
Oncolytic Vaccinia Virus in Lung Cancer Vaccines



Abstract
:1. Introduction
2. Cancer Vaccines in NSCLC
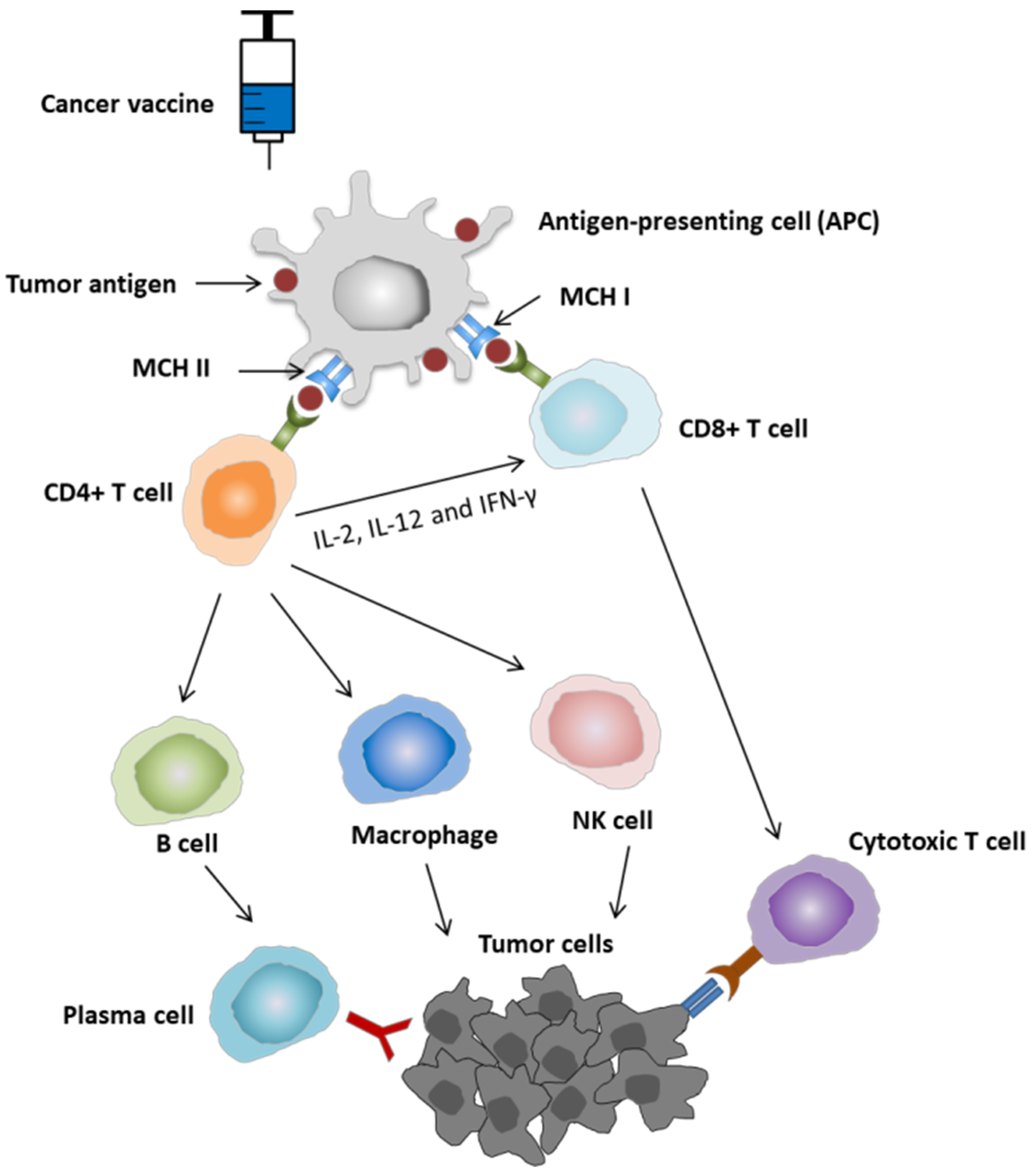
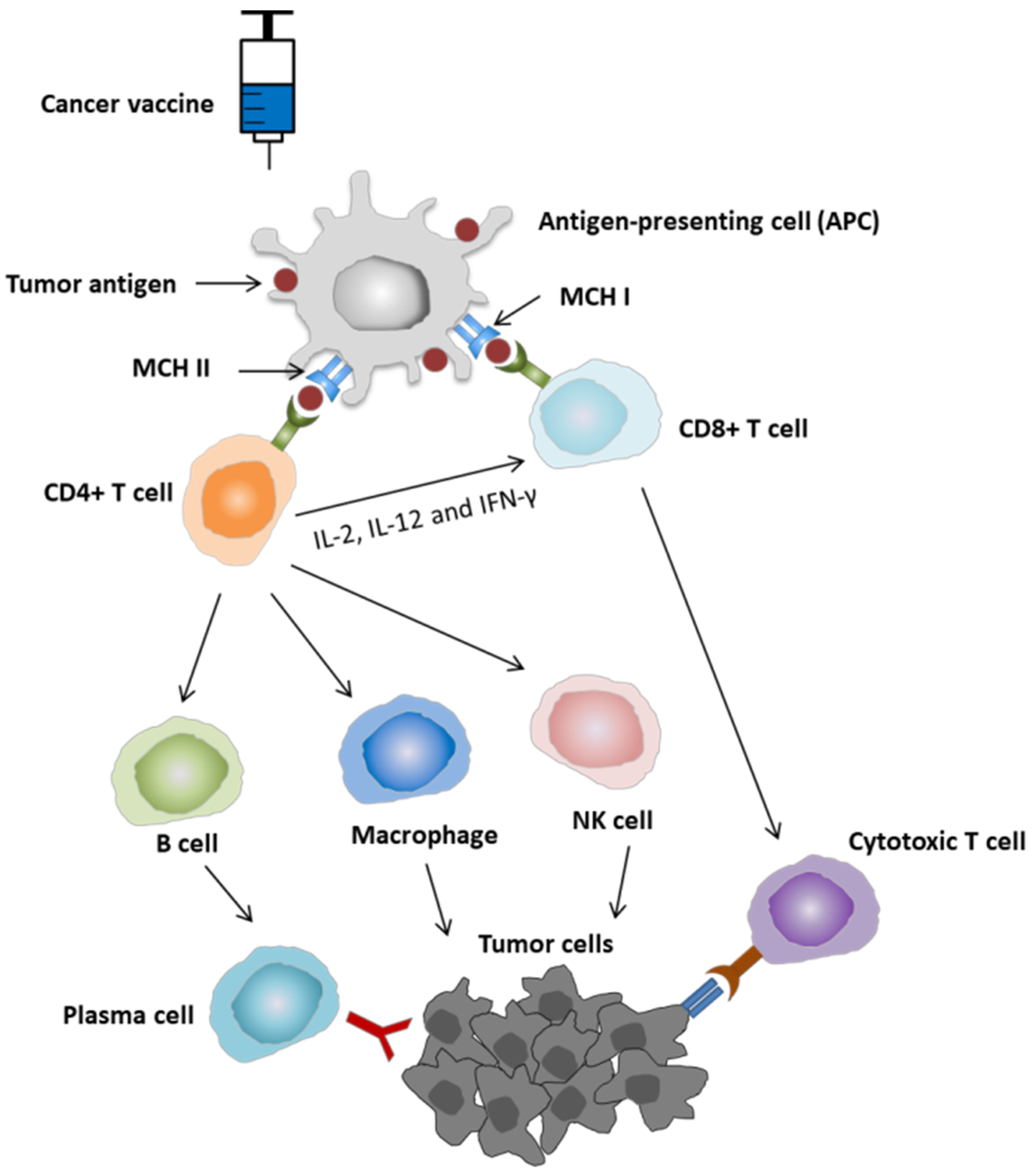
2.1. Mechanism of Action of Cancer Vaccine
2.2. Why Are Cancer Vaccines Needed in the Treatment of NSCLC?
2.3. Current Cancer Vaccines for NSCLC

{kind=link}
{kind=link}
{kind=link}
| Type of Vaccine | Vaccine | Tumor Stage | Phase Trial | Patients | Time | Results | Adverse Events (AEs) | Reference |
|---|---|---|---|---|---|---|---|---|
| Allogeneic vaccines | Belagenpumatucel-L | NSCLC II, IIIA, IIIB and IV | II | 75 | 2006 | Belagenpumatucel-L is well tolerated, and the survival advantage justifies further phase III evaluation. | Non-significant. | [12] |
| NSCLC IV | II | 21 | 2009 | Overall survival was 562 days. | Non-significant. | [13] | ||
| NSCLC III/IV | III | 532 | 2015 | No difference in survival between the arms. No differences in progression-free survival. | No serious AEs. | [30] | ||
| Autologous or allogeneic NSCLC cells plus GM.CD40L-expressing K562 cells | NSCLC IV | I | 21 | 2007 | There was no tumor regression after vaccination, but many patients had stable disease. | No toxicity. | [31] | |
| Refractory advanced stage | II | 24 | 2013 | The primary endpoint, inducing radiologic tumor regression, was not reached. Median OS was 7.9 months and median PFS was only 1.7 months. | Common toxicities were headache and site reaction. | [32] | ||
| Peptide or protein vaccines | CIMAvax-EGF | IIIB/IV | II | 80 | 2008 | Good anti-EGF antibody response was obtained in 51.3% of vaccinated patients. | Less than 25% of cases and were grade 1 or 2 | [34] |
| IIIB/IV | III | 405 | 2016 | Survival benefit was significant: Median survival time (MST) was 12.43 months for the vaccine arm versus 9.43 months for the control arm. MST was higher (14.66 months) for vaccinated patients with high EGF concentration at baseline. | Long-term vaccination is safe. Most frequent adverse reactions were grade 1 or 2 injection-site pain, fever, vomiting, and headache. | [35] | ||
| MAGE-A3 | IB, II, and IIIA MAGE-A3-positive NSCLC | III (MAGRIT) | 13.849 | 2016 | Adjuvant treatment with the MAGE-A3 immunotherapeutic did not increase disease-free survival. Further development of the MAGE-A3 immunotherapeutic for use in NSCLC has been stopped. | The most frequently reported grade 3 or higher adverse events were infections and infestations, vascular disorders, and neoplasm. | [38] | |
| Racotumomab (IE10) | NSCLC IIIB/IV | II/III | 176 | 2014 | Median progression-free survival (PFS) in vaccinated patients was 5.33 versus 3.90 months for placebo. | The most common adverse events in the racotumomab-alum arm were burning and pain at the injection site, bone pain, and asthenia. | [43] | |
| BLP25 liposome vaccine | NSCLC IIIB/IV | IIB | 171 | 2005 | The survival difference of 4.4 months observed with the vaccine did not reach statistical significance. | Non-significant. | [44] | |
| NSCLC III | III (START) | 1239 | 2014 | No significant difference in overall survival. | Serious adverse events with a greater than 2% frequency with tecemotide were dyspnea, metastases to central nervous system and pneumonia. | [45] | ||
| DNA vaccines | Elenagen | Advanced solid tumors | I/IIA | 27 | 2017 | Most of the patients achieved stable disease for at least 8 weeks. | No severe AEs. | [46] |
| Vector-based vaccines | TG4010 | Different solid tumors | I | 13 | 2003 | A total of 4 of the 13 patients achieved stable disease. One lung cancer patient who was initially progressing after the first injections later showed a marked decrease in the size of his metastases that lasted for 14 months. | Injection site pain and influenza-like symptoms. | [47] |
| NSCLC III/IV | II | 65 | 2008 | The median overall survival was 12.7 months for arm 1 (combined TG4010 with chemotherapy) and 14.9 for arm 2 (vaccine alone). | Mild–moderate injection site reactions, flu-like symptoms, and fatigue being the most frequent adverse reactions. | [48] | ||
| NSCLC IIIB/IV | IIB | 148 | 2011 | 6-month progression-free survival (PFS) was 43.2% in the TG4010 plus chemotherapy group, and 35.1% in the chemotherapy alone group | Common AEs include fever, abdominal pain, and injection-site pain. The most common grade 3–4 AEs were neutropenia, and fatigue. Anorexia and pleural effusion were grade 3–4 AEs that differed significantly between groups. | [49] | ||
| NSCLC IV | IIB/III | 222 | 2016 | The combination of TG4010 with chemotherapy seems to improve PFS relative to placebo plus chemotherapy. | No grade 3–4 or serious AEs deemed to be related to TG4010 only; 4 (4%) patients presented grade 3 or 4 AEs related to TG4010 and other study treatments. The most frequent severe AEs were neutropenia, anemia, and fatigue. | [50] |
3. Recent Applications of Vaccinia Virus for NSCLC Treatment
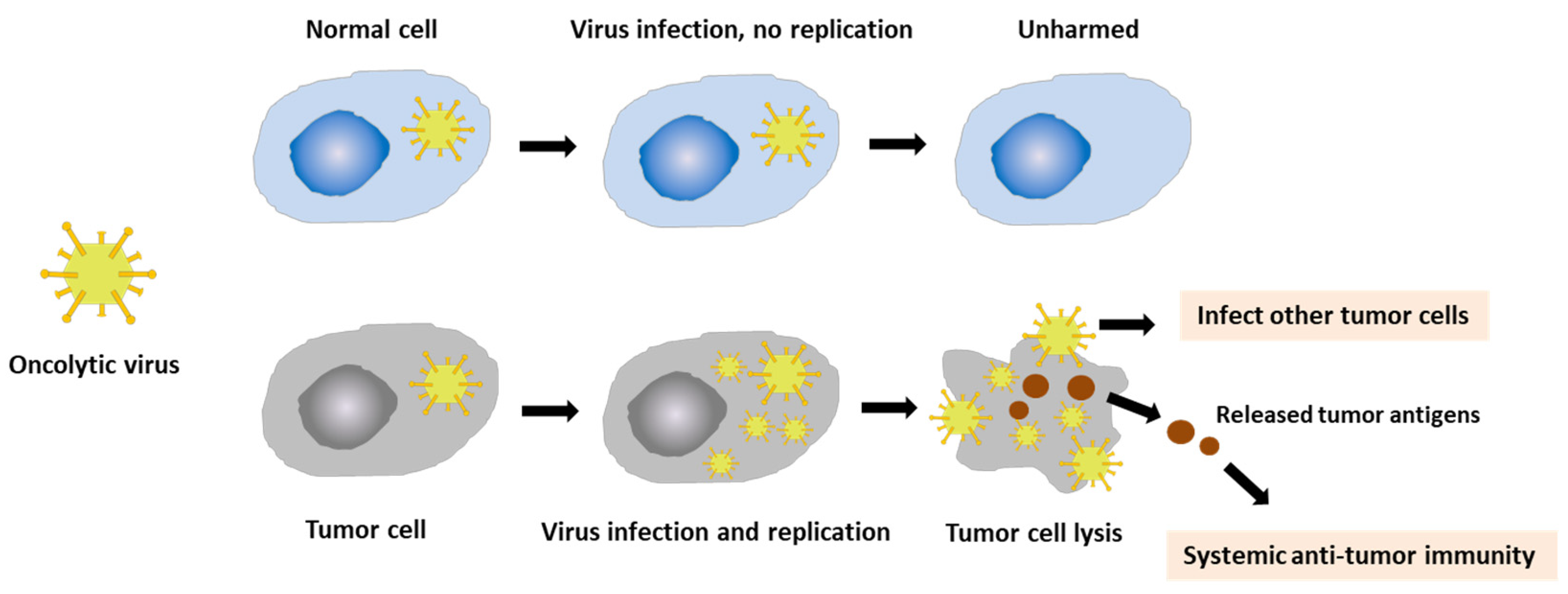
3.1. Vaccinia Virus
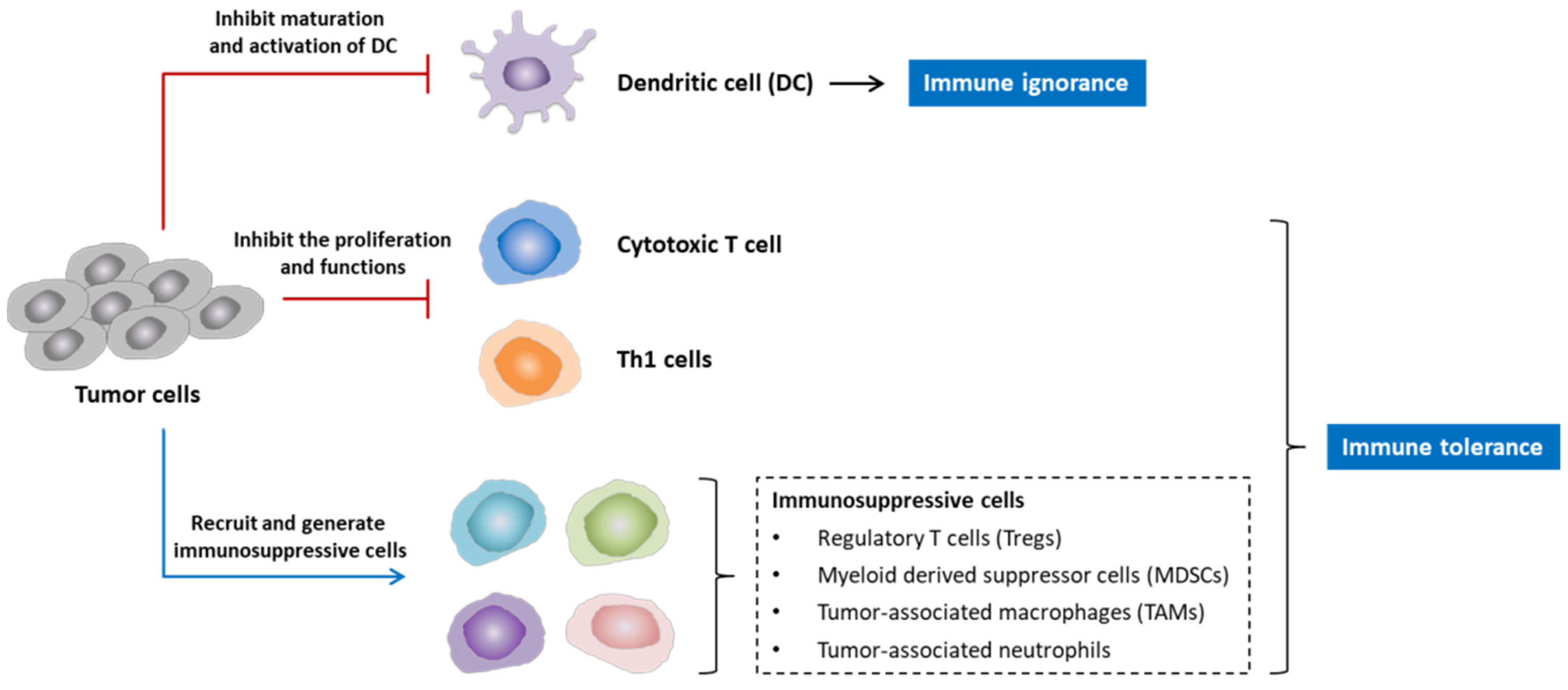
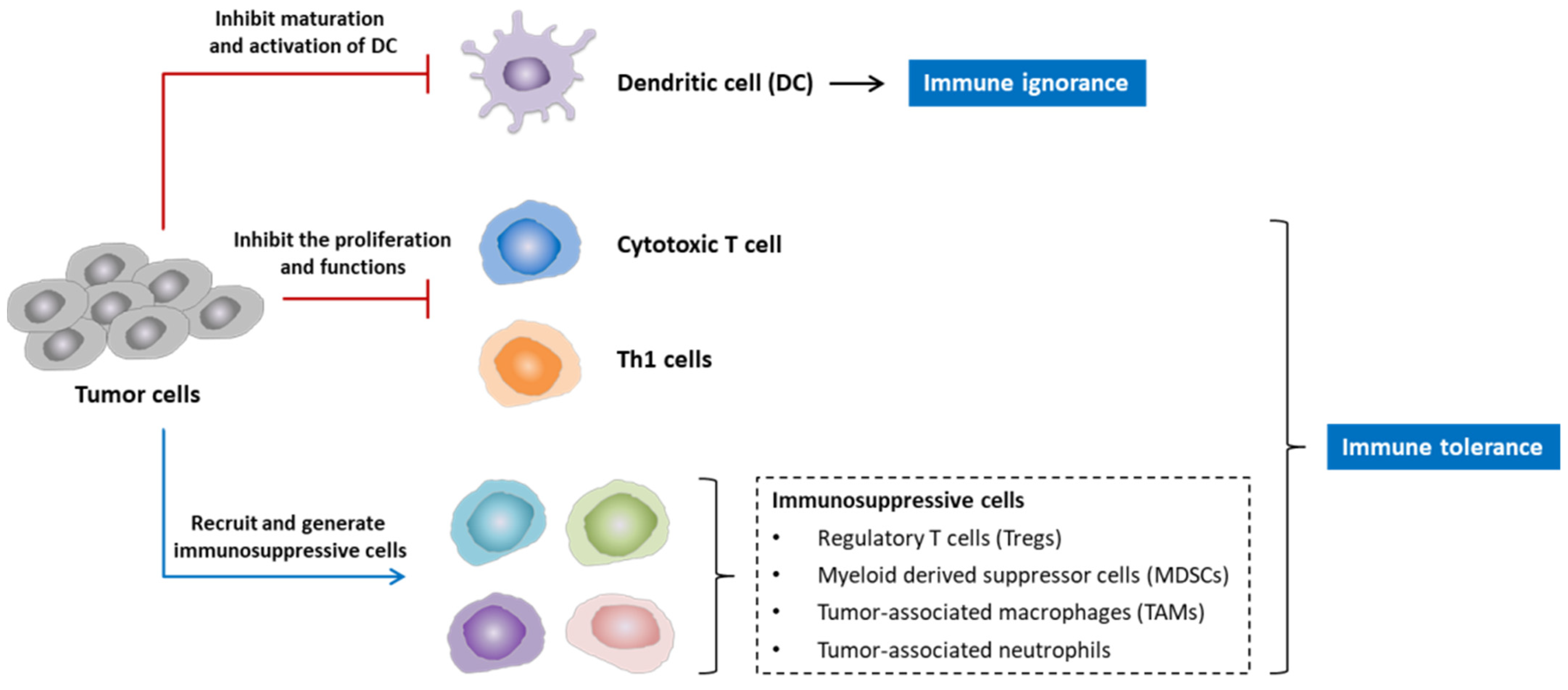
3.2. Vaccinia Virus for the Modulation of Tumor Microenvironment
3.3. Vaccinia Virus for Cancer Treatment
3.4. Vaccinia Virus for Cancer Vaccines in NSCLC
3.5. Adverse Effects of Vaccinia Virus in Cancer Treatment
3.6. Future Directions for Using Vaccinia Virus as Lung Cancer Vaccines
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Wang, C.; Long, Y.; Li, W.; Dai, W.; Xie, S.; Liu, Y.; Zhang, Y.; Liu, M.; Tian, Y.; Li, Q. Exploratory study on classification of lung cancer subtypes through a combined K-nearest neighbor classifier in breathomics. Sci. Rep. 2020, 10, 5880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, M.; Huang, L.-L.; Chen, J.-H.; Wu, J.; Xu, Q. The emerging treatment landscape of targeted therapy in non-small-cell lung cancer. Signal Transduct. Target. Ther. 2019, 4, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortés-Jofré, M.; Uranga, R.; Pombert, A.T.; Prado, M.d.C.A.; Aguirrechu, I.C.; Pacheco, C.; Reyes, R.M.O.; Chuecas, F.; Bermejo, P.I.M. Therapeutic vaccines for advanced non-small cell lung cancer. Cochrane Database Syst. Rev. 2019, 2019, CD013377. [Google Scholar] [CrossRef]
- Cáceres-Lavernia, H.H.; Nenínger-Vinageras, E.; Varona-Rodríguez, L.M.; Olivares-Romero, Y.A.; Sánchez-Rojas, I.; Mazorra-Herrera, Z.; Basanta-Bergolla, D.; Duvergel-Calderín, D.; Torres-Cuevas, B.L.; del Castillo-Carrillo, C. Racotumomab in Non-Small Cell Lung Cancer as Maintenance and Second-Line Treatment. MEDICC Rev. 2021, 23, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Losanno, T.; Gridelli, C. Recent advances in targeted advanced lung cancer therapy in the elderly. Expert Rev. Anticancer Ther. 2017, 17, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Hammerschmidt, S.; Wirtz, H. Lung cancer: Current diagnosis and treatment. Dtsch. Ärzteblatt Int. 2009, 106, 809. [Google Scholar]
- Stevens, D.; Ingels, J.; Van Lint, S.; Vandekerckhove, B.; Vermaelen, K. Dendritic Cell-Based Immunotherapy in Lung Cancer. Front. Immunol. 2020, 11, 620374. [Google Scholar] [CrossRef]
- Schirrmacher, V. Cancer Vaccines and Oncolytic Viruses Exert Profoundly Lower Side Effects in Cancer Patients than Other Systemic Therapies: A Comparative Analysis. Biomedicines 2020, 8, 61. [Google Scholar] [CrossRef] [Green Version]
- van der Hoorn, I.A.E.; Flórez-Grau, G.; van den Heuvel, M.M.; de Vries, I.J.M.; Piet, B. Recent Advances and Future Perspective of DC-Based Therapy in NSCLC. Front. Immunol. 2021, 12, 704776. [Google Scholar] [CrossRef]
- Badrinath, N.; Yoo, S.Y. Recent Advances in Cancer Stem Cell-Targeted Immunotherapy. Cancers 2019, 11, 310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cascone, T.; Fradette, J.; Pradhan, M.; Gibbons, D.L. Tumor Immunology and Immunotherapy of Non-Small-Cell Lung Cancer. Cold Spring Harb. Perspect. Med. 2021, 12, a037895. [Google Scholar] [CrossRef]
- Jung, C.Y.; Antonia, S.J. Tumor immunology and immune checkpoint inhibitors in non-small cell lung cancer. Tuberc. Respir. Dis. 2018, 81, 29–41. [Google Scholar] [CrossRef]
- Nemunaitis, J.; Dillman, R.O.; Schwarzenberger, P.O.; Senzer, N.; Cunningham, C.; Cutler, J.; Tong, A.; Kumar, P.; Pappen, B.; Hamilton, C. Phase II study of belagenpumatucel-L, a transforming growth factor beta-2 antisense gene-modified allogeneic tumor cell vaccine in non-small-cell lung cancer. J. Clin. Oncol. 2006, 24, 4721–4730. [Google Scholar] [CrossRef] [PubMed]
- Nemunaitis, J.; Nemunaitis, M.; Senzer, N.; Snitz, P.; Bedell, C.; Kumar, P.; Pappen, B.; Maples, P.; Shawler, D.; Fakhrai, H. Phase II trial of Belagenpumatucel-L, a TGF-β2 antisense gene modified allogeneic tumor vaccine in advanced non small cell lung cancer (NSCLC) patients. Cancer Gene Ther. 2009, 16, 620–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveres, H.; Caglevic, C.; Passiglia, F.; Taverna, S.; Smits, E.; Rolfo, C. Vaccine and immune cell therapy in non-small cell lung cancer. J. Thorac. Dis. 2018, 10, S1602. [Google Scholar] [CrossRef]
- Zhang, Z.; Lu, S.; Dunmall, L.S.C.; Wang, Z.; Cheng, Z.; Zhang, Z.; Yan, W.; Chu, Y.; Gao, D.; Wang, N. Treatment and prevention of lung cancer using a Virus-Infected Reprogrammed Somatic cell-derived Tumor cell vaccination (VIReST) regime. Front. Immunol. 2020, 11, 1996. [Google Scholar] [CrossRef]
- Cuzzubbo, S.; Mangsbo, S.; Nagarajan, D.; Habra, K.; Pockley, A.G.; McArdle, S.E. Cancer vaccines: Adjuvant potency, importance of age, lifestyle, and treatments. Front. Immunol. 2021, 11, 3850. [Google Scholar] [CrossRef]
- Rodriguez, C.P.; Sanchez, B. Challenges and opportunities for cancer vaccines in the current NSCLC clinical scenario. Curr. Top. Med. Chem. 2013, 13, 2551–2561. [Google Scholar] [CrossRef] [Green Version]
- Roy, D.; Geoffroy, K.; Marguerie, M.; Khan, S.; Martin, N.; Kmiecik, J.; Bobbala, D.; Aitken, A.; de Souza, C.; Stephenson, K. Adjuvant oncolytic virotherapy for personalized anti-cancer vaccination. Nat. Commun. 2021, 12, 2626. [Google Scholar] [CrossRef]
- Jeong, S.-N.; Yoo, S.Y. Novel oncolytic virus armed with cancer suicide gene and normal vasculogenic gene for improved anti-tumor activity. Cancers 2020, 12, 1070. [Google Scholar] [CrossRef]
- Decoster, L.; Wauters, I.; Vansteenkiste, J. Vaccination therapy for non-small-cell lung cancer: Review of agents in phase III development. J. Ann. Oncol. 2012, 23, 1387–1393. [Google Scholar] [CrossRef] [PubMed]
- Page, A.; Hubert, J.; Fusil, F.; Cosset, F.-L. Exploiting B Cell transfer for cancer therapy: Engineered B cells to eradicate tumors. Int. J. Mol. Sci. 2021, 22, 9991. [Google Scholar] [CrossRef] [PubMed]
- Saxena, M.; van der Burg, S.H.; Melief, C.J.; Bhardwaj, N. Therapeutic cancer vaccines. Nat. Rev. Cancer 2021, 21, 360–378. [Google Scholar] [CrossRef] [PubMed]
- Fusco, M.J.; West, H.J.; Walko, C.M. Tumor Mutation Burden and Cancer Treatment. JAMA Oncol. 2021, 7, 316. [Google Scholar] [CrossRef] [PubMed]
- Shao, C.; Li, G.; Huang, L.; Pruitt, S.; Castellanos, E.; Frampton, G.; Carson, K.R.; Snow, T.; Singal, G.; Fabrizio, D. Prevalence of high tumor mutational burden and association with survival in patients with less common solid tumors. JAMA Netw. Open 2020, 3, e2025109. [Google Scholar] [CrossRef]
- Wu, Y.; Xu, J.; Xu, J.; Wang, Y.; Wang, L.; Lv, W.; Hu, J. The predictive value of tumor mutation burden for immune checkpoint inhibitors therapy in non-small cell lung cancer is affected by patients’ age. Biomark. Res. 2020, 8, 9. [Google Scholar] [CrossRef] [Green Version]
- Mellstedt, H.; Vansteenkiste, J.; Thatcher, N. Vaccines for the treatment of non-small cell lung cancer: Investigational approaches and clinical experience. Lung Cancer 2011, 73, 11–17. [Google Scholar] [CrossRef]
- Dessureault, S.; Noyes, D.; Lee, D.; Dunn, M.; Janssen, W.; Cantor, A.; Sotomayor, E.; Messina, J.; Antonia, S.J. A phase-I trial using a universal GM-CSF-producing and CD40L-expressing bystander cell line (GM. CD40L) in the formulation of autologous tumor cell-based vaccines for cancer patients with stage IV disease. Ann. Surg. Oncol. 2007, 14, 869–884. [Google Scholar] [CrossRef]
- Shepherd, F.A.; Douillard, J.-Y.; Blumenschein, G.R., Jr. Immunotherapy for non-small cell lung cancer: Novel approaches to improve patient outcome. J. Thorac. Oncol. 2011, 6, 1763–1773. [Google Scholar] [CrossRef]
- Melief, C.J.; van Hall, T.; Arens, R.; Ossendorp, F.; van der Burg, S.H. Therapeutic cancer vaccines. J. Clin. Investig. 2015, 125, 3401–3412. [Google Scholar] [CrossRef]
- Shi, T.; Song, X.; Wang, Y.; Liu, F.; Wei, J. Combining oncolytic viruses with cancer immunotherapy: Establishing a new generation of cancer treatment. Front. Immunol. 2020, 11, 683. [Google Scholar] [CrossRef]
- Giaccone, G.; Bazhenova, L.; Nemunaitis, J.; Tan, M.; Juhász, E.; Ramlau, R.; Van den Heuvel, M.; Lal, R.; Kloecker, G.; Eaton, K. A phase III study of belagenpumatucel-L, an allogeneic tumour cell vaccine, as maintenance therapy for non-small cell lung cancer. Eur. J. Cancer 2015, 51, 2321–2329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Creelan, B.C.; Antonia, S.; Noyes, D.; Hunter, T.B.; Simon, G.R.; Bepler, G.; Williams, C.C.; Tanvetyanon, T.; Haura, E.B.; Schell, M.J. Phase II trial of a GM-CSF-producing and CD40L-expressing bystander cell line combined with an allogeneic tumor cell-based vaccine for refractory lung adenocarcinoma. J. Immunother. 2013, 36, 442–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, J.E.; Chiappori, A.; Williams, C.C.; Tanvetyanon, T.; Haura, E.B.; Creelan, B.C.; Kim, J.; Boyle, T.A.; Pinder-Schenck, M.; Khalil, F. A phase I/randomized phase II study of GM. CD40L vaccine in combination with CCL21 in patients with advanced lung adenocarcinoma. Cancer Immunol. Immunother. 2018, 67, 1853–1862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinageras, E.N.; de la Torre, A.; Rodríguez, M.O.; Ferrer, M.C.; Bravo, I.; del Pino, M.M.; Abreu, D.A.; Brooks, S.A.; Rives, R.; Carrillo, D.C. Phase II randomized controlled trial of an epidermal growth factor vaccine in advanced non-small-cell lung cancer. J. Clin. Oncol. 2008, 26, 1452–1458. [Google Scholar] [CrossRef]
- Rodriguez, P.C.; Popa, X.; Martínez, O.; Mendoza, S.; Santiesteban, E.; Crespo, T.; Amador, R.M.; Fleytas, R.; Acosta, S.C.; Otero, Y. A phase III clinical trial of the epidermal growth factor vaccine CIMAvax-EGF as switch maintenance therapy in advanced non–small cell lung cancer patients. Clin. Cancer Res. 2016, 22, 3782–3790. [Google Scholar] [CrossRef] [Green Version]
- Saavedra, D.; Neninger, E.; Rodriguez, C.; Viada, C.; Mazorra, Z.; Lage, A.; Crombet, T. CIMAvax-EGF: Toward long-term survival of advanced NSCLC. Semin. Oncol. 2018, 45, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Grah, J.J.; Katalinic, D.; Juretic, A.; Santek, F.; Samarzija, M. Clinical significance of immunohistochemical expression of cancer/testis tumor-associated antigens (MAGE-A1, MAGE-A3/4, NY-ESO-1) in patients with non-small cell lung cancer. Tumori J. 2014, 100, 60–68. [Google Scholar] [CrossRef]
- Vansteenkiste, J.F.; Cho, B.C.; Vanakesa, T.; De Pas, T.; Zielinski, M.; Kim, M.S.; Jassem, J.; Yoshimura, M.; Dahabreh, J.; Nakayama, H. Efficacy of the MAGE-A3 cancer immunotherapeutic as adjuvant therapy in patients with resected MAGE-A3-positive non-small-cell lung cancer (MAGRIT): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2016, 17, 822–835. [Google Scholar] [CrossRef]
- Raza, A.; Merhi, M.; Inchakalody, V.P.; Krishnankutty, R.; Relecom, A.; Uddin, S.; Dermime, S. Unleashing the immune response to NY-ESO-1 cancer testis antigen as a potential target for cancer immunotherapy. J. Transl. Med. 2020, 18, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- John, T.; Starmans, M.H.; Chen, Y.-T.; Russell, P.A.; Barnett, S.A.; White, S.C.; Mitchell, P.L.; Walkiewicz, M.; Azad, A.; Lambin, P. The role of Cancer-Testis antigens as predictive and prognostic markers in non-small cell lung cancer. PLoS ONE 2013, 8, e67876. [Google Scholar] [CrossRef] [PubMed]
- Gure, A.O.; Chua, R.; Williamson, B.; Gonen, M.; Ferrera, C.A.; Gnjatic, S.; Ritter, G.; Simpson, A.J.; Chen, Y.-T.; Old, L.J. Cancer-testis genes are coordinately expressed and are markers of poor outcome in non-small cell lung cancer. Clin. Cancer Res. 2005, 11, 8055–8062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, X.; Sun, X.; Liu, Z.; He, Y. A novel era of cancer/testis antigen in cancer immunotherapy. Int. Immunopharmacol. 2021, 98, 107889. [Google Scholar] [CrossRef]
- Alfonso, S.; Valdés-Zayas, A.; Santiesteban, E.R.; Flores, Y.I.; Areces, F.; Hernández, M.; Viada, C.E.; Mendoza, I.C.; Guerra, P.P.; García, E. A randomized, multicenter, placebo-controlled clinical trial of racotumomab-alum vaccine as switch maintenance therapy in advanced non–small cell lung cancer patients. Clin. Cancer Res. 2014, 20, 3660–3671. [Google Scholar] [CrossRef] [Green Version]
- Butts, C.; Murray, N.; Maksymiuk, A.; Goss, G.; Marshall, E.; Soulières, D.; Cormier, Y.; Ellis, P.; Price, A.; Sawhney, R. Randomized phase IIB trial of BLP25 liposome vaccine in stage IIIB and IV non–small-cell lung cancer. J. Clin. Oncol. 2005, 23, 6674–6681. [Google Scholar] [CrossRef] [Green Version]
- Butts, C.; Socinski, M.A.; Mitchell, P.L.; Thatcher, N.; Havel, L.; Krzakowski, M.; Nawrocki, S.; Ciuleanu, T.-E.; Bosquée, L.; Trigo, J.M. Tecemotide (L-BLP25) versus placebo after chemoradiotherapy for stage III non-small-cell lung cancer (START): A randomised, double-blind, phase 3 trial. Lancet Oncol. 2014, 15, 59–68. [Google Scholar] [CrossRef] [Green Version]
- Ponomarenko, D.M.; Klimova, I.D.; Chapygina, Y.A.; Dvornichenko, V.V.; Zhukova, N.V.; Orlova, R.V.; Manikhas, G.M.; Zyryanov, A.V.; Burkhanova, L.A.; Badrtdinova, I.I. Safety and efficacy of p62 DNA vaccine ELENAGEN in a first-in-human trial in patients with advanced solid tumors. Oncotarget 2017, 8, 53730. [Google Scholar] [CrossRef] [Green Version]
- Rochlitz, C.; Figlin, R.; Squiban, P.; Salzberg, M.; Pless, M.; Herrmann, R.; Tartour, E.; Zhao, Y.; Bizouarne, N.; Baudin, M. Phase I immunotherapy with a modified vaccinia virus (MVA) expressing human MUC1 as antigen-specific immunotherapy in patients with MUC1-positive advanced cancer. J. Gene Med. Cross-Discip. J. Res. Sci. Gene Transf. Clin. Appl. 2003, 5, 690–699. [Google Scholar] [CrossRef]
- Ramlau, R.; Quoix, E.; Rolski, J.; Pless, M.; Lena, H.; Lévy, E.; Krzakowski, M.; Hess, D.; Tartour, E.; Chenard, M.-P. A phase II study of Tg4010 (Mva-Muc1-Il2) in association with chemotherapy in patients with stage III/IV Non-small cell lung cancer. J. Thorac. Oncol. 2008, 3, 735–744. [Google Scholar] [CrossRef]
- Quoix, E.; Ramlau, R.; Westeel, V.; Papai, Z.; Madroszyk, A.; Riviere, A.; Koralewski, P.; Breton, J.-L.; Stoelben, E.; Braun, D. Therapeutic vaccination with TG4010 and first-line chemotherapy in advanced non-small-cell lung cancer: A controlled phase 2B trial. Lancet Oncol. 2011, 12, 1125–1133. [Google Scholar] [CrossRef]
- Quoix, E.; Lena, H.; Losonczy, G.; Forget, F.; Chouaid, C.; Papai, Z.; Gervais, R.; Ottensmeier, C.; Szczesna, A.; Kazarnowicz, A. TG4010 immunotherapy and first-line chemotherapy for advanced non-small-cell lung cancer (TIME): Results from the phase 2b part of a randomised, double-blind, placebo-controlled, phase 2b/3 trial. Lancet Oncol. 2016, 17, 212–223. [Google Scholar] [CrossRef]
- Mondal, M.; Guo, J.; He, P.; Zhou, D. Recent advances of oncolytic virus in cancer therapy. Hum. Vaccines Immunother. 2020, 16, 2389–2402. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic viruses: A new class of immunotherapy drugs. Nat. Rev. Drug Discov. 2015, 14, 642–662. [Google Scholar] [CrossRef] [PubMed]
- Badrinath, N.; Heo, J.; Yoo, S.Y. Viruses as nanomedicine for cancer. Int. J. Nanomed. 2016, 11, 4835. [Google Scholar]
- Shen, Y.; Nemunaitis, J. Fighting cancer with vaccinia virus: Teaching new tricks to an old dog. Mol. Ther. 2005, 11, 180–195. [Google Scholar] [CrossRef]
- Yoo, S.Y.; Badrinath, N.; Woo, H.Y.; Heo, J. Oncolytic Virus-Based Immunotherapies for Hepatocellular Carcinoma. Mediat. Inflamm. 2017, 2017, 5198798. [Google Scholar] [CrossRef] [Green Version]
- Yoo, S.Y.; Bang, S.Y.; Jeong, S.-N.; Kang, D.H.; Heo, J. A cancer-favoring oncolytic vaccinia virus shows enhanced suppression of stem-cell like colon cancer. Oncotarget 2016, 7, 16479–16489. [Google Scholar] [CrossRef]
- Yoo, S.Y.; Jeong, S.-N.; Kang, D.H.; Heo, J. Evolutionary cancer-favoring engineered vaccinia virus for metastatic hepatocellular carcinoma. Oncotarget 2017, 8, 71489–71499. [Google Scholar] [CrossRef] [Green Version]
- Yoo, S.Y.; Badrinath, N.; Lee, H.L.; Heo, J.; Kang, D.-H. A Cancer-Favoring, Engineered Vaccinia Virus for Cholangiocarcinoma. Cancers 2019, 11, 1667. [Google Scholar] [CrossRef] [Green Version]
- Haddad, D. Genetically engineered vaccinia viruses as agents for cancer treatment, imaging, and transgene delivery. Front. Oncol. 2017, 7, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Z.S.; Lu, B.; Guo, Z.; Giehl, E.; Feist, M.; Dai, E.; Liu, W.; Storkus, W.J.; He, Y.; Liu, Z. Vaccinia virus-mediated cancer immunotherapy: Cancer vaccines and oncolytics. J. Immunother. Cancer 2019, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, E.; Weibel, S.; Szalay, A.A. Combination treatment with oncolytic Vaccinia virus and cyclophosphamide results in synergistic antitumor effects in human lung adenocarcinoma bearing mice. J. Transl. Med. 2014, 12, 197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaghchi, C.A.; Zhang, Z.; Alusi, G.; Lemoine, N.R.; Wang, Y. Vaccinia virus, a promising new therapeutic agent for pancreatic cancer. Immunotherapy 2015, 7, 1249–1258. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Jiang, P.; Wei, S.; Xu, X.; Wang, J. Regulatory T cells in tumor microenvironment: New mechanisms, potential therapeutic strategies and future prospects. Mol. Cancer 2020, 19, 116. [Google Scholar] [CrossRef]
- Vignali, D.A.; Collison, L.W.; Workman, C.J. How regulatory T cells work. Nat. Rev. Immunol. 2008, 8, 523–532. [Google Scholar] [CrossRef] [Green Version]
- Yoo, S.Y.; Badrinath, N.; Jeong, S.-N.; Woo, H.Y.; Heo, J. Overcoming Tumor Resistance to Oncolyticvaccinia Virus with Anti-PD-1-Based Combination Therapy by Inducing Antitumor Immunity in the Tumor Microenvironment. Vaccines 2020, 8, 321. [Google Scholar] [CrossRef]
- Smith, S.A.; Kotwal, G. Virokines: Novel immunomodulatory agents. Expert Opin. Biol. Ther. 2001, 1, 343–357. [Google Scholar] [CrossRef]
- Sharp, D.W.; Lattime, E.C. Recombinant poxvirus and the tumor microenvironment: Oncolysis, immune regulation and immunization. Biomedicines 2016, 4, 19. [Google Scholar] [CrossRef] [Green Version]
- Weibel, S.; Raab, V.; Yong, A.Y.; Worschech, A.; Wang, E.; Marincola, F.M.; Szalay, A.A. Viral-mediated oncolysis is the most critical factor in the late-phase of the tumor regression process upon vaccinia virus infection. BMC Cancer 2011, 11, 68. [Google Scholar] [CrossRef] [Green Version]
- Nakatake, M.; Kuwano, N.; Kaitsurumaru, E.; Kurosaki, H.; Nakamura, T. Fusogenic oncolytic vaccinia virus enhances systemic antitumor immune response by modulating the tumor microenvironment. Mol. Ther. 2021, 29, 1782–1793. [Google Scholar] [CrossRef] [PubMed]
- DePeaux, K.; Vignali, P.; Sarkar, S.; Delgoffe, G. 589 Efficacy of oncolytic vaccinia virus requires infection of suppressive immune cells in the tumor microenvironment leading to their reprogramming and deletion. J. ImmunoTher. Cancer 2020, 8, A352–A353. [Google Scholar] [CrossRef]
- Chen, N.G.; Szalay, A.A. Oncolytic vaccinia virus: A theranostic agent for cancer. Future Virol. 2010, 5, 763–784. [Google Scholar] [CrossRef]
- Yu, F.; Wang, X.; Guo, Z.S.; Bartlett, D.L.; Gottschalk, S.M.; Song, X.-T. T-cell engager-armed oncolytic vaccinia virus significantly enhances antitumor therapy. Mol. Ther. 2014, 22, 102–111. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Yong, A.Y.; Wang, E.; Chen, N.; Danner, R.L.; Munson, P.J.; Marincola, F.M.; Szalay, A.A. Eradication of solid human breast tumors in nude mice with an intravenously injected light-emitting oncolytic vaccinia virus. Cancer Res. 2007, 67, 10038–10046. [Google Scholar] [CrossRef] [Green Version]
- Yong, A.Y.; Galanis, C.; Woo, Y.; Chen, N.; Zhang, Q.; Fong, Y.; Szalay, A.A. Regression of human pancreatic tumor xenografts in mice after a single systemic injection of recombinant vaccinia virus GLV-1h68. Mol. Cancer Ther. 2009, 8, 141–151. [Google Scholar]
- Seubert, C.; Stritzker, J.; Hess, M.; Donat, U.; Sturm, J.; Chen, N.; Von Hof, J.; Krewer, B.; Tietze, L.; Gentschev, I. Enhanced tumor therapy using vaccinia virus strain GLV-1h68 in combination with a β-galactosidase-activatable prodrug seco-analog of duocarmycin SA. Cancer Gene Ther. 2011, 18, 42–52. [Google Scholar] [CrossRef]
- Sturm, J.B.; Hess, M.; Weibel, S.; Chen, N.G.; Yong, A.Y.; Zhang, Q.; Donat, U.; Reiss, C.; Gambaryan, S.; Krohne, G. Functional hyper-IL-6 from vaccinia virus-colonized tumors triggers platelet formation and helps to alleviate toxicity of mitomycin C enhanced virus therapy. J. Transl. Med. 2012, 10, 9. [Google Scholar] [CrossRef] [Green Version]
- Advani, S.J.; Buckel, L.; Chen, N.G.; Scanderbeg, D.J.; Geissinger, U.; Zhang, Q.; Yong, A.Y.; Aguilar, R.J.; Mundt, A.J.; Szalay, A.A. Preferential replication of systemically delivered oncolytic vaccinia virus in focally irradiated glioma xenografts. Clin. Cancer Res. 2012, 18, 2579–2590. [Google Scholar] [CrossRef] [Green Version]
- Dai, M.; Liu, S.; Chen, N.; Zhang, T.; You, L.; Zhang, F.; Chou, T.; Szalay, A.; Fong, Y.; Zhao, Y. Oncolytic vaccinia virus in combination with radiation shows synergistic antitumor efficacy in pancreatic cancer. Cancer Lett. 2014, 344, 282–290. [Google Scholar] [CrossRef]
- Lu, S.; Zhang, Z.; Du, P.; Chard, L.S.; Yan, W.; El Khouri, M.; Wang, Z.; Zhang, Z.; Chu, Y.; Gao, D. A virus-infected, reprogrammed somatic cell–derived tumor cell (VIReST) vaccination regime can prevent initiation and progression of pancreatic cancer. Clin. Cancer Res. 2020, 26, 465–476. [Google Scholar] [CrossRef] [Green Version]
- Tysome, J.R.; Li, X.; Wang, S.; Wang, P.; Gao, D.; Du, P.; Chen, D.; Gangeswaran, R.; Chard, L.S.; Yuan, M. A novel therapeutic regimen to eradicate established solid tumors with an effective induction of tumor-specific immunity. Clin. Cancer Res. 2012, 18, 6679–6689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemay, C.G.; Rintoul, J.L.; Kus, A.; Paterson, J.M.; Garcia, V.; Falls, T.J.; Ferreira, L.; Bridle, B.W.; Conrad, D.P.; Tang, V.A. Harnessing oncolytic virus-mediated antitumor immunity in an infected cell vaccine. Mol. Ther. 2012, 20, 1791–1799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mell, L.K.; Brumund, K.T.; Daniels, G.A.; Advani, S.J.; Zakeri, K.; Wright, M.E.; Onyeama, S.-J.; Weisman, R.A.; Sanghvi, P.R.; Martin, P.J. Phase I trial of intravenous oncolytic vaccinia virus (GL-ONC1) with cisplatin and radiotherapy in patients with locoregionally advanced head and neck carcinoma. Clin. Cancer Res. 2017, 23, 5696–5702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, J.; Shalabi, A.; Hubbard-Lucey, V. Comprehensive analysis of the clinical immuno-oncology landscape. Ann. Oncol. 2018, 29, 84–91. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Truong, C.-S.; Yoo, S.Y. Oncolytic Vaccinia Virus in Lung Cancer Vaccines. Vaccines 2022, 10, 240. https://doi.org/10.3390/vaccines10020240
Truong C-S, Yoo SY. Oncolytic Vaccinia Virus in Lung Cancer Vaccines. Vaccines. 2022; 10(2):240. https://doi.org/10.3390/vaccines10020240
Chicago/Turabian StyleTruong, Cao-Sang, and So Young Yoo. 2022. "Oncolytic Vaccinia Virus in Lung Cancer Vaccines" Vaccines 10, no. 2: 240. https://doi.org/10.3390/vaccines10020240
APA StyleTruong, C.-S., & Yoo, S. Y. (2022). Oncolytic Vaccinia Virus in Lung Cancer Vaccines. Vaccines, 10(2), 240. https://doi.org/10.3390/vaccines10020240