
Delineating the SARS-CoV-2 Induced Interplay between the Host Immune System and the DNA Damage Response Network

 , , and
, , and
Abstract
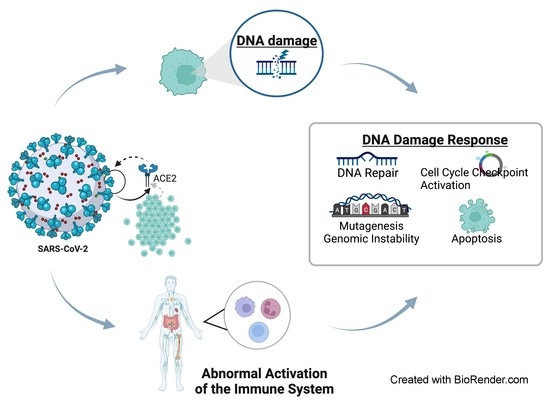
{kind=link}
{kind=link}
{kind=link}
1. Introduction
1.1. Immune Dysregulation
1.2. Impaired Interferon Induction
1.3. Hyper-Inflammation
1.4. Delayed Adaptive Immune Response
2. The Impact of COVID-19 on Cancer Patients
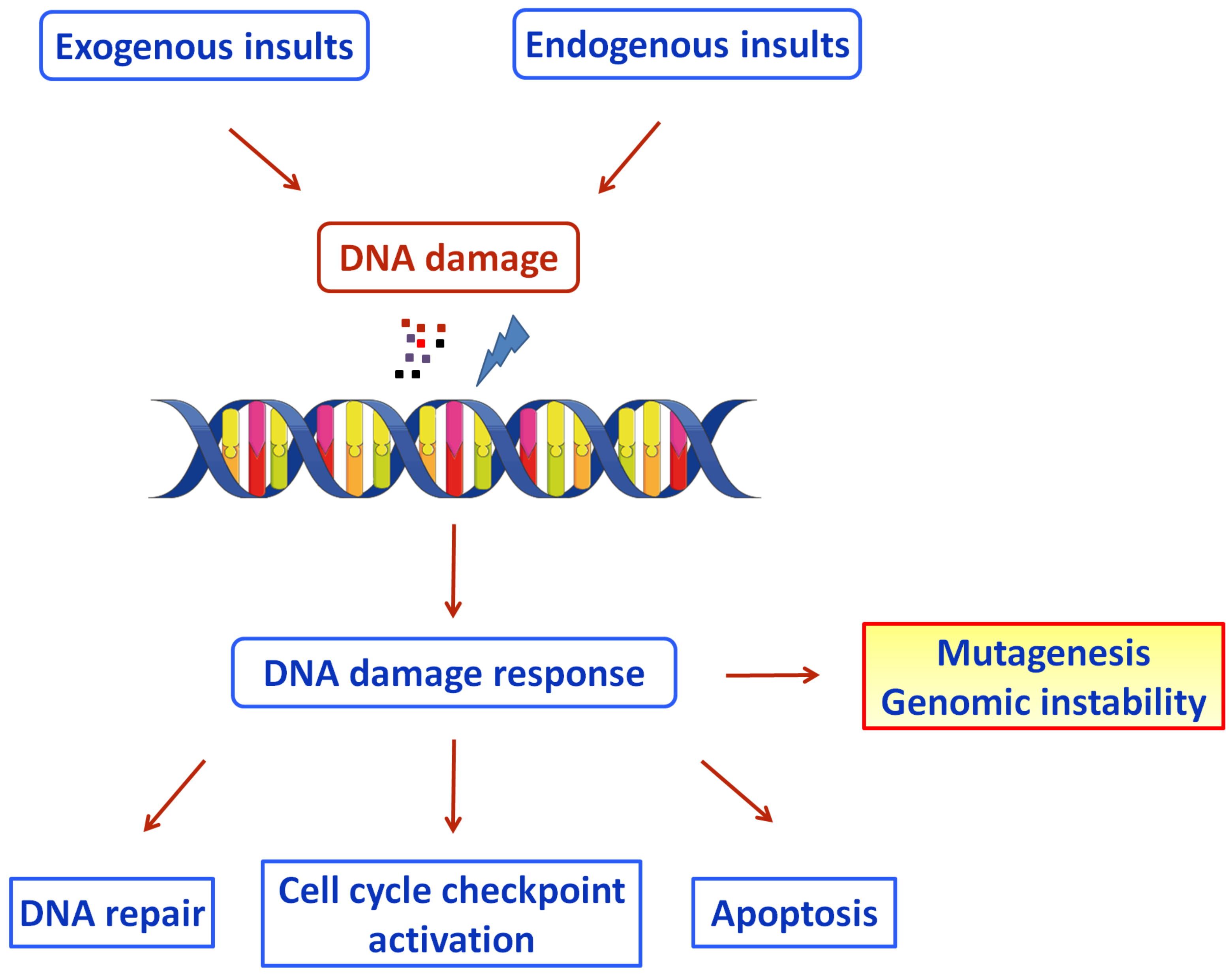
3. DNA Damage Response and COVID-19
3.1. DNA Repair Mechanisms
- (a)
- Nucleotide excision repair (NER). This mechanism repairs lesions that disrupt the DNA double-helix, such as bulky base adducts [111]. NER detects helix-distorting base lesions via two sub-pathways with different lesion detection mechanisms: transcription-coupled repair (TCR), which identifies lesions that inhibit transcription, and global-genome repair (GGR), which removes lesions throughout the genome.
- (b)
- Base excision repair (BER). This is a commonly used DNA repair process that identifies and repairs damaged DNA bases that do not alter the structure of the DNA helix. The cell uses BER to repair abnormal DNA bases, simple base-adducts, oxidative DNA damage and single-strand breaks (SSBs) [112]. There are two BER sub-pathways: the short-patch and the long-patch pathway. The activation of one or both of these two BER sub-pathways is determined by the origin of the damage and the cell cycle phase in which the damage occurs.
- (c)
- Mismatch repair (MMR). This pathway eliminates base substitution and insertion/deletion mismatches that occur when replication errors escape DNA polymerases’ proofreading function [113].
- (d)
- Homologous recombination repair (HRR). This is an error-free DSB repair mechanism that works throughout the S and G2 phases of the cell cycle to find a sister chromatid, which serves as a template to direct the repair of the damaged sequence [114].
- (e)
- Non-homologous end-joining (NHEJ). This mechanism repairs radiation- or chemically-induced double-strand breaks (DSBs), as well as intermediates of the V(D)J recombination and class-switch recombination (CSR) processes [115,116,117]. It is prone to errors and can function at any stage of the cell cycle. There are two subtypes of NHEJ: the canonical (c-NHEJ) and the alternative non-homologous end-joining (alt-NHEJ).
- (f)
- Interstrand cross-link (ICL) repair. This pathway repairs cross-links between the two strands of DNA, a critical event that usually results in cell cycle and replication arrest and eventually cell death [118]. In non-replicating cells, ICL repair is mediated by the NER mechanism, while in the S phase it is coupled to DNA replication and depends on the homologous recombination machinery [119].
- (g)
- Direct repair pathway. The only protein that is implicated in this mechanism is the O6-methylguanine-DNA methyltransferase (MGMT), which removes alkyl groups from the O6 position of guanine to a cysteine residue on itself and undergoes the degradation process [120].
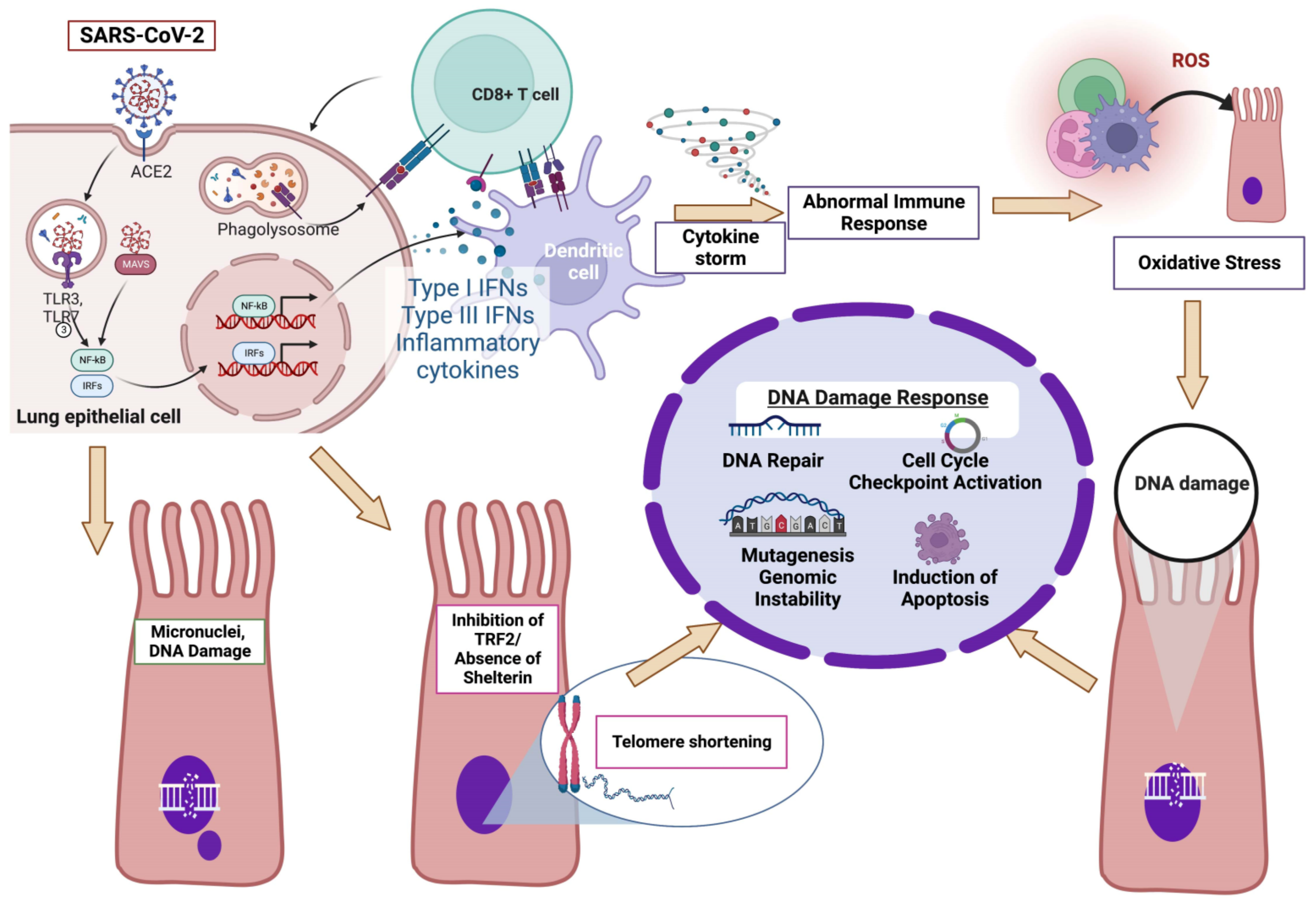
3.2. COVID-19 and DDR
3.3. COVID-19 and Oxidative Stress
3.4. SARS-CoV-2 Vaccination, the Immune System and the DDR Network
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cucinotta, D.; Vanelli, M. WHO Declares COVID-19 a Pandemic. Acta Biomed. 2020, 91, 157–160. [Google Scholar]
- Hu, T.; Liu, Y.; Zhao, M.; Zhuang, Q.; Xu, L.; He, Q. A comparison of COVID-19, SARS and MERS. PeerJ 2020, 8, e9725. [Google Scholar] [CrossRef]
- Boechat, J.L.; Chora, I.; Morais, A.; Delgado, L. The immune response to SARS-CoV-2 and COVID-19 immunopathology—Current perspectives. Pulmonology 2021, 27, 423–437. [Google Scholar] [CrossRef]
- Gupta, A.; Madhavan, M.V.; Sehgal, K.; Nair, N.; Mahajan, S.; Sehrawat, T.S.; Bikdeli, B.; Ahluwalia, N.; Ausiello, J.C.; Wan, E.Y.; et al. Extrapulmonary manifestations of COVID-19. Nat. Med. 2020, 26, 1017–1032. [Google Scholar] [CrossRef]
- Pánico, P.; Ostrosky-Wegman, P.; Salazar, A.M. The potential role of COVID-19 in the induction of DNA damage. Mutat. Res. Rev. Mutat. Res. 2022, 789, 108411. [Google Scholar] [CrossRef]
- Wong, L.R.; Perlman, S. Immune dysregulation and immunopathology induced by SARS-CoV-2 and related coronaviruses—Are we our own worst enemy? Nat. Rev. Immunol. 2022, 22, 47–56. [Google Scholar] [CrossRef]
- Tan, L.Y.; Komarasamy, T.V.; Rmt Balasubramaniam, V. Hyperinflammatory Immune Response and COVID-19: A Double Edged Sword. Front. Immunol. 2021, 12, 742941. [Google Scholar] [CrossRef]
- Sen, G.C. Viruses and interferons. Annu. Rev. Microbiol. 2001, 55, 255–281. [Google Scholar] [CrossRef]
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’Garra, A. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015, 15, 87–103. [Google Scholar] [CrossRef]
- Galbraith, M.D.; Kinning, K.T.; Sullivan, K.D.; Araya, P.; Smith, K.P.; Granrath, R.E.; Shaw, J.R.; Baxter, R.; Jordan, K.R.; Russell, S.; et al. Specialized interferon action in COVID-19. Proc. Natl. Acad. Sci. USA 2022, 119, e2116730119. [Google Scholar] [CrossRef]
- Hadjadj, J.; Yatim, N.; Barnabei, L.; Corneau, A.; Boussier, J.; Smith, N.; Péré, H.; Charbit, B.; Bondet, V.; Chenevier-Gobeaux, C.; et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 2020, 369, 718–724. [Google Scholar] [CrossRef] [PubMed]
- Galani, I.E.; Rovina, N.; Lampropoulou, V.; Triantafyllia, V.; Manioudaki, M.; Pavlos, E.; Koukaki, E.; Fragkou, P.C.; Panou, V.; Rapti, V.; et al. Untuned antiviral immunity in COVID-19 revealed by temporal type I/III interferon patterns and flu comparison. Nat. Immunol. 2021, 22, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, C.G.K.; Miao, V.N.; Owings, A.H.; Navia, A.W.; Tang, Y.; Bromley, J.D.; Lotfy, P.; Sloan, M.; Laird, H.; Williams, H.B.; et al. Impaired local intrinsic immunity to SARS-CoV-2 infection in severe COVID-19. Cell 2021, 184, 4713–4733. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bastard, P.; Liu, Z.; Le Pen, J.; Moncada-Velez, M.; Chen, J.; Ogishi, M.; Sabli, I.K.D.; Hodeib, S.; Korol, C.; et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 2020, 370, eabd4570. [Google Scholar] [CrossRef] [PubMed]
- Pairo-Castineira, E.; Clohisey, S.; Klaric, L.; Bretherick, A.D.; Rawlik, K.; Pasko, D.; Walker, S.; Parkinson, N.; Fourman, M.H.; Russell, C.D.; et al. Genetic mechanisms of critical illness in COVID-19. Nature 2021, 591, 92–98. [Google Scholar] [CrossRef]
- Bastard, P.; Rosen, L.B.; Zhang, Q.; Michailidis, E.; Hoffmann, H.H.; Zhang, Y.; Dorgham, K.; Philippot, Q.; Rosain, J.; Béziat, V.; et al. Autoantibodies against type I IFNsin patients with life-threatening COVID-19. Science 2020, 370, eabd4585. [Google Scholar] [CrossRef]
- Wang, E.Y.; Mao, T.; Klein, J.; Dai, Y.; Huck, J.D.; Jaycox, J.R.; Liu, F.; Zhou, T.; Israelow, B.; Wong, P.; et al. Diverse functional autoantibodies in patients with COVID-19. Nature 2021, 595, 283–288. [Google Scholar] [CrossRef]
- Bastard, P.; Orlova, E.; Sozaeva, L.; Lévy, R.; James, A.; Schmitt, M.M.; Ochoa, S.; Kareva, M.; Rodina, Y.; Gervais, A.; et al. Preexisting autoantibodies to type I IFNs underlie critical COVID-19 pneumonia in patients with APS-1. J. Exp. Med. 2021, 218, e20210554. [Google Scholar] [CrossRef]
- Bastard, P.; Gervais, A.; Le Voyer, T.; Rosain, J.; Philippot, Q.; Manry, J.; Michailidis, E.; Hoffmann, H.H.; Eto, S.; Garcia-Prat, M.; et al. Autoantibodies neutralizing type I IFNs are present in ~4% of uninfected individuals over 70 years old and account for ~20% of COVID-19 deaths. Sci. Immunol. 2021, 6, eabl4340. [Google Scholar] [CrossRef]
- Bastard, P.; Vazquez, S.; Liu, J.; Laurie, M.T.; Wang, C.Y.; Gervais, A.; Le Voyer, T.; Bizien, L.; Zamecnik, C.; Philippot, Q.; et al. Vaccine breakthrough hypoxemic COVID-19 pneumonia in patients with auto-Abs neutralizing type I IFNs. Sci. Immunol. 2022, eabp8966. [Google Scholar] [CrossRef]
- Eskandarian Boroujeni, M.; Sekrecka, A.; Antonczyk, A.; Hassani, S.; Sekrecki, M.; Nowicka, H.; Lopacinska, N.; Olya, A.; Kluzek, K.; Wesoly, J.; et al. Dysregulated Interferon Response and Immune Hyperactivation in Severe COVID-19: Targeting STATs as a Novel Therapeutic Strategy. Front. Immunol. 2022, 13, 888897. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Fan, Y.; Lai, Y.; Han, T.; Li, Z.; Zhou, P.; Pan, P.; Wang, W.; Hu, D.; Liu, X.; et al. Coronavirus infections and immune responses. J. Med. Virol. 2020, 92, 424–432. [Google Scholar] [CrossRef] [PubMed]
- Konno, Y.; Kimura, I.; Uriu, K.; Fukushi, M.; Irie, T.; Koyanagi, Y.; Sauter, D.; Gifford, R.J.; USFQ-COVID19 Consortium; Nakagawa, S.; et al. SARS-CoV-2 ORF3b Is a Potent Interferon Antagonist Whose Activity Is Increased by a Naturally Occurring Elongation Variant. Cell Rep. 2020, 32, 108185. [Google Scholar] [CrossRef]
- Lei, X.; Dong, X.; Ma, R.; Wang, W.; Xiao, X.; Tian, Z.; Wang, C.; Wang, Y.; Li, L.; Ren, L.; et al. Activation and evasion of type I interferon responses by SARS-CoV-2. Nat. Commun. 2020, 11, 3810. [Google Scholar] [CrossRef]
- Wu, Y.; Ma, L.; Zhuang, Z.; Cai, S.; Zhao, Z.; Zhou, L.; Zhang, J.; Wang, P.H.; Zhao, J.; Cui, J. Main protease of SARS-CoV-2 serves as a bifunctional molecule in restricting type I interferon antiviral signaling. Signal Transduct. Target. Ther. 2020, 5, 221. [Google Scholar] [CrossRef]
- Zheng, Y.; Zhuang, M.W.; Han, L.; Zhang, J.; Nan, M.L.; Zhan, P.; Kang, D.; Liu, X.; Gao, C.; Wang, P.H. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) membrane (M) protein inhibits type I and III interferon production by targeting RIG-I/MDA-5 signaling. Signal Transduct. Target. Ther. 2020, 5, 299. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.; Mukherjee, R.; Grewe, D.; Bojkova, D.; Baek, K.; Bhattacharya, A.; Schulz, L.; Widera, M.; Mehdipour, A.R.; Tascher, G.; et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 2020, 587, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Thoms, M.; Buschauer, R.; Ameismeier, M.; Koepke, L.; Denk, T.; Hirschenberger, M.; Kratzat, H.; Hayn, M.; Mackens-Kiani, T.; Cheng, J.; et al. Structural basis for translational shutdown and immune evasion by the Nsp1 protein of SARS-CoV-2. Science 2020, 369, 1249–1255. [Google Scholar] [CrossRef]
- Xia, H.; Cao, Z.; Xie, X.; Zhang, X.; Chen, J.Y.; Wang, H.; Menachery, V.D.; Rajsbaum, R.; Shi, P.Y. Evasion of Type I Interferon by SARS-CoV-2. Cell Rep. 2020, 33, 108234. [Google Scholar] [CrossRef]
- Zhang, Q.; Chen, Z.; Huang, C.; Sun, J.; Xue, M.; Feng, T.; Pan, W.; Wang, K.; Dai, J. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Membrane (M) and Spike (S) Proteins Antagonize Host Type I Interferon Response. Front. Cell Infect. Microbiol. 2021, 11, 766922. [Google Scholar] [CrossRef]
- Zhang, J.; Zhao, C.; Zhao, W. Virus Caused Imbalance of Type I IFN Responses and Inflammation in COVID-19. Front. Immunol. 2021, 12, 633769. [Google Scholar] [CrossRef] [PubMed]
- Lowery, S.A.; Sariol, A.; Perlman, S. Innate immune and inflammatory responses to SARS-CoV-2: Implications for COVID-19. Cell Host Microbe 2021, 29, 1052–1062. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Park, S.; Jeong, H.W.; Ahn, J.Y.; Choi, S.J.; Lee, H.; Choi, B.; Nam, S.K.; Sa, M.; Kwon, J.S.; et al. Immunophenotyping of COVID-19 and influenza highlights the role of type I interferons in development of severe COVID-19. Sci. Immunol. 2020, 5, eabd1554. [Google Scholar] [CrossRef] [PubMed]
- Fajgenbaum, D.C.; June, C.H. Cytokine Storm. N. Engl. J. Med. 2020, 383, 2255–2273. [Google Scholar] [CrossRef] [PubMed]
- Ragab, D.; Salah Eldin, H.; Taeimah, M.; Khattab, R.; Salem, R. The COVID-19 Cytokine Storm; What We Know So Far. Front. Immunol. 2020, 11, 1446. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Zhu, Z.; Cai, T.; Fan, L.; Lou, K.; Hua, X.; Huang, Z.; Gao, G. Clinical value of immune-inflammatory parameters to assess the severity of coronavirus disease 2019. Int. J. Infect. Dis. 2020, 95, 332–339. [Google Scholar] [CrossRef]
- Tay, M.Z.; Poh, C.M.; Rénia, L.; MacAry, P.A.; Ng, L.F.P. The trinity of COVID-19: Immunity, inflammation and intervention. Nat. Rev. Immunol. 2020, 20, 363–374. [Google Scholar] [CrossRef]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J. HLH Across Speciality Collaboration, UK. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034. [Google Scholar] [CrossRef]
- Shi, Y.; Wang, Y.; Shao, C.; Huang, J.; Gan, J.; Huang, X.; Bucci, E.; Piacentini, M.; Ippolito, G.; Melino, G. COVID-19 infection: The perspectives on immune responses. Cell Death Differ. 2020, 27, 1451–1454. [Google Scholar] [CrossRef]
- Choudhary, S.; Sharma, K.; Silakari, O. The interplay between inflammatory pathways and COVID-19: A critical review on pathogenesis and therapeutic options. Microb. Pathog. 2021, 150, 104673. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, L.; Zhang, D.; Xu, J.; Dai, H.; Tang, N.; Su, X.; Cao, B. SARS-CoV-2 and viral sepsis: Observations and hypotheses. Lancet 2020, 395, 1517–1520. [Google Scholar] [CrossRef]
- Wang, J.; Li, Q.; Qiu, Y.; Lu, H. COVID-19: Imbalanced cell-mediated immune response drives to immunopathology. Emerg. Microbes Infect. 2022, 11, 2393–2404. [Google Scholar] [CrossRef] [PubMed]
- Knoll, R.; Schultze, J.L.; Schulte-Schrepping, J. Monocytes and Macrophages in COVID-19. Front. Immunol. 2021, 12, 720109. [Google Scholar] [CrossRef]
- Merad, M.; Martin, J.C. Pathological inflammation in patients with COVID-19: A key role for monocytes and macrophages. Nat. Rev. Immunol. 2020, 20, 355–362. [Google Scholar] [CrossRef]
- Zhou, Y.; Fu, B.; Zheng, X.; Wang, D.; Zhao, C.; Qi, Y.; Sun, R.; Tian, Z.; Xu, X.; Wei, H. Pathogenic T-cells and inflammatory monocytes incite inflammatory storms in severe COVID-19 patients. Natl. Sci. Rev. 2020, 7, 998–1002. [Google Scholar] [CrossRef]
- Gatti, A.; Radrizzani, D.; Viganò, P.; Mazzone, A.; Brando, B. Decrease of Non-Classical and Intermediate Monocyte Subsets in Severe Acute SARS-CoV-2 Infection. Cytometry A 2020, 97, 887–890. [Google Scholar] [CrossRef]
- Giamarellos-Bourboulis, E.J.; Netea, M.G.; Rovina, N.; Akinosoglou, K.; Antoniadou, A.; Antonakos, N.; Damoraki, G.; Gkavogianni, T.; Adami, M.E.; Katsaounou, P.; et al. Complex Immune Dysregulation in COVID-19 Patients with Severe Respiratory Failure. Cell Host Microbe. 2020, 27, 992–1000.e3. [Google Scholar] [CrossRef]
- Spinetti, T.; Hirzel, C.; Fux, M.; Walti, L.N.; Schober, P.; Stueber, F.; Luedi, M.M.; Schefold, J.C. Reduced Monocytic Human Leukocyte Antigen-DR Expression Indicates Immunosuppression in Critically Ill COVID-19 Patients. Anesth. Analg. 2020, 131, 993–999. [Google Scholar] [CrossRef]
- Wang, F.; Hou, H.; Yao, Y.; Wu, S.; Huang, M.; Ran, X.; Zhou, H.; Liu, Z.; Sun, Z. Systemically comparing host immunity between survived and deceased COVID-19 patients. Cell. Mol. Immunol. 2020, 17, 875–877. [Google Scholar] [CrossRef]
- Kvedaraite, E.; Hertwig, L.; Sinha, I.; Ponzetta, A.; HedMyrberg, I.; Lourda, M.; Dzidic, M.; Akber, M.; Klingström, J.; Folkesson, E.; et al. Major alterations in the mononuclear phagocyte landscape associated with COVID-19 severity. Proc. Natl. Acad. Sci. USA 2021, 118, e2018587118. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Kuba, K.; Neely, G.G.; Yaghubian-Malhami, R.; Perkmann, T.; van Loo, G.; Ermolaeva, M.; Veldhuizen, R.; Leung, Y.H.; Wang, H.; et al. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell 2008, 133, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Crozat, K.; Beutler, B. TLR7: A new sensor of viral infection. Proc. Natl. Acad. Sci. USA 2004, 101, 6835–6836. [Google Scholar] [CrossRef] [PubMed]
- Moss, P. The T cell immune response against SARS-CoV-2. Nat. Immunol. 2022, 23, 186–193. [Google Scholar] [CrossRef]
- Rydyznski Moderbacher, C.; Ramirez, S.I.; Dan, J.M.; Grifoni, A.; Hastie, K.M.; Weiskopf, D.; Belanger, S.; Abbott, R.K.; Kim, C.; Choi, J.; et al. Antigen-Specific Adaptive Immunity to SARS-CoV-2 in Acute COVID-19 and Associations with Age and Disease Severity. Cell 2020, 183, 996–1012.e19. [Google Scholar] [CrossRef]
- Notarbartolo, S.; Ranzani, V.; Bandera, A.; Gruarin, P.; Bevilacqua, V.; Putignano, A.R.; Gobbini, A.; Galeota, E.; Manara, C.; Bombaci, M.; et al. Integrated longitudinal immunophenotypic, transcriptional and repertoire analyses delineate immune responses in COVID-19 patients. Sci. Immunol. 2021, 6, eabg5021. [Google Scholar] [CrossRef]
- Bergamaschi, L.; Mescia, F.; Turner, L.; Hanson, A.L.; Kotagiri, P.; Dunmore, B.J.; Ruffieux, H.; De Sa, A.; Huhn, O.; Morgan, M.D.; et al. Longitudinal analysis reveals that delayed bystander CD8+ T cell activation and early immune pathology distinguish severe COVID-19 from mild disease. Immunity 2021, 54, 1257–1275.e8. [Google Scholar] [CrossRef]
- Chen, G.; Wu, D.; Guo, W.; Cao, Y.; Huang, D.; Wang, H.; Wang, T.; Zhang, X.; Chen, H.; Yu, H.; et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J. Clin. Investig. 2020, 130, 2620–2629. [Google Scholar] [CrossRef]
- Diao, B.; Wang, C.; Tan, Y.; Chen, X.; Liu, Y.; Ning, L.; Chen, L.; Li, M.; Liu, Y.; Wang, G.; et al. Reduction and Functional Exhaustion of T Cells in Patients with Coronavirus Disease 2019 (COVID-19). Front. Immunol. 2020, 11, 827. [Google Scholar] [CrossRef]
- Le Bert, N.; Clapham, H.E.; Tan, A.T.; Chia, W.N.; Tham, C.Y.L.; Lim, J.M.; Kunasegaran, K.; Tan, L.W.L.; Dutertre, C.A.; Shankar, N.; et al. Highly functional virus-specific cellular immune response in asymptomatic SARS-CoV-2 infection. J. Exp. Med. 2021, 218, e20202617. [Google Scholar] [CrossRef]
- Schulien, I.; Kemming, J.; Oberhardt, V.; Wild, K.; Seidel, L.M.; Killmer, S.; Sagar; Daul, F.; Salvat Lago, M.; Decker, A.; et al. Characterization of pre-existing and induced SARS-CoV-2-specific CD8+ T cells. Nat. Med. 2021, 27, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.T.; Linster, M.; Tan, C.W.; Le Bert, N.; Chia, W.N.; Kunasegaran, K.; Zhuang, Y.; Tham, C.Y.L.; Chia, A.; Smith, G.J.D.; et al. Early induction of functional SARS-CoV-2-specific T cells associates with rapid viral clearance and mild disease in COVID-19 patients. Cell Rep. 2021, 34, 108728. [Google Scholar] [CrossRef] [PubMed]
- Szabo, P.A.; Dogra, P.; Gray, J.I.; Wells, S.B.; Connors, T.J.; Weisberg, S.P.; Krupska, I.; Matsumoto, R.; Poon, M.M.L.; Idzikowski, E.; et al. Longitudinal profiling of respiratory and systemic immune responses reveals myeloid cell-driven lung inflammation in severe COVID-19. Immunity 2021, 54, 797–814. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Chen, D.; Yuan, D.; Lausted, C.; Choi, J.; Dai, C.L.; Voillet, V.; Duvvuri, V.R.; Scherler, K.; Troisch, P.; et al. Multi-Omics Resolves a Sharp Disease-State Shift between Mild and Moderate COVID-19. Cell 2020, 183, 1479–1495.e20. [Google Scholar] [CrossRef] [PubMed]
- Mathew, D.; Giles, J.R.; Baxter, A.E.; Oldridge, D.A.; Greenplate, A.R.; Wu, J.E.; Alanio, C.; Kuri-Cervantes, L.; Pampena, M.B.; D’Andrea, K.; et al. Deep immune profiling of COVID-19 patients reveals distinct immunotypes with therapeutic implications. Science 2020, 369, eabc8511. [Google Scholar] [CrossRef] [PubMed]
- Kusnadi, A.; Ramírez-Suástegui, C.; Fajardo, V.; Chee, S.J.; Meckiff, B.J.; Simon, H.; Pelosi, E.; Seumois, G.; Ay, F.; Vijayanand, P.; et al. Severely ill COVID-19 patients display impaired exhaustion features in SARS-CoV-2-reactive CD8+ T cells. Sci. Immunol. 2021, 6, eabe4782. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, L.; Sang, L.; Ye, F.; Ruan, S.; Zhong, B.; Song, T.; Alshukairi, A.N.; Chen, R.; Zhang, Z.; et al. Kinetics of viral load and antibody response in relation to COVID-19 severity. J. Clin. Investig. 2020, 130, 5235–5244. [Google Scholar] [CrossRef] [PubMed]
- Dogan, M.; Kozhaya, L.; Placek, L.; Gunter, C.; Yigit, M.; Hardy, R.; Plassmeyer, M.; Coatney, P.; Lillard, K.; Bukhari, Z.; et al. SARS-CoV-2 specific antibody and neutralization assays reveal the wide range of the humoral immune response to virus. Commun. Biol. 2021, 4, 129. [Google Scholar] [CrossRef]
- Loos, C.; Atyeo, C.; Fischinger, S.; Burke, J.; Slein, M.D.; Streeck, H.; Lauffenburger, D.; Ryan, E.T.; Charles, R.C.; Alter, G. Evolution of Early SARS-CoV-2 and Cross-Coronavirus Immunity. mSphere 2020, 5, e00622-20. [Google Scholar] [CrossRef]
- Sun, B.; Feng, Y.; Mo, X.; Zheng, P.; Wang, Q.; Li, P.; Peng, P.; Liu, X.; Chen, Z.; Huang, H.; et al. Kinetics of SARS-CoV-2 specific IgM and IgG responses in COVID-19 patients. Emerg. Microbes Infect. 2020, 9, 940–948. [Google Scholar] [CrossRef]
- Kuri-Cervantes, L.; Pampena, M.B.; Meng, W.; Rosenfeld, A.M.; Ittner, C.A.G.; Weisman, A.R.; Agyekum, R.S.; Mathew, D.; Baxter, A.E.; Vella, L.A.; et al. Comprehensive mapping of immune perturbations associated with severe COVID-19. Sci. Immunol. 2020, 5, eabd7114. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, C.J.; Swadling, L.; Gibbons, J.M.; Pade, C.; Jensen, M.P.; Diniz, M.O.; Schmidt, N.M.; Butler, D.K.; Amin, O.E.; Bailey, S.N.L.; et al. Discordant neutralizing antibody and T cell responses in asymptomatic and mild SARS-CoV-2 infection. Sci. Immunol. 2020, 5, eabf3698. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yu, J.; McMahan, K.; Jacob-Dolan, C.; He, X.; Giffin, V.; Wu, C.; Sciacca, M.; Powers, O.; Nampanya, F.; et al. CD8 T Cells Contribute to Vaccine Protection Against SARS-CoV-2 in Macaques. Sci. Immunol. 2022, eabq7647. [Google Scholar] [CrossRef] [PubMed]
- Newson, L.; Manyonda, I.; Lewis, R.; Preissner, R.; Preissner, S.; Seeland, U. Sensitive to Infection but Strong in Defense-Female Sex and the Power of Oestradiol in the COVID-19 Pandemic. Front. Glob. Womens Health 2021, 11, 651752. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Chen, X.; Cai, Y.; Xia, J.; Zhou, X.; Xu, S.; Huang, H.; Zhang, L.; Zhou, X.; Du, C.; et al. Risk Factors Associated With Acute Respiratory Distress Syndrome and Death in Patients with Coronavirus Disease 2019 Pneumonia in Wuhan, China. JAMA Intern. Med. 2020, 180, 934–943. [Google Scholar] [CrossRef] [PubMed]
- Passaro, A.; Bestvina, C.; Velez Velez, M.; Garassino, M.C.; Garon, E.; Peters, S. Severity of COVID-19 in patients with lung cancer: Evidence and challenges. J. Immunother. Cancer 2021, 9, e002266. [Google Scholar] [CrossRef]
- Bakouny, Z.; Hawley, J.E.; Choueiri, T.K.; Peters, S.; Rini, B.I.; Warner, J.L.; Painter, C.A. COVID-19 and cancer: Current challenges and perspectives. Cancer Cell 2020, 38, 629–646. [Google Scholar] [CrossRef]
- Zhang, L.; Zhu, F.; Xie, L.; Wang, C.; Wang, J.; Chen, R.; Jia, P.; Guan, H.Q.; Peng, L.; Chen, Y.; et al. Clinical characteristics of COVID-19-infected cancer patients: A retrospective case study in three hospitals within Wuhan, China. Ann. Oncol. 2020, 31, 894–901. [Google Scholar] [CrossRef]
- Saini, K.S.; Tagliamento, M.; Lambertini, M.; McNally, R.; Romano, M.; Leone, M.; Curigliano, G.; de Azambuja, E. Mortality in patients with cancer and coronavirus disease 2019: A systematic review and pooled analysis of 52 studies. Eur. J. Cancer 2020, 139, 43–50. [Google Scholar] [CrossRef]
- Dai, M.; Liu, D.; Liu, M.; Zhou, F.; Li, G.; Chen, Z.; Zhang, Z.; You, H.; Wu, M.; Zheng, Q.; et al. Patients with cancer appear more vulnerable to SARS-CoV-2: A multicenter study during the COVID-19 outbreak. Cancer Discov. 2020, 10, 783–791. [Google Scholar] [CrossRef]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Kulkarni, A.A.; Wilson, G.; Fujioka, N.; Patel, M.R. Mortality from COVID-19 in patients with lung cancer. J. Cancer Metastasis Treat. 2021, 7, 31. [Google Scholar] [CrossRef]
- Passaro, A.; Peters, S.; Mok, T.S.K.; Attili, I.; Mitsudomi, T.; de Marinis, F. Testing for COVID-19 in lung cancer patients. Ann. Oncol. 2020, 31, 832–834. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Guan, W.; Chen, R.; Wang, W.; Li, J.; Xu, K.; Li, C.; Ai, Q.; Lu, W.; Liang, H.; et al. Cancer patients in SARS-CoV-2 infection: A nationwide analysis in China. Lancet Oncol. 2020, 21, 335–337. [Google Scholar] [CrossRef]
- Yu, J.; Ouyang, W.; Chua, M.L.K.; Xie, C. SARS-CoV-2 transmission in patients with cancer at a tertiary care hospital in Wuhan, China. JAMA Oncol. 2020, 6, 1108. [Google Scholar] [CrossRef] [PubMed]
- Al-Quteimat, O.M.; Amer, A.M. The Impact of the COVID-19 Pandemic on Cancer Patients. Am. J. Clin. Oncol. 2020, 43, 452–455. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the immune system in cancer: From tumor initiation to metastatic progression. Genes Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef]
- Kos, K.; de Visser, K.E. The multifaceted role of regulatory T cells in breast cancer. Annu. Rev. Cancer Biol. 2021, 5, 291–310. [Google Scholar] [CrossRef]
- Ohue, Y.; Nishikawa, H. Regulatory T (Treg) cells in cancer: Can Treg cells be a new therapeutic target? Cancer Sci. 2019, 110, 2080–2089. [Google Scholar] [CrossRef]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef]
- Fornetti, J.; Welm, A.L.; Stewart, S.A. Understanding the Bone in Cancer Metastasis. J. Bone Miner. Res. 2018, 33, 2099–2113. [Google Scholar] [CrossRef] [PubMed]
- Ménétrier-Caux, C.; Ray-Coquard, I.; Blay, J.Y.; Caux, C. Lymphopenia in Cancer Patients and its Effects on Response to Immunotherapy: An opportunity for combination with Cytokines? J. Immunother. Cancer 2019, 7, 85. [Google Scholar] [CrossRef] [PubMed]
- Ghanavat, M.; Ebrahimi, M.; Rafieemehr, H.; Maniati, M.; Behzad, M.M.; Shahrabi, S. Thrombocytopenia in solid tumors: Prognostic significance. Oncol. Rev. 2019, 13, 413. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, B.L.; Cooksley, T.; Johnson, D.B.; Anderson, R.; Shannon, V.R. Treatment of infections in cancer patients: An update from the neutropenia, infection and myelosuppression study group of the Multinational Association for Supportive Care in Cancer (MASCC). Expert Rev. Clin. Pharmacol. 2021, 14, 295–313. [Google Scholar] [CrossRef] [PubMed]
- Yeoh, C.B.; Lee, K.J.; Rieth, E.F.; Mapes, R.; Tchoudovskaia, A.V.; Fischer, G.W.; Tollinche, L.E. COVID-19 in the Cancer Patient. Anesth. Analg. 2020, 131, 16–23. [Google Scholar] [CrossRef]
- Kasymjanova, G.; Anwar, A.; Cohen, V.; Sultanem, K.; Pepe, C.; Sakr, L.; Friedmann, J.; Agulnik, J.S. The Impact of COVID-19 on the Diagnosis and Treatment of Lung Cancer at a Canadian Academic Center: A Retrospective Chart Review. Curr. Oncol. 2021, 28, 360. [Google Scholar] [CrossRef]
- Buti, S.; Perrone, F.; Zielli, T.; Mazzaschi, G.; Casartelli, C.; Leonetti, A.; Milanese, G.; Silva, M.; Eufrasia Ledda, R.; Musolino, A.; et al. Clinical Impact of COVID-19 Outbreak on Cancer Patients: A Retrospective Study. Clin. Med. Insights Oncol. 2021, 15, 11795549211043427. [Google Scholar] [CrossRef]
- Cantini, L.; Mentrasti, G.; Russo, G.L.; Signorelli, D.; Pasello, G.; Rijavec, E.; Russano, M.; Antonuzzo, L.; Rocco, D.; Giusti, R.; et al. Evaluation of COVID-19 impact on DELAYing diagnostic-therapeutic pathways of lung cancer patients in Italy (COVID-DELAY study): Fewer cases and higher stages from a real-world scenario. ESMO Open 2022, 7, 100406. [Google Scholar] [CrossRef]
- Werner, R.S.; Lörtscher, A.; Kirschner, M.B.; Lauk, O.; Furrer, K.; Caviezel, C.; Schneiter, D.; Inci, I.; Hillinger, S.; Curioni-Fontecedro, A.; et al. Surgical management of lung cancer during the COVID-19 pandemic—A narrative review and single-centre report. Swiss Med. Wkly. 2022, 152, w30109. [Google Scholar]
- Gourd, E. Lung cancer control in the UK hit badly by COVID-19 pandemic. Lancet Oncol. 2020, 21, 1559. [Google Scholar] [CrossRef]
- United Kingdom Lung Cancer Coalition. COVID-19 Matters. Available online: https://www.uklcc.org.uk/wp-content/uploads/2020/10/UKLCC-COVID-19-Matters-Report-Oct-2020.pdf (accessed on 10 December 2020).
- Wilkinson, E. Dramatic drop in new cancer drug trials during the COVID-19 pandemic. Lancet Oncol. 2021, 22, 305. [Google Scholar] [CrossRef]
- Moujaess, E.; Kourie, H.R.; Ghosn, M. Cancer patients and research during COVID-19 pandemic: A systematic review of current evidence. Crit. Rev. Oncol. Hematol. 2020, 150, 102972. [Google Scholar] [CrossRef] [PubMed]
- Upadhaya, S.; Yu, J.X.; Oliva, C.; Hooton, M.; Hodge, J.; Hubbard-Lucey, V.M. Impact of COVID-19 on oncology clinical trials. Nat. Rev. Drug Discov. 2020, 19, 376–377. [Google Scholar] [CrossRef] [PubMed]
- Tarantino, P.; Trapani, D.; Curigliano, G. Conducting phase 1 cancer clinical trials during the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)-related disease pandemic. Eur. J. Cancer 2020, 132, 8–10. [Google Scholar] [CrossRef] [PubMed]
- Bakouny, Z.; Labaki, C.; Bhalla, S.; Schmidt, A.L.; Steinharter, J.A.; Cocco, J.; Tremblay, D.A.; Awad, M.M.; Kessler, A.; Haddad, R.I.; et al. Oncology clinical trial disruption during the COVID-19 pandemic: A COVID-19 and cancer outcomes study. Ann. Oncol. 2022, 33, 836–844. [Google Scholar] [CrossRef]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef]
- Hakem, R. DNA-damage repair; the good, the bad, and the ugly. EMBO J. 2008, 27, 589–605. [Google Scholar] [CrossRef]
- Harper, J.W.; Elledge, S.J. The DNA damage response: Ten years after. Mol. Cell 2007, 14, 739–745. [Google Scholar] [CrossRef]
- Pateras, I.S.; Havaki, S.; Nikitopoulou, X.; Vougas, K.; Townsend, P.A.; Panayiotidis, M.I.; Georgakilas, A.G.; Gorgoulis, V.G. The DNA damage response and immune signaling alliance: Is it good or bad? Nature decides when and where. Pharmacol. Ther. 2015, 154, 36–56. [Google Scholar] [CrossRef]
- Shuck, S.C.; Short, E.A.; Turchi, J.J. Eukaryotic nucleotide excision repair: From understanding mechanisms to influencing biology. Cell Res. 2008, 18, 64–72. [Google Scholar] [CrossRef]
- Krokan, H.E.; Bjørås, M. Base excision repair. Cold Spring Harb. Perspect. Biol. 2013, 5, 012583. [Google Scholar] [CrossRef] [PubMed]
- Jiricny, J. The multifaceted mismatch-repair system. Nat. Rev. Mol. Cell Biol. 2006, 7, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T. Homologous recombination in cancer development, treatment and development of drug resistance. Carcinogenesis 2010, 6, 955–960. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, A.; Tapryal, N.; Venkova, T.; Horikoshi, N.; Pandita, R.K.; Sarker, A.H.; Sarkar, P.S.; Pandita, T.K.; Hazra, T.K. Classical non-homologous end-joining pathway utilizes nascent RNA for error-free double-strand break repair of transcribed genes. Nat. Commun. 2016, 7, 13049. [Google Scholar] [CrossRef] [PubMed]
- Iliakis, G.; Murmann, T.; Soni, A. Alternative end-joining repair pathways are the ultimate backup for abrogated classical non-homologous end-joining and homologous recombination repair: Implications for the formation of chromosome translocations. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2015, 793, 166–175. [Google Scholar] [CrossRef]
- Lieber, M.R. The mechanism of human nonhomologous DNA end-joining. J. Biol. Chem. 2008, 283, 1–5. [Google Scholar] [CrossRef]
- Hashimoto, S.; Anai, H.; Hanada, K. Mechanisms of interstrand DNA crosslink repair and human disorders. Genes Environ. 2016, 38, 9. [Google Scholar] [CrossRef]
- Wang, Y.; Leung, J.W.; Jiang, Y.; Lowery, M.G.; Do, H.; Vasquez, K.M.; Chen, J.; Wang, W.; Li, L. FANCM and FAAP24 maintain genome stability via cooperative as well as unique functions. Mol. Cell 2013, 49, 997–1009. [Google Scholar] [CrossRef]
- Hiddinga, B.I.; Pauwels, P.; Janssens, A.; van Meerbeeck, J.P. O6-Methylguanine-DNA methyltransferase (MGMT): A drugable target in lung cancer? Lung Cancer 2017, 107, 91–99. [Google Scholar] [CrossRef]
- Ryan, E.L.; Hollingworth, R.; Grand, R.J. Activation of the DNA damage response by RNA viruses. Biomolecules 2016, 6, 2. [Google Scholar] [CrossRef]
- Chambers, B.S.; Heaton, B.E.; Rausch, K.; Dumm, R.E.; Hamilton, J.R.; Cherry, S.; Heaton, N.S. DNA mismatch repair is required for the host innate response and controls cellular fate after influenza virus infection. Nat. Microbiol. 2019, 4, 1964–1977. [Google Scholar] [CrossRef] [PubMed]
- Shubina, M.; Tummers, B.; Boyd, D.F.; Zhang, T.; Yin, C.; Gautam, A.; Guo, X.J.; Rodriguez, D.A.; Kaiser, W.J.; Vogel, P.; et al. Necroptosis restricts influenza A virus as a stand-alone cell death mechanism. J. Exp. Med. 2020, 217, e20191259. [Google Scholar] [CrossRef] [PubMed]
- Downey, J.; Pernet, E.; Coulombe, F.; Divangahi, M. Dissecting host cell death programs in the pathogenesis of influenza. Microbes Infect. 2018, 20, 560–569. [Google Scholar] [CrossRef] [PubMed]
- Van de Sandt, C.E.; Kreijtz, J.H.; Rimmelzwaan, G.F. Evasion of influenza A viruses from innate and adaptive immune responses. Viruses 2012, 4, 1438–1476. [Google Scholar] [CrossRef]
- Haque, F.; Lillie, P.; Haque, F.; Maraveyas, A. Deficient DNA mismatch repair and persistence of SARS-CoV-2 RNA shedding: A case report of hereditary nonpolyposis colorectal cancer with COVID-19 infection. BMC Infect. Dis. 2021, 21, 854. [Google Scholar] [CrossRef] [PubMed]
- Dash, S.; Dash, C.; Pandhare, J. Therapeutic Significance of microRNA-Mediated Regulation of PARP-1 in SARS-CoV-2 Infection. Non-Coding RNA 2021, 7, 60. [Google Scholar] [CrossRef]
- Challa, S.; Stokes, M.S.; Kraus, W.L. MARTs and MARylation in the Cytosol: Biological Functions, Mechanisms of Action, and Therapeutic Potential. Cells 2021, 10, 313. [Google Scholar] [CrossRef]
- Heer, C.D.; Sanderson, D.J.; Voth, L.S.; Alhammad, Y.M.O.; Schmidt, M.S.; Trammell, S.A.J.; Perlman, S.; Cohen, M.S.; Fehr, A.R.; Brenner, C. Coronavirus infection and PARP expression dysregulate the NAD metabolome: An actionable component of innate immunity. J. Biol. Chem. 2020, 295, 17986–17996. [Google Scholar] [CrossRef]
- Fehr, A.R.; Singh, S.A.; Kerr, C.M.; Mukai, S.; Higashi, H.; Aikawa, M. The impact of PARPs and ADP-ribosylation on inflammation and host–pathogen interactions. Genes Dev. 2020, 34, 341–359. [Google Scholar] [CrossRef]
- Bai, P. Biology of poly (ADP-ribose) polymerases: The factotums of cell maintenance. Mol. Cell 2015, 58, 947–958. [Google Scholar] [CrossRef]
- Fatokun, A.A.; Dawson, V.L.; Dawson, T.M. Parthanatos: Mitochondrial-linked mechanisms and therapeutic opportunities. Br. J. Pharmacol. 2014, 171, 2000–2016. [Google Scholar] [PubMed]
- Victor, J.; Deutsch, J.; Whitaker, A.; Lamkin, E.N.; March, A.; Zhou, P.; Botten, J.W.; Chatterjee, N. SARS-CoV-2 triggers DNA damage response in Vero E6 cells. Biochem. Biophys. Res. Commun. 2021, 579, 141–145. [Google Scholar] [CrossRef] [PubMed]
- De Lange, T. Shelterin: The protein complex that shapes and safeguards human telomeres. Genes Dev. 2005, 19, 2100–2110. [Google Scholar] [CrossRef] [PubMed]
- Sepe, S.; Rossiello, F.; Cancila, V.; Iannelli, F.; Matti, V.; Cicio, G.; Cabrini, M.; Marinelli, E.; Alabi, B.R.; di Lillo, A.; et al. DNA damage response at telomeres boosts the transcription of SARS-CoV-2 receptor ACE2 during aging. EMBO Rep. 2022, 23, 53658. [Google Scholar] [CrossRef]
- Ren, H.; Ma, C.; Peng, H.; Zhang, B.; Zhou, L.; Su, Y.; Gao, X.; Huang, H. Micronucleus production, activation of DNA damage response and cGAS-STING signaling in syncytia induced by SARS-CoV-2 infection. Biol. Direct. 2021, 16, 20. [Google Scholar] [CrossRef]
- Gonçalves, S.O.; Luz, T.M.D.; Silva, A.M.; de Souza, S.S.; Montalvão, M.F.; Guimarães, A.T.B.; Ahmed, M.A.I.; Araújo, A.P.D.C.; Karthi, S.; Malafaia, G. Can spike fragments of SARS-CoV-2 induce genomic instability and DNA damage in the guppy, Poecilia reticulate? An unexpected effect of the COVID-19 pandemic. Sci. Total Environ. 2022, 825, 153988. [Google Scholar] [CrossRef]
- Amatore, D.; Sgarbanti, R.; Aquilano, K.; Baldelli, S.; Limongi, D.; Civitelli, L.; Nencioni, L.; Garaci, E.; Ciriolo, M.R.; Palamara, A.T. Influenza virus replication in lung epithelial cells depends on redox-sensitive pathways activated by NOX4-derived ROS. Cell Microbiol. 2015, 17, 131–145. [Google Scholar] [CrossRef]
- Garcia, G., Jr.; Sharma, A.; Ramaiah, A.; Sen, C.; Purkayastha, A.; Kohn, D.B.; Parcells, M.S.; Beck, S.; Kim, H.; Bakowski, M.A.; et al. Antiviral drug screen identifies DNA-damage response inhibitor as potent blocker of SARS-CoV-2 replication. Cell Rep. 2021, 35, 108940. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Cheng, S.C.; Wahner Hendrickson, A.E.; Penson, R.T.; Schumer, S.T.; Doyle, L.A.; Lee, E.K.; Kohn, E.C.; Duska, L.R.; Crispens, M.A.; et al. Berzosertib plus gemcitabine versus gemcitabine alone in platinum-resistant high-grade serous ovarian cancer: A multicentre, open-label, randomised, phase 2 trial. Lancet Oncol. 2020, 21, 957–968. [Google Scholar] [CrossRef]
- Kulasinghe, A.; Liu, N.; Tan, C.W.; Monkman, J.; Sinclair, J.E.; Bhuva, D.D.; Godbolt, D.; Pan, L.; Nam, A.; Sadeghirad, H.; et al. Transcriptomic profiling of cardiac tissues from SARS-CoV-2 patients identifies DNA damage. Immunology 2022, in press. [CrossRef]
- Souliotis, V.L.; Vlachogiannis, N.I.; Pappa, M.; Argyriou, A.; Ntouros, P.A.; Sfikakis, P.P. DNA Damage Response and Oxidative Stress in Systemic Autoimmunity. Int. J. Mol. Sci. 2019, 21, 55. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; Prather, E.R.; Stetskiv, M.; Garrison, D.E.; Meade, J.R.; Peace, T.I.; Zhou, T. Inflammaging and Oxidative Stress in Human Diseases: From Molecular Mechanisms to Novel Treatments. Int. J. Mol. Sci. 2019, 20, 4472. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [PubMed]
- Ntyonga-Pono, M.P. COVID-19 infection and oxidative stress: An under-explored approach for prevention and treatment? Pan. Afr. Med. J. 2020, 35, 12. [Google Scholar] [CrossRef] [PubMed]
- Schönrich, G.; Raftery, M.J.; Samstag, Y. Devilishly radical NETwork in COVID-19: Oxidative stress, neutrophil extracellular traps (NETs), and T cell suppression. Adv. Biol. Regul. 2020, 77, 100741. [Google Scholar]
- Laforge, M.; Elbim, C.; Frère, C.; Hémadi, M.; Massaad, C.; Nuss, P.; Benoliel, J.J.; Becker, C. Tissue damage from neutrophil-induced oxidative stress in COVID-19. Nat. Rev. Immunol. 2020, 20, 515–516. [Google Scholar] [CrossRef]
- Checconi, P.; De Angelis, M.; Marcocci, M.E.; Fraternale, A.; Magnani, M.; Palamara, A.T.; Nencioni, L. Redox-Modulating Agents in the Treatment of Viral Infections. Int. J. Mol. Sci. 2020, 21, 4084. [Google Scholar] [CrossRef]
- Hati, S.; Bhattacharyya, S. Impact of Thiol-Disulfide Balance on the Binding of COVID-19 Spike Protein with Angiotensin-Converting Enzyme 2 Receptor. ACS Omega 2020, 5, 16292–16298. [Google Scholar] [CrossRef]
- Busse, L.W.; Chow, J.H.; McCurdy, M.T.; Khanna, A.K. COVID-19 and the RAAS-a potential role for angiotensin II? Crit. Care 2020, 24, 136. [Google Scholar] [CrossRef]
- Suhail, S.; Zajac, J.; Fossum, C.; Lowater, H.; McCracken, C.; Severson, N.; Laatsch, B.; Narkiewicz-Jodko, A.; Johnson, B.; Liebau, J.; et al. Role of Oxidative Stress on SARS-CoV (SARS) and SARS-CoV-2 (COVID-19) Infection: A Review. Protein J. 2020, 39, 644–656. [Google Scholar]
- Fraternale, A.; Paoletti, M.F.; Casabianca, A.; Oiry, J.; Clayette, P.; Vogel, J.U.; Cinatl, J.J.; Palamara, A.T.; Sgarbanti, R.; Garaci, E.; et al. Antiviral and immunomodulatory properties of new pro-glutathione (GSH) molecules. Curr. Med. Chem. 2006, 13, 1749–1755. [Google Scholar] [CrossRef] [PubMed]
- Polonikov, A. Endogenous Deficiency of Glutathione as the Most Likely Cause of Serious Manifestations and Death in COVID-19 Patients. ACS Infect. Dis. 2020, 6, 1558–1562. [Google Scholar] [CrossRef] [PubMed]
- Kalvandi, F.; Azarbayjani, M.A.; Azizbeigi, R.; Azizbeigi, K. Elastic resistance training is more effective than vitamin D3 supplementation in reducing oxidative stress and strengthen antioxidant enzymes in healthy men. Eur. J. Clin. Nutr. 2022, 76, 610–615. [Google Scholar] [CrossRef] [PubMed]
- Razdan, K.; Singh, K.; Singh, D. Vitamin D Levels and COVID-19 Susceptibility: Is there any Correlation? Med. Drug Discov. 2020, 7, 100051. [Google Scholar] [CrossRef]
- Saso, L.; Firuzi, O. Pharmacological applications of antioxidants: Lights and shadows. Curr. Drug Targets 2014, 15, 1177–1799. [Google Scholar] [CrossRef]
- Sgarbanti, R.; Amatore, D.; Celestino, I.; Marcocci, M.E.; Fraternale, A.; Ciriolo, M.R.; Magnani, M.; Saladino, R.; Garaci, E.; Palamara, A.T.; et al. Intracellular redox state as target for anti-influenza therapy: Are antioxidants always effective? Curr. Top. Med. Chem. 2014, 14, 2529–2541. [Google Scholar] [CrossRef]
- De Flora, S.; Balansky, R.; La Maestra, S. Rationale for the use of N-acetylcysteine in both prevention and adjuvant therapy of COVID-19. FASEB J. 2020, 34, 13185–13193. [Google Scholar] [CrossRef]
- Poe, F.L.; Corn, J. N-Acetylcysteine: A potential therapeutic agent for SARS-CoV-2. Med. Hypotheses 2020, 143, 109862. [Google Scholar]
- Palamara, A.T.; Brandi, G.; Rossi, L.; Millo, E.; Benatti, U.; Nencioni, L.; Iuvara, A.; Garaci, E.; Magnani, M. New synthetic glutathione derivatives with increased antiviral activities. Antivir. Chem. Chemother. 2004, 15, 83–91. [Google Scholar] [CrossRef]
- Sgarbanti, R.; Nencioni, L.; Amatore, D.; Coluccio, P.; Fraternale, A.; Sale, P.; Mammola, C.L.; Carpino, G.; Gaudio, E.; Magnani, M.; et al. Redox regulation of the influenza hemagglutinin maturation process: A new cell-mediated strategy for anti-influenza therapy. Antioxid. Redox. Signal. 2011, 15, 593–606. [Google Scholar] [CrossRef]
- Musarra-Pizzo, M.; Pennisi, R.; Ben-Amor, I.; Smeriglio, A.; Mandalari, G.; Sciortino, M.T. In vitro anti-HSV-1 activity of polyphenol-rich extracts and pure polyphenol compounds derived from pistachios kernels (Pistacia vera L.). Plants 2020, 9, 267. [Google Scholar] [CrossRef] [PubMed]
- Annunziata, G.; Maisto, M.; Schisano, C.; Ciampaglia, R.; Narciso, V.; Tenore, G.C.; Novellino, E. Resveratrol as a novel anti-herpes simplex virus nutraceutical agent: An overview. Viruses 2018, 10, 473. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lin, Z.; Guo, M.; Zhao, M.; Xia, Y.; Wang, C.; Xu, T.; Zhu, B. Inhibition of H1N1 influenza virus-induced apoptosis by functionalized selenium nanoparticles with amantadine through ROS-mediated AKT signaling pathways. Int. J. Nanomed. 2018, 13, 2005–2016. [Google Scholar] [CrossRef] [PubMed]
- Kosanovic, T.; Sagic, D.; Djukic, V.; Pljesa-Ercegovac, M.; Savic-Radojevic, A.; Bukumiric, Z.; Lalosevic, M.; Djordjevic, M.; Coric, V.; Simic, T. Time Course of Redox Biomarkers in COVID-19 Pneumonia: Relation with Inflammatory, Multiorgan Impairment Biomarkers and CT Findings. Antioxidants 2021, 10, 1126. [Google Scholar] [CrossRef]
- Lorente, L.; Martín, M.M.; González-Rivero, A.F.; Pérez-Cejas, A.; Cáceres, J.J.; Perez, A.; Ramos-Gómez, L.; Solé-Violán, J.; Marcos, Y.; Ramos, J.A.; et al. DNA and RNA Oxidative Damage and Mortality of Patients with COVID-19. Am. J. Med. Sci. 2021, 361, 585–590. [Google Scholar] [CrossRef]
- Härtlova, A.; Erttmann, S.F.; Raffi, F.A.; Schmalz, A.M.; Resch, U.; Anugula, S.; Lienenklaus, S.; Nilsson, L.M.; Kröger, A.; Nilsson, J.A.; et al. DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti-microbial innate immunity. Immunity 2015, 42, 332–343. [Google Scholar] [CrossRef]
- Günther, C.; Kind, B.; Reijns, M.A.; Berndt, N.; Martinez-Bueno, M.; Wolf, C.; Tüngler, V.; Chara, O.; Lee, Y.A.; Hübner, N.; et al. Defective removal of ribonucleotides from DNA promotes systemic autoimmunity. J. Clin. Investig. 2015, 125, 413–424. [Google Scholar] [CrossRef]
- Gehrke, N.; Mertens, C.; Zillinger, T.; Wenzel, J.; Bald, T.; Zahn, S.; Tüting, T.; Hartmann, G.; Barchet, W. Oxidative damage of DNA confers resistance to cytosolic nuclease TREX1 degradation and potentiates STING-dependent immune sensing. Immunity 2013, 39, 482–495. [Google Scholar] [CrossRef]
- Jaiswal, M.; LaRussom, N.F.; Burgart, L.J.; Gores, G.J. Inflammatory cytokines induce DNA damage and inhibit DNA repair in cholangiocarcinoma cells by a nitric oxide-dependent mechanism. Cancer Res. 2000, 60, 184–190. [Google Scholar]
- Meira, L.B.; Bugni, J.M.; Green, S.L.; Lee, C.W.; Pang, B.; Borenshtein, D.; Rickman, B.H.; Rogers, A.B.; Moroski-Erkul, C.A.; McFaline, J.L.; et al. DNA damage induced by chronic inflammation contributes to colon carcinogenesis in mice. J. Clin. Investig. 2008, 118, 2516–2525. [Google Scholar] [CrossRef]
- Ntouros, P.A.; Vlachogiannis, N.I.; Pappa, M.; Nezos, A.; Mavragani, C.P.; Tektonidou, M.G.; Souliotis, V.L.; Sfikakis, P.P. Effective DNA damage response after acute but not chronic immune challenge: SARS-CoV-2 vaccine versus Systemic Lupus Erythematosus. Clin. Immunol. 2021, 229, 108765. [Google Scholar] [CrossRef] [PubMed]
- Bergamaschi, C.; Terpos, E.; Rosati, M.; Angel, M.; Bear, J.; Stellas, D.; Karaliota, S.; Apostolakou, F.; Bagratuni, T.; Patseas, D.; et al. Systemic IL-15, IFN-γ, and IP-10/CXCL10 signature associated with effective immune response to SARS-CoV-2 in BNT162b2 mRNA vaccine recipients. Cell Rep. 2021, 36, 109504. [Google Scholar] [CrossRef] [PubMed]
- Hyams, C.; Marlow, R.; Maseko, Z.; King, J.; Ward, L.; Fox, K.; Heath, R.; Tuner, A.; Friedrich, Z.; Morrison, L.; et al. Effectiveness of BNT162b2 and ChAdOx1 nCoV-19 COVID-19 vaccination at preventing hospitalisations in people aged at least 80 years: A test-negative, case-control study. Lancet Infect. Dis. 2021, 21, 1539–1548. [Google Scholar] [PubMed]
- Ntouros, P.A.; Kravvariti, E.; Vlachogiannis, N.I.; Pappa, M.; Trougakos, I.P.; Terpos, E.; Tektonidou, M.G.; Souliotis, V.L.; Sfikakis, P.P. Oxidative stress and endogenous DNA damage in blood mononuclear cells may predict anti-SARS-CoV-2 antibody titers after vaccination in older adults. Biochim. Biophys. Acta Mol. Basis Dis. 2022, 1868, 166393. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papanikolaou, C.; Rapti, V.; Stellas, D.; Stefanou, D.T.; Syrigos, K.; Pavlakis, G.N.; Souliotis, V.L. Delineating the SARS-CoV-2 Induced Interplay between the Host Immune System and the DNA Damage Response Network. Vaccines 2022, 10, 1764. https://doi.org/10.3390/vaccines10101764
Papanikolaou C, Rapti V, Stellas D, Stefanou DT, Syrigos K, Pavlakis GN, Souliotis VL. Delineating the SARS-CoV-2 Induced Interplay between the Host Immune System and the DNA Damage Response Network. Vaccines. 2022; 10(10):1764. https://doi.org/10.3390/vaccines10101764
Chicago/Turabian StylePapanikolaou, Christina, Vasiliki Rapti, Dimitris Stellas, Dimitra T. Stefanou, Konstantinos Syrigos, George N. Pavlakis, and Vassilis L. Souliotis. 2022. "Delineating the SARS-CoV-2 Induced Interplay between the Host Immune System and the DNA Damage Response Network" Vaccines 10, no. 10: 1764. https://doi.org/10.3390/vaccines10101764
APA StylePapanikolaou, C., Rapti, V., Stellas, D., Stefanou, D. T., Syrigos, K., Pavlakis, G. N., & Souliotis, V. L. (2022). Delineating the SARS-CoV-2 Induced Interplay between the Host Immune System and the DNA Damage Response Network. Vaccines, 10(10), 1764. https://doi.org/10.3390/vaccines10101764