
mDrop-Seq: Massively Parallel Single-Cell RNA-Seq of Saccharomyces cerevisiae and Candida albicans

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
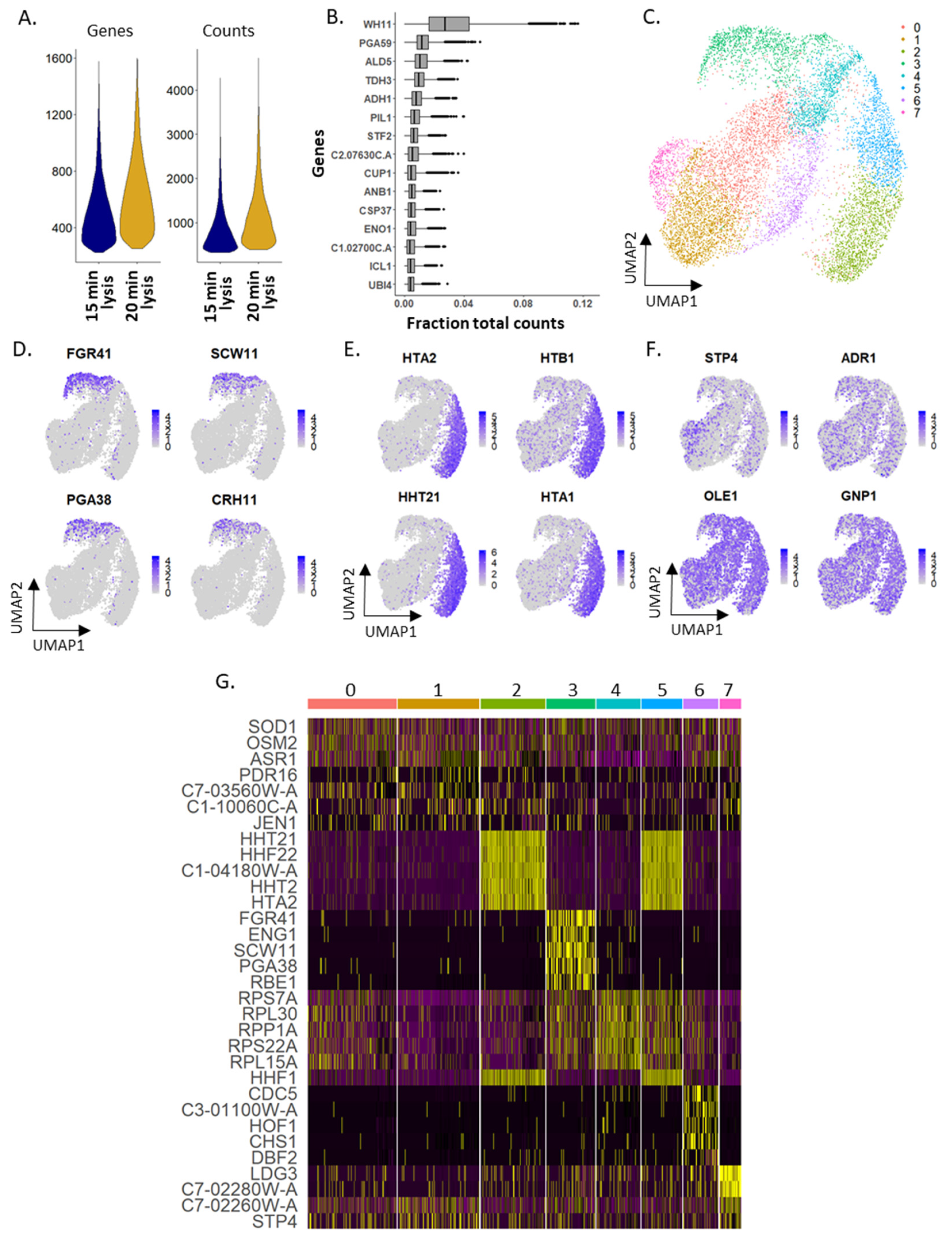
2.1. Optimizing Single Yeast Cell Lysis in Droplets
2.2. Single-Cell Specificity of mDrop-Seq Confirmed by Species Mixing Experiments
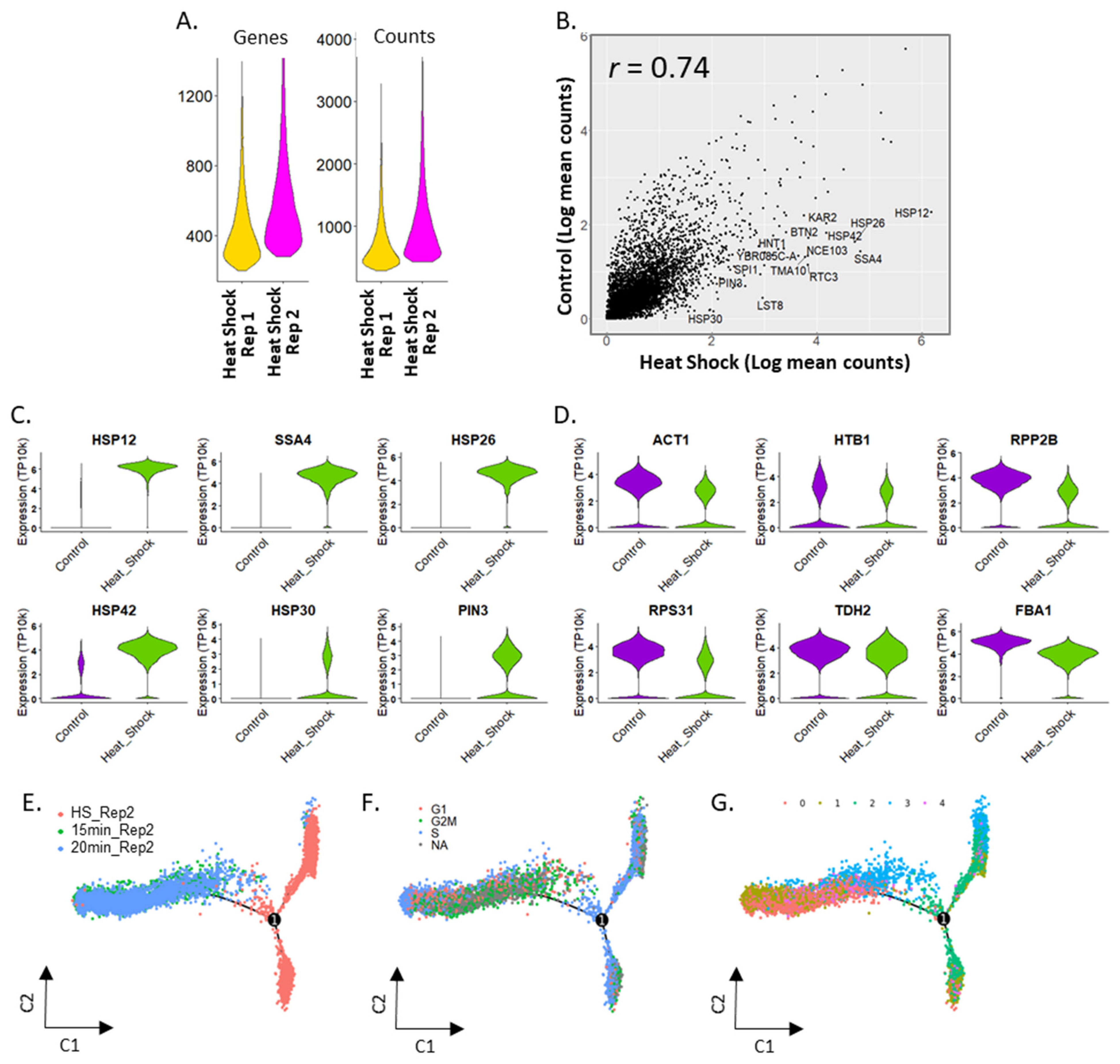
2.3. Heterogeneity in Heat Shock Response of Saccharomyces cerevisiae
2.4. Activation of Stress Response in S. cerevisiae under Heat Shock
2.5. Characterizing Expression Heterogeneity in Candida albicans Cells
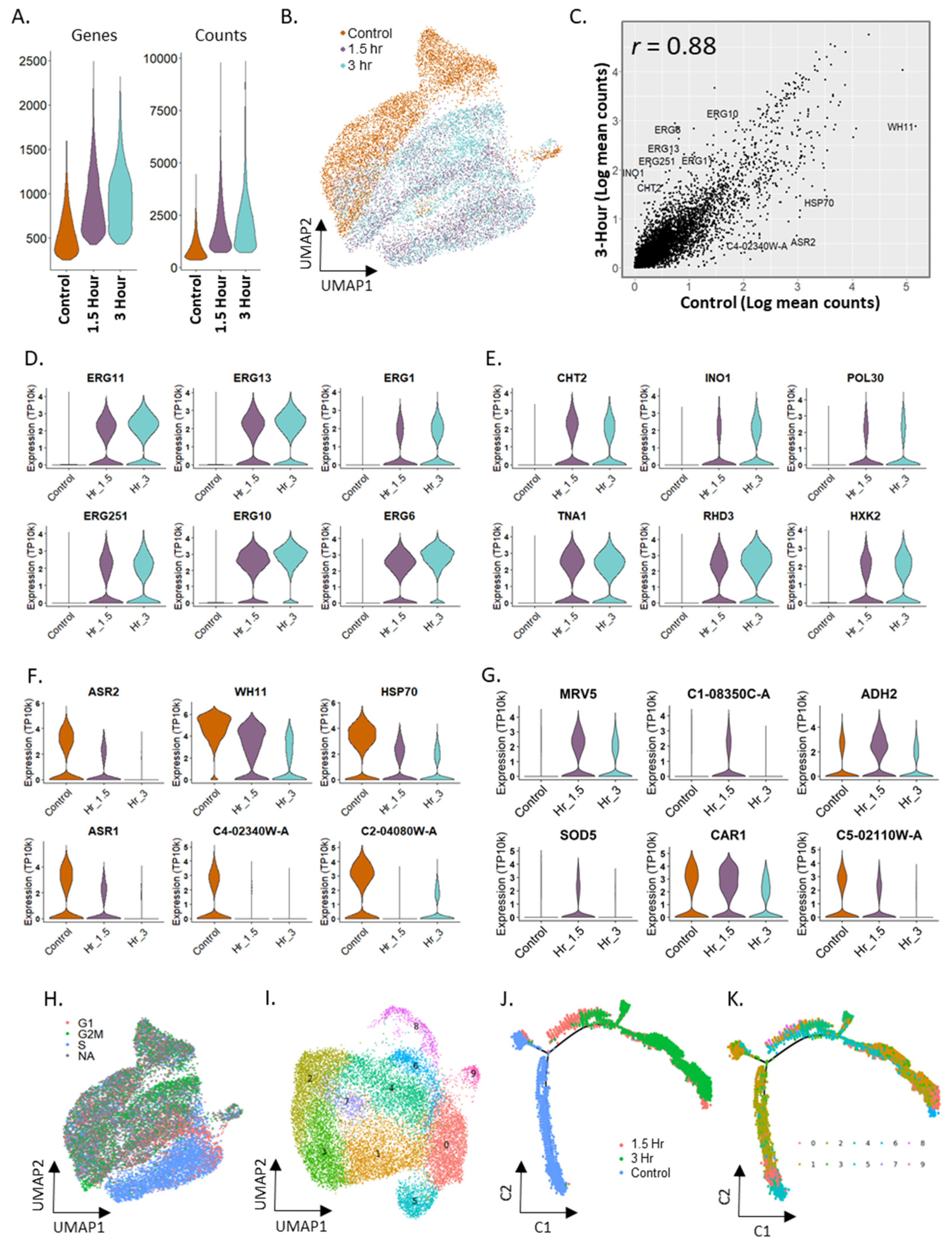
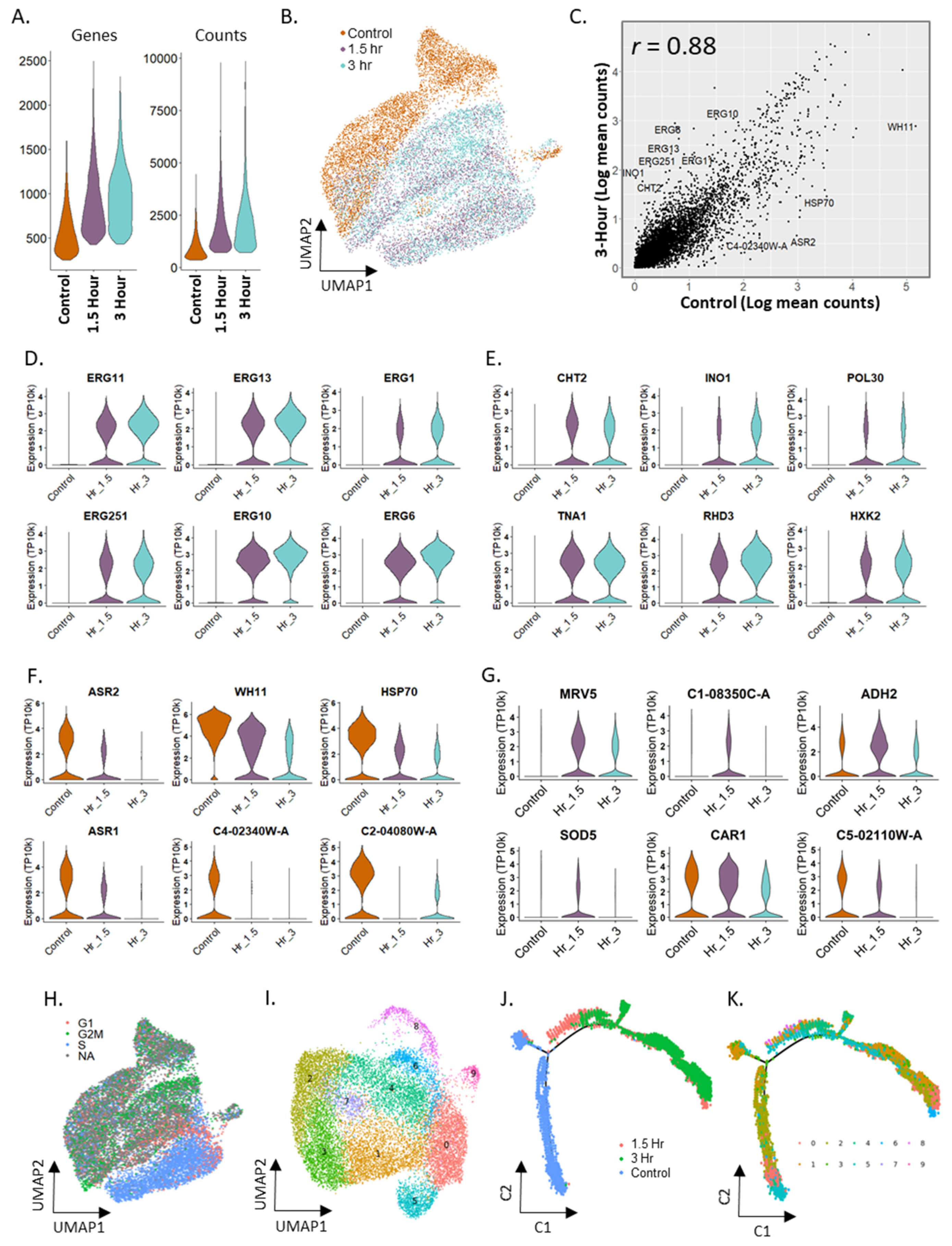
2.6. Differential Expression in Candida albicans in Response to Fluconazole Exposure
2.7. Trajectory Inference in Fluconazole Stimulation in Candida albicans
3. Discussion
3.1. Droplet Content and Stability
3.2. Sequencing and Alignment
3.3. Analysis of Stress, Heat Shock and Response to Anti-Fungal Agent in Yeast Cells
3.4. Scalability as a Technology
4. Materials and Methods
4.1. Yeast Strains and Cell Culture
4.2. Heat Shock Stimulation of S. cerevisiae
4.3. Fluconazole Stimulation of C. albicans
4.4. mDrop-seq Cell Preparation and Co-Encapsulation in Droplets
4.5. Cell Lysis, Reverse Transcription, cDNA Amplification and Next Era Library Generation for mDrop-Seq
4.6. Population-Level RNA-Seq Library Preparation of S. cerevisiae and C. albicans
4.7. Sequencing
4.8. mDrop-Seq Data Preprocessing, Alignment and Quality Control
4.9. Bulk RNA-Seq Data Processing
4.10. Clustering Cells and Generating UMAP
4.11. Calculating Doublet Rates from Species Mixing Experiments
4.12. Batch Correction
4.13. Cell Cycle Analysis
4.14. Hierarchical Clustering of Cells
4.15. Trajectory Analysis on Single-Cell Data
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Macosko, E.Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas, A.R.; Kamitaki, N.; Martersteck, E.M.; et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 2015, 161, 1202–1214. [Google Scholar] [CrossRef] [Green Version]
- Andrews, T.S.; Hemberg, M. Identifying cell populations with scRNASeq. Mol. Aspects Med. 2018, 59, 114–122. [Google Scholar] [CrossRef]
- Szabo, P.A.; Levitin, H.M.; Miron, M.; Snyder, M.E.; Senda, T.; Yuan, J.; Cheng, Y.L.; Bush, E.C.; Dogra, P.; Thapa, P.; et al. Single-cell transcriptomics of human T cells reveals tissue and activation signatures in health and disease. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef]
- Raj, A.; van Oudenaarden, A. Nature, Nurture, or Chance: Stochastic Gene Expression and Its Consequences. Cell 2008, 135, 216–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, G.X.Y.; Terry, J.M.; Belgrader, P.; Ryvkin, P.; Bent, Z.W.; Wilson, R.; Ziraldo, S.B.; Wheeler, T.D.; McDermott, G.P.; Zhu, J.; et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017, 8, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Gao, J.; Huang, Y.; Wang, J. Recent Developments in Single-Cell RNA-Seq of Microorganisms. Biophys. J. 2018, 115, 173–180. [Google Scholar] [CrossRef] [Green Version]
- Saliba, A.-E.; Santos, S.C.; Vogel, J. New RNA-seq approaches for the study of bacterial pathogens. Curr. Opin. Microbiol. 2017, 35, 78–87. [Google Scholar] [CrossRef]
- Gow, N.A.R.; Hube, B. Importance of the Candida albicans cell wall during commensalism and infection. Curr. Opin. Microbiol. 2012, 15, 406–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lesage, G.; Bussey, H. Cell wall assembly in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 2006, 70, 317–343. [Google Scholar] [CrossRef] [Green Version]
- Silhavy, T.J.; Kahne, D.; Walker, S. The bacterial cell envelope. Cold Spring Harb. Perspect. Biol. 2010, 2, a000414. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Norris, M.H.; Zarzycki-Siek, J.; Nierman, W.C.; Donachie, S.P.; Hoang, T.T. Transcript amplification from single bacterium for transcriptome analysis. Genome Res. 2011, 21, 925–935. [Google Scholar] [CrossRef] [Green Version]
- Zenklusen, D.; Larson, D.R.; Singer, R.H. Single-RNA counting reveals alternative modes of gene expression in yeast. Nat. Struct. Mol. Biol. 2008, 15, 1263–1271. [Google Scholar] [CrossRef] [PubMed]
- Nadal-ribelles, M.; Islam, S.; Wei, W.; Latorre, P. Sensitive high-throughput single-cell RNA-Seq reveals within- clonal transcript-correlations in yeast populations. Nat. Microbiol. 2019, 4, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Jariani, A.; Vermeersch, L.; Cerulus, B.; Perez-Samper, G.; Voordeckers, K.; Van Brussel, T.; Thienpont, B.; Lambrechts, D.; Verstrepen, K.J. A new protocol for single-cell RNA-seq reveals stochastic gene expression during lag phase in budding yeast. Elife 2020, 9, e55320. [Google Scholar] [CrossRef]
- Jackson, C.A.; Castro, D.M.; Saldi, G.; Bonneau, R.; Gresham, D. Gene regulatory network reconstruction using single-cell RNA sequencing of barcoded genotypes in diverse environments. Elife 2020, 9, e51254. [Google Scholar] [CrossRef]
- Urbonaite, G.; Lee, J.T.H.; Liu, P.; Parada, G.E.; Hemberg, M.; Acar, M. A yeast-optimized single-cell transcriptomics platform elucidates how mycophenolic acid and guanine alter global mRNA levels. Commun. Biol. 2021, 4, 822. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, J.F.; Delorey, T.; Ford, C.B.; Li, B.Y.; Thompson, D.A.; Rao, R.P.; Cuomo, C.A. Coordinated host-pathogen transcriptional dynamics revealed using sorted subpopulations and single macrophages infected with Candida albicans. Nat. Commun. 2019, 10, 1607. [Google Scholar] [CrossRef] [Green Version]
- Pfaller, M.A.; Diekema, D.J. Epidemiology of invasive candidiasis: A persistent public health problem. Clin. Microbiol. Rev. 2007, 20, 133–163. [Google Scholar] [CrossRef] [Green Version]
- Muñoz, P.; Bouza, E.; Cuenca-Estrella, M.; Eiros, J.M.; Pérez, M.J.; Sánchez-Somolinos, M.; Rincón, C.; Hortal, J.; Peláez, T. Saccharomyces cerevisiae Fungemia: An Emerging Infectious Disease. Clin. Infect. Dis. 2005, 40, 1625–1634. [Google Scholar] [CrossRef]
- Mayer, F.L.; Wilson, D.; Hube, B. Candida albicans pathogenicity mechanisms. Virulence 2013, 4, 119–128. [Google Scholar] [CrossRef] [Green Version]
- Centers for Disease Control and Prevention: Antibiotic Resistance Threats in the United States. 2019. Available online: https://www.cdc.gov/drugresistance/pdf/threatsreport/2019-ar-threats-report-508.pdf (accessed on 25 May 2021).
- Levin, B.R.; Rozen, D.E. Non-inherited antibiotic resistance. Nat. Rev. Microbiol. 2006, 4, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Habib, N.; Avraham-Davidi, I.; Basu, A.; Burks, T.; Shekhar, K.; Hofree, M.; Choudhury, S.R.; Aguet, F.; Gelfand, E.; Ardlie, K.; et al. Massively parallel single-nucleus RNA-seq with DroNc-seq. Nat. Methods 2017, 14, 955–958. [Google Scholar] [CrossRef] [Green Version]
- Klis, F.M.; de Koster, C.G.; Brul, S. Cell wall-related bionumbers and bioestimates of Saccharomyces cerevisiae and Candida albicans. Eukaryot. Cell 2014, 13, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Miura, F.; Kawaguchi, N.; Yoshida, M.; Uematsu, C.; Kito, K.; Sakaki, Y.; Ito, T. Absolute quantification of the budding yeast transcriptome by means of competitive PCR between genomic and complementary DNAs. BMC Genom. 2008, 9, 574. [Google Scholar] [CrossRef] [Green Version]
- McInnes, L.; Healy, J.; Saul, N.; Großberger, L. UMAP: Uniform Manifold Approximation and Projection. J. Open Source Softw. 2018, 3, 861. [Google Scholar] [CrossRef]
- Silva, A.; Cavero, S.; Sarah, V.; Solé, C.; Böttcher, R.; Chávez, S.; Posas, F.; de Nadal, E. Regulation of transcription elongation in response to osmostress. PLoS Genet. 2017, 13, e1007090. [Google Scholar] [CrossRef] [PubMed]
- Richter, K.; Haslbeck, M.; Buchner, J. The Heat Shock Response: Life on the Verge of Death. Mol. Cell 2010, 40, 253–266. [Google Scholar] [CrossRef]
- Qiu, X.; Mao, Q.; Tang, Y.; Wang, L.; Chawla, R.; Pliner, H.A.; Trapnell, C. Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 2017, 14, 979–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Sudbery, P. Candida albicans, a major human fungal pathogen. J. Microbiol. 2011, 49, 171–177. [Google Scholar] [CrossRef]
- Slutsky, B.; Staebell, M.; Anderson, J.; Risen, L.; Pfaller, M.; Soll, D.R. “White-opaque transition”: A second high-frequency switching system in Candida albicans. J. Bacteriol. 1987, 169, 189–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, C.-Y.; Newport, G.; Murillo, L.A.; Jones, T.; Scherer, S.; Davis, R.W.; Agabian, N. Metabolic specialization associated with phenotypic switching in Candidaalbicans. Proc. Natl. Acad. Sci. USA 2002, 99, 14907–14912. [Google Scholar] [CrossRef] [Green Version]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M., 3rd; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive Integration of Single-Cell Data. Cell 2019, 177, 1888–1902.e21. [Google Scholar] [CrossRef] [PubMed]
- Issi, L.; Farrer, R.A.; Pastor, K.; Landry, B.; Delorey, T.; Bell, G.W.; Thompson, D.A.; Cuomo, C.A.; Rao, R.P. Zinc Cluster Transcription Factors Alter Virulence in Candida albicans. Genetics 2017, 205, 559–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cottier, F.; Tan, A.S.M.; Yurieva, M.; Liao, W.; Lum, J.; Poidinger, M.; Zolezzi, F.; Pavelka, N. The transcriptional response of Candida albicans to weak organic acids, carbon source, and MIG1 inactivation unveils a role for HGT16 in mediating the fungistatic effect of acetic acid. G3 Genes Genomes Genet. 2017, 7, 3597–3604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrow, J.D. Fluconazole: A new triazole antifungal agent. Am. J. Med. Sci. 1991, 302, 129–132. [Google Scholar] [CrossRef]
- Berkow, E.L.; Lockhart, S.R. Fluconazole resistance in Candida species: A current perspective. Infect. Drug Resist. 2017, 10, 237–245. [Google Scholar] [CrossRef] [Green Version]
- Pfizer. DIFLUCAN (Fluconazole Tablets) (Fluconazole Injection—For Intravenous Infusion Only) (Fluconazole for Oral Suspension). FDA Drug Label 2011, 1–37. Available online: http://www.accessdata.fda.gov/drugsatfda_docs/label/2014/019949s058,019950s062,020090s042lbl.pdf (accessed on 2 February 2021).
- Bandara, H.M.H.N.; Wood, D.L.A.; Vanwonterghem, I.; Hugenholtz, P.; Cheung, B.P.K.; Samaranayake, L.P. Fluconazole resistance in Candida albicans is induced by Pseudomonas aeruginosa quorum sensing. Sci. Rep. 2020, 10, 7769. [Google Scholar] [CrossRef]
- Henry, K.W.; Nickels, J.T.; Edlind, T.D. Upregulation of ERG genes in Candida species by azoles and other sterol biosynthesis inhibitors. Antimicrob. Agents Chemother. 2000, 44, 2693–2700. [Google Scholar] [CrossRef] [Green Version]
- Leber, R.; Fuchsbichler, S.; Klobucníková, V.; Schweighofer, N.; Pitters, E.; Wohlfarter, K.; Lederer, M.; Landl, K.; Ruckenstuhl, C.; Hapala, I.; et al. Molecular mechanism of terbinafine resistance in Saccharomyces cerevisiae. Antimicrob. Agents Chemother. 2003, 47, 3890–3900. [Google Scholar] [CrossRef] [Green Version]
- Fahrner, K.; Yarger, J.; Hereford, L. Yeast histone mRNA is polyadenylated. Nucleic Acids Res. 1980, 8, 5725–5737. [Google Scholar] [CrossRef] [Green Version]
- Castrillo, J.I.; Zeef, L.A.; Hoyle, D.C.; Zhang, N.; Hayes, A.; Gardner, D.C.; Cornell, M.J.; Petty, J.; Hakes, L.; Wardleworth, L.; et al. Growth control of the eukaryote cell: A systems biology study in yeast. J. Biol. 2007, 6, 4. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Esquivel, B.D.; White, T.C. Overexpression or Deletion of Ergosterol Biosynthesis Genes Alters Doubling Time, Response to Stress Agents, and Drug Susceptibility in Saccharomyces cerevisiae. MBio 2018, 9, e01291-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tzur, Y.B.; Winter, E.; Gao, J.; Hashimshony, T.; Yanai, I.; Colaiácovo, M.P. Spatiotemporal Gene Expression Analysis of the Caenorhabditis elegans Germline Uncovers a Syncytial Expression Switch. Genetics 2018, 210, 587–605. [Google Scholar] [CrossRef] [Green Version]
- Farrell, J.A.; Wang, Y.; Riesenfeld, S.J.; Shekhar, K.; Regev, A.; Schier, A.F. Single-cell reconstruction of developmental trajectories during zebrafish embryogenesis. Science 2018, 360, 6392. [Google Scholar] [CrossRef] [Green Version]
- Ariss, M.M.; Islam, A.B.M.M.K.; Critcher, M.; Zappia, M.P.; Frolov, M. V Single cell RNA-sequencing identifies a metabolic aspect of apoptosis in Rbf mutant. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Matejuk, A.; Leng, Q.; Begum, M.D.; Woodle, M.C.; Scaria, P.; Chou, S.-T.; Mixson, A.J. Peptide-based Antifungal Therapies against Emerging Infections. Drugs Future 2010, 35, 197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, V.A.; Graves-lindsay, T.; Howe, K.; Bouk, N.; Chen, H.; Kitts, P.A.; Murphy, T.D.; Pruitt, K.D.; Thibaud-nissen, F.; Albracht, D.; et al. Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res. 2017, 27, 849–864. [Google Scholar] [CrossRef] [Green Version]
- Saccharomyces Cerevisiae (ID 15)—Genome—NCBI. Available online: https://www.ncbi.nlm.nih.gov/genome/?term=s+cerevisiae (accessed on 21 May 2020).
- Candida Albicans (ID 21)—Genome—NCBI. Available online: https://www.ncbi.nlm.nih.gov/genome/?term=Candida%20albicans (accessed on 21 May 2020).
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Costa-de-Oliveira, S.; Rodrigues, A.G. Candida albicans Antifungal Resistance and Tolerance in Bloodstream Infections: The Triad Yeast-Host-Antifungal. Microorganisms 2020, 8, 154. [Google Scholar] [CrossRef] [Green Version]
- Cowen, L.E.; Steinbach, W.J. Stress, drugs, and evolution: The role of cellular signaling in fungal drug resistance. Eukaryot. Cell 2008, 7, 747–764. [Google Scholar] [CrossRef] [Green Version]
- Köster, J.; Rahmann, S. Snakemake-a scalable bioinformatics workflow engine. Bioinformatics 2012, 28, 2520–2522. [Google Scholar] [CrossRef] [Green Version]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 12 December 2021).
- Smith, T.; Heger, A.; Sudbery, I. UMI-tools: Modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res. 2017, 27, 491–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. FeatureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spellman, P.T.; Sherlock, G.; Zhang, M.Q.; Iyer, V.R.; Anders, K.; Eisen, M.B.; Brown, P.O.; Botstein, D.; Futcher, B. Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol. Biol. Cell 1998, 9, 3273–3297. [Google Scholar] [CrossRef] [PubMed]
- Côte, P.; Herve, H.; Whiteway, M. Transcriptional Analysis of the Candida albicansCell Cycle. Mol. Biol. Cell 2009, 20, 3363–3373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stuart, T.; Satija, R. Integrative single-cell analysis. Nat. Rev. Genet. 2019, 20, 257–272. [Google Scholar] [CrossRef]
- Mao, Q.; Wang, L.; Goodison, S.; Sun, Y. Dimensionality Reduction Via Graph Structure Learning. In Proceedings of the 21th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, Sydney, Australia, 10–14 August 2015; Association for Computing Machinery: New York, NY, USA, 2015; pp. 765–774. [Google Scholar]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dohn, R.; Xie, B.; Back, R.; Selewa, A.; Eckart, H.; Rao, R.P.; Basu, A. mDrop-Seq: Massively Parallel Single-Cell RNA-Seq of Saccharomyces cerevisiae and Candida albicans. Vaccines 2022, 10, 30. https://doi.org/10.3390/vaccines10010030
Dohn R, Xie B, Back R, Selewa A, Eckart H, Rao RP, Basu A. mDrop-Seq: Massively Parallel Single-Cell RNA-Seq of Saccharomyces cerevisiae and Candida albicans. Vaccines. 2022; 10(1):30. https://doi.org/10.3390/vaccines10010030
Chicago/Turabian StyleDohn, Ryan, Bingqing Xie, Rebecca Back, Alan Selewa, Heather Eckart, Reeta Prusty Rao, and Anindita Basu. 2022. "mDrop-Seq: Massively Parallel Single-Cell RNA-Seq of Saccharomyces cerevisiae and Candida albicans" Vaccines 10, no. 1: 30. https://doi.org/10.3390/vaccines10010030
APA StyleDohn, R., Xie, B., Back, R., Selewa, A., Eckart, H., Rao, R. P., & Basu, A. (2022). mDrop-Seq: Massively Parallel Single-Cell RNA-Seq of Saccharomyces cerevisiae and Candida albicans. Vaccines, 10(1), 30. https://doi.org/10.3390/vaccines10010030