
High-Titer Hepatitis C Virus Production in a Scalable Single-Use High Cell Density Bioreactor

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Maintenance of Huh7.5 Cells
2.2. HCV Virus Stocks
2.3. CelCradle™ Culture and Virus Production
2.3.1. Cell Seed, Attachment, and Expansion in the CelCradle™
2.3.2. HCV Infection of the CelCradle™ Culture in SCM
2.3.3. Virus Harvests under SF Culture Conditions
2.3.4. Maintenance and Monitoring of the CelCradle™ Cultures and Evaluation of Spread of Infection
2.4. Shake Flask Cell Cultures
2.5. Immunostaining of HCV NS5A
2.6. HCV Infectivity Titers, RNA Titers, and Core Titers
2.7. HCV Neutralization Assays
2.8. Equilibrium Density Gradient Centrifugation
2.9. Virus Genome Sequencing
3. Results
3.1. Attachment and Expansion of Huh7.5 Cells Cultured on BioNOCII™ Carriers
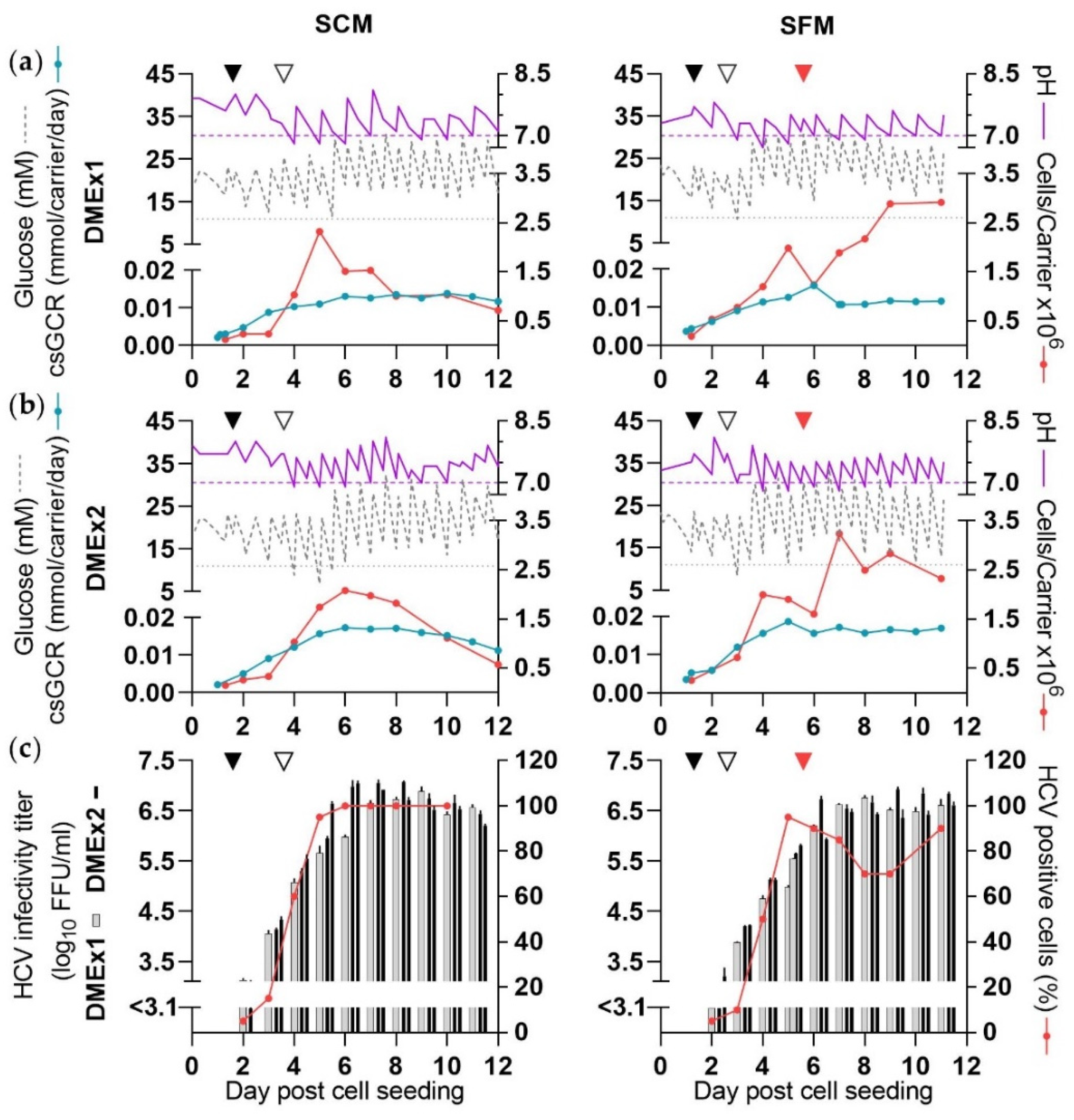
3.2. Production of HCV in SCM (CC-DMEx1-SCM)
3.3. Production of HCV in SFM (CC-DMEx1-SFM1)
3.4. Increased Cell Numbers Did Not Enhance HCV Production under SF Conditions (CC*-DMEx2-SFM)
3.5. Evaluation of the Influence of DME Frequency on HCV Yield in Shake Flasks (ShF-DMEx1-SCM, ShF-DMEx2-SCM, ShF-DMEx1-SFM, ShF-DMEx2-SFM)
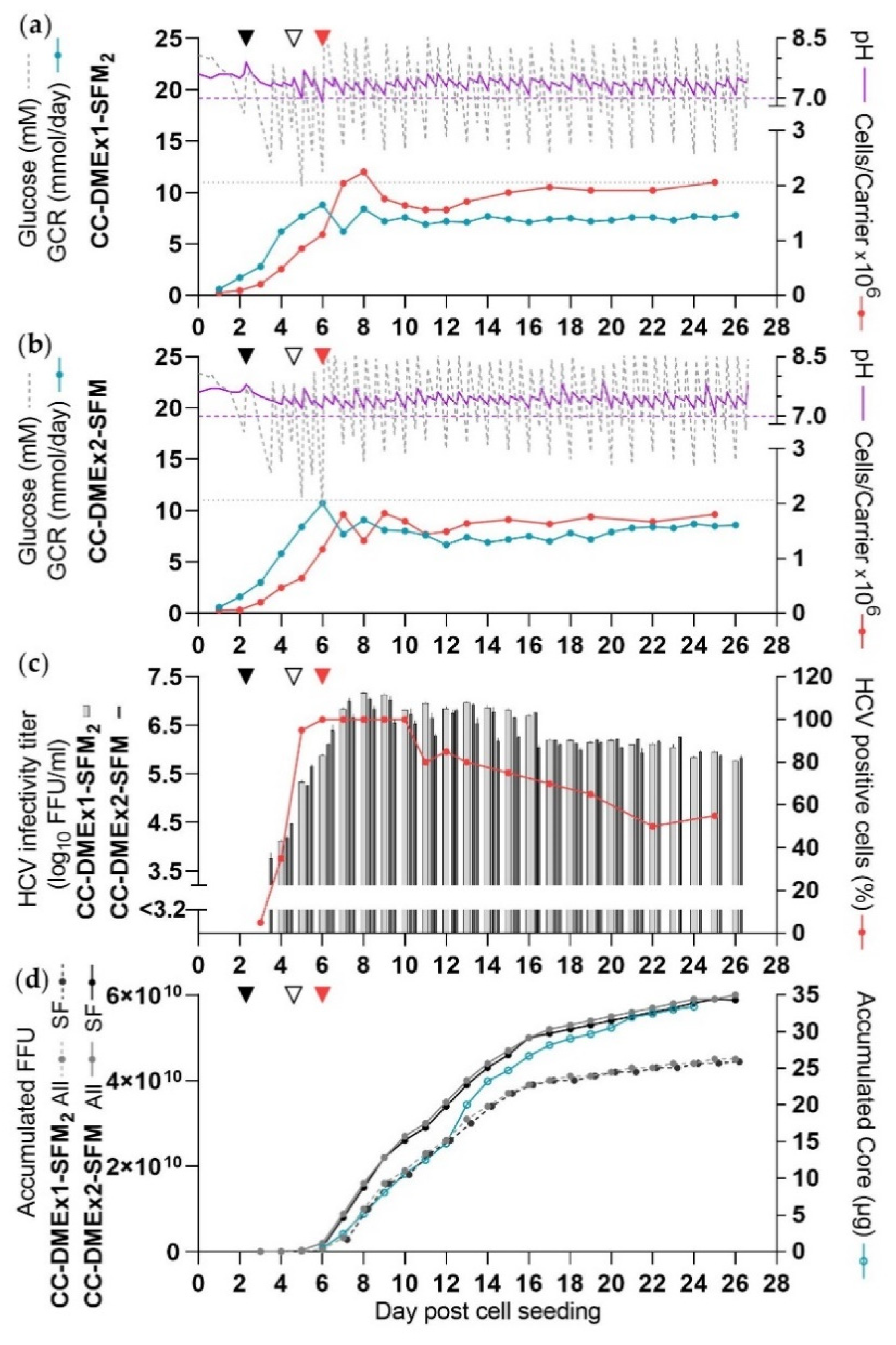
3.6. Two DME Improved HCV Yields in SFM (CC-DMEx1-SFM2, CC-DMEx2-SFM)
3.7. Selected Characteristics Were Similar for HCV Produced in the CelCradle™ and in Monolayer Cell Cultures
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dubuisson, J.; Cosset, F.-L. Virology and cell biology of the hepatitis C virus life cycle–An update. J. Hepatol. 2014, 61, S3–S13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, D.B.; Bukh, J.; Kuiken, C.; Muerhoff, A.S.; Rice, C.M.; Stapleton, J.T.; Simmonds, P. Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: Updated criteria and genotype assignment web resource. Hepatology 2014, 59, 318–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bukh, J. The history of hepatitis C virus (HCV): Basic research reveals unique features in phylogeny, evolution and the viral life cycle with new perspectives for epidemic control. J. Hepatol. 2016, 65, S2–S21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borgia, S.M.; Hedskog, C.; Parhy, B.; Hyland, R.H.; Stamm, L.M.; Brainard, D.M.; Subramanian, M.G.; McHutchison, J.G.; Mo, H.; Svarovskaia, E.; et al. Identification of a Novel Hepatitis C Virus Genotype From Punjab, India: Expanding Classification of Hepatitis C Virus Into 8 Genotypes. J. Infect. Dis. 2018, 218, 1722–1729. [Google Scholar] [CrossRef] [Green Version]
- Confirmed HCV Genotypes/Subtypes (May 2019). Available online: https://talk.ictvonline.org/ictv_wikis/flaviviridae/w/sg_flavi/634/table-1---confirmed-hcv-genotypes-subtypes-may-2019 (accessed on 28 October 2021).
- Polaris Observatory HCV Collaborators. Global prevalence and genotype distribution of hepatitis C virus infection in 2015: A modelling study. Lancet Gastroenterol. Hepatol. 2017, 2, 161–176. [Google Scholar] [CrossRef] [Green Version]
- Bailey, J.R.; Barnes, E.; Cox, A.L. Approaches, Progress, and Challenges to Hepatitis C Vaccine Development. Gastroenterology 2019, 156, 418–430. [Google Scholar] [CrossRef] [Green Version]
- Micallef, J.M.; Kaldor, J.M.; Dore, G.J. Spontaneous viral clearance following acute hepatitis C infection: A systematic review of longitudinal studies. J. Viral Hepat. 2006, 13, 34–41. [Google Scholar] [CrossRef]
- Hepatitis C Fact Sheet. Available online: https://www.who.int/news-room/fact-sheets/detail/hepatitis-c (accessed on 28 October 2021).
- Thrift, A.P.; El-Serag, H.B.; Kanwal, A.P.T.H.B.E.-S.F. Global epidemiology and burden of HCV infection and HCV-related disease. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 122–132. [Google Scholar] [CrossRef]
- WHO. Global Hepatitis Report, 2017; WHO: Geneva, Switzerland, 2017. [Google Scholar]
- Sarrazin, C. The importance of resistance to direct antiviral drugs in HCV infection in clinical practice. J. Hepatol. 2016, 64, 486–504. [Google Scholar] [CrossRef]
- Page, K.; Melia, M.T.; Veenhuis, R.T.; Winter, M.; Rousseau, K.E.; Massaccesi, G.; Osburn, W.O.; Forman, M.; Thomas, E.; Thornton, K.; et al. Randomized Trial of a Vaccine Regimen to Prevent Chronic HCV Infection. N. Engl. J. Med. 2021, 384, 541–549. [Google Scholar] [CrossRef]
- Chen, F.; Nagy, K.; Chavez, D.; Willis, S.; McBride, R.; Giang, E.; Honda, A.; Bukh, J.; Ordoukhanian, P.; Zhu, J.; et al. Antibody Responses to Immunization With HCV Envelope Glycoproteins as a Baseline for B-Cell–Based Vaccine Development. Gastroenterology 2020, 158, 1058–1071.e1056. [Google Scholar] [CrossRef] [PubMed]
- Frey, S.E.; Houghton, M.; Coates, S.; Abrignani, S.; Chien, D.; Rosa, D.; Pileri, P.; Ray, R.; Di Bisceglie, A.M.; Rinella, P.; et al. Safety and immunogenicity of HCV E1E2 vaccine adjuvanted with MF59 administered to healthy adults. Vaccine 2010, 28, 6367–6373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plotkin, S.A. Correlates of Protection Induced by Vaccination. Clin. Vaccine Immunol. 2010, 17, 1055–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plotkin, S.A.; Plotkin, S.L. The development of vaccines: How the past led to the future. Nat. Rev. Genet. 2011, 9, 889–893. [Google Scholar] [CrossRef]
- Fauvelle, C.; Colpitts, C.; Keck, Z.-Y.; Pierce, B.; Foung, S.K.H.; Baumert, T.F. Hepatitis C virus vaccine candidates inducing protective neutralizing antibodies. Expert Rev. Vaccines 2016, 15, 1535–1544. [Google Scholar] [CrossRef]
- Akazawa, D.; Moriyama, M.; Yokokawa, H.; Omi, N.; Watanabe, N.; Date, T.; Morikawa, K.; Aizaki, H.; Ishii, K.; Kato, T.; et al. Neutralizing Antibodies Induced by Cell Culture–Derived Hepatitis C Virus Protect Against Infection in Mice. Gastroenterol. 2013, 145, 447–455.e4. [Google Scholar] [CrossRef]
- Kinchen, V.J.; Massaccesi, G.; Flyak, A.I.; Mankowski, M.; Colbert, M.D.; Osburn, W.O.; Ray, S.C.; Cox, A.; Jr, J.E.C.; Bailey, J.R. Plasma deconvolution identifies broadly neutralizing antibodies associated with hepatitis C virus clearance. J. Clin. Investig. 2019, 129, 4786–4796. [Google Scholar] [CrossRef]
- Colbert, M.D.; Flyak, A.I.; Ogega, C.O.; Kinchen, V.; Massaccesi, G.; Hernandez, M.; Davidson, E.; Doranz, B.J.; Cox, A.; Crowe, J.E.; et al. Broadly Neutralizing Antibodies Targeting New Sites of Vulnerability in Hepatitis C Virus E1E2. J. Virol. 2019, 93, 21. [Google Scholar] [CrossRef] [Green Version]
- Lindenbach, B.D.; Evans, M.J.; Syder, A.J.; Wölk, B.; Tellinghuisen, T.L.; Liu, C.C.; Maruyama, T.; Hynes, R.O.; Burton, D.R.; McKeating, J.A.; et al. Complete Replication of Hepatitis C Virus in Cell Culture. Science 2005, 309, 623–626. [Google Scholar] [CrossRef] [Green Version]
- Wakita, T.; Pietschmann, T.; Kato, T.; Date, T.; Miyamoto, M.; Zhao, Z.; Murthy, K.; Habermann, A.; Kräusslich, H.-G.; Mizokami, M.; et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 2005, 11, 791–796. [Google Scholar] [CrossRef] [Green Version]
- Gottwein, J.M.; Scheel, T.K.H.; Jensen, T.B.; Lademann, J.B.; Prentoe, J.C.; Knudsen, M.L.; Hoegh, A.M.; Bukh, J. Development and characterization of hepatitis C virus genotype 1-7 cell culture systems: Role of CD81 and scavenger receptor class B type I and effect of antiviral drugs. Hepatology 2009, 49, 364–377. [Google Scholar] [CrossRef] [PubMed]
- Scheel, T.K.H.; Gottwein, J.M.; Carlsen, T.H.R.; Li, Y.-P.; Jensen, T.B.; Spengler, U.; Weis, N.; Bukh, J. Efficient Culture Adaptation of Hepatitis C Virus Recombinants with Genotype-Specific Core-NS2 by Using Previously Identified Mutations. J. Virol. 2011, 85, 2891–2906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheel, T.; Gottwein, J.; Jensen, T.B.; Prentoe, J.; Hoegh, A.M.; Alter, H.J.; Eugen-Olsen, J.; Bukh, J. Development of JFH1-based cell culture systems for hepatitis C virus genotype 4a and evidence for cross-genotype neutralization. Proc. Natl. Acad. Sci. USA 2008, 105, 997–1002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, T.B.; Gottwein, J.; Scheel, T.; Hoegh, A.M.; Eugen-Olsen, J.; Bukh, J. Highly Efficient JFH1-Based Cell-Culture System for Hepatitis C Virus Genotype 5a: Failure of Homologous Neutralizing-Antibody Treatment to Control Infection. J. Infect. Dis. 2008, 198, 1756–1765. [Google Scholar] [CrossRef] [PubMed]
- Mathiesen, C.K.; Prentoe, J.; Meredith, L.W.; Jensen, T.B.; Krarup, H.; McKeating, J.A.; Gottwein, J.M.; Bukh, J. Adaptive Mutations Enhance Assembly and Cell-to-Cell Transmission of a High-Titer Hepatitis C Virus Genotype 5a Core-NS2 JFH1-Based Recombinant. J. Virol. 2015, 89, 7758–7775. [Google Scholar] [CrossRef] [Green Version]
- Lesch, H.P.; Valonen, P.; Karhinen, M. Evaluation of the Single-Use Fixed-Bed Bioreactors in Scalable Virus Production. Biotechnol. J. 2021, 16, 2000020. [Google Scholar] [CrossRef]
- Barrett, P.N.; Mundt, W.; Kistner, O.; Howard, M.K. Vero cell platform in vaccine production: Moving towards cell culture-based viral vaccines. Expert Rev. Vaccines 2009, 8, 607–618. [Google Scholar] [CrossRef]
- Merten, O.-W. Advances in cell culture: Anchorage dependence. Philos. Trans. R. Soc. B Biol. Sci. 2015, 370, 20140040. [Google Scholar] [CrossRef] [Green Version]
- Pihl, A.F.; Offersgaard, A.F.; Mathiesen, C.K.; Prentoe, J.; Fahnøe, U.; Krarup, H.; Bukh, J.; Gottwein, J.M. High density Huh7.5 cell hollow fiber bioreactor culture for high-yield production of hepatitis C virus and studies of antivirals. Sci. Rep. 2018, 8, 17505. [Google Scholar] [CrossRef] [Green Version]
- Toriniwa, H.; Komiya, T. Japanese encephalitis virus production in Vero cells with serum-free medium using a novel oscillating bioreactor. Biologicals 2007, 35, 221–226. [Google Scholar] [CrossRef]
- Chen, Y.-H.; Wu, J.-C.; Wang, K.-C.; Chiang, Y.-W.; Lai, C.-W.; Chung, Y.-C.; Hu, Y.-C. Baculovirus-mediated production of HDV-like particles in BHK cells using a novel oscillating bioreactor. J. Biotechnol. 2005, 118, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.-C.; Weng, T.-C.; Tseng, Y.-F.; Chiang, J.-R.; Lee, M.-S.; Hu, A.Y.-C. Evaluation of novel disposable bioreactors on pandemic influenza virus production. PLoS ONE 2019, 14, e0220803. [Google Scholar] [CrossRef] [Green Version]
- Offersgaard, A.; Hernandez, C.D.; Pihl, A.; Costa, R.; Venkatesan, N.; Lin, X.; Van Pham, L.; Feng, S.; Fahnøe, U.; Scheel, T.; et al. SARS-CoV-2 Production in a Scalable High Cell Density Bioreactor. Vaccines 2021, 9, 706. [Google Scholar] [CrossRef] [PubMed]
- Rhazi, H.; Safini, N.; Mikou, K.; Alhyane, M.; Tadlaoui, K.O.; Lin, X.; Venkatesan, N.P.; Elharrak, M. Production of small ruminant morbillivirus, rift valley fever virus and lumpy skin disease virus in CelCradle™ -500A bioreactors. BMC Veter-Res. 2021, 17, 93. [Google Scholar] [CrossRef]
- Esco VacciXcell. TideXcell The Gentle Giant of Adherent Bioprocessing. Available online: https://escovaccixcell.com/assets/central_download/document/9010289_Vaccixcell_Product%20Guide_A4_Brochure_vE_LR.pdf (accessed on 28 October 2021).
- Mathiesen, C.K.; Jensen, T.B.; Prentoe, J.; Krarup, H.; Nicosia, A.; Law, M.; Bukh, J.; Gottwein, J.M. Production and characterization of high-titer serum-free cell culture grown hepatitis C virus particles of genotype 1–6. Virology 2014, 458-459, 190–208. [Google Scholar] [CrossRef] [Green Version]
- Esco VacciXcell. CelCradle Operation Manual; Esco VacciXcell: Singapore, 2016. [Google Scholar]
- Gottwein, J.; Scheel, T.; Hoegh, A.M.; Lademann, J.B.; Eugen-Olsen, J.; Lisby, G.; Bukh, J. Robust Hepatitis C Genotype 3a Cell Culture Releasing Adapted Intergenotypic 3a/2a (S52/JFH1) Viruses. Gastroenterology 2007, 133, 1614–1626. [Google Scholar] [CrossRef]
- Scheel, T.K.; Gottwein, J.M.; Mikkelsen, L.S.; Jensen, T.B.; Bukh, J. Recombinant HCV Variants With NS5A From Genotypes 1–7 Have Different Sensitivities to an NS5A Inhibitor but Not Interferon-α. Gastroenterology 2011, 140, 1032–1042.e6. [Google Scholar] [CrossRef]
- Gottwein, J.M.; Scheel, T.K.H.; Callendret, B.; Li, Y.-P.; Eccleston, H.B.; Engle, R.E.; Govindarajan, S.; Satterfield, W.; Purcell, R.H.; Walker, C.M.; et al. Novel Infectious cDNA Clones of Hepatitis C Virus Genotype 3a (Strain S52) and 4a (Strain ED43): Genetic Analyses and In Vivo Pathogenesis Studies. J. Virol. 2010, 84, 5277–5293. [Google Scholar] [CrossRef] [Green Version]
- Serre, S.B.N.; Krarup, H.B.; Bukh, J.; Gottwein, J.M. Identification of Alpha Interferon-Induced Envelope Mutations of Hepatitis C Virus In Vitro Associated with Increased Viral Fitness and Interferon Resistance. J. Virol. 2013, 87, 12776–12793. [Google Scholar] [CrossRef] [Green Version]
- Prentoe, J.; Velázquez-Moctezuma, R.; Augestad, E.H.; Galli, A.; Wang, R.; Law, M.; Alter, H.; Bukh, J. Hypervariable region 1 and N-linked glycans of hepatitis C regulate virion neutralization by modulating envelope conformations. Proc. Natl. Acad. Sci. USA 2019, 116, 10039–10047. [Google Scholar] [CrossRef] [Green Version]
- Law, M.; Maruyama, T.; Lewis, J.T.; Giang, E.; Tarr, A.; Stamataki, Z.; Gastaminza, P.; Chisari, F.; Jones, I.M.; Fox, I.R.; et al. Broadly neutralizing antibodies protect against hepatitis C virus quasispecies challenge. Nat. Med. 2008, 14, 25–27. [Google Scholar] [CrossRef] [PubMed]
- Giang, E.; Dorner, M.; Prentoe, J.; Dreux, M.; Evans, M.; Bukh, J.; Rice, C.M.; Ploss, A.; Burton, D.R.; Law, M. Human broadly neutralizing antibodies to the envelope glycoprotein complex of hepatitis C virus. Proc. Natl. Acad. Sci. USA 2012, 109, 6205–6210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prentoe, J.; Jensen, T.B.; Meuleman, P.; Serre, S.B.N.; Scheel, T.; Leroux-Roels, G.; Gottwein, J.; Bukh, J. Hypervariable Region 1 Differentially Impacts Viability of Hepatitis C Virus Strains of Genotypes 1 to 6 and Impairs Virus Neutralization. J. Virol. 2010, 85, 2224–2234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, S.B.; Fahnøe, U.; Pham, L.V.; Serre, S.B.N.; Tang, Q.; Ghanem, L.; Pedersen, M.S.; Ramirez, S.; Humes, D.; Pihl, A.F.; et al. Evolutionary Pathways to Persistence of Highly Fit and Resistant Hepatitis C Virus Protease Inhibitor Escape Variants. Hepatology 2019, 70, 771–787. [Google Scholar] [CrossRef]
- Fahnoe, U.; Bukh, J. Full-Length Open Reading Frame Amplification of Hepatitis C Virus. Methods Mol. Biol. 2019, 1911, 85–91. [Google Scholar] [CrossRef]
- Falcon, V.; Acosta-Rivero, N.; González, S.; Dueñas-Carrera, S.; Martinez-Donato, G.; Menéndez, I.; Garateix, R.; Silva, J.A.; Acosta, E.; Kourı, J. Ultrastructural and biochemical basis for hepatitis C virus morphogenesis. Virus Genes 2017, 53, 151–164. [Google Scholar] [CrossRef]
- WHO. Annex 2 WHO Good Manufacturing Practices for Biological Products Replacement of Annex 1 of WHO Technical Report Series, No. 822; WHO: Geneva, Switzerland, 2016. [Google Scholar]
- Lehmann-Grube, F.; Groth, S.F.D.S. Influenza viruses in cell cultures. Arch. Virol. 1965, 17, 534–548. [Google Scholar] [CrossRef]
- Harcourt, J.; Tamin, A.; Lu, X.; Kamili, S.; Sakthivel, S.K.; Murray, J.; Queen, K.; Tao, Y.; Paden, C.R.; Zhang, J.; et al. Severe Acute Respiratory Syndrome Coronavirus 2 from Patient with Coronavirus Disease, United States. Emerg. Infect. Dis. 2020, 26, 1266–1273. [Google Scholar] [CrossRef]
- Zhong, J.; Gastaminza, P.; Chung, J.; Stamataki, Z.; Isogawa, M.; Cheng, G.; McKeating, J.; Chisari, F.V. Persistent Hepatitis C Virus Infection In Vitro: Coevolution of Virus andHost. J. Virol. 2006, 80, 11082–11093. [Google Scholar] [CrossRef] [Green Version]
- Tsai, P.; Lin, C.-C.; Sun, H.-Y.; Lee, J.-C.; Chang, T.-T.; Young, K.-C. Viral dynamics of persistent hepatitis C virus infection in high-sensitive reporter cells resemble patient’s viremia. J. Microbiol. Immunol. Infect. 2018, 51, 446–455. [Google Scholar] [CrossRef]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. China Novel Coronavirus Investigating and Research Team. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Esco VacciXcell. CelCradle-500 Technical Report XII Cultivation of Huh Cells. Available online: https://escovaccixcell.com/assets/downloadables/CelCradle-500%20Technical%20Report%20XII.pdf (accessed on 22 November 2021).
- Jardon, M.; Garnier, A. pH, pCO2, and Temperature Effect on R-Adenovirus Production. Biotechnol. Prog. 2003, 19, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Dill, V.; Ehret, J.; Zimmer, A.; Beer, M.; Eschbaumer, M. Cell Density Effects in Different Cell Culture Media and Their Impact on the Propagation of Foot-And-Mouth Disease Virus. Viruses 2019, 11, 511. [Google Scholar] [CrossRef] [Green Version]
- Dill, V.; Zimmer, A.; Beer, M.; Eschbaumer, M. Investigation of cell culture conditions for optimal foot-and-mouth disease virus production. BMC Biotechnol. 2019, 19, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vázquez-Ramírez, D.; Genzel, Y.; Jordan, I.; Sandig, V.; Reichl, U. High-cell-density cultivations to increase MVA virus production. Vaccine 2018, 36, 3124–3133. [Google Scholar] [CrossRef] [PubMed]
- Tapia, F.; Vázquez-Ramírez, D.; Genzel, Y.; Reichl, U. Bioreactors for high cell density and continuous multi-stage cultivations: Options for process intensification in cell culture-based viral vaccine production. Appl. Microbiol. Biotechnol. 2016, 100, 2121–2132. [Google Scholar] [CrossRef] [Green Version]
- Law, J.L.M.; Logan, M.; Wong, J.; Kundu, J.; Hockman, D.; Landi, A.; Chen, C.; Crawford, K.; Wininger, M.; Johnson, J.; et al. Role of the E2 Hypervariable Region (HVR1) in the Immunogenicity of a Recombinant Hepatitis C Virus Vaccine. J. Virol. 2018, 92, e02141-17. [Google Scholar] [CrossRef] [Green Version]
- Wrensch, F.; Crouchet, E.; Ligat, G.; Zeisel, M.B.; Keck, Z.-Y.; Foung, S.K.H.; Schuster, C.; Baumert, T.F. Hepatitis C Virus (HCV)–Apolipoprotein Interactions and Immune Evasion and Their Impact on HCV Vaccine Design. Front. Immunol. 2018, 9, 1436. [Google Scholar] [CrossRef]
- Tao, W.; Xu, C.; Ding, Q.; Li, R.; Xiang, Y.; Chung, J.; Zhong, J. A single point mutation in E2 enhances hepatitis C virus infectivity and alters lipoprotein association of viral particles. Virology 2009, 395, 67–76. [Google Scholar] [CrossRef]
- Grove, J.; Nielsen, S.; Zhong, J.; Bassendine, M.F.; Drummer, H.E.; Balfe, P.; McKeating, J. Identification of a Residue in Hepatitis C Virus E2 Glycoprotein That Determines Scavenger Receptor BI and CD81 Receptor Dependency and Sensitivity to Neutralizing Antibodies. J. Virol. 2008, 82, 12020–12029. [Google Scholar] [CrossRef] [Green Version]
- Sainz, B.; TenCate, V.; Uprichard, S.L. Three-dimensional Huh7 cell culture system for the study of Hepatitis C virus infection. Virol. J. 2009, 6, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, K.; Ishii, K.; Ishihara, Y.; Yoshizaki, S.; Tanaka, K.; Gotoh, Y.; Aizaki, H.; Kohara, M.; Yoshioka, H.; Mori, Y.; et al. Production of infectious hepatitis C virus particles in three-dimensional cultures of the cell line carrying the genome-length dicistronic viral RNA of genotype 1b. Virology 2006, 351, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Lothert, K.; Offersgaard, A.F.; Pihl, A.F.; Mathiesen, C.K.; Jensen, T.B.; Alzua, G.P.; Fahnøe, U.; Bukh, J.; Gottwein, J.M.; Wolff, M.W. Development of a downstream process for the production of an inactivated whole hepatitis C virus vaccine. Sci. Rep. 2020, 10, 16261. [Google Scholar] [CrossRef] [PubMed]
- Yokokawa, H.; Higashino, A.; Suzuki, S.; Moriyama, M.; Nakamura, N.; Suzuki, T.; Suzuki, R.; Ishii, K.; Kobiyama, K.; Ishii, K.; et al. Induction of humoural and cellular immunity by immunisation with HCV particle vaccine in a non-human primate model. Gut 2016, 67, 372–379. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experiment | Cells Only SCM | CC-DMEx1-SCM | CC-DMEx1-SFM1 | CC*-DMEx2-SFM | CC-DMEx1-SFM2 | CC-DMEx2-SFM |
|---|---|---|---|---|---|---|
| SF virus production a | na | no | yes | yes | yes | yes |
| DME/harvests per day | 1 | 1 | 1 | 2 | 1 b | 2 b |
| BioNOCII™ carriers, g | 5.5 | 5.5 | 5.5 | 10.7 | 5.5 | 5.5 |
| Peak cell numbers | ||||||
| Day of peak total cell number, dpcs | 9 | 8 | 8 | 12 | 8 | 7 |
| Peak total cell number | 1.6 × 109 | 0.96 × 109 | 1.2 × 109 | 3.6 × 109 | 1.8 × 109 | 1.5 × 109 |
| cells/mL | 3.2 × 106 | 1.9 × 106 | 2.4 × 106 | 7.1 × 106 | 3.6 × 106 | 2.9 × 106 |
| cells/carrier | 2.0 × 106 | 1.2 × 106 | 1.5 × 106 | 2.3 × 106 | 2.3 × 106 | 1.8 × 106 |
| Cell culture supernatant parameters | ||||||
| Peak GCR, mmol/day (dpcs) | 9 (8) | 10 (6) | 11 (7) | 23 (7) | 9 (6) | 11 (6) |
| Glucose range, mM | 5–24 | 10–25 | 11–25 | 5–29 | 11–27 | 10–27 |
| Lactate peak concentration, mM (dpcs) c | nd | nd | nd | 48 (7) | 37 (6) | 26 (6) |
| Glutamine range, mM c | nd | nd | nd | 1.7–4.3 | 2.1–3.6 | 2.7–3.6 |
| Ammonia range, mM c | nd | nd | nd | 0.3–1.0 | 0.3–1.0 | 0.3–1.0 |
| pH range | 6.8–8.3 | 6.9–7.7 | 6.9–8.3 | 6.9–7.9 | 6.9–7.9 | 7.1–7.9 |
| Virus yield | ||||||
| Total harvest volume, L d | na | 2.3 | 5.9 | 10.6 | 8.6 | 18.5 |
| Peak infectivity titer, log10 FFU/mL | na | 6.8 | 6.9 | 6.9 | 7.2 | 7.0 |
| Days with high- or moderate yield | na | 9–13 | 7–19 | 8–18 e | 7–25 | 6–26 f |
| Total accumulated FFU g | na | 0.81 × 1010 | 2.2 × 1010 | 1.7 × 1010 | 4.4 × 1010 | 5.9 × 1010 |
| CSVY, FFU/cell | na | 8.5 | 18.5 | 4.8 | 24.5 | 41.0 |
| Yield compared to CC-DMEx2-SFM h | na | 0.14 | 0.37 | 0.29 | 0.74 | 1.0 |
| Culture | ShF-DMEx1-SCM | ShF-DMEx2-SCM | ShF-DMEx1-SFM | ShF-DMEx2-SFM |
|---|---|---|---|---|
| SF virus production a | no | no | yes | yes |
| DME/harvests per day | 1 | 2 | 1 | 2 |
| BioNOCII™ carriers, g | 0.39 | 0.39 | 0.39 | 0.39 |
| Peak cell numbers | ||||
| Day of peak total cell number, dpcs | 5 | 6 | 9 | 7 |
| Peak total cell number | 1.1 × 108 | 0.94 × 108 | 0.95× 108 | 1.4 × 108 |
| cells/mL | 3.9 × 106 | 3.5 × 106 | 4.9 × 106 | 5.5 × 106 |
| cells/carrier | 2.3 × 106 | 2.1 × 106 | 2.9 × 106 | 3.2 × 106 |
| Cell culture supernatant | ||||
| Peak csGCR, mmol/carrier/day (dpcs) | 0.014 (6) | 0.017 (6) | 0.016 (6) | 0.019 (5) |
| Glucose range, mM | 12–31 | 7–32 | 11–32 | 9–34 |
| Lactate peak concentration, mM (dpcs) | 43 (8) | 33 (7) | 39 (5) | 36 (5) |
| Glutamine range, mM b | 1.2–3.6 | 1.8–4.0 | 1.7–3.6 | 2.0–3.6 |
| Ammonia range, mM b | 0.4–1.5 | 0.3–1.4 | 0.3–1.2 | 0.3–1.0 |
| pH range | 6.8–8.1 | 6.9–8.1 | 6.7–7.8 | 6.8–8.1 |
| Virus yield | ||||
| Peak infectivity titer, log10 FFU/mL | 6.9 | 7.1 | 6.8 | 6.9 |
| Days with high- or moderate yield | 7–12 c | 5–12 c | 7–11 c | 6–11 c |
| CSVY, FFU/cell | 4.8 | 20.0 | 4.7 | 7.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Offersgaard, A.; Duarte Hernandez, C.R.; Pihl, A.F.; Venkatesan, N.P.; Krarup, H.; Lin, X.; Reichl, U.; Bukh, J.; Genzel, Y.; Gottwein, J.M. High-Titer Hepatitis C Virus Production in a Scalable Single-Use High Cell Density Bioreactor. Vaccines 2022, 10, 249. https://doi.org/10.3390/vaccines10020249
Offersgaard A, Duarte Hernandez CR, Pihl AF, Venkatesan NP, Krarup H, Lin X, Reichl U, Bukh J, Genzel Y, Gottwein JM. High-Titer Hepatitis C Virus Production in a Scalable Single-Use High Cell Density Bioreactor. Vaccines. 2022; 10(2):249. https://doi.org/10.3390/vaccines10020249
Chicago/Turabian StyleOffersgaard, Anna, Carlos Rene Duarte Hernandez, Anne Finne Pihl, Nandini Prabhakar Venkatesan, Henrik Krarup, Xiangliang Lin, Udo Reichl, Jens Bukh, Yvonne Genzel, and Judith Margarete Gottwein. 2022. "High-Titer Hepatitis C Virus Production in a Scalable Single-Use High Cell Density Bioreactor" Vaccines 10, no. 2: 249. https://doi.org/10.3390/vaccines10020249
APA StyleOffersgaard, A., Duarte Hernandez, C. R., Pihl, A. F., Venkatesan, N. P., Krarup, H., Lin, X., Reichl, U., Bukh, J., Genzel, Y., & Gottwein, J. M. (2022). High-Titer Hepatitis C Virus Production in a Scalable Single-Use High Cell Density Bioreactor. Vaccines, 10(2), 249. https://doi.org/10.3390/vaccines10020249