1938–1950. [Google Scholar] [CrossRef]
- Rotiroti, M.C.; Buracchi, C.; Arcangeli, S.; Galimberti, S.; Valsecchi, M.G.; Perriello, V.M.; Rasko, T.; Alberti, G.; Magnani, C.F.; Cappuzello, C.; et al. Targeting CD33 in Chemoresistant AML Patient-Derived Xenografts by CAR-CIK Cells Modified with an Improved SB Transposon System. Mol. Ther. J. Am. Soc. Gene Ther. 2020, 28, 1974–1986. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, M.; Xiao, X.; Lv, H.; Jiang, Y.; Li, X.; Yuan, T.; Zhao, M. A combination of humanized anti-BCMA and murine anti-CD38 CAR-T cell therapy in patients with relapsed or refractory multiple myeloma. Leuk. Lymphoma 2022, 1–10. [Google Scholar] [CrossRef]
- Wang, X.; Urak, R.; Walter, M.; Guan, M.; Han, T.; Vyas, V.; Chien, S.; Gittins, B.; Clark, M.C.; Mokhtari, S.; et al. Large-scale manufacturing and characterization of CMV-CD19CAR T cells. J. Immunother. Cancer 2022, 10, e003461. [Google Scholar] [CrossRef]
- Kochenderfer, J.N.; Rosenberg, S.A. Treating B-cell cancer with T cells expressing anti-CD19 chimeric antigen receptors. Nat. Rev. Clin. Oncol. 2013, 10, 267–276. [Google Scholar] [CrossRef]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef]
- Brentjens, R.J.; Davila, M.L.; Riviere, I.; Park, J.; Wang, X.; Cowell, L.G.; Bartido, S.; Stefanski, J.; Taylor, C.; Olszewska, M.; et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci. Transl. Med. 2013, 5, 177ra38. [Google Scholar] [CrossRef]
- Kansagra, A.J.; Frey, N.V.; Bar, M.; Laetsch, T.W.; Carpenter, P.A.; Savani, B.N.; Heslop, H.E.; Bollard, C.M.; Perales, M.A.; Hudecek, M.; et al. Clinical Utilization of Chimeric Antigen Receptor T Cells in B Cell Acute Lymphoblastic Leukemia: An Expert Opinion from the European Society for Blood and Marrow Transplantation and the American Society for Blood and Marrow Transplantation. Biol. Blood Marrow Transplant. J. Am. Soc. Blood Marrow Transplant. 2019, 25, e76–e85. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, X.; Han, W.; Zhang, Y. Tisagenlecleucel, an approved anti-CD19 chimeric antigen receptor T-cell therapy for the treatment of leukemia. Drugs Today 2017, 53, 597–608. [Google Scholar] [CrossRef]
- O’Leary, M.C.; Lu, X.; Huang, Y.; Lin, X.; Mahmood, I.; Przepiorka, D.; Gavin, D.; Lee, S.; Liu, K.; George, B.; et al. FDA Approval Summary: Tisagenlecleucel for Treatment of Patients with Relapsed or Refractory B-cell Precursor Acute Lymphoblastic Leukemia. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 1142–1146. [Google Scholar] [CrossRef]
- Sharma, P.; Kanapuru, B.; George, B.; Lin, X.; Xu, Z.; Bryan, W.W.; Pazdur, R.; Theoret, M.R. FDA Approval Summary: Idecabtagene Vicleucel for Relapsed or Refractory Multiple Myeloma. Clin. Cancer Res. 2022. [Google Scholar] [CrossRef]
- Munshi, N.C.; Anderson, L.D.; Shah, N.; Madduri, D.; Berdeja, J.; Lonial, S.; Raje, N.; Lin, Y.; Siegel, D.; Oriol, A.; et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N. Engl. J. Med. 2021, 384, 705–716. [Google Scholar] [CrossRef]
- Raje, N.; Berdeja, J.; Lin, Y.; Siegel, D.; Jagannath, S.; Madduri, D.; Liedtke, M.; Rosenblatt, J.; Maus, M.V.; Turka, A.; et al. Anti-BCMA CAR T-Cell Therapy bb2121 in Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2019, 380, 1726–1737. [Google Scholar] [CrossRef]
- Abramson, J.S.; Palomba, M.L.; Gordon, L.I.; Lunning, M.A.; Wang, M.; Arnason, J.; Mehta, A.; Purev, E.; Maloney, D.G.; Andreadis, C.; et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): A multicentre seamless design study. Lancet 2020, 396, 839–852. [Google Scholar] [CrossRef]
- Wang, M.; Munoz, J.; Goy, A.; Locke, F.L.; Jacobson, C.A.; Hill, B.T.; Timmerman, J.M.; Holmes, H.; Jaglowski, S.; Flinn, I.W.; et al. KTE-X19 CAR T-Cell Therapy in Relapsed or Refractory Mantle-Cell Lymphoma. N. Engl. J. Med. 2020, 382, 1331–1342. [Google Scholar] [CrossRef]
- Newick, K.; O’Brien, S.; Moon, E.; Albelda, S.M. CAR T Cell Therapy for Solid Tumors. Annu. Rev. Med. 2017, 68, 139–152. [Google Scholar] [CrossRef]
- Krenciute, G.; Krebs, S.; Torres, D.; Wu, M.F.; Liu, H.; Dotti, G.; Li, X.N.; Lesniak, M.S.; Balyasnikova, I.V.; Gottschalk, S. Characterization and Functional Analysis of scFv-based Chimeric Antigen Receptors to Redirect T Cells to IL13Ralpha2-positive Glioma. Mol. Ther. J. Am. Soc. Gene Ther. 2016, 24, 354–363. [Google Scholar] [CrossRef]
- Ahmed, N.; Brawley, V.; Hegde, M.; Bielamowicz, K.; Kalra, M.; Landi, D.; Robertson, C.; Gray, T.L.; Diouf, O.; Wakefield, A.; et al. HER2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol. 2017, 3, 1094–1101. [Google Scholar] [CrossRef]
- Ahmed, N.; Brawley, V.S.; Hegde, M.; Robertson, C.; Ghazi, A.; Gerken, C.; Liu, E.; Dakhova, O.; Ashoori, A.; Corder, A.; et al. Human Epidermal Growth Factor Receptor 2 (HER2) -Specific Chimeric Antigen Receptor-Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 1688–1696. [Google Scholar] [CrossRef]
- Lo, A.S.; Ma, Q.; Liu, D.L.; Junghans, R.P. Anti-GD3 chimeric sFv-CD28/T-cell receptor zeta designer T cells for treatment of metastatic melanoma and other neuroectodermal tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010, 16, 2769–2780. [Google Scholar] [CrossRef]
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.E.; Badie, B.; Barish, M.E.; Weng, L.; Ostberg, J.R.; Chang, W.C.; Naranjo, A.; Starr, R.; Wagner, J.; Wright, C.; et al. Bioactivity and Safety of IL13Ralpha2-Redirected Chimeric Antigen Receptor CD8+ T Cells in Patients with Recurrent Glioblastoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 4062–4072. [Google Scholar] [CrossRef] [PubMed]
- Haas, A.R.; Tanyi, J.L.; O’Hara, M.H.; Gladney, W.L.; Lacey, S.F.; Torigian, D.A.; Soulen, M.C.; Tian, L.; McGarvey, M.; Nelson, A.M.; et al. Phase I Study of Lentiviral-Transduced Chimeric Antigen Receptor-Modified T Cells Recognizing Mesothelin in Advanced Solid Cancers. Mol. Ther. J. Am. Soc. Gene Ther. 2019, 27, 1919–1929. [Google Scholar] [CrossRef] [PubMed]
- Klampatsa, A.; Dimou, V.; Albelda, S.M. Mesothelin-targeted CAR-T cell therapy for solid tumors. Expert Opin. Biol. Ther. 2020, 21, 473–486. [Google Scholar] [CrossRef]
- Lamers, C.H.; Klaver, Y.; Gratama, J.W.; Sleijfer, S.; Debets, R. Treatment of metastatic renal cell carcinoma (mRCC) with CAIX CAR-engineered T-cells-a completed study overview. Biochem. Soc. Trans. 2016, 44, 951–959. [Google Scholar] [CrossRef]
- Lo, A.S.; Xu, C.; Murakami, A.; Marasco, W.A. Regression of established renal cell carcinoma in nude mice using lentivirus-transduced human T cells expressing a human anti-CAIX chimeric antigen receptor. Mol. Ther. Oncolytics 2014, 1, 14003. [Google Scholar] [CrossRef]
- Maher, J.; Wilkie, S.; Davies, D.M.; Arif, S.; Picco, G.; Julien, S.; Foster, J.; Burchell, J.M.; Taylor-Papadimitriou, J. Targeting of Tumor-Associated Glycoforms of MUC1 with CAR T Cells. Immunity 2016, 45, 945–946. [Google Scholar] [CrossRef]
- Posey, A.D., Jr.; Schwab, R.D.; Boesteanu, A.C.; Steentoft, C.; Mandel, U.; Engels, B.; Stone, J.D.; Madsen, T.D.; Schreiber, K.; Haines, K.M.; et al. Engineered CAR T Cells Targeting the Cancer-Associated Tn-Glycoform of the Membrane Mucin MUC1 Control Adenocarcinoma. Immunity 2016, 44, 1444–1454. [Google Scholar] [CrossRef]
- Supimon, K.; Sangsuwannukul, T.; Sujjitjoon, J.; Phanthaphol, N.; Chieochansin, T.; Poungvarin, N.; Chieochansin, T.; Poungvarin, N.; Wongkham, S.; Junking, M.; et al. Anti-mucin 1 chimeric antigen receptor T cells for adoptive T cell therapy of cholangiocarcinoma. Sci. Rep. 2021, 11, 6276. [Google Scholar] [CrossRef]
- Wilkie, S.; Picco, G.; Foster, J.; Davies, D.M.; Julien, S.; Cooper, L.; Arif, S.; Mather, S.J.; Taylor-Papadimitriou, J.; Burchell, J.M.; et al. Retargeting of human T cells to tumor-associated MUC1, the evolution of a chimeric antigen receptor. J. Immunol. 2008, 180, 4901–4909. [Google Scholar] [CrossRef]
- Wilkie, S.; van Schalkwyk, M.C.; Hobbs, S.; Davies, D.M.; van der Stegen, S.J.; Pereira, A.C.; Burbridge, S.E.; Box, C.; Eccles, S.A.; Maher, J. Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J. Clin. Immunol. 2012, 32, 1059–1070. [Google Scholar] [CrossRef]
- Budi, H.S.; Ahmad, F.N.; Achmad, H.; Ansari, M.J.; Mikhailova, M.V.; Suksatan, W.; Chupradit, S.; Shomali, N.; Marofi, F. Human epidermal growth factor receptor 2 (HER2)-specific chimeric antigen receptor (CAR) for tumor immunotherapy; recent progress. Stem Cell Res. Ther. 2022, 13, 40. [Google Scholar] [CrossRef]
- Brown, C.E.; Rodriguez, A.; Palmer, J.; Ostberg, J.R.; Naranjo, A.; Wagner, J.; Aguilar, B.; Starr, R.; Weng, L.; Synold, T.W.; et al. Off-the-shelf, Steroid Resistant, IL13Rα2-Specific CAR T Cells for Treatment of Glioblastoma. Neuro-Oncology 2022, noac024. [Google Scholar] [CrossRef]
- Xia, A.L.; Wang, X.C.; Lu, Y.J.; Lu, X.J.; Sun, B. Chimeric-antigen receptor T (CAR-T) cell therapy for solid tumors: Challenges and opportunities. Oncotarget 2017, 8, 90521–90531. [Google Scholar] [CrossRef]
- Brentjens, R.J.; Riviere, I.; Park, J.H.; Davila, M.L.; Wang, X.; Stefanski, J.; Taylor, C.; Yeh, R.; Bartido, S.; Borquez-Ojeda, O.; et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood 2011, 118, 4817–4828. [Google Scholar] [CrossRef] [PubMed]
- Brudno, J.N.; Kochenderfer, J.N. Recent advances in CAR T-cell toxicity: Mechanisms, manifestations and management. Blood Rev. 2019, 34, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Yang, X.F.; Xue, S.L.; Tan, J.W.; Li, M.H.; Ye, J.; Lou, X.Y.; Yu, Z.; Kang, L.Q.; Yan, Z.Q.; et al. Ruxolitinib reduces severe CRS response by suspending CAR-T cell function instead of damaging CAR-T cells. Biochem. Biophys. Res. Commun. 2022, 595, 54–61. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Trial Number and Title | Status | Phase | T-Cell Source | Location |
|---|---|---|---|---|
| NCT00338377: Lymphodepletion Plus Adoptive Cell Transfer with or without Dendritic Cell Immunization in Patients with Metastatic Melanoma | Not recruiting | II | TIL | Texas, United States |
| NCT00604136: Treatment of Metastatic Melanoma with Tumor Infiltrating Lymphocytes and IL-2 Following Lympho-Depleting Chemotherapy | Unknown | II | TIL | Israel |
| NCT01740557: Genetically Modified Therapeutic Autologous Lymphocytes Followed by Aldesleukin in Treating Patients with Stage III or Metastatic Melanoma | Not recruiting | I-II | Nerve Growth Factor Receptor and CXCR2 Transduced TIL | Texas, United States |
| NCT01883323: Tumor-Infiltrating Lymphocytes and Low-Dose Interleukin-2 Therapy Following Cyclophosphamide and Fludarabine in Patients with Melanoma | Completed | II | TIL | Ontario, Canada |
| NCT01946373: T Cell Transfer with or without Dendritic Cell Vaccination in Patients with Melanoma | Recruiting | I | TIL | Sweden |
| NCT02278887: Study Comparing TIL to Standard Ipilimumab in Patients with Metastatic Melanoma (TIL) | Recruiting | III | TIL | Denmark and Netherlands |
| NCT02354690: Vemurafenib and TIL Therapy for Metastatic Melanoma | Completed | I/II | TIL | Denmark |
| NCT02379195: Peginterferon and TIL Therapy for Metastatic Melanoma | Completed | I/II | TIL | Denmark |
| NCT02424916: Adoptive Transfer of Specific Melanoma Antigens CD8+ T Cells in Metastatic Melanoma Patients | Completed | I/II | Melan-A and MELO-1 Antigen Specific T Cells | France |
| NCT02959905: Treatment of Advanced Solid Tumor with TSA-CTL | Unknown | I | Tumor-Specific Antigen (TSA) Induced Cytotoxic T Lymphocytes | China |
| NCT02568748: Evaluation of Cytokine-induced Killer (CIK) Cells as Therapy or Adjuvant Treatment for Advanced HCC | Unknown | III | Cytokine-Induced Killer Cells | Egypt |
| NCT02498756: Cytokine-Induced Killer Study for Patients with Stage II Melanoma | Not yet recruiting | II | Cytokine-Induced Killer Cells | China |
| NCT00779337: Epstein-Barr Virus (EBV)-Specific T Cells as Therapy for Relapsed/Refractory EBV-Positive Lymphomas (EPL) | Completed | I | EBV-Specific Cytotoxic T Lymphocytes | Australia |
| NCT02408016: Genetically Modified T Cells in Treating Patients with Stage III-IV Non-Small Cell Lung Cancer or Mesothelioma | Terminated | I/II | WT-1 TCR Transduced PBL T Cells | Washington, United States |
| NCT02457650: T Cell Receptor-Transduced T Cells Targeting NY-ESO-1 for Treatment of Patients With NY-ESO-1- Expressing Malignancies | Unknown | I | NY-ESO-1 Specific TCR Transduced PBL T Cells | China |
| NCT02770820: Laboratory-Treated (Central Memory/Naive) CD8+ T Cells in Treating Patients with Newly Diagnosed or Relapsed Acute Myeloid Leukemia | Terminated | I/II | WT-1 TCR Transduced PBL T Cells | Washington, United States |
| NCT02774291: Anti-ESO mTCR-transduced Autologous Peripheral Blood Lymphocytes and Combination Chemotherapy in Treating Patients with Metastatic Cancer | Unknown | I | NY-ESO-1 Specific Murine TCR Transduced PBL T Cells | New York, United States |
| NCT02858310: E7 TCR T Cells for Human Papillomavirus-Associated Cancers | Recruiting | I/II | E7 Specific TCR Transduced PBL T Cells | Maryland, United States |
| NCT03354390: HERV-E TCR Transduced Autologous T Cells in People with Metastatic Clear Cell Renal Cell Carcinoma | Recruiting | I | HERV-E Specific TCR Transduced PBL T Cells | Maryland, United States |
| NCT00910650: Study of Gene Modified Immune Cells in Patients with Advanced Melanoma (F5) | Completed | II | MART-1 F5 TCR-Transduced PBL T Cells | California, United States |
| NCT01967823: T Cell Receptor Immunotherapy Targeting NY-ESO-1 for Patients with NY-ESO-1 Expressing Cancer | Completed | II | NY-ESO-1 Specific TCR Transduced PBL T Cells | Maryland, United States |
| NCT02096614: Investigator Initiated Phase 1 Study of TBI-1201 | Completed | I | MAGE A4-Specific TCR Transduced PBL T Cells | Japan |
| NCT02111850: T Cell Receptor Immunotherapy Targeting MAGE-A3 for Patients with Metastatic Cancer Who Are HLA-DP0401 Positive | Completed | I/II | MAGE A3-Specific TCR Transduced PBL T Cells | Maryland, United States |
| NCT02830724: Administering Peripheral Blood Lymphocytes Transduced with a CD70-Binding Chimeric Antigen Receptor to People with CD70 Expressing Cancers | Recruiting | I/II | CD70-Specific CAR Transduced PBL T Cells | Maryland, United States |
| NCT03851146: A Study of Anti-Lewis Y Chimeric Antigen Receptor-T Cells (LeY-CAR-T) in Patients with Solid Tumours (LeY-CAR-T) | Not yet recruiting | I | Lewis Y-Specific CAR Transduced PBL T Cells | Australia |
| NCT05063682: The Efficacy and Safety of Brain-Targeting Immune Cells (EGFRvIII-CAR T Cells) in Treating Patients with Leptomeningeal Disease From Glioblastoma. Administering Patients EGFRvIII -CAR T Cells May Help to Recognize and Destroy Brain Tumor Cells in Patients (CARTREMENDOUS) | Not yet recruiting | I | EGFRvIII-Specific 4-1BB CAR Transduced PBL T Cells | Finland and India |
| NCT04206943: Study of CD19 Specific Chimeric Antigen Receptor Positive T Cells (CAR-T) in ALL and NHL (ISIKOK-19) | Unknown | I/II | CD19-Specific CAR Transduced PBL T Cells | Turkey |
| NCT03937544: Intravenous Autologous CD19 CAR-T Cells for R/R B-ALL | Recruiting | II/III | CD19-Specific CAR Transduced PBL T Cells | Malaysia |
| NCT02482532: Vaccine Enriched, Autologous, Activated T-Cells Directed to Tumor in Patients with Relapsed/Refractory Melanoma | Completed | I | GD2-CAR Transduced PBL T Cells | Kansas, United States |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quinn, S.; Lenart, N.; Dronzek, V.; Scurti, G.M.; Hossain, N.M.; Nishimura, M.I. Genetic Modification of T Cells for the Immunotherapy of Cancer. Vaccines 2022, 10, 457. https://doi.org/10.3390/vaccines10030457
Quinn S, Lenart N, Dronzek V, Scurti GM, Hossain NM, Nishimura MI. Genetic Modification of T Cells for the Immunotherapy of Cancer. Vaccines. 2022; 10(3):457. https://doi.org/10.3390/vaccines10030457
Chicago/Turabian StyleQuinn, Suzanne, Natasha Lenart, Victoria Dronzek, Gina M. Scurti, Nasheed M. Hossain, and Michael I. Nishimura. 2022. "Genetic Modification of T Cells for the Immunotherapy of Cancer" Vaccines 10, no. 3: 457. https://doi.org/10.3390/vaccines10030457
APA StyleQuinn, S., Lenart, N., Dronzek, V., Scurti, G. M., Hossain, N. M., & Nishimura, M. I. (2022). Genetic Modification of T Cells for the Immunotherapy of Cancer. Vaccines, 10(3), 457. https://doi.org/10.3390/vaccines10030457