
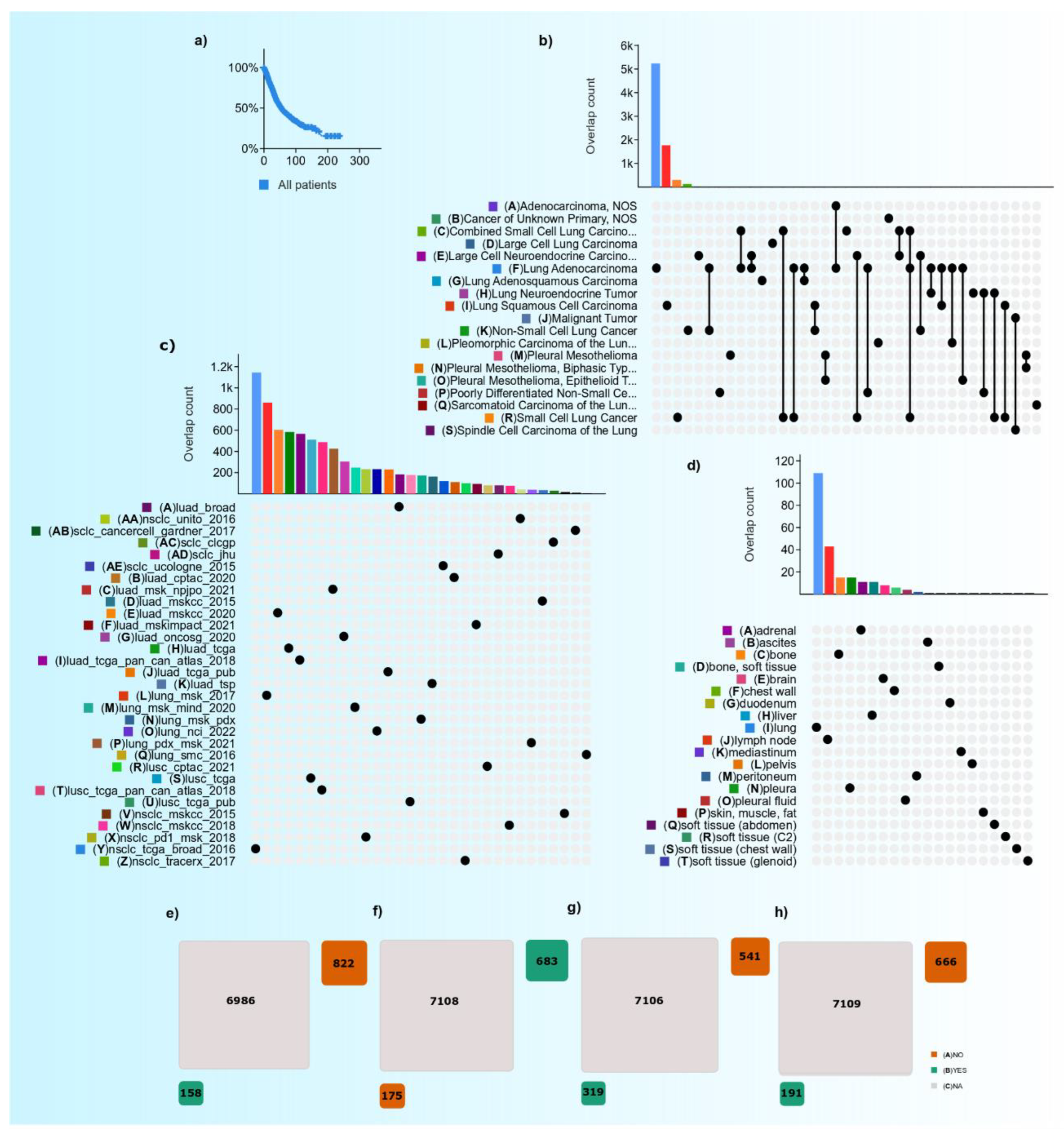
Advances in the Lung Cancer Immunotherapy Approaches
 , , ,
, , ,  , ,
, ,  ,
,  ,
, 

Abstract
1. Introduction
2. Current Lung Cancer Epidemiology
3. Five Pillars of Lung Cancer Therapy
4. Lung Cancer Agonistic and Antagonistic Immune Cells
5. Immunophenotyping of Lung Cancer
Impact of Immune Profiling and Scoring on Lung Cancer Prognosis
6. Immunotherapy-Based Clinical Studies in Lung Cancer
6.1. Immunotherapy in Non-Small Cell Lung Cancer
6.2. Immunotherapy in Small Cell Lung Cancer
7. Cancer Immunotherapy Approaches
7.1. Checkpoint Inhibitors
7.2. Monoclonal Antibodies
7.3. CAR-T Cell Therapy
7.4. Lung Cancer Vaccines
7.4.1. Belagenpumatucel-L Vaccine (Lucanix)
7.4.2. MAGE-A3
7.4.3. CIMAvax-EGF
7.4.4. Racotumomab
7.4.5. TG4010
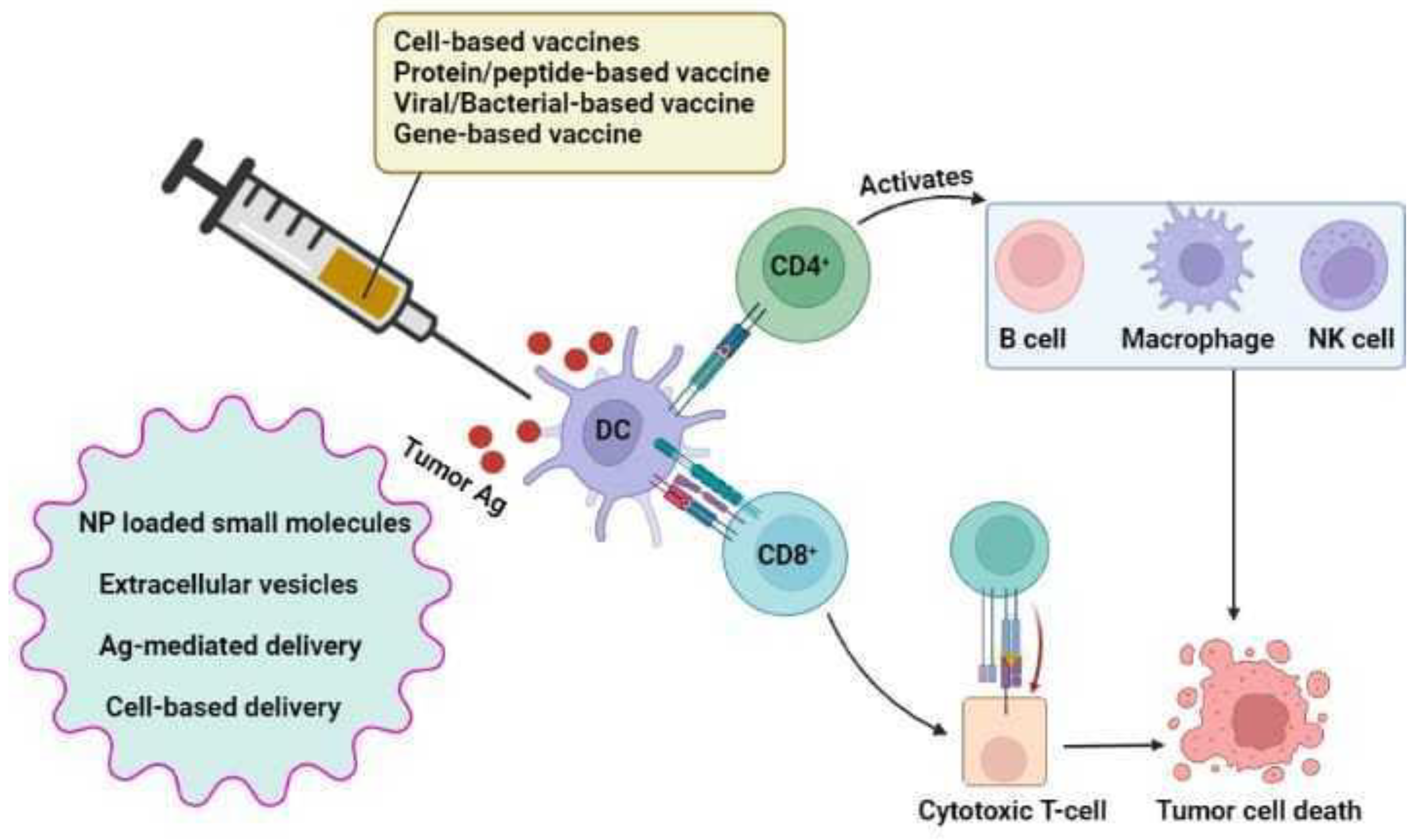
7.4.6. Vaccine Delivery Vehicles
Nanoparticle-Based Delivery
NP-Loaded Small Molecules
Extracellular Vesicles
Antigen-Mediated Delivery
Cell-Based Delivery
8. Combinatorial Approaches to Lung Cancer-Immunotherapy
8.1. a. Targeted therapy
8.2. b. Chemotherapy
8.3. c. Radiotherapy
8.4. Emerging Combination Strategies in Adoptive Cell Therapy
9. Lung Cancer Preclinical Models in Testing Immunotherapy
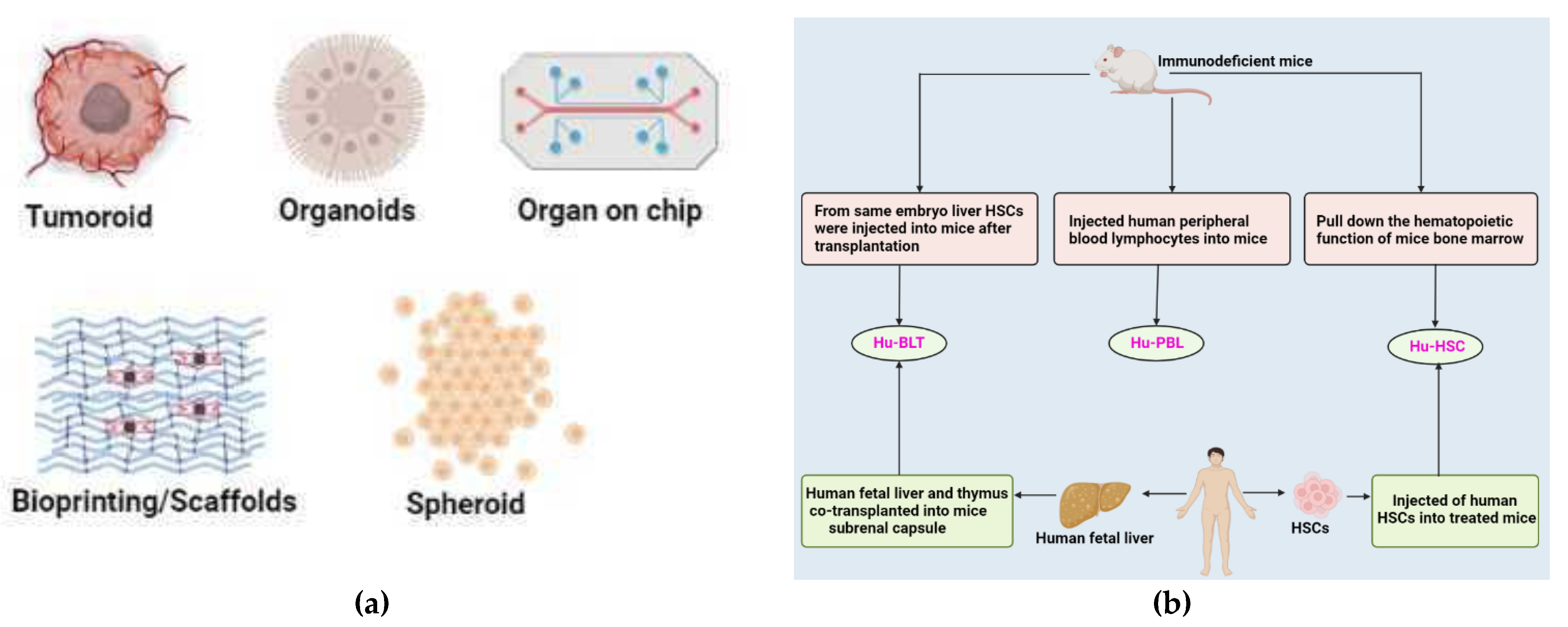
9.1. 3D Models
9.1.1. Cell-Lines
9.1.2. Patient-Derived Lung Cancer Organoid
9.1.3. Patient-Derived 3D Models

{kind=link}
{kind=link}
{kind=link}
| Models | Source | Application | Reference |
|---|---|---|---|
| spheroids | Resection | Lung cancer stem cells’ identification and description; production of Xenografts that replicate the parental tumor’s histology | [265] |
| Resection, pleural effusion | Technique to expand cancer cells from the lungs of patients | [266] | |
| Core needle biopsy, surgical biopsy, pleural effusion | Drug testing | [267] | |
| Organoids | biopsy | Drug testing and evaluation of immune cell populations penetrating cultured tissues | [268] |
| Resection/PDX | LCOs’ long-term growth, confirmation, and drug screening | [269,270] | |
| biopsy | For use in immuno-oncology research and testing for customized immunotherapy, a new approach for maintaining endogenous tumor-infiltrating cells has been developed. | [271] | |
| Resection/biopsy | Biobanking, drug testing | [272,273,274] | |
| Resection/biopsy | Examining and blocking regulators of mitochondrial fission in various tumor organoids | [275] | |
| Pleural effusion | drug testing and the establishment of an LCO culture system from pleural effusions | [276] | |
| Resection/biopsy | Analyzing several techniques to determine the tumor purity of organoids created from intrapulmonary tumors | [277] | |
| PDX derived from biopsies | Drug testing and organoid creation using PDXs from SCLC biopsies | [278] | |
| Resection/biopsy | Analyses of pathway inhibitors found by single-cell proteomics | [279] | |
| pleural effusion | Targeted drug testing and LCO production and characterization | [280] |
| Tumor Type | Model | Application | Reference |
|---|---|---|---|
| NSCLC (Non-small cell lung carcinoma) | Lung cancer organoids | Drug screening | [269] |
| NSCLC organoids | Drug screening | [270] | |
| Patient-derived organoids model | Genomics, production of treatment outcomes | [274] | |
| Lung ADK (LADC)-derived organoid model | Drug screening, biomarker development, and living biobank | [273,274] | |
| Patient-derived tumoroids (PDTs) | PDTs are developed to be used in microfluidic devices for drug screening and mimic the cancer vascular network. | [281] | |
| Patient-derived lung cancer organoids | Patient-specific medication screening and support for the xenograft model from a living biobank | [272] | |
| Patient-derived tumoroids (PDTs) | creating new cell lines | [282] | |
| Patient lung-derived tumoroids (PLDTs) | Drug screening | [283] | |
| Lung cancer organoids | Personalized medicine | [277] |
9.1.4. Organ-On-Chip
9.1.5. 3D Bioprinting
9.2. In Vivo Animal Models
9.2.1. Zebrafish Model
9.2.2. Patient-Derived Tumor Xenograft (PDX) Model
9.2.3. Patient-Derived Orthotropic Xenograft (PDOX) Model
9.2.4. Mini Patient-Derived Xenograft (Mini-PDX) Model
10. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Howlader, N.; Forjaz, G.; Mooradian, M.J.; Meza, R.; Kong, C.Y.; Cronin, K.A.; Mariotto, A.B.; Lowy, D.R.; Feuer, E.J. The Effect of Advances in Lung-Cancer Treatment on Population Mortality. N. Engl. J. Med. 2020, 383, 640–649. [Google Scholar] [CrossRef] [PubMed]
- Chevallier, M.; Borgeaud, M.; Addeo, A.; Friedlaender, A. Oncogenic Driver Mutations in Non-Small Cell Lung Cancer: Past, Present and Future. World J. Clin. Oncol. 2021, 12, 217. [Google Scholar] [CrossRef] [PubMed]
- Pawelczyk, K.; Piotrowska, A.; Ciesielska, U.; Jablonska, K.; Gletzel-Plucinska, N.; Grzegrzolka, J.; Podhorska-Okolow, M.; Dziegiel, P.; Nowinska, K. Role of PD-L1 Expression in Non-Small Cell Lung Cancer and Their Prognostic Significance According to Clinicopathological Factors and Diagnostic Markers. Int. J. Mol. Sci. 2019, 20, 824. [Google Scholar] [CrossRef] [PubMed]
- Small Cell Lung Cancer Stages | Stages of Small Cell Lung Cancer. Available online: https://www.cancer.org/cancer/lung-cancer/detection-diagnosis-staging/staging-sclc.html (accessed on 13 October 2022).
- Wang, C.; Li, J.; Zhang, Q.; Wu, J.; Xiao, Y.; Song, L.; Gong, H.; Li, Y. The Landscape of Immune Checkpoint Inhibitor Therapy in Advanced Lung Cancer. BMC Cancer 2021, 21, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Mathers, C.D.; Loncar, D. Projections of Global Mortality and Burden of Disease from 2002 to 2030. PLoS Med. 2006, 3, 2011–2030. [Google Scholar] [CrossRef]
- Padinharayil, H.; Varghese, J.; John, M.C.; Rajanikant, G.K.; Wilson, C.M.; Al-Yozbaki, M.; Renu, K.; Dewanjee, S.; Sanyal, R.; Dey, A.; et al. Non-Small Cell Lung Carcinoma (NSCLC): Implications on Molecular Pathology and Advances in Early Diagnostics and Therapeutics. Genes Dis. 2022, 2352–3042. [Google Scholar] [CrossRef]
- Registry, P.C. Global Cancer Observatory. Available online: https://gco.iarc.fr/ (accessed on 10 October 2022).
- Conger, A.B. Cancer Statistics. Available online: https://seer.cancer.gov/statistics/ (accessed on 10 October 2022).
- Gao CBioPortal for Cancer Genomics. Available online: https://www.cbioportal.org/%0Ahttps://www.cbioportal.org/%0Ahttp://www.cbioportal.org/public-portal/sci_signal_reprint.jsp (accessed on 10 October 2022).
- Siamof, C.M.; Goel, S.; Cai, W. Moving Beyond the Pillars of Cancer Treatment: Perspectives From Nanotechnology. Front. Chem. 2020, 8, 598100. [Google Scholar] [CrossRef]
- Hoy, H.; Lynch, T.; Beck, M. Surgical Treatment of Lung Cancer. Crit. Care Nurs. Clin. N. Am. 2019, 31, 303–313. [Google Scholar] [CrossRef]
- Bogart, J.A.; Waqar, S.N.; Mix, M.D. Radiation and Systemic Therapy for Limited-Stage Small-Cell Lung Cancer. J. Clin. Oncol. 2022, 40, 661–670. [Google Scholar] [CrossRef]
- Lung Cancer-Non-Small Cell: Types of Treatment | Cancer.Net. Available online: https://www.cancer.net/cancer-types/lung-cancer-non-small-cell/types-treatment (accessed on 13 October 2022).
- Nagasaka, M.; Gadgeel, S.M. Role of Chemotherapy and Targeted Therapy in Early-Stage Non-Small Cell Lung Cancer. Expert Rev. Anticancer. Ther. 2018, 18, 63–70. [Google Scholar] [CrossRef]
- Ruiz-Cordero, R.; Devine, W.P. Targeted Therapy and Checkpoint Immunotherapy in Lung Cancer. Surg. Pathol. Clin. 2020, 13, 17–33. [Google Scholar] [CrossRef] [PubMed]
- Doroshow, D.B.; Sanmamed, M.F.; Hastings, K.; Politi, K.; Rimm, D.L.; Chen, L.; Melero, I.; Schalper, K.A.; Herbst, R.S. Immunotherapy in Non-Small Cell Lung Cancer: Facts and Hopes. Clin Cancer Res 2019, 25, 4592. [Google Scholar] [CrossRef] [PubMed]
- Mok, T.S.K.; Wu, Y.L.; Kudaba, I. Pembrolizumab versus Chemotherapyfor Previously Untreated, PD-L1-expressing, Locally Advancedor Metastatic Non-small-cell Lung Cancer (KEYNOTE-042): Arandomised, Open-label, Controlled, Phase 3 Trial. Lancet 2019, 393, 1819–1830. [Google Scholar] [CrossRef]
- Non-Small Cell Lung Cancer Chemotherapy | Chemo Side Effects. Available online: https://www.cancer.org/cancer/lung-cancer/treating-non-small-cell/chemotherapy.html (accessed on 13 November 2022).
- Chen, L.; Smith, D.A.; Somarouthu, B.; Gupta, A.; Gilani, K.A.; Ramaiya, N.H. A Radiologist’s Guide to the Changing Treatment Paradigm of Advanced Non–Small Cell Lung Cancer: The ASCO 2018 Molecular Testing Guidelines and Targeted Therapies. Am. J. Roentgenol. 2019, 213, 1047–1058. [Google Scholar] [CrossRef]
- Pakkala, S.; Ramalingam, S.S. Personalized Therapy for Lung Cancer: Striking a Moving Target. JCI Insight 2018, 3, e120858. [Google Scholar] [CrossRef]
- Soria, J.-C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. N Engl J Med 2018, 378, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, S.S.; Vansteenkiste, J.; Planchard, D.; Cho, B.C.; Gray, J.E.; Ohe, Y.; Zhou, C.; Reungwetwattana, T.; Cheng, Y.; Chewaskulyong, B.; et al. Overall Survival with Osimertinib in Untreated, EGFR -Mutated Advanced NSCLC. N. Engl. J. Med. 2020, 382, 41–50. [Google Scholar] [CrossRef]
- Kong, X.; Zhang, K.; Wang, X.; Yang, X.; Li, Y.; Zhai, J.; Xing, Z.; Qi, Y.; Gao, R.; Feng, X.; et al. Mechanism of Trastuzumab Resistance Caused by HER-2 Mutation in Breast Carcinomas. Cancer Manag. Res. 2019, 11, 5971–5982. [Google Scholar] [CrossRef]
- Home-ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ (accessed on 4 March 2022).
- Camidge, D.R.; Dziadziuszko, R.; Peters, S.; Mok, T.; Noe, J.; Nowicka, M.; Gadgeel, S.M.; Cheema, P.; Pavlakis, N.; de Marinis, F.; et al. Updated Efficacy and Safety Data and Impact of the EML4-ALK Fusion Variant on the Efficacy of Alectinib in Untreated ALK-Positive Advanced Non–Small Cell Lung Cancer in the Global Phase III ALEX Study. J. Thorac. Oncol. 2019, 14, 1233–1243. [Google Scholar] [CrossRef]
- Peters, S.; Mok, T.S.K.; Gadgeel, S.M.; Rosell, R.; Dziadziuszko, R.; Kim, D.-W.; Perol, M.; Ou, S.-H.I.; Shaw, A.T.; Bordogna, W.; et al. Updated Overall Survival (OS) and Safety Data from the Randomized, Phase III ALEX Study of Alectinib (ALC) versus Crizotinib (CRZ) in Untreated Advanced ALK + NSCLC. J. Clin. Oncol. 2020, 38, 9518. [Google Scholar] [CrossRef]
- Camidge, R.; Kim, H.; Ahn, M.; Al, E.; Al, E.; Al, E. Brigatinib versus Crizotinib in ALK-Positive Non–Small-Cell Lung Cancer [Published Online September 25, 2018]. N. Engl. J. Med. 2018, 379, 2027–2039. [Google Scholar] [CrossRef] [PubMed]
- Soria, J.C.; Tan, D.S.W.; Chiari, R.; Wu, Y.L.; Paz-Ares, L.; Wolf, J.; Geater, S.L.; Orlov, S.; Cortinovis, D.; Yu, C.J.; et al. First-Line Ceritinib versus Platinum-Based Chemotherapy in Advanced ALK-Rearranged Non-Small-Cell Lung Cancer (ASCEND-4): A Randomised, Open-Label, Phase 3 Study. Lancet 2017, 389, 917–929. [Google Scholar] [CrossRef]
- Kumar, M.; Ernani, V.; Owonikoko, T.K. Biomarkers and Targeted Systemic Therapies in Advanced Non-Small Cell Lung Cancer. Mol. Asp. Med. 2015, 45, 55–66. [Google Scholar] [CrossRef]
- Drilon, A.; Siena, S.; Dziadziuszko, R. Entrectinib in ROS1 Fusion-Positive Non-Small-Cell Lung Cancer: Integrated Analysis of Three Phase 1–2 Trials. Lancet Oncol. 2020, 21, 261–270. [Google Scholar] [CrossRef]
- Shaw, A.T.; Ou, S.H.; Bang, Y.J. Crizotinib in ROS1-Rearranged Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2014, 371, 1963–1971. [Google Scholar] [CrossRef]
- Shaw, A.T.; Riely, G.J.; Bang, Y.J.; Kim, D.W.; Camidge, D.R.; Solomon, B.J.; Varella-Garcia, M.; Iafrate, A.J.; Shapiro, G.I.; Usari, T.; et al. Crizotinib in ROS1-Rearranged Advanced Non-Small-Cell Lung Cancer (NSCLC): Updated Results, Including Overall Survival, from PROFILE 1001. Ann. Oncol. 2019, 30, 1121–1126. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, T.M.; Kim, D.W.; Kim, S.; Kim, M.; Ahn, Y.O.; Keam, B.; Heo, D.S. Acquired Resistance of MET-Amplified Non-Small Cell Lung Cancer Cells to the MET Inhibitor Capmatinib. Cancer Res. Treat. 2019, 51, 951–962. [Google Scholar] [CrossRef]
- Wolf, J.; Seto, T.; Han, J.-Y.; Reguart, N.; Garon, E.B.; Groen, H.J.M.; Tan, D.S.W.; Hida, T.; de Jonge, M.; Orlov, S.V.; et al. Capmatinib in MET Exon 14–Mutated or MET -Amplified Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2020, 383, 944–957. [Google Scholar] [CrossRef]
- Drilon, A.; Oxnard, G.R.; Tan, D.S.W. Efficacy of Selpercatinib in RET Fusion-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2020, 383, 813–824. [Google Scholar] [CrossRef]
- Gainor, J.F.; Curigliano, G.; Kim, D.-W.; Lee, D.H.; Besse, B.; Baik, C.S.; Doebele, R.C.; Cassier, P.A.; Lopes, G.; Tan, D.S.-W.; et al. Registrational Dataset from the Phase I/II ARROW Trial of Pralsetinib (BLU-667) in Patients (Pts) with Advanced RET Fusion+ Non-Small Cell Lung Cancer (NSCLC). J. Clin. Oncol. 2020, 38, 9515. [Google Scholar] [CrossRef]
- Nowicki, T.S.; Hu-Lieskovan, S.; Ribas, A. Mechanisms of Resistance to PD-1 and PD-L1 Blockade. Cancer J. 2018, 24, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Skov, B.G.; Rørvig, S.B.; Jensen, T.H.L.; Skov, T. The Prevalence of Programmed Death Ligand-1 (PD-L1) Expression in Non-Small Cell Lung Cancer in an Unselected, Consecutive Population. Mod. Pathol. 2019, 33, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csoszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Updated Analysis of KEYNOTE-024: Pembrolizumab versus Platinum-Based Chemotherapy for Advanced Non–Small-Cell Lung Cancer with PD-L1 Tumor Proportion Score of 50% or Greater. J. Clin. Oncol. 2019, 37, 537–546. [Google Scholar] [CrossRef]
- Herbst, R.; de Marinis, F.; Giaccone, G.; Reinmuth, N.; Vergnenegre, A.; Barrios, C.; Morise, M.; Font, E.; Andric, Z.; Geater, S.; et al. O81 IMpower110: Interim Overall Survival (OS) Analysis of a Phase III Study of Atezolizumab (ATEZO) Monotherapy vs Platinum-Based Chemotherapy (CHEMO) as First-Line (1L) Treatment in PD-L1–Selected NSCLC. Ann. Oncol. 2020, 30, v915. [Google Scholar] [CrossRef]
- Tony, S.K.; Mok, M.; Yi-Long Wu, M.; Iveta Kudaba, M.; Dariusz, M.; Kowalski, M.; Byoung Chul Cho, M.; Hande, Z.; Turna, M.; Gilberto Castro, P., Jr.; et al. Pembrolizumab versus Chemotherapy for Previously Untreated, PD-L1-Expressing, Locally Advanced or Metastatic Non-Small-Cell Lung Cancer (KEYNOTE-042): A Randomised, Open-Label, Controlled, Phase 3 Trial. Lancet 2019, 393, 1819–1830. [Google Scholar]
- Hellmann, M.D.; Paz-Ares, L.; Bernabe Caro, R.; Zurawski, B.; Kim, S.-W.; Carcereny Costa, E.; Park, K.; Alexandru, A.; Lupinacci, L.; de la Mora Jimenez, E.; et al. Nivolumab plus Ipilimumab in Advanced Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2019, 381, 2020–2031. [Google Scholar] [CrossRef]
- Qin, H.; Wang, F.; Liu, H.; Zeng, Z.; Wang, S.; Pan, X.; Gao, H. New Advances in Immunotherapy for Non-Small Cell Lung Cancer. Am. J. Transl. Res. 2018, 10, 2234. [Google Scholar] [CrossRef]
- Galon, J.; Bruni, D. Tumor Immunology and Tumor Evolution: Intertwined Histories. Immunity 2020, 52, 55–81. [Google Scholar] [CrossRef]
- Domagala-Kulawik, J. The Role of the Immune System in Non-Small Cell Lung Carcinoma and Potential for Therapeutic Intervention. Transl. Lung Cancer Res. 2015, 4, 177–190. [Google Scholar] [CrossRef]
- Franceschi, C.; Bonafè, M.; Valensin, S.; Olivieri, F.; de Luca, M.; Ottaviani, E.; de Benedictis, G. Inflamm-Aging. An Evolutionary Perspective on Immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Elements of Cancer Immunity and the Cancer-Immune Set Point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer Immunoediting: From Immunosurveillance to Tumor Escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer Immunoediting: Integrating Immunity’s Roles in Cancer Suppression and Promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef]
- Bonaventura, P.; Shekarian, T.; Alcazer, V.; Valladeau-Guilemond, J.; Valsesia-Wittmann, S.; Amigorena, S.; Caux, C.; Depil, S. Cold Tumors: A Therapeutic Challenge for Immunotherapy. Front. Immunol. 2019, 10, 168. [Google Scholar] [CrossRef]
- Chi, A.; He, X.; Hou, L.; Nguyen, N.P.; Zhu, G.; Cameron, R.B.; Lee, J.M. Classification of Non-Small Cell Lung Cancer’s Tumor Immune Micro-Environment and Strategies to Augment Its Response to Immune Checkpoint Blockade. Cancers 2021, 13, 2924. [Google Scholar] [CrossRef]
- Gajewski, T.F.; Corrales, L.; Williams, J.; Horton, B.; Sivan, A.; Spranger, S. Cancer Immunotherapy Targets Based on Understanding the t Cell-Inflamed versus Non-t Cell-Inflamed Tumor Microenvironment. Adv. Exp. Med. Biol. 2017, 1036, 19–31. [Google Scholar] [CrossRef]
- Pai, S.I.; Cesano, A.; Marincola, F.M. The Paradox of Cancer Immune Exclusion: Immune Oncology Next Frontier. Cancer Treat Res. 2020, 180, 173–195. [Google Scholar] [CrossRef]
- Li, Y.L.; Zhao, H.; Ren, X.B. Relationship of VEGF/VEGFR with Immune and Cancer Cells: Staggering or Forward? Cancer Biol. Med. 2016, 13, 206–214. [Google Scholar] [CrossRef]
- Yang, J.; Yan, J.; Liu, B. Targeting VEGF/VEGFR to Modulate Antitumor Immunity. Front. Immunol. 2018, 9, 978. [Google Scholar] [CrossRef]
- Dillon, L.; Miller, T. Therapeutic Targeting of Cancers with Loss of PTEN Function. Curr. Drug Targets 2014, 15, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN Promotes Resistance to T Cell–Mediated Immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef] [PubMed]
- Gil-Julio, H.; Perea, F.; Rodriguez-Nicolas, A.; Cozar, J.M.; González-Ramirez, A.R.; Concha, A.; Garrido, F.; Aptsiauri, N.; Ruiz-Cabello, F. Tumor Escape Phenotype in Bladder Cancer Is Associated with Loss of HLA Class I Expression, T-Cell Exclusion and Stromal Changes. Int. J. Mol. Sci. 2021, 22, 7248. [Google Scholar] [CrossRef] [PubMed]
- Sim, S.H.; Ahn, Y.O.; Yoon, J.; Kim, T.M.; Lee, S.H.; Kim, D.W.; Heo, D.S. Influence of Chemotherapy on Nitric Oxide Synthase, Indole-Amine-2,3-Dioxygenase and CD124 Expression in Granulocytes and Monocytes of Non-Small Cell Lung Cancer. Cancer Sci. 2012, 103, 155–160. [Google Scholar] [CrossRef]
- Su, P.L.; Chen, J.Y.; Chu, C.Y.; Chen, Y.L.; Chen, W.L.; Lin, K.Y.; Ho, C.L.; Tsai, J.S.; Yang, S.C.; Chen, C.W.; et al. The Impact of Driver Mutation on the Treatment Outcome of Early-Stage Lung Cancer Patients Receiving Neoadjuvant Immunotherapy and Chemotherapy. Sci. Rep. 2022, 12, 1–9. [Google Scholar] [CrossRef]
- Broz, M.L.; Binnewies, M.; Boldajipour, B.; Nelson, A.E.; Pollack, J.L.; Erle, D.J.; Barczak, A.; Rosenblum, M.D.; Daud, A.; Barber, D.L.; et al. Dissecting the Tumor Myeloid Compartment Reveals Rare Activating Antigen-Presenting Cells Critical for T Cell Immunity. Cancer Cell 2014, 26, 938. [Google Scholar] [CrossRef]
- Gocher, A.M.; Workman, C.J.; Vignali, D.A.A. Interferon-γ: Teammate or Opponent in the Tumour Microenvironment? Nat. Rev. Immunol. 2022, 22, 158–172. [Google Scholar] [CrossRef]
- Zhang, Y.; Guan, X.Y.; Jiang, P. Cytokine and Chemokine Signals of T-Cell Exclusion in Tumors. Front. Immunol. 2020, 11, 594609. [Google Scholar] [CrossRef]
- Jensen, H.K.; Donskov, F.; Nordsmark, M.; Marcussen, N.; von Maase, H. der Increased Intratumoral FOXP3-Positive Regulatory Immune Cells during Interleukin-2 Treatment in Metastatic Renal Cell Carcinoma. Clin. Cancer Res. 2009, 15, 1052–1058. [Google Scholar] [CrossRef]
- Weber, R.; Fleming, V.; Hu, X.; Nagibin, V.; Groth, C.; Altevogt, P.; Utikal, J.; Umansky, V. Myeloid-Derived Suppressor Cells Hinder the Anti-Cancer Activity of Immune Checkpoint Inhibitors. Front. Immunol. 2018, 9, 1310. [Google Scholar] [CrossRef]
- Maby, P.; Bindea, G.; Mlecnik, B.; Galon, J. License to Kill: Microsatellite Instability and Immune Contexture. Oncoimmunology 2021, 10, 1905935. [Google Scholar] [CrossRef] [PubMed]
- Jayasingam, S.D.; Citartan, M.; Thang, T.H.; Mat Zin, A.A.; Ang, K.C.; Ch’ng, E.S. Evaluating the Polarization of Tumor-Associated Macrophages Into M1 and M2 Phenotypes in Human Cancer Tissue: Technicalities and Challenges in Routine Clinical Practice. Front. Oncol. 2020, 9, 1512. [Google Scholar] [CrossRef]
- Najafi, M.; Hashemi Goradel, N.; Farhood, B.; Salehi, E.; Nashtaei, M.S.; Khanlarkhani, N.; Khezri, Z.; Majidpoor, J.; Abouzaripour, M.; Habibi, M.; et al. Macrophage Polarity in Cancer: A Review. J. Cell. Biochem. 2019, 120, 2756–2765. [Google Scholar] [CrossRef] [PubMed]
- Allavena, P.; Mantovani, A. Immunology in the Clinic Review Series; Focus on Cancer: Tumour-Associated Macrophages: Undisputed Stars of the Inflammatory Tumour Microenvironment. Clin. Exp. Immunol. 2012, 167, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Través, P.G.; Luque, A.; Hortelano, S. Macrophages, Inflammation, and Tumor Suppressors: ARF, a New Playerin the Game. Mediators. Inflamm. 2012, 2012, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, I.; Yano, T.; Murata, M.; Miyamoto, M.; Ishida, T.; Sugimachi, K.; Kimura, G.; Nomoto, K. Phenotypes of Lymphocytes Infiltrating Non-Small Cell Lung Cancer Tissues and Its Variation with Histological Types of Cancer. Lung Cancer 1993, 10, 13–19. [Google Scholar] [CrossRef]
- Al-Shibli, K.I.; Donnem, T.; Al-Saad, S.; Persson, M.; Bremnes, R.M.; Busund, L.T. Prognostic Effect of Epithelial and Stromal Lymphocyte Infiltration in Non-Small Cell Lung Cancer. Clin. Cancer Res. 2008, 14, 5220–5227. [Google Scholar] [CrossRef]
- Gatault, S.; Legrand, F.; Delbeke, M. Involvement Ofeosinophils in the Anti-Tumor Response. Cancer Immunol. Immunother. 2012, 61, 1527–1534. [Google Scholar] [CrossRef]
- Fridlender, Z.G.; Albelda, S.M. Tumor-Associated Neutrophils: Friend or Foe? Carcinogenesis 2012, 33, 949–955. [Google Scholar] [CrossRef]
- Aerts, J.G.; Hegmans, J.P. Tumor-Specific Cytotoxic t Cells Are Crucial for Efficacy of Immunomodulatory Antibodies in Patients with Lung Cancer. Cancer Res. 2013, 73, 2381–2388. [Google Scholar] [CrossRef]
- Salagianni, M.; Baxevanis, C.N.; Papamichail, M.; Perez, S.A. New Insights into the Role of NK Cells in Cancer Immunotherapy. Oncoimmunology 2012, 1, 205–207. [Google Scholar] [CrossRef] [PubMed]
- Motohashi, S.; Okamoto, Y.; Yoshino, I. Antitumorimmune Responses Induced by INKT Cell-Basedimmunotherapy for Lung Cancer and Head and Neck Cancer. Clin. Immunol. 2011, 140, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Cook, D.N.; Nakano, H. Pulmonary Dendritic Cells. Comp. Biol. Norm. Lung Second. Ed. 2015, 172, 651–664. [Google Scholar] [CrossRef]
- Montenegro, G.B.; Farid, S.; Liu, S.V. Immunotherapy in Lung Cancer. J. Surg. Oncol. 2021, 123, 718–729. [Google Scholar] [CrossRef]
- Tartour, E.; Zitvogel, L. Lung Cancer: Potential Targets Forimmunotherapy. Lancet Respir. Med. 2013, 1, 551–563. [Google Scholar] [CrossRef]
- Brahmer, J.R. PD-1-Targeted Immunotherapy: Recentclinical Findings. Clin. Adv. Hematol. Oncol. 2012, 10, 674–675. [Google Scholar] [PubMed]
- Dasanu, C.A.; Sethi, N.; Ahmed, N. Immune Alterations Andemerging Immunotherapeutic Approaches in Lung Cancer. Expert Opin. Biol. Ther. 2012, 12, 923–937. [Google Scholar] [CrossRef] [PubMed]
- Declerck, S.; Vansteenkiste, J. Immunotherapy for Lung Cancer: Ongoing Clinical Trials. Future Oncol. 2014, 10, 91–105. [Google Scholar] [CrossRef]
- Shih, K.; Arkenau, H.T.; Infante, J.R. Clinical Impact of Checkpoint Inhibitors as Novel Cancer Therapies. Drugs 2014, 74, 1993–2013. [Google Scholar] [CrossRef]
- Hoser, G.; Wasilewska, D.; Domagała-Kulawik, J. Expression of FAs Receptor on Peripheral Blood Lymphocytes from Patients with Non-Small Cell Lung Cancer. Folia Histochem. Cytobiol. 2004, 42, 249–252. [Google Scholar]
- Gajewski, T.F.; Meng, Y.; Harlin, H. Immune Suppression in the Tumor Microenvironment. J. Immunother. 2006, 29, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Erfani, N.; Khademi, B.; Haghshenas, M.R.; Mojtahedi, Z.; Khademi, B.; Ghaderi, A. Intracellular CTLA4 and Regulatory T Cells in Patients with Laryngeal Squamous Cell Carcinoma. Immunol. Invest. 2013, 42, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Erfani, N.; Mehrabadi, S.M.; Ghayumi, M.A.; Haghshenas, M.R.; Mojtahedi, Z.; Ghaderi, A.; Amani, D. Increase of Regulatory T Cells in Metastatic Stage and CTLA-4 over Expression in Lymphocytes of Patients with Non-Small Cell Lung Cancer (NSCLC). Lung Cancer 2012, 77, 306–311. [Google Scholar] [CrossRef] [PubMed]
- Mocellin, S.; Nitti, D. CTLA-4 Blockade and the Renaissance of Cancer Immunotherapy. Biochim. Biophys. Acta Rev. Cancer 2013, 1836, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Baecher-Allan, C.; Viglietta, V.; Hafler, D.A. Human CD4+CD25+ Regulatory T Cells. Semin. Immunol. 2004, 16, 89–98. [Google Scholar] [CrossRef]
- Chambers, C.A.; Kuhns, M.S.; Egen, J.G. CTLA-4- Mediated Inhibition in Regulation of T Cell Responses. Mech. Manip. Tumor Immunotherapy. Annu. Rev. Immunol. 2001, 19, 565–594. [Google Scholar]
- Egen, J.G.; Kuhns, M.S.; Allison, J.P. CTLA-4: New Insights into Its Biological Function and Use in Tumor Immunotherapy. Nat. Immunol. 2002, 3, 611–618. [Google Scholar] [CrossRef]
- Iida, T.; Ohno, H.; Nakaseko, C.; Sakuma, M.; Takeda-Ezaki, M.; Arase, H.; Kominami, E.; Fujisawa, T.; Saito, T. Regulation of Cell Surface Expression of CTLA-4 by Secretion of CTLA-4-Containing Lysosomes Upon Activation of CD4 + T Cells. J. Immunol. 2000, 165, 5062–5068. [Google Scholar] [CrossRef]
- Wang, X.B.; Zheng, C.Y.; Giscombe, R.; Lefvert, A.K. Regulation of Surface and Intracellular Expression of CTLA-4 on Human Peripheral T Cells. Scand. J. Immunol. 2001, 54, 453–458. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the Tumor Immune Microenvironment (TIME) for Effective Therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Ness, N.; Andersen, S.; Valkov, A.; Nordby, Y.; Donnem, T.; Al-Saad, S.; Busund, L.T.; Bremnes, R.M.; Richardsen, E. Infiltration of CD8+ Lymphocytes Is an Independent Prognostic Factor of Biochemical Failure-Free Survival in Prostate Cancer. Prostate 2014, 74, 1452–1461. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, S.A.; Frigola, X.; Bonne-Annee, S.; Mercader, M.; Kuntz, S.M.; Krambeck, A.E.; Sengupta, S.; Dong, H.; Cheville, J.C.; Lohse, C.M.; et al. Tumor-Infiltrating Foxp3-CD4+CD25+ T Cells Predict Poor Survival in Renal Cell Carcinoma. Clin. Cancer Res. 2007, 13, 2075–2081. [Google Scholar] [CrossRef]
- Chen, D.; Wang, Y.; Zhang, X.; Ding, Q.; Wang, X.; Xue, Y.; Wang, W.; Mao, Y.; Chen, C.; Chen, Y. Characterization of Tumor Microenvironment in Lung Adenocarcinoma Identifies Immune Signatures to Predict Clinical Outcomes and Therapeutic Responses. Front. Oncol. 2021, 11, 356. [Google Scholar] [CrossRef] [PubMed]
- Galli, F.; Aguilera, J.V.; Palermo, B.; Markovic, S.N.; Nisticò, P.; Signore, A. Relevance of Immune Cell and Tumor Microenvironment Imaging in the New Era of Immunotherapy. J. Exp. Clin. Cancer Res. 2020, 39, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.S.; Kim, A.; Shin, J.Y.; Kim, Y.T. Comprehensive Analysis of the Tumor Immune Micro-Environment in Non-Small Cell Lung Cancer for Efficacy of Checkpoint Inhibitor. Sci. Rep. 2018, 8, 14576. [Google Scholar] [CrossRef]
- Wojas-Krawczyk, K.; Paśnik, I.; Kucharczyk, T.; Wieleba, I.; Krzyżanowska, N.; Gil, M.; Krawczyk, P.; Milanowski, J. Immunoprofiling: An Encouraging Method for Predictive Factors Examination in Lung Cancer Patients Treated with Immunotherapy. Int. J. Mol. Sci. 2021, 22, 9133. [Google Scholar] [CrossRef]
- Mella, M.; Kauppila, J.H.; Karihtala, P.; Lehenkari, P.; Jukkola-Vuorinen, A.; Soini, Y.; Auvinen, P.; Vaarala, M.H.; Ronkainen, H.; Kauppila, S.; et al. Tumor Infiltrating CD8+ T Lymphocyte Count Is Independent of Tumor TLR9 Status in Treatment Na�ve Triple Negative Breast Cancer and Renal Cell Carcinoma. Oncoimmunology 2015, 4, 1002726. [Google Scholar] [CrossRef]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT: Co-Inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef]
- Brambilla, E.; le Teuff, G.; Marguet, S.; Lantuejoul, S.; Dunant, A.; Graziano, S.; Pirker, R.; Douillard, J.Y.; le Chevalier, T.; Filipits, M.; et al. Prognostic Effect of Tumor Lymphocytic Infiltration in Resectable Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2016, 34, 1223–1230. [Google Scholar] [CrossRef]
- Kinoshita, T.; Kudo-Saito, C.; Muramatsu, R.; Fujita, T.; Saito, M.; Nagumo, H.; Sakurai, T.; Noji, S.; Takahata, E.; Yaguchi, T.; et al. Determination of Poor Prognostic Immune Features of Tumour Microenvironment in Non-Smoking Patients with Lung Adenocarcinoma. Eur. J. Cancer 2017, 86, 15–27. [Google Scholar] [CrossRef]
- Lizotte, P.H.; Ivanova, E.V.; Awad, M.M.; Jones, R.E.; Keogh, L.; Liu, H.; Dries, R.; Almonte, C.; Herter-Sprie, G.S.; Santos, A.; et al. Multiparametric Profiling of Non–Small-Cell Lung Cancers Reveals Distinct Immunophenotypes. JCI Insight 2016, 1, 89014. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Kadota, K.; Sima, C.S.; Nitadori, J.I.; Rusch, V.W.; Travis, W.D.; Sadelain, M.; Adusumilli, P.S. Clinical Impact of Immune Microenvironment in Stage i Lung Adenocarcinoma: Tumor Interleukin-12 Receptor Β2 (IL-12Rβ2), IL-7R, and Stromal FoxP3/CD3 Ratio Are Independent Predictors of Recurrence. J. Clin. Oncol. 2013, 31, 490–498. [Google Scholar] [CrossRef]
- Riaz, N.; Havel, J.J.; Makarov, V.; Desrichard, A.; Urba, W.J.; Sims, J.S.; Hodi, F.S.; Martín-Algarra, S.; Mandal, R.; Sharfman, W.H.; et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 2017, 171, 934–949.e15. [Google Scholar] [CrossRef]
- Kümpers, C.; Jokic, M.; Haase, O.; Offermann, A.; Vogel, W.; Grätz, V.; Langan, E.A.; Perner, S.; Terheyden, P. Immune Cell Infiltration of the Primary Tumor, Not PD-L1 Status, Is Associated With Improved Response to Checkpoint Inhibition in Metastatic Melanoma. Front. Med. 2019, 6, 27. [Google Scholar] [CrossRef]
- Lam, V.K.; Zhang, J. Blood-Based Tumor Mutation Burden: Continued Progress toward Personalizing Immunotherapy in Non-Small Cell Lung Cancer. J. Thorac. Dis. 2019, 11, 2208–2211. [Google Scholar] [CrossRef]
- van Campenhout, C.; Meléndez, B.; Remmelink, M.; Salmon, I.; D’Haene, N. Blood Tumor Mutational Burden: Are We Ready for Clinical Implementation? J. Thorac. Dis. 2019, 11, S1906–S1908. [Google Scholar] [CrossRef]
- Kooshkaki, O.; Derakhshani, A.; Hosseinkhani, N.; Torabi, M.; Safaei, S.; Brunetti, O.; Racanelli, V.; Silvestris, N.; Baradaran, B. Combination of Ipilimumab and Nivolumab in Cancers: From Clinical Practice to Ongoing Clinical Trials. Int J Mol Sci 2020, 21, 4427. [Google Scholar] [CrossRef]
- Hwang, S.; Kwon, A.Y.; Jeong, J.Y.; Kim, S.; Kang, H.; Park, J.; Kim, J.H.; Han, O.J.; Lim, S.M.; An, H.J. Immune Gene Signatures for Predicting Durable Clinical Benefit of Anti-PD-1 Immunotherapy in Patients with Non-Small Cell Lung Cancer. Sci. Rep. 2020, 10, 1–10. [Google Scholar] [CrossRef]
- Althammer, S.; Tan, T.H.; Spitzmüller, A.; Rognoni, L.; Wiestler, T.; Herz, T.; Widmaier, M.; Rebelatto, M.C.; Kaplon, H.; Damotte, D.; et al. Automated Image Analysis of NSCLC Biopsies to Predict Response to Anti-PD-L1 Therapy. J. Immunother. Cancer 2019, 7, 121. [Google Scholar] [CrossRef]
- Gettinger, S.; Horn, L.; Jackman, D.; Spigel, D.; Antonia, S.; Hellmann, M.; Powderly, J.; Heist, R.; Sequist, L.V.; Smith, D.C.; et al. Five-Year Follow-up of Nivolumab in Previously Treated Advanced Non–Small-Cell Lung Cancer: Results from the CA209-003 Study. J. Clin. Oncol. 2018, 36, 1675–1684. [Google Scholar] [CrossRef]
- Garon, E.B.; Rizvi, N.A.; Hui, R. Pembrolizumab for the Treatmentof Non-small-cell Lung Cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Gordon, M.S.; Fine, G.D. A Study of MPDL3280A, Anengineered PD-L1 Antibody in Patients with Locally Advanced Ormetastatic Tumors. J. Clin. Oncol. 2013, 31, 3000. [Google Scholar] [CrossRef]
- Brahmer, J.; Reckamp, K.L.; Baas, P. Nivolumab versus Docetaxelin Advanced Squamous-cell Non-small-cell Lung Cancer. N. Engl. J. Med. 2015, 373, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Vansteenkiste, J.; Wauters, E.; Park, K.; Rittmeyer, A.; Sandler, A.; Spira, A. Prospects and Progress of Atezolizumab in Non-Small Cell Lung Cancer. Expert Opin. Biol. Ther. 2017, 17, 781–789. [Google Scholar] [CrossRef]
- Barlesi, F.; Vansteenkiste, J.; Spigel, D. Avelumab versus Docetaxelin Patients with Platinum-treated Advanced Non-small-celllung Cancer (JAVELIN Lung 200): An Open-label, Randomised, Phase3 Study. Lancet Oncol. 2018, 19, 1468–1479. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Cho, B.C.; Reinmuth, N. Durvalumab with or Withouttremelimumab vs Standard Chemotherapy in First-line Treatment Ofmetastatic Non-small Cell Lung Cancer: The MYSTIC Phase 3 Randomizedclinical Trial. JAMA Oncol. 2020, 6, 661–674. [Google Scholar] [CrossRef]
- Araz, M.; Karakurt Eryilmaz, M. Immunotherapy Era in the Treatment of Small Cell Lung Cancer. Med. Oncol. 2021, 38, 1–7. [Google Scholar] [CrossRef]
- Peters, S.; Pujol, J.-L.; Dafni, U. Consolidation Ipilimumab Andnivolumab vs Observation in Limited Stage SCLC after Chemoradiotherapy:Results from the ETOP/IFCT 4–12. STIMULI Trial. Ann. Oncol. 2021, 10, 1016. [Google Scholar]
- Paz-Ares, L.; Dvorkin, M.; Chen, Y. Durvalumab plus Platinum-Etoposide versus Platinum-Etoposide in First-Line Treatmentof Extensive-Stage Small-Cell Lung Cancer (CASPIAN):A Randomised, Controlled, Open-Label, Phase 3 Trial. Lancet 2019, 394, 1929–1939. [Google Scholar] [CrossRef]
- Leal, T.; Wang, Y.; Afshin Dowlati, A. Randomized Phase IIclinical Trial of Cisplatin/Carboplatin and Etoposide (CE) Alone Orin Combination with Nivolumab as Frontline Therapy for Extensivestagesmall Cell Lung Cancer (ES-SCLC): ECOG-ACRIN EA5161. J. Clin. Oncol. 2020, 38. Available online: https://ascopubs.org/doi/abs/10.1200/JCO.2020.38.15_suppl.9000 (accessed on 1 July 2022). [CrossRef]
- Ready, N.; Owonikoko, T.K.; Postmus, P.E.; Reck, M.; Peters, S.; Pieters, A.; Selvaggi, G.; Fairchild, J.P.; Govindan, R. CheckMate 451: A Randomized, Double-Blind, Phase III Trial of Nivolumab (Nivo), Nivo plus Ipilimumab (Ipi), or Placebo as Maintenance Therapy in Patients (Pts) with Extensive-Stage Disease Small Cell Lung Cancer (ED-SCLC) after First-Line Platinum-Based d. J. Clin. Oncol. 2016, 34, TPS8579. [Google Scholar] [CrossRef]
- Antonia, S.J.; Lopez-Martin, J.A.; Bendell, J. Nivolumab Aloneand Nivolumab plus Ipilimumab in Recurrent Small-Cell Lung Cancer(CheckMate 032): A Multicentre, Open-Label, Phase 1/2 Trial. Lancet Oncol. 2016, 17, 883–895. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, J. Current Status and Future Directions of Cancer Immunotherapy. J. Cancer 2018, 9, 1773–1781. [Google Scholar] [CrossRef] [PubMed]
- Taefehshokr, S.; Parhizkar, A.; Hayati, S.; Mousapour, M.; Mahmoudpour, A.; Eleid, L.; Rahmanpour, D.; Fattahi, S.; Shabani, H.; Taefehshokr, N. Cancer Immunotherapy: Challenges and Limitations. Pathol. Res. Pract. 2022, 229, 153723. [Google Scholar] [CrossRef]
- Home-ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/results?cond=Non+Small+Cell+Lung+Cancer (accessed on 14 October 2022).
- Qu, J. Mechanism and Potential Predictive Biomarkers of Immune Checkpoint Inhibitors in NSCLC. Biomed. Pharmacother. 2020, 127, 109996. [Google Scholar] [CrossRef]
- Jain, P. Role of Immune-Checkpoint Inhibitors in Lung Cancer. Ther. Adv. Respir. Dis. 2018, 12, 1753465817750075. [Google Scholar] [CrossRef]
- Sharon, E. Immune Checkpoint Inhibitors in Clinical Trials. Chin. J. Cancer 2014, 33, 434–444. [Google Scholar] [CrossRef]
- Sławiński, G.; Wrona, A.; Dabrowska-Kugacka, A.; Raczak, G.; Lewicka, E. Immune Checkpoint Inhibitors and Cardiac Toxicity in Patients Treated for Non-Small Lung Cancer: A Review. Int. J. Mol. Sci. 2020, 21, 7195. [Google Scholar] [CrossRef]
- Carbone, D.P. First-Line Nivolumab in Stage IV or Recurrent Non-Small Cell Lung Cancer. Oncol. Times 2017, 39, 28–29. [Google Scholar] [CrossRef]
- Marabelle, A.; Fakih, M.; Lopez, J.; Shah, M.; Shapira-Frommer, R.; Nakagawa, K.; Chung, H.C.; Kindler, H.L.; Lopez-Martin, J.A.; Miller, W.H.; et al. Association of Tumour Mutational Burden with Outcomes in Patients with Advanced Solid Tumours Treated with Pembrolizumab: Prospective Biomarker Analysis of the Multicohort, Open-Label, Phase 2 KEYNOTE-158 Study. Lancet Oncol. 2020, 21, 1353–1365. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer Immunology. Mutational Landscape Determines Sensitivity to PD-1 Blockade in Non-Small Cell Lung Cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Akinleye, A.; Rasool, Z. Immune Checkpoint Inhibitors of PD-L1 as Cancer Therapeutics. J. Hematol. Oncol. 2019, 12, 92. [Google Scholar] [CrossRef] [PubMed]
- Rosenblatt, J.; Avigan, D. Targeting the PD-1/PD-L1 Axis in Multiple Myeloma: A Dream or a Reality? Blood 2017, 129, 275–279. [Google Scholar] [CrossRef]
- Brahmer, J.R. Safety and Activity of Anti–PD-L1 Antibody in Patients with Advanced Cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- LoConte, N.; Sahasrabudhe, K. Faculty Opinions Recommendation of Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. Fac. Opin.-Post-Publ. Peer Rev. Biomed. Lit. 2018, 375, 1823–1833. [Google Scholar] [CrossRef]
- Mamdani, H. Immunotherapy in Lung Cancer: Current Landscape and Future Directions. Front. Immunol. 2022, 13, 823618. [Google Scholar] [CrossRef]
- Anagnostou, V.K.; Brahmer, J.R. Cancer Immunotherapy: A Future Paradigm Shift in the Treatment of Non-Small Cell Lung Cancer. Clin. Cancer Res. 2015, 21, 976–984. [Google Scholar] [CrossRef]
- Yang, J. Management of Adverse Events in Cancer Patients Treated With PD-1/PD-L1 Blockade: Focus on Asian Populations. Front. Pharmacol. 2019, 10, 726. [Google Scholar] [CrossRef]
- Theelen, W.S.M.E.; Baas, P. Pembrolizumab Monotherapy for PD-L1 ≥50% Non-Small Cell Lung Cancer, Undisputed First Choice? Ann. Transl. Med. 2019, 7, S140. [Google Scholar] [CrossRef]
- Chen, R. Emerging Therapeutic Agents for Advanced Non-Small Cell Lung Cancer. J. Hematol. Oncol. 2020, 13, 58. [Google Scholar] [CrossRef]
- Cabel, L. Clinical Potential of Circulating Tumour DNA in Patients Receiving Anticancer Immunotherapy. Nat. Rev. Clin. Oncol. 2018, 15, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Brueckl, W.M.; Ficker, J.H.; Zeitler, G. Clinically Relevant Prognostic and Predictive Markers for Immune-Checkpoint-Inhibitor (ICI) Therapy in Non-Small Cell Lung Cancer (NSCLC). BMC Cancer 2020, 20, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Zulato, E. Early Assessment of KRAS Mutation in CfDNA Correlates with Risk of Progression and Death in Advanced Non-Small-Cell Lung Cancer. Br. J. Cancer 2020, 123, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Reungwetwattana, T.; Dy, G.K. Targeted Therapies in Development for Non-Small Cell Lung Cancer. J. Carcinog. 2013, 12, 22. [Google Scholar] [PubMed]
- Bonomi, P.D.; MacE, J.; Mandanas, R.A.; Min, M.; Olsen, M.; Youssoufian, H.; Katz, T.L.; Sheth, G.; Lee, H.J. Randomized Phase II Study of Cetuximab and Bevacizumab in Combination with Two Regimens of Paclitaxel and Carboplatin in Chemonaive Patients with Stage IIIB/IV Non-Small-Cell Lung Cancer. J. Thorac. Oncol. 2013, 8, 338–345. [Google Scholar] [CrossRef]
- Garrett, C.R.; Siu, L.L.; El-Khoueiry, A.; Buter, J.; Rocha-Lima, C.M.; Marshall, J.; Lorusso, P.; Major, P.; Chemidlin, J.; Mokliatchouk, O.; et al. Phase I Dose-Escalation Study to Determine the Safety, Pharmacokinetics and Pharmacodynamics of Brivanib Alaninate in Combination with Full-Dose Cetuximab in Patients with Advanced Gastrointestinal Malignancies Who Have Failed Prior Therapy. Br. J. Cancer 2011, 105, 44–52. [Google Scholar] [CrossRef]
- Janjigian, Y.Y.; Azzoli, C.G.; Krug, L.M.; Pereira, L.K.; Rizvi, N.A.; Pietanza, M.C.; Kris, M.G.; Ginsberg, M.S.; Pao, W.; Miller, V.A.; et al. Phase I/II Trial of Cetuximab and Erlotinib in Patients with Lung Adenocarcinoma and Acquired Resistance to Erlotinib. Clin. Cancer Res. 2011, 17, 2521–2527. [Google Scholar] [CrossRef]
- Hilbe, W.; Pall, G.; Kocher, F.; Pircher, A.; Zabernigg, A.; Schmid, T.; Schumacher, M.; Jamnig, H.; Fiegl, M.; Gächter, A.; et al. Multicenter Phase II Study Evaluating Two Cycles of Docetaxel, Cisplatin and Cetuximab as Induction Regimen Prior to Surgery in Chemotherapy-Naive Patients with NSCLC Stage IB-IIIA (INN06-Study). PLoS ONE 2015, 10, e0125364. [Google Scholar] [CrossRef]
- Silva, A.P.S.; Coelho, P.V.; Anazetti, M.; Simioni, P.U. Targeted Therapies for the Treatment of Non-Small-Cell Lung Cancer: Monoclonal Antibodies and Biological Inhibitors. Hum. Vaccin. Immunother. 2017, 13, 843–853. [Google Scholar] [CrossRef]
- Sul, J. FDA Approval Summary: Pembrolizumab for the Treatment of Patients With Metastatic Non-Small Cell Lung Cancer Whose Tumors Express Programmed Death-Ligand 1. Oncologist 2016, 21, 643–650. [Google Scholar] [CrossRef]
- Langer, C.J.; Olszanski, A.J.; Karp, D.D.; Benner, R.J.; Scranton, J.R.; Novello, S.; Park, K.; Krzakowski, M.; Jassem, J.; Mok, T. Randomized, Phase III Trial of First-Line Figitumumab in Combination With Paclitaxel and Carboplatin Versus Paclitaxel and Carboplatin Alone in Patients With Advanced Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2014, 32, 2059. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Riddell, S.R. Engineering CAR-T Cells: Design Concepts. Trends Immunol. 2015, 36, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Ying, Z.; Huang, X.F.; Xiang, X.; Liu, Y.; Kang, X.; Song, Y.; Guo, X.; Liu, H.; Ding, N.; Zhang, T.; et al. A Safe and Potent Anti-CD19 CAR T Cell Therapy. Nat. Med. 2019, 25, 947–953. [Google Scholar] [CrossRef]
- Kim, D.W.; Cho, J.Y. Recent Advances in Allogeneic CAR-T Cells. Biomolecules 2020, 10, 263. [Google Scholar] [CrossRef] [PubMed]
- Tokarew, N.; Ogonek, J.; Endres, S.; von Bergwelt-Baildon, M.; Kobold, S. Teaching an Old Dog New Tricks: Next-Generation CAR T Cells. Br. J. Cancer 2019, 120, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Qu, J. Chimeric Antigen Receptor (CAR)-T-Cell Therapy in Non-Small-Cell Lung Cancer (NSCLC): Current Status and Future Perspectives. Cancer Immunol. Immunother. 2020, 70, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Kachala, S.S. Mesothelin Overexpression Is a Marker of Tumor Aggressiveness and Is Associated with Reduced Recurrence-Free and Overall Survival in Early-Stage Lung Adenocarcinoma. Clin. Cancer Res. 2014, 20, 1020–1028. [Google Scholar] [CrossRef]
- Wei, X. PSCA and MUC1 in Non-Small-Cell Lung Cancer as Targets of Chimeric Antigen Receptor T Cells. Oncoimmunology 2017, 6, 1284722. [Google Scholar] [CrossRef]
- Wallstabe, L. ROR1-CAR T Cells Are Effective against Lung and Breast Cancer in Advanced Microphysiologic 3D Tumor Models. JCI Insight 2019, 4, 18. [Google Scholar] [CrossRef]
- Feldmann, A.; Arndt, C.; Koristka, S.; Berndt, N.; Bergmann, R.; Bachmann, M.P. Conventional CARs versus Modular CARs. Cancer Immunol. Immunother. 2019, 68, 1713–1719. [Google Scholar] [CrossRef]
- Rafiq, S.; Hackett, C.S.; Brentjens, R.J. Engineering Strategies to Overcome the Current Roadblocks in CAR T Cell Therapy. Nat. Rev. Clin. Oncol. 2020, 17, 147–167. [Google Scholar] [CrossRef] [PubMed]
- MacKay, M.; Afshinnekoo, E.; Rub, J.; Hassan, C.; Khunte, M.; Baskaran, N.; Owens, B.; Liu, L.; Roboz, G.J.; Guzman, M.L.; et al. The Therapeutic Landscape for Cells Engineered with Chimeric Antigen Receptors. Nat. Biotechnol. 2020, 38, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Lindner, S.E.; Johnson, S.M.; Brown, C.E.; Wang, L.D. Chimeric Antigen Receptor Signaling: Functional Consequences and Design Implications. Sci. Adv. 2020, 6, eaaz3223. [Google Scholar] [CrossRef] [PubMed]
- Mokhtari, R.B.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination Therapy in Combating Cancer. Oncotarget 2017, 8, 38022. [Google Scholar] [CrossRef]
- Fischer, J.R. Constitutive Secretion of Bioactive Transforming Growth Factor Beta 1 by Small Cell Lung Cancer Cell Lines. Eur. J. Cancer 1994, 30, 2125–2129. [Google Scholar] [CrossRef]
- Simmons, O.; Magee, M.; Nemunaitis, J. Current Vaccine Updates for Lung Cancer. Expert Rev. Vaccines 2010, 9, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Fakhrai, H.; Mantil, J.C.; Liu, L.; Nicholson, G.L.; Murphy-Satter, C.S.; Ruppert, J.; Shawler, D.L. Phase I Clinical Trial of a TGF-Beta Antisense-Modified Tumor Cell Vaccine in Patients with Advanced Glioma. Cancer Gene Ther. 2006, 13, 1052–1060. [Google Scholar] [CrossRef]
- Nemunaitis, J.; Dillman, R.O.; Schwarzenberger, P.O.; Senzer, N.; Cunningham, C.; Cutler, J.; Tong, A.; Kumar, P.; Pappen, B.; Hamilton, C.; et al. Phase II Study of Belagenpumatucel-L, a Transforming Growth Factor Beta-2 Antisense Gene-Modified Allogeneic Tumor Cell Vaccine in Non-Small-Cell Lung Cancer. J Clin Oncol 2006, 24, 4721–4730. [Google Scholar] [CrossRef]
- Kelly, R.J.; Giaccone, G. Lung Cancer Vaccines. Cancer J. 2011, 17, 302–308. [Google Scholar] [CrossRef]
- Giaccone, G. A Phase III Study of Belagenpumatucel-L, an Allogeneic Tumour Cell Vaccine, as Maintenance Therapy for Non-Small Cell Lung Cancer. Eur. J. Cancer 2015, 51, 2321–2329. [Google Scholar] [CrossRef]
- Nemunaitis, J.; Nemunaitis, M.; Senzer, N.; Snitz, P.; Bedell, C.; Kumar, P.; Pappen, B.; Maples, P.B.; Shawler, D.; Fakhrai, H. Phase II Trial of Belagenpumatucel-L, a TGF-Β2 Antisense Gene Modified Allogeneic Tumor Vaccine in Advanced Non Small Cell Lung Cancer (NSCLC) Patients. Cancer Gene Ther. 2009, 16, 620–624. [Google Scholar] [CrossRef] [PubMed]
- Esfandiary, A.; Ghafouri-Fard, S. MAGE-A3: An Immunogenic Target Used in Clinical Practice. Immunotherapy 2015, 7, 683–704. [Google Scholar] [CrossRef] [PubMed]
- Vansteenkiste, J.F.; Cho, B.C.; Vanakesa, T.; de Pas, T.; Zielinski, M.; Kim, M.S.; Jassem, J.; Yoshimura, M.; Dahabreh, J.; Nakayama, H.; et al. Efficacy of the MAGE-A3 Cancer Immunotherapeutic as Adjuvant Therapy in Patients with Resected MAGE-A3-Positive Non-Small-Cell Lung Cancer (MAGRIT): A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet Oncol. 2016, 17, 822–835. [Google Scholar] [CrossRef]
- Takahashi, K.; Shichijo, S.; Noguchi, M.; Hirohata, M.; Itoh, K. Identification of MAGE-1 and MAGE-4 Proteins in Spermatogonia and Primary Spermatocytes of Testis. Cancer Res. 1995, 55, 3478–3482. [Google Scholar]
- Tomita, Y.; Kimura, M.; Tanikawa, T.; Nishiyama, T.; Morishita, H.; Takeda, M.; Fujiwara, M.; Sato, S. Immunohistochemical Detection of Intercellular Adhesion Molecule-1 (ICAM- 1) and Major Histocompatibility Complex Class I Antigens in Seminoma. J. Urol. 1993, 149, 659–663. [Google Scholar] [CrossRef]
- Weon, J.L.; Potts, P.R. The MAGE Protein Family and Cancer. Curr. Opin. Cell Biol. 2015, 37, 1. [Google Scholar] [CrossRef]
- Koop, A.; Sellami, N.; Adam-Klages, S.; Lettau, M.; Kabelitz, D.; Janssen, O.; Heidebrecht, H.J. Down-Regulation of the Cancer/Testis Antigen 45 (CT45) Is Associated with Altered Tumor Cell Morphology, Adhesion and Migration. Cell Commun. Signal. 2013, 11, 4. [Google Scholar] [CrossRef]
- Caballero, O.L.; Cohen, T.; Gurung, S.; Chua, R.; Lee, P.; Chen, Y.T.; Jat, P.; Simpson, A.J.G. Effects of CT-Xp Gene Knock down in Melanoma Cell Lines. Oncotarget 2013, 4, 531–541. [Google Scholar] [CrossRef][Green Version]
- Vansteenkiste, J.; Zielinski, M.; Linder, A.; Dahabreh, J.; Gonzalez, E.E.; Malinowski, W.; Lopez-Brea, M.; Vanakesa, T.; Jassem, J.; Kalofonos, H.; et al. Adjuvant MAGE-A3 Immunotherapy in Resected Non–Small-Cell Lung Cancer: Phase II Randomized Study Results. J. Clin. Oncol. 2013, 31, 2396–2403. [Google Scholar] [CrossRef]
- Vansteenkiste, J.F.; Zielinski, M.; Dahabreh, I.J.; Linder, A.; Lehmann, F.; Gruselle, O.; Therasse, P.; Louahed, J.; Brichard, V.G. Association of Gene Expression Signature and Clinical Efficacy of MAGE-A3 Antigen-Specific Cancer Immunotherapeutic (ASCI) as Adjuvant Therapy in Resected Stage IB/II Non-Small Cell Lung Cancer (NSCLC). J. Clin. Oncol. 2008, 26, 7501. [Google Scholar] [CrossRef]
- Tyagi, P.; Mirakhur, B. MAGRIT: The Largest-Ever Phase III Lung Cancer Trial Aims to Establish a Novel Tumor-Specific Approach to Therapy. Clin. Lung Cancer 2009, 10, 371–374. [Google Scholar] [CrossRef] [PubMed]
- Ramos, T.C.; Vinageras, E.N.; Ferrer, M.C.; Verdecia, B.G.; Rupalé, I.L.; Pérez, L.M.; Marinello, G.G.; Rodríguez, R.P.; Dávila, A.L. Treatment of NSCLC Patients with an EGF-Based Cancer Vaccine: Report of a Phase I Trial. Cancer Biol. Ther. 2006, 5, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Massarelli, E.; Papadimitrakopoulou, V.; Welsh, J.; Tang, C.; Tsao, A.S. Immunotherapy in Lung Cancer. Transl. Lung Cancer Res. 2014, 3, 53. [Google Scholar] [CrossRef] [PubMed]
- Saavedra, D.; Crombet, T. CIMAvax-EGF: A New Therapeutic Vaccine for Advanced Non-Small Cell Lung Cancer Patients. Front. Immunol. 2017, 8, 269. [Google Scholar] [CrossRef] [PubMed]
- Crombet Ramos, T.; Rodríguez, P.C.; Neninger Vinageras, E.; Garcia Verdecia, B.; Lage Davila, A. CIMAvax EGF (EGF-P64K) Vaccine for the Treatment of Non-Small-Cell Lung Cancer. Expert Rev. Vaccines 2015, 14, 1303–1311. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, P.C.; Rodríguez, G.; González, G.; Lage, A. Clinical Development and Perspectives of CIMAvax EGF, Cuban Vaccine for Non-Small-Cell Lung Cancer Therapy. MEDICC Rev. 2010, 12, 17–23. [Google Scholar] [CrossRef]
- González, G.; Crombet, T.; Catalá, M.; Mirabal, V.; Hernández, J.C.; González, Y.; Marinello, P.; Guillén, G.; Lage, A. A Novel Cancer Vaccine Composed of Human-Recombinant Epidermal Growth Factor Linked to a Carrier Protein: Report of a Pilot Clinical Trial. Ann. Oncol. 1998, 9, 431–435. [Google Scholar] [CrossRef]
- Gonzalez, G.; Crombet, T.; Torres, F.; Catala, M.; Alfonso, L.; Osorio, M.; Neninger, E.; Garcia, B.; Mulet, A.; Perez, R.; et al. Epidermal Growth Factor-Based Cancer Vaccine for Non-Small-Cell Lung Cancer Therapy. Ann. Oncol. 2003, 14, 461–466. [Google Scholar] [CrossRef]
- Neninger, E.; Verdecia, B.G.; Crombet, T.; Viada, C.; Pereda, S.; Leonard, I.; Mazorra, Z.; Fleites, G.; González, M.; Wilkinson, B.; et al. Combining an EGF-Based Cancer Vaccine with Chemotherapy in Advanced Nonsmall Cell Lung Cancer. J. Immunother. 2009, 32, 92–99. [Google Scholar] [CrossRef]
- Rodriguez, P.C.; Popa, X.; Martínez, O.; Mendoza, S.; Santiesteban, E.; Crespo, T.; Amador, R.M.; Fleytas, R.; Acosta, S.C.; Otero, Y.; et al. A Phase III Clinical Trial of the Epidermal Growth Factor Vaccine CIMAvax-EGF as Switch Maintenance Therapy in Advanced Non-Small Cell Lung Cancer Patients. Clin. Cancer Res. 2016, 22, 3782–3790. [Google Scholar] [CrossRef]
- Gabri, M.R.; Cacciavillano, W.; Chantada, G.L.; Alonso, D.F. Racotumomab for Treating Lung Cancer and Pediatric Refractory Malignancies. Expert Opin. Biol. Ther. 2016, 16, 573–578. [Google Scholar] [CrossRef] [PubMed]
- Samraj, A.N.; Pearce, O.M.T.; Läubli, H.; Critten-den, A.N.; Bergfeld, A.K.; Banda, K. A Red Meat-Derived Glycan Promotes Infl Ammation and Cancer Progression. Proc. Natl. Acad. Sci. USA 2015, 112, 542–547. [Google Scholar] [CrossRef] [PubMed]
- Segatori, V.I.; Cuello, H.A.; Gulino, C.A.; Albertó, M.; Venier, C.; Guthmann, M.D.; Demarco, I.A.; Alonso, D.F.; Gabri, M.R. Antibody-Dependent Cell-Mediated Cytotoxicity Induced by Active Immunotherapy Based on Racotumomab in Non-Small Cell Lung Cancer Patients. Cancer Immunol. Immunother. 2018, 67, 1285–1296. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Huang, H.; Zeng, X.; Ma, Y.; Zhao, X.; Huang, M. Single-Agent Maintenance Therapy for Advanced Non-Small Cell Lung Cancer (NSCLC): A Systematic Review and Bayesian Network Meta-Analysis of 26 Randomized Controlled Trials. PeerJ 2016, 2016, e2550. [Google Scholar] [CrossRef]
- Alfonso, S.; Valdés-Zayas, A.; Santiesteban, E.R.; Flores, Y.I.; Areces, F.; Hernández, M.; Viada, C.E.; Mendoza, I.C.; Guerra, P.P.; García, E.; et al. A Randomized, Multicenter, Placebo-Controlled Clinical Trial of Racotumomab-Alum Vaccine as Switch Maintenance Therapy in Advanced Non-Small Cell Lung Cancer Patients. Clin. Cancer Res. 2014, 20, 3660–3671. [Google Scholar] [CrossRef]
- Cáceres-Lavernia, H.H.; Nenínger-Vinageras, E.; Varona-Rodríguez, L.M.; Olivares-Romero, Y.A.; Sánchez-Rojas, I.; Mazorra-Herrera, Z.; Basanta-Bergolla, D.; Duvergel-Calderín, D.; Torres-Cuevas, B.L.; del Castillo-Carrillo, C. Racotumomab in Non-Small Cell Lung Cancer as Maintenance and Second-Line Treatment. MEDICC Rev. 2021, 23, 21–28. [Google Scholar] [CrossRef]
- Limacher, J.M.; Quoix, E. TG4010: A Therapeutic Vaccine against MUC1 Expressing Tumors. Oncoimmunology 2012, 1, 791–792. [Google Scholar] [CrossRef][Green Version]
- Ramlau, R.; Quoix, E.; Rolski, J.; Pless, M.; Lena, H.; Lévy, E.; Krzakowski, M.; Hess, D.; Tartour, E.; Chenard, M.P.; et al. A Phase II Study of Tg4010 (Mva-Muc1-Il2) in Association with Chemotherapy in Patients with Stage III/IV Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2008, 3, 735–744. [Google Scholar] [CrossRef]
- Rosenblum, D.; Joshi, N.; Tao, W.; Karp, J.M.; Peer, D. Progress and Challenges towards Targeted Delivery of Cancer Therapeutics. Nat. Commun. 2018, 9, 9. [Google Scholar] [CrossRef]
- Kudling, T.V.; Clubb, J.H.A.; Quixabeira, D.C.A.; Santos, J.M.; Havunen, R.; Kononov, A.; Heiniö, C.; Cervera-Carrascon, V.; Pakola, S.; Basnet, S.; et al. Local Delivery of Interleukin 7 with an Oncolytic Adenovirus Activates Tumor-Infiltrating Lymphocytes and Causes Tumor Regression. Oncoimmunology 2022, 11, 20965–20972. [Google Scholar] [CrossRef]
- Zhang, P.; Meng, J.; Li, Y.; Yang, C.; Hou, Y.; Tang, W.; McHugh, K.J.; Jing, L. Nanotechnology-Enhanced Immunotherapy for Metastatic Cancer. Innovation 2021, 2, 100174. [Google Scholar] [CrossRef] [PubMed]
- Patra, J.K.; Das, G.; Fraceto, L.F.; Campos, E.V.R.; Rodriguez-Torres, M.D.P.; Acosta-Torres, L.S.; Diaz-Torres, L.A.; Grillo, R.; Swamy, M.K.; Sharma, S.; et al. Nano Based Drug Delivery Systems: Recent Developments and Future Prospects. J. Nanobiotechnology 2018, 16, 71. [Google Scholar] [CrossRef]
- Li, L.; Zhang, Y.; Zhou, Y.; Hu, H.; Hu, Y.; Georgiades, C.; Mao, H.Q.; Selaru, F.M. Quaternary Nanoparticles Enable Sustained Release of Bortezomib for Hepatocellular Carcinoma. Hepatology 2022, 76, 1660–1672. [Google Scholar] [CrossRef] [PubMed]
- Reda, M.; Ngamcherdtrakul, W.; Nelson, M.A.; Siriwon, N.; Wang, R.; Zaidan, H.Y.; Bejan, D.S.; Reda, S.; Hoang, N.H.; Crumrine, N.A.; et al. Development of a Nanoparticle-Based Immunotherapy Targeting PD-L1 and PLK1 for Lung Cancer Treatment. Nat. Commun. 2022, 13, 4261. [Google Scholar] [CrossRef] [PubMed]
- Carrasco-Esteban, E.; Domínguez-Rullán, J.A.; Barrionuevo-Castillo, P.; Pelari-Mici, L.; Leaman, O.; Sastre-Gallego, S.; López-Campos, F. Current Role of Nanoparticles in the Treatment of Lung Cancer. J. Clin. Transl. Res. 2021, 7, 140. [Google Scholar] [CrossRef]
- Wiklander, O.P.B.; Brennan, M.; Lötvall, J.; Breakefield, X.O.; Andaloussi, S.E.L. Advances in Therapeutic Applications of Extracellular Vesicles. Sci. Transl. Med. 2019, 11, 8521. [Google Scholar] [CrossRef]
- Yang, P.; Peng, Y.; Feng, Y.; Xu, Z.; Feng, P.; Cao, J.; Chen, Y.; Chen, X.; Cao, X.; Yang, Y.; et al. Immune Cell-Derived Extracellular Vesicles—New Strategies in Cancer Immunotherapy. Front. Immunol. 2021, 12, 771551. [Google Scholar] [CrossRef]
- Ma, Y.; Dong, S.; Li, X.; Kim, B.Y.S.; Yang, Z.; Jiang, W. Extracellular Vesicles: An Emerging Nanoplatform for Cancer Therapy. Front. Oncol. 2021, 10, 606906. [Google Scholar] [CrossRef]
- Kageyama, S.; Wada, H.; Muro, K.; Niwa, Y.; Ueda, S.; Miyata, H.; Takiguchi, S.; Sugino, S.H.; Miyahara, Y.; Ikeda, H.; et al. Dose-Dependent Effects of NY-ESO-1 Protein Vaccine Complexed with Cholesteryl Pullulan (CHP-NY-ESO-1) on Immune Responses and Survival Benefits of Esophageal Cancer Patients. J. Transl. Med. 2013, 11, 246. [Google Scholar] [CrossRef]
- Morotti, M.; Albukhari, A.A.; Artibani, A.M.; Brenton, J.D. Promises and Challenges of Adoptive T-Cell Therapies for Solid Tumours. Br. J. Cancer 2021, 124, 1759–1776. [Google Scholar] [CrossRef]
- Cellular & Gene Therapy Products | FDA. Available online: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products (accessed on 12 October 2022).
- Santarpia, M. Recent Developments in the Use of Immunotherapy in Non-Small Cell Lung Cancer. Expert Rev. Respir. Med. 2016, 10, 781–798. [Google Scholar] [CrossRef] [PubMed]
- Kwak, E.L. Anaplastic Lymphoma Kinase Inhibition in Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2010, 363, 1693–1703. [Google Scholar] [CrossRef] [PubMed]
- Gainor, J.F. ALK Rearrangements Are Mutually Exclusive with Mutations in EGFR or KRAS: An Analysis of 1,683 Patients with Non–Small Cell Lung Cancer. Clin. Cancer Res. 2013, 19, 4273–4281. [Google Scholar] [CrossRef] [PubMed]
- Bekaii-Saab, T.S.; Roda, J.M.; Guenterberg, K.D.; Ramaswamy, B.; Young, D.C.; Ferketich, A.K.; Lamb, T.A.; Grever, M.R.; Shapiro, C.L.; Carson, W.E. A Phase I Trial of Paclitaxel and Trastuzumab in Combination with Interleukin-12 in Patients with HER2/Neu-Expressing Malignancies. Mol. Cancer Ther. 2009, 8, 2983–2991. [Google Scholar] [CrossRef]
- Gerber, D.E.; Schiller, J.H. Maintenance Chemotherapy for Advanced Non-Small-Cell Lung Cancer: New Life for an Old Idea. J. Clin. Oncol. 2013, 31, 1009–1020. [Google Scholar] [CrossRef]
- Paz-Ares, L. First-Line Nivolumab plus Ipilimumab Combined with Two Cycles of Chemotherapy in Patients with Non-Small-Cell Lung Cancer (CheckMate 9LA): An International, Randomised, Open-Label, Phase 3 Trial. Lancet Oncol. 2021, 22, 198–211. [Google Scholar] [CrossRef]
- Pabani, A.; Butts, C.A. Current Landscape of Immunotherapy for the Treatment of Metastatic Non-Small-Cell Lung Cancer. Curr. Oncol. 2018, 25, 94–102. [Google Scholar] [CrossRef]
- Lynch, T.J. Ipilimumab in Combination with Paclitaxel and Carboplatin as First-Line Treatment in Stage IIIB/IV Non-Small-Cell Lung Cancer: Results from a Randomized, Double-Blind, Multicenter Phase II Study. J. Clin. Oncol. 2012, 30, 2046–2054. [Google Scholar] [CrossRef]
- Gadgeel, S.M. Pembrolizumab (Pembro) plus Chemotherapy as Front-Line Therapy for Advanced NSCLC: KEYNOTE-021 Cohorts A-C. J. Clin. Oncol. 2016, 34, 9016. [Google Scholar] [CrossRef]
- Juergens, R. MA09.03 Cisplatin/Pemetrexed Durvalumab /- Tremelimumab in Pts with Advanced Non-Squamous NSCLC: A CCTG Phase IB Study-IND.226. J. Thorac. Oncol. 2017, 12, 392–393. [Google Scholar] [CrossRef][Green Version]
- Reck, M. Atezolizumab plus Bevacizumab and Chemotherapy in Non-Small-Cell Lung Cancer (IMpower150): Key Subgroup Analyses of Patients with EGFR Mutations or Baseline Liver Metastases in a Randomised, Open-Label Phase 3 Trial. Lancet Respir. Med. 2019, 7, 387–401. [Google Scholar] [CrossRef]
- Azghadi, S.; Daly, M.E. Radiation and Immunotherapy Combinations in Non-Small Cell Lung Cancer. Cancer Treat. Res. Commun. 2020, 26, 100298. [Google Scholar] [CrossRef] [PubMed]
- Jabbour, S.K.; Lee, K.H.; Frost, N.; Breder, V.; Kowalski, D.M.; Pollock, T.; Levchenko, E.; Reguart, N.; Martinez-Marti, A.; Houghton, B.; et al. Pembrolizumab plus Concurrent Chemoradiation Therapy in Patients with Unresectable, Locally Advanced, Stage III Non-Small Cell Lung Cancer: The Phase 2 KEYNOTE-799 Nonrandomized Trial. JAMA Oncol. 2021, 7, 1351–1359. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Liang, H.; Burnette, B.; Beckett, M.; Darga, T.; Weichselbaum, R.R.; Fu, Y.X. Irradiation and Anti-PD-L1 Treatment Synergistically Promote Antitumor Immunity in Mice. J. Clin. Investig. 2014, 124, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Victor, T.-S.; Rech, A.J.; Maity, A.; Rengan, R.; Pauken, K.E.; Stelekati, E.; Benci, J.L.; Xu, B.; Dada, H.; Odorizzi, P.M.; et al. Radiation and Dual Checkpoint Blockade Activate Non-Redundant Immune Mechanisms in Cancer. Nature 2015, 520, 373–377. [Google Scholar] [CrossRef]
- Ott, P.A.; Hodi, F.S.; Kaufman, H.L.; Wigginton, J.M.; Wolchok, J.D. Combination Immunotherapy: A Road Map. J. Immunother. Cancer 2017, 5, 16. [Google Scholar] [CrossRef]
- Hung, L.V.M.; Ngo, H.T.; van Pham, P. Clinical Trials with Cytokine-Induced Killer Cells and CAR-T Cell Transplantation for Non-Small Cell Lung Cancer Treatment. Adv. Exp. Med. Biol. 2020, 1292, 113–130. [Google Scholar] [CrossRef]
- Vera, J.F.; Brenner, L.J.; Gerdemann, U.; Ngo, M.C.; Sili, U.; Liu, H.; Wilson, J.; Dotti, G.; Heslop, H.E.; Leen, A.M.; et al. Accelerated Production of Antigen-Specific T Cells for Preclinical and Clinical Applications Using Gas-Permeable Rapid Expansion Cultureware (G-Rex). J. Immunother. 2010, 33, 305–315. [Google Scholar] [CrossRef]
- Barrett, J.; Bollard, C.M. T-Cell Therapy for Cancer. Immunotherapy 2012, 4, 347–350. [Google Scholar] [CrossRef]
- Zheng, Y.W.; Li, R.M.; Zhang, X.W.; Ren, X.B. Current Adoptive Immunotherapy in Non-Small Cell Lung Cancer and Potential Influence of Therapy Outcome. Cancer Investig. 2013, 31, 197–205. [Google Scholar] [CrossRef]
- Rosenberg, S.A. Durable Complete Responses in Heavily Pretreated Patients with Metastatic Melanoma Using T-Cell Transfer Immunotherapy. Clin. Cancer Res. 2011, 17, 4550–4557. [Google Scholar] [CrossRef] [PubMed]
- Zhukova, O.S. Combined Effect of Cisplatin and Lymphokine-Activated Killer Cells on A549 Cells of Non-Small Cell Lung Cancer. Bull. Exp. Biol. Med. 2007, 144, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Choe, E.-A. Dynamic Changes in PD-L1 Expression and CD8 T Cell Infiltration in Non-Small Cell Lung Cancer Following Chemoradiation Therapy. Lung Cancer 2019, 136, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Ratto, G.B. A Randomized Trial of Adoptive Immunotherapy with Tumor-Infiltrating Lymphocytes and Interleukin-2 versus Standard Therapy in the Postoperative Treatment of Resected Nonsmall Cell Lung Carcinoma. Cancer 1996, 78, 244–251. [Google Scholar] [CrossRef]
- Yoneda, K. Alteration in Tumoural PD-L1 Expression and Stromal CD8-Positive Tumour-Infiltrating Lymphocytes after Concurrent Chemo-Radiotherapy for Non-Small Cell Lung Cancer. Br. J. Cancer 2019, 121, 490–496. [Google Scholar] [CrossRef]
- Mesiano, G. Cytokine-Induced Killer (CIK) Cells as Feasible and Effective Adoptive Immunotherapy for the Treatment of Solid Tumors. Expert Opin. Biol. Ther. 2012, 12, 673–684. [Google Scholar] [CrossRef]
- Li, H.; Wang, C.; Yu, J.; Cao, S.; Wei, F.; Zhang, W.; Han, Y.; Ren, X.B. Dendritic Cell-Activated Cytokine-Induced Killer Cells Enhance the Anti-Tumor Effect of Chemotherapy on Non-Small Cell Lung Cancer in Patients after Surgery. Cytotherapy 2009, 11, 1076–1083. [Google Scholar] [CrossRef]
- Wu, C. Prospective Study of Chemotherapy in Combination with Cytokine-Induced Killer Cells in Patients Suffering from Advanced Non-Small Cell Lung Cancer. Anticancer Res. 2009, 28, 3997–4002. [Google Scholar]
- Zhou, J. Anti-Γδ TCR Antibody-Expanded Γδ T Cells: A Better Choice for the Adoptive Immunotherapy of Lymphoid Malignancies. Cell Mol. Immunol. 2012, 9, 34–44. [Google Scholar] [CrossRef]
- Murakami, T. Novel Human NK Cell Line Carrying CAR Targeting EGFRvIII Induces Antitumor Effects in Glioblastoma Cells. Anticancer Res. 2018, 38, 5049–5056. [Google Scholar] [CrossRef]
- Iliopoulou, E.G. A Phase I Trial of Adoptive Transfer of Allogeneic Natural Killer Cells in Patients with Advanced Non-Small Cell Lung Cancer. Cancer Immunol. Immunother. 2010, 59, 1781–1789. [Google Scholar] [CrossRef] [PubMed]
- Zhong, R. Dendritic Cells Combining with Cytokine-Induced Killer Cells Synergize Chemotherapy in Patients with Late-Stage Non-Small Cell Lung Cancer. Cancer Immunol. Immunother. 2011, 60, 1497–1502. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, A. Immune Cell Engineering: Opportunities in Lung Cancer Therapeutics. Drug Deliv. Transl. Res. 2020, 10, 1203–1227. [Google Scholar] [CrossRef] [PubMed]
- Stephan, M.T.; Moon, J.J. Therapeutic Cell Engineering with Surface-Conjugated Synthetic Nanoparticles. Nat. Med. 2010, 16, 1035–1041. [Google Scholar] [CrossRef]
- Stephan, M.T.; Stephan, S.B. Synapse-Directed Delivery of Immunomodulators Using T-Cell-Conjugated Nanoparticles. Biomaterials 2012, 33, 5776–5787. [Google Scholar] [CrossRef] [PubMed]
- Boucherit, N.; Gorvel, L.; Olive, D. 3D Tumor Models and Their Use for the Testing of Immunotherapies. Front. Immunol. 2020, 11, 603640. [Google Scholar] [CrossRef]
- Weiswald, L.-B.; Bellet, D.; Dangles-Marie, V. Spherical Cancer Models in Tumor Biology. Neoplasia 2015, 17, 1–15. [Google Scholar] [CrossRef]
- Nunes, A.S.; Barros, A.S.; Costa, E.C.; Moreira, A.F.; Correia, I.J. 3D Tumor Spheroids as in Vitro Models to Mimic In Vivo Human Solid Tumors Resistance to Therapeutic Drugs. Biotechnol. Bioeng. 2019, 116, 206–226. [Google Scholar] [CrossRef]
- Rozenberg, J.M.; Filkov, G.I.; Trofimenko, A.V.; Karpulevich, E.A.; Parshin, V.D.; Royuk, V.V.; Sekacheva, M.I.; Durymanov, M.O. Biomedical Applications of Non-Small Cell Lung Cancer Spheroids. Front Oncol 2021, 11. [Google Scholar] [CrossRef]
- Huo, K.-G.; D’Arcangelo, E.; Tsao, M.-S. Patient-Derived Cell Line, Xenograft and Organoid Models in Lung Cancer Therapy. Transl. Lung Cancer Res. 2020, 9, 2214–2232. [Google Scholar] [CrossRef]
- Lê, H.; Seitlinger, J.; Lindner, V.; Olland, A.; Falcoz, P.-E.; Benkirane-Jessel, N.; Quéméneur, E. Patient-Derived Lung Tumoroids-An Emerging Technology in Drug Development and Precision Medicine. Biomedicines 2022, 10, 1677. [Google Scholar] [CrossRef]
- Shi, R.; Radulovich, N.; Ng, C.; Liu, N.; Notsuda, H.; Cabanero, M.; Martins-Filho, S.N. Organoid Cultures as Preclinical Models of Non-Small Cell Lung Cancer. Clin. Cancer Res. 2020, 26, 1162–1174. [Google Scholar] [CrossRef]
- Liu, L.; Yu, L.; Li, Z.; Li, W.; Huang, W. Patient-Derived Organoid (PDO) Platforms to Facilitate Clinical Decision Making. J. Transl. Med. 2021, 19, 40. [Google Scholar] [CrossRef]
- Peng, D.; Gleyzer, R.; Tai, W.-H.; Kumar, P.; Bian, Q.; Isaacs, B.; Rocha, E.L. Evaluating the Transcriptional Fidelity of Cancer Models. Genome Med. 2021, 13, 73. [Google Scholar] [CrossRef]
- Ma, X.; Yang, S.; Jiang, H.; Wang, Y.; Xiang, Z. Transcriptomic Analysis of Tumor Tissues and Organoids Reveals the Crucial Genes Regulating the Proliferation of Lung Adenocarcinoma. J. Transl. Med. 2021, 19, 368. [Google Scholar] [CrossRef]
- Eramo, A. Identification and Expansion of the Tumorigenic Lung Cancer Stem Cell Population. Cell Death Differ. 2007, 15, 504–514. [Google Scholar] [CrossRef]
- Endo, H. Spheroid Culture of Primary Lung Cancer Cells with Neuregulin 1/HER3 Pathway Activation. J. Thorac. Oncol. 2013, 8, 131–139. [Google Scholar] [CrossRef]
- Ivanova, E. Use of Ex Vivo Patient-Derived Tumor Organotypic Spheroids to Identify Combination Therapies for HER2 Mutant Non–Small Cell Lung Cancer. Clin. Cancer Res. 2020, 26, 2393–2403. [Google Scholar] [CrossRef]
- Finnberg, N.K.; Gokare, P.; Lev, A.; Grivennikov, S.I.; MacFarlane, A.W.; Campbell, K.S.; Winters, R.M.; Kaputa, K.; Farma, J.M.; Abbas, A.E.-S.; et al. Application of 3D Tumoroid Systems to Define Immune and Cytotoxic Therapeutic Responses Based on Tumoroid and Tissue Slice Culture Molecular Signatures. Oncotarget 2017, 8, 66747–66757. [Google Scholar] [CrossRef]
- Sachs, N. Long-Term Expanding Human Airway Organoids for Disease Modeling. EMBO J. 2019, 38, e100300. [Google Scholar] [CrossRef]
- Shi, J.; Hua, X.; Zhu, B.; Ravichandran, S.; Wang, M.; Nguyen, C.; Brodie, S.A.; Palleschi, A.; Alloisio, M.; Pariscenti, G.; et al. Somatic Genomics and Clinical Features of Lung Adenocarcinoma: A Retrospective Study. PLoS Med. 2016, 13, e1002162. [Google Scholar] [CrossRef] [PubMed]
- Neal, J.T.; Li, X.; Zhu, J.; Giangarra, V.; Grzeskowiak, C.L.; Ju, J.; Liu, I.H. Organoid Modeling of the Tumor Immune Microenvironment. Cell 2018, 175, 1972–1988.e16. [Google Scholar] [CrossRef] [PubMed]
- Kim, M. Patient-Derived Lung Cancer Organoids as in Vitro Cancer Models for Therapeutic Screening. Nat. Commun. 2019, 10, 3991. [Google Scholar] [CrossRef] [PubMed]
- Li, Z. Human Lung Adenocarcinoma-Derived Organoid Models for Drug Screening. iScience 2020, 23, 101411. [Google Scholar] [CrossRef]
- Chen, J. Genomic Characteristics and Drug Screening among Organoids Derived from Non-small Cell Lung Cancer Patients. Thorac. Cancer 2020, 11, 2279–2290. [Google Scholar] [CrossRef]
- Seo, J.H. MFF Regulation of Mitochondrial Cell Death Is a Therapeutic Target in Cancer. Cancer Res. 2019, 79, 6215–6226. [Google Scholar] [CrossRef]
- Mazzocchi, A.; Devarasetty, M.; Herberg, S.; Petty, W.J.; Marini, F.; Miller, L.; Kucera, G. Pleural Effusion Aspirate for Use in 3D Lung Cancer Modeling and Chemotherapy Screening. ACS Biomater. Sci. Eng. 2019, 5, 1937–1943. [Google Scholar] [CrossRef]
- Dijkstra, K.K. Challenges in Establishing Pure Lung Cancer Organoids Limit Their Utility for Personalized Medicine. Cell Rep. 2020, 31, 107588. [Google Scholar] [CrossRef]
- Gmeiner, W.H. Dysregulated Pyrimidine Biosynthesis Contributes to 5-FU Resistance in SCLC Patient-Derived Organoids but Response to a Novel Polymeric Fluoropyrimidine, CF10. Cancers 2020, 12, 788. [Google Scholar] [CrossRef]
- Taverna, J.A.; Hung, C.N.; DeArmond, D.T.; Chen, M.; Lin, C.L.; Osmulski, P.A.; Gaczynska, M.E.; Wang, C.M.; Lucio, N.D.; Chou, C.W.; et al. Single-Cell Proteomic Profiling Identifies Combined AXL and JAK1 Inhibition as a Novel Therapeutic Strategy for Lung Cancer. Cancer Res 2020, 80, 1551–1563. [Google Scholar] [CrossRef]
- Kim, S.-Y. Modeling Clinical Responses to Targeted Therapies by Patient-Derived Organoids of Advanced Lung Adenocarcinoma. Clin. Cancer Res. 2021, 27, 4397–4409. [Google Scholar] [CrossRef] [PubMed]
- Seitlinger, J. Vascularization of Patient-Derived Tumoroid from Non-Small-Cell Lung Cancer and Its Microenvironment. Biomedicines 2022, 10, 1103. [Google Scholar] [CrossRef] [PubMed]
- Yokota, E. Clinical Application of a Lung Cancer Organoid (Tumoroid) Culture System. NPJ Precis. Oncol. 2021, 5, 29. [Google Scholar] [CrossRef]
- Delom, F. Patients Lung Derived Tumoroids (PLDTs) to Model Therapeutic Response. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118808. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, D.; Nuala, V.S.S.; Ye Bi, S.P.G.; Gholami, S.; Hughes, C.C.W.; Fields, R.C.; George, S.C. Tumor-on-Chip Modeling of Organ-Specific Cancer and Metastasis. Adv. Drug Deliv. Rev. 2021, 175, 113798. [Google Scholar] [CrossRef]
- Ying, L.; Zhu, Z.; Xu, Z.; He, T.; Li, E.; Guo, Z.; Liu, F.; Jiang, C.; Wang, Q. Cancer Associated Fibroblast-Derived Hepatocyte Growth Factor Inhibits the Paclitaxel-Induced Apoptosis of Lung Cancer A549 Cells by Up-Regulating the PI3K/Akt and GRP78 Signaling on a Microfluidic Platform. PLoS ONE 2015, 10, 129593. [Google Scholar] [CrossRef]
- Moore, N.; Doty, D.; Zielstorff, M.; Kariv, I.; Moy, L.Y.; Gimbel, A.; Chevillet, J.R. A Multiplexed Microfluidic System for Evaluation of Dynamics of Immune-Tumor Interactions. Lab Chip 2018, 18, 1844–1858. [Google Scholar] [CrossRef]
- Beckwith, A.L.; Velásquez-García, L.F.; Borenstein, J.T. Microfluidic Model for Evaluation of Immune Checkpoint Inhibitors in Human Tumors. Adv. Healthc. Mater. 2019, 8, 1900289. [Google Scholar] [CrossRef]
- Augustine, R.; Kalva, S.N.; Ahmad, R.; Zahid, A.A.; Hasan, S.; Nayeem, A.; McClements, L.; Hasan, A. 3D Bioprinted Cancer Models: Revolutionizing Personalized Cancer Therapy. Transl. Oncol. 2021, 14, 101015. [Google Scholar] [CrossRef]
- Mondal, A.; Gebeyehu, A.; Miranda, M.; Bahadur, D.; Patel, N.; Ramakrishnan, S.; Rishi, A.K.; Singh, M. Characterization and Printability of Sodium Alginate -Gelatin Hydrogel for Bioprinting NSCLC Co-Culture. Sci. Rep. 2019, 9, 19914. [Google Scholar] [CrossRef]
- Wang, X. Tumor-like Lung Cancer Model Based on 3D Bioprinting. 3 Biotech 2018, 8, 501. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zheng, W.; Wang, H.; Cheng, Y.; Fang, Y.; Wu, F.; Sun, G.; Sun, G.; Lv, C.; Hui, B. Application of Animal Models in Cancer Research: Recent Progress and Future Prospects. Cancer Manag. Res. 2021, 13, 2455–2475. [Google Scholar] [CrossRef]
- Shultz, L.D.; Brehm, M.A.; Garcia-Martinez, J.V.; Greiner, D.L. Humanized Mice for Immune System Investigation: Progress, Promise and Challenges. Nat. Rev. Immunol. 2012, 12, 786–798. [Google Scholar] [CrossRef] [PubMed]
- Konantz, M.; Balci, T.B.; Hartwig, U.F.; Dellaire, G.; André, M.C.; Berman, J.N.; Lengerke, C. Zebrafish Xenografts as a Tool for in Vivo Studies on Human Cancer. Ann. New York Acad. Sci. 2012, 1266, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Pu, J.; Sun, J.; Tan, B.; Wang, W.; Wang, L.; Cheng, J.; Zuo, Y. Zebrafish Xenograft Model of Human Lung Cancer for Studying the Function of LINC00152 in Cell Proliferation and Invasion. Cancer Cell Int. 2020, 20, 1–11. [Google Scholar] [CrossRef]
- Hason, H.; Bartůněk, P. Zebrafish Models of Cancer—New Insights on Modeling Human Cancer in a Non-Mammalian Vertebrate. Genes 2019, 10, 935. [Google Scholar] [CrossRef]
- Lin, S.; Huang, G.; Cheng, L.; Li, Z.; Xiao, Y.; Deng, Q.; Jiang, Y. Establishment of Peripheral Blood Mononuclear Cell-Derived Humanized Lung Cancer Mouse Models for Studying Efficacy of PD-L1/PD-1 Targeted Immunotherapy. MAbs 2018, 10, 1301–1311. [Google Scholar] [CrossRef]
- Sanmamed, M.F.; Rodriguez, I.; Schalper, K.A.; Oñate, C.; Azpilikueta, A.; Rodriguez-Ruiz, M.E.; Morales-Kastresana, A. Nivolumab and Urelumab Enhance Antitumor Activity of Human T Lymphocytes Engrafted in Rag2-/-IL2Rγnull Immunodeficient Mice. Cancer Res. 2015, 75, 3466–3478. [Google Scholar] [CrossRef]
- Sicklick, J.K.; Leonard, S.Y.; Babicky, M.L.; Tang, C.-M.; Mose, E.S.; French, R.P.; Jaquish, D.V. Generation of Orthotopic Patient-Derived Xenografts from Gastrointestinal Stromal Tumor. J. Transl. Med. 2014, 12, 1–11. [Google Scholar] [CrossRef]
- Hiroshima, Y.; Zhang, Y.; Zhang, N.; Maawy, A.; Mii, S.; Yamamoto, M.; Uehara, F. Establishment of a Patient-Derived Orthotopic Xenograft (PDOX) Model of HER-2-Positive Cervical Cancer Expressing the Clinical Metastatic Pattern. PLoS ONE 2015, 10, 117417. [Google Scholar] [CrossRef]
- Igarashi, K.; Kawaguchi, K.; Kiyuna, T.; Murakami, T.; Miwa, S.; Nelson, S.D.; Dry, S.M. Patient-Derived Orthotopic Xenograft (PDOX) Mouse Model of Adult Rhabdomyosarcoma Invades and Recurs after Resection in Contrast to the Subcutaneous Ectopic Model. Cell Cycle 2017, 16, 91–94. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Wang, W.; Long, Y.; Liu, H.; Cheng, J.; Guo, L.; Li, R.; Meng, C.; Yu, S.; Zhao, Q.; et al. Characterization of Drug Responses of Mini Patient-Derived Xenografts in Mice for Predicting Cancer Patient Clinical Therapeutic Response. Cancer Commun. 2018, 38, 1–12. [Google Scholar] [CrossRef] [PubMed]



| Therapy | No. of Patients Undergone Therapy | No. of Patients Does Not Undergo Therapy | Not Applicable | The Ratio between No. of Patients Undergone to Do Not Undergone (%) | Total Number of Cases with LC Therapy-Data Availability | The Proportion of Subjects with Data Availability and ‘Not Applicable’ (%) |
|---|---|---|---|---|---|---|
| Radiotherapy | 158 | 822 | 6986 | 19.2 | 920 | 13.2% |
| Chemotherapy | 683 | 175 | 7108 | _ | 858 | 12.1% |
| Targeted therapy | 319 | 541 | 7106 | 58.9 | 860 | 12.1% |
| Immunotherapy | 191 | 666 | 7109 | 28.6 | 857 | 12.0% |
| Therapy | Drugs | Target | Estimated Frequency of Mutation in LC (%) | References |
|---|---|---|---|---|
| Chemotherapy | Carboplatin, cisplatin, docetaxel, etoposide, gemcitabine, nab-paclitaxel, paclitaxel, pemetrexed, vinorelbine | [19] | ||
| Targeted therapy | Afatinib, dacomitinib, entrectinib, erlotinib, gefitinib, osimertinib | EGFR (receptor protein) | 15 | [20,21,22,23] |
| Amivantamab, mobocertinib | EGFR (exon 20 insertion) | 15 | [20,21,22,23] | |
| Fam-trastuzumab deruxtecan-nxki | HER2 | 2 | [21,24,25] | |
| Alectinib, brigatinib, ceritinib, crizotinib, loralitinib | ALK | 5 | [20,21,26,27,28,29] | |
| Ceritinib, crizotinib, entrectinib | ROS1 | 2 | [21,30,31,32,33] | |
| Sotorasib | KRAS G12C | 25–33 | [20,21,25] | |
| Larotrectinib | NTRK | |||
| Dabrafenib, trametinib | BRAF V600E | 2 | [20,21,25] | |
| Capmatinib, tepotinib | MET (exon 14 skipping) | 3 | [21,34,35] | |
| Pralsetinib, selpercatinib | RET | 2 | [20,21,36,37] | |
| Immunotherapy | Atezolizumab, durvalumab, cemiplimab, nivolumab, pembrolizumab | PD1/PDL1 pathway | 33 | [21,38,39,40,41,42,43] |
| Ipilimumab | CTLA4 pathway | [25,44] |
| Agent | Phase | Study Population | Design and Description | Primary Endpoint | Enrolment | NCT |
|---|---|---|---|---|---|---|
| Durvalumab | II | Advanced NSCLC | Evaluating efficacy and safety of the PD-L1 inhibitor durvalumab as first-line therapy | OS | 50 | NCT02879617 |
| Niraparib | II | NSCLC | Niraparib + Pembrolizumab Niraparib alone Niraparib + Dostarlimab | ORR | 53 | NCT03308942 |
| Ipilimumab | III | Stage IV/Recurrent NSCLC | Ipilimumab + Paclitaxel/Carboplatin Placebo + Paclitaxel/Carboplatin | OS | 1289 | NCT01285609 |
| Pembrolizumab | II | Advanced NSCLC | Pembrolizumab + Physician’s choice chemotherapy | PFS | 35 | NCT03083808 |
| AK104 | II | Advanced NSCLC | AK104 +Docetaxel | ORR | 40 | NCT05215067 |
| Pembrolizumab | II | Metastatic NSCLC | Pembrolizumab + chemotherapy vs. Placebo + chemotherapy | PFS | 98 | NCT03656094 |
| AK105 | III | Metastatic Nonsquamous NSCLC Stage IV | AK105 + Carboplatin and Pemetrexed vs. Placebo + Carboplatin and Pemetrexed | PFS | 360 | NCT03866980 |
| ONC-392 | I & II | advanced or metastatic solid tumors and NSCLC | ONC-392 Treatment as a single agent vs. ONC-392 in combination with pembrolizumab | DLT, MTD, RP2D, TRAE | 468 | NCT04140526 |
| mRNA Vaccine | I & II | Metastatic NSCLC | BI 1361849 mRNA Vaccine + durvalumab BI 1361849 mRNA Vaccine + durvalumab + tremelimumab | TEAE | 61 | NCT03164772 |
| KN046 | II | Advanced NSCLC | KN046 + Axitinib | ORR | 54 | NCT05420220 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Padinharayil, H.; Alappat, R.R.; Joy, L.M.; Anilkumar, K.V.; Wilson, C.M.; George, A.; Valsala Gopalakrishnan, A.; Madhyastha, H.; Ramesh, T.; Sathiyamoorthi, E.; et al. Advances in the Lung Cancer Immunotherapy Approaches. Vaccines 2022, 10, 1963. https://doi.org/10.3390/vaccines10111963
Padinharayil H, Alappat RR, Joy LM, Anilkumar KV, Wilson CM, George A, Valsala Gopalakrishnan A, Madhyastha H, Ramesh T, Sathiyamoorthi E, et al. Advances in the Lung Cancer Immunotherapy Approaches. Vaccines. 2022; 10(11):1963. https://doi.org/10.3390/vaccines10111963
Chicago/Turabian StylePadinharayil, Hafiza, Reema Rose Alappat, Liji Maria Joy, Kavya V. Anilkumar, Cornelia M. Wilson, Alex George, Abilash Valsala Gopalakrishnan, Harishkumar Madhyastha, Thiyagarajan Ramesh, Ezhaveni Sathiyamoorthi, and et al. 2022. "Advances in the Lung Cancer Immunotherapy Approaches" Vaccines 10, no. 11: 1963. https://doi.org/10.3390/vaccines10111963
APA StylePadinharayil, H., Alappat, R. R., Joy, L. M., Anilkumar, K. V., Wilson, C. M., George, A., Valsala Gopalakrishnan, A., Madhyastha, H., Ramesh, T., Sathiyamoorthi, E., Lee, J., & Ganesan, R. (2022). Advances in the Lung Cancer Immunotherapy Approaches. Vaccines, 10(11), 1963. https://doi.org/10.3390/vaccines10111963