Implications of Oxidative Stress and Potential Role of Mitochondrial Dysfunction in COVID-19: Therapeutic Effects of Vitamin D

 ,
, 
{kind=link}
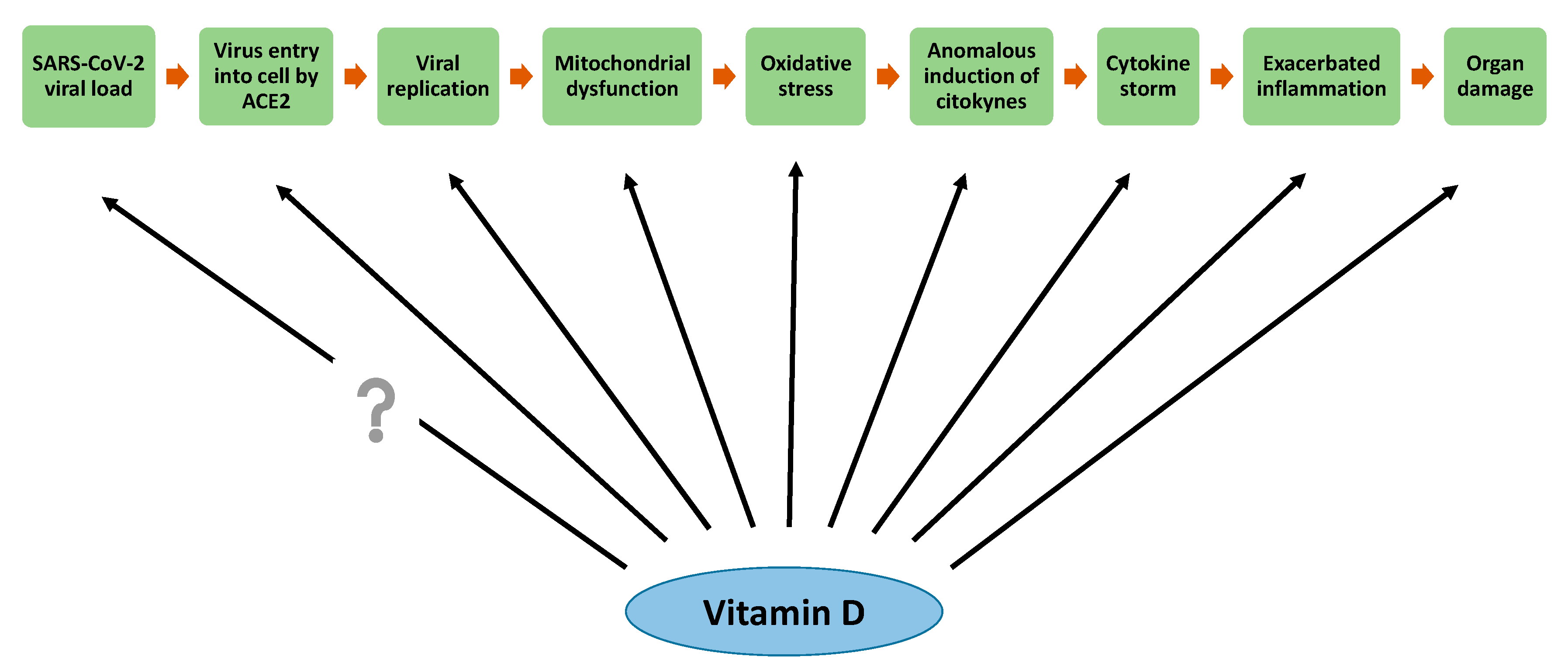
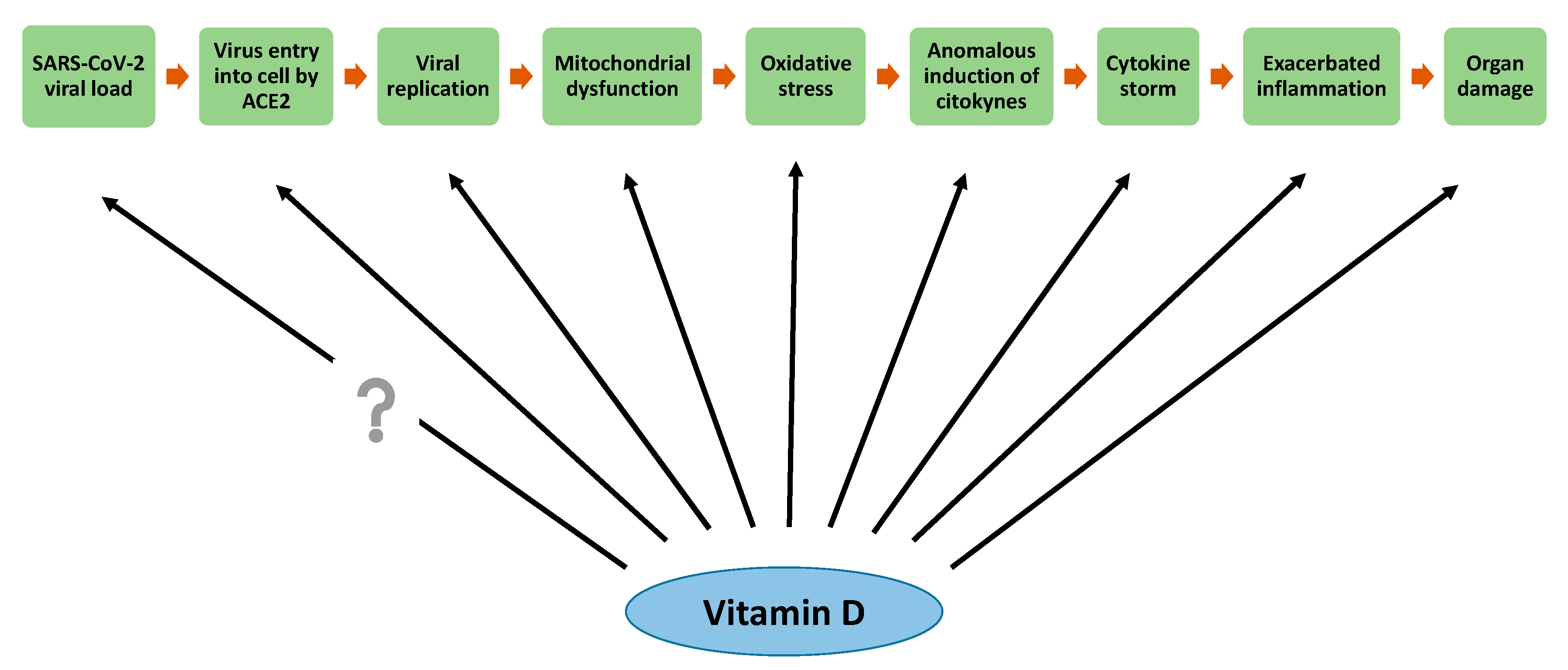{kind=link}
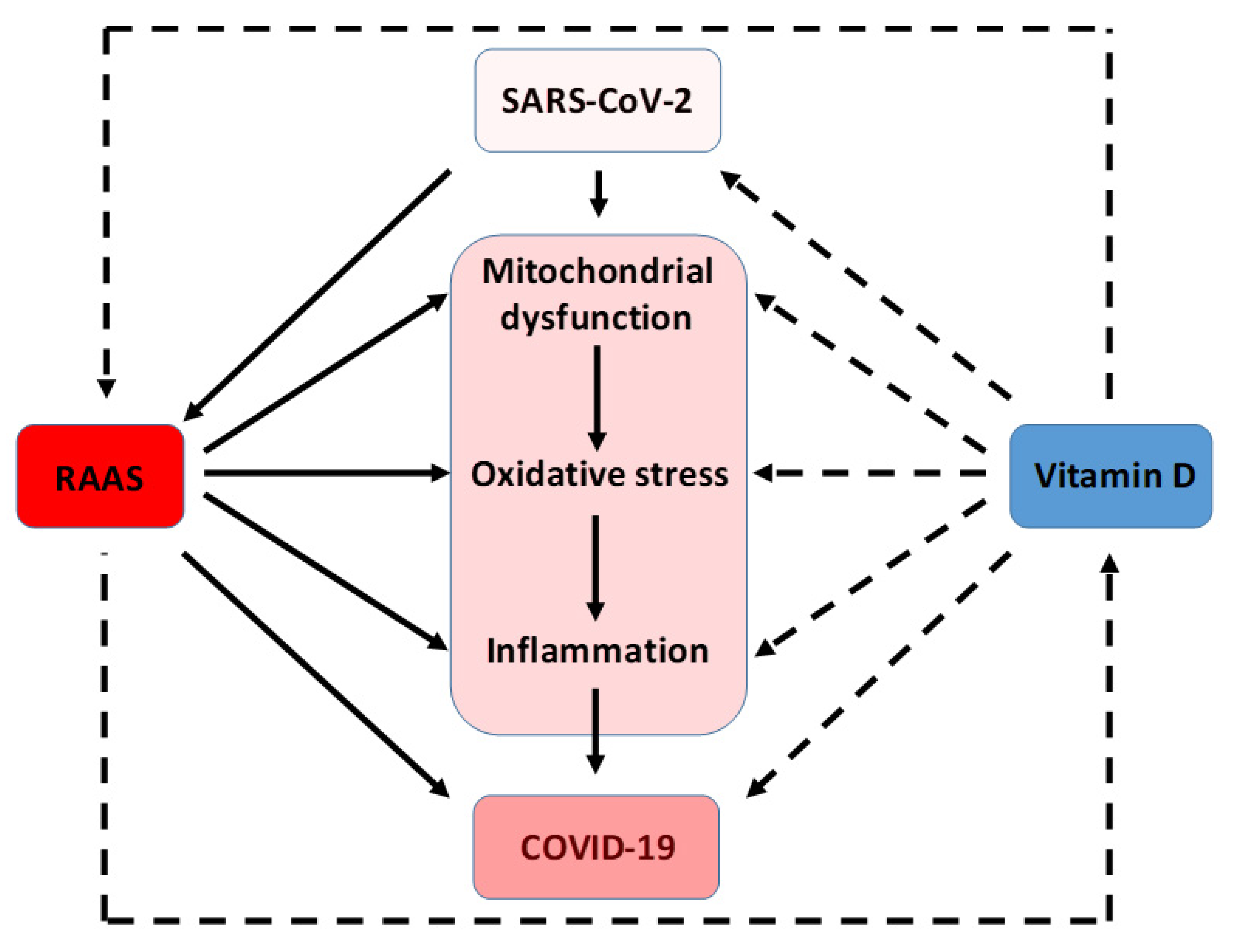{kind=link}
Abstract
:1. Introduction
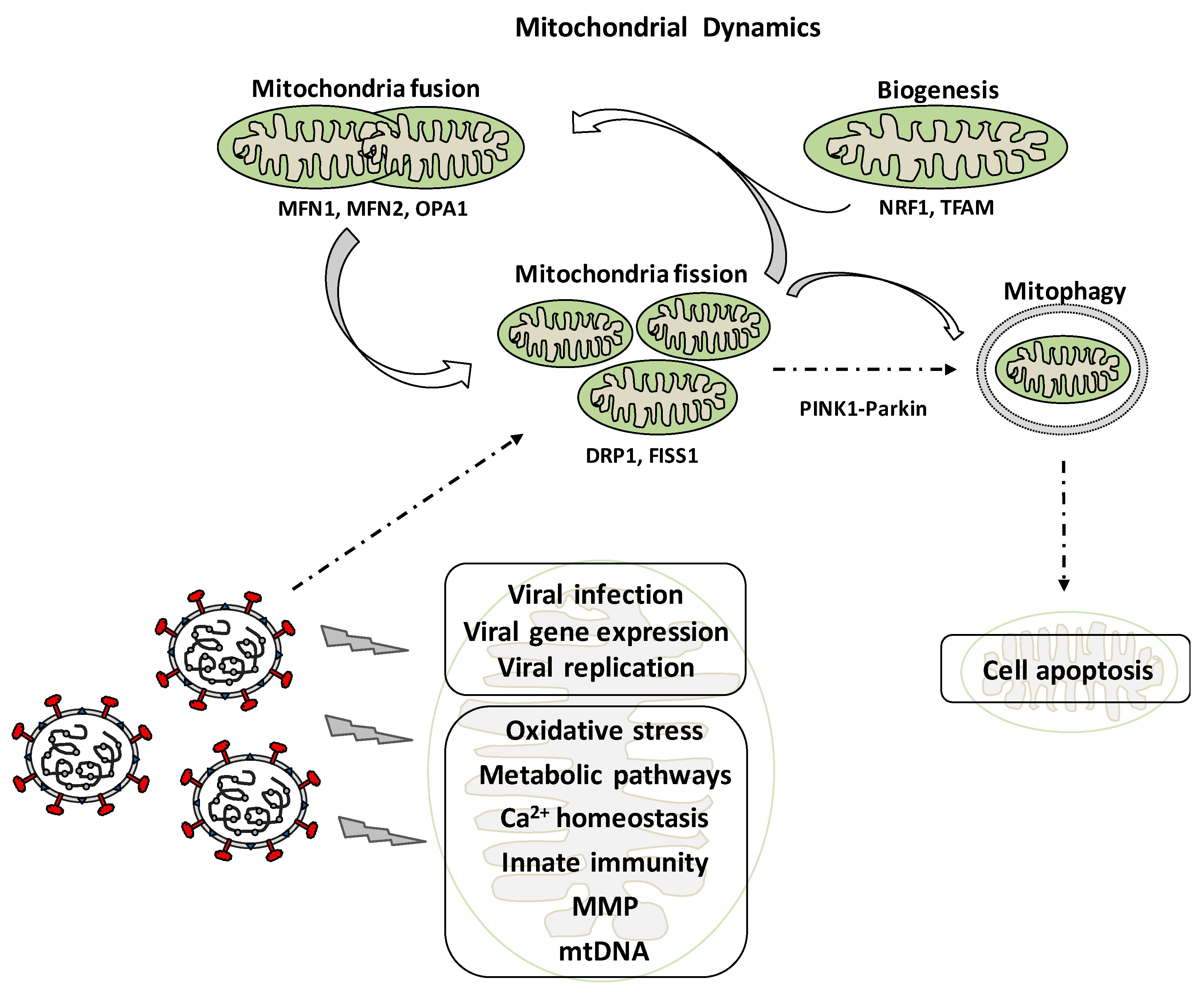
2. Mitochondrial Dynamics: Role in Cell Physiology
3. Coronaviruses and Acute Respiratory Syndromes
4. Mitochondrial Dynamics and Viral Infection
5. Oxidative Stress and Interaction with the Immune System in the Worsening of Viral Infection Symptoms
6. Effects of Vitamin D in the Attenuation of Mitochondrial Oxidative Stress
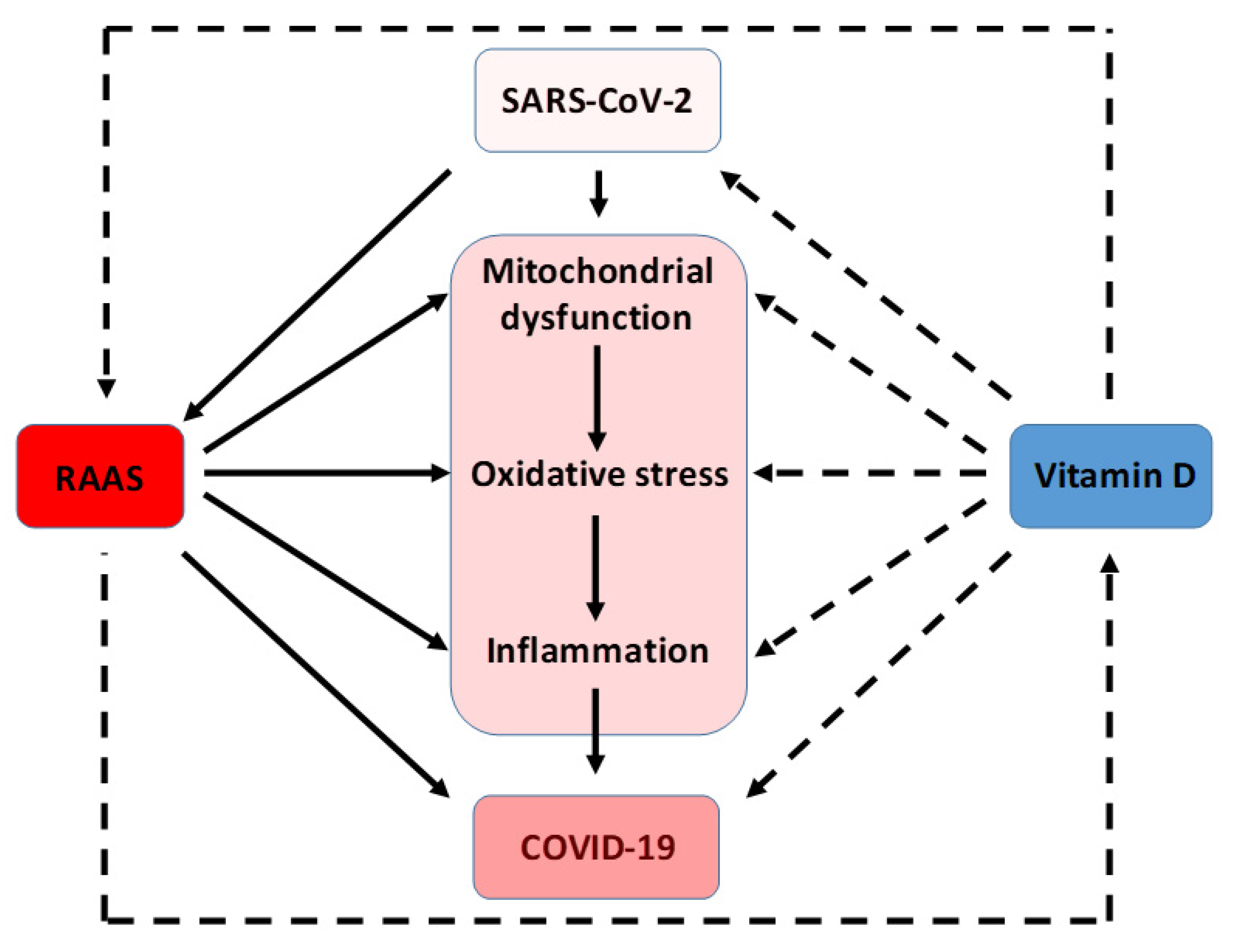
7. Interrelation among Oxidative Stress, RAAS, and SARS-CoV-2 Infection
8. Vitamin D Antioxidative Actions against SARS-CoV-2 Infection
9. Conclusions and Prospect
Author Contributions
Funding
Conflicts of Interest
References
- Liu, Y.C.; Kuo, R.L.; Shih, S.R. COVID-19: The first documented coronavirus pandemic in history. Biomed. J. 2020. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. COVID-19 vaccine development pipeline gears up. Lancet 2020, 395, 1751–1752. [Google Scholar] [CrossRef]
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X.; et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B 2020, 10, 766–788. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Hou, Y.; Shen, J.; Huang, Y.; Martin, W.; Cheng, F. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. 2020, 6, 14. [Google Scholar] [CrossRef] [Green Version]
- Delgado-Roche, L.; Mesta, F. Oxidative Stress as Key Player in Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV) Infection. Arch. Med. Res. 2020, 51, 384–387. [Google Scholar] [CrossRef]
- Ntyonga-Pono, M.P. COVID-19 infection and oxidative stress: An under-explored approach for prevention and treatment? Pan Afr. Med. J. 2020, 35, 12. [Google Scholar] [CrossRef]
- Martín Giménez, V.M.; Inserra, F.; Ferder, L.; García, J.; Manucha, W. Vitamin D deficiency in African Americans is associated with a high risk of severe disease and mortality by SARS-CoV-2. J. Hum. Hypertens. 2020. [Google Scholar] [CrossRef]
- Martín Giménez, V.M.; Inserra, F.; Tajer, C.D.; Mariani, J.; Ferder, L.; Reiter, R.J.; Manucha, W. Lungs as target of COVID-19 infection: Protective common molecular mechanisms of vitamin D and melatonin as a new potential synergistic treatment. Life Sci. 2020, 254, 117808. [Google Scholar] [CrossRef]
- Meltzer, D.O.; Best, T.J.; Zhang, H.; Vokes, T.; Arora, V.; Solway, J. Association of Vitamin D Status and Other Clinical Characteristics With COVID-19 Test Results. JAMA Netw. Open 2020, 3, e2019722. [Google Scholar] [CrossRef]
- Glingston, R.S.; Deb, R.; Kumar, S.; Nagotu, S. Organelle dynamics and viral infections: At cross roads. Microbes Infect. 2019, 21, 20–32. [Google Scholar] [CrossRef]
- Lahera, V.; de Las Heras, N.; López-Farré, A.; Manucha, W.; Ferder, L. Role of Mitochondrial Dysfunction in Hypertension and Obesity. Curr. Hypertens. Rep. 2017, 19, 11. [Google Scholar] [CrossRef]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [Green Version]
- Malka, F.; Guillery, O.; Cifuentes-Diaz, C.; Guillou, E.; Belenguer, P.; Lombès, A.; Rojo, M. Separate fusion of outer and inner mitochondrial membranes. EMBO Rep. 2005, 6, 853–859. [Google Scholar] [CrossRef] [Green Version]
- Meyer, J.N.; Leuthner, T.C.; Luz, A.L. Mitochondrial fusion, fission, and mitochondrial toxicity. Toxicology 2017, 391, 42–53. [Google Scholar] [CrossRef]
- Kagan, J.C. Signaling organelles of the innate immune system. Cell 2012, 151, 1168–1178. [Google Scholar] [CrossRef] [Green Version]
- Ploumi, C.; Daskalaki, I.; Tavernarakis, N. Mitochondrial biogenesis and clearance: A balancing act. FEBS J. 2017, 284, 183–195. [Google Scholar] [CrossRef]
- Vives-Bauza, C.; Zhou, C.; Huang, Y.; Cui, M.; de Vries, R.L.; Kim, J.; May, J.; Tocilescu, M.A.; Liu, W.; Ko, H.S.; et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. USA 2010, 107, 378–383. [Google Scholar] [CrossRef] [Green Version]
- Paules, C.I.; Marston, H.D.; Fauci, A.S. Coronavirus Infections-More than Just the Common Cold. JAMA 2020. [Google Scholar] [CrossRef] [Green Version]
- Hung, L.S. The SARS epidemic in Hong Kong: What lessons have we learned? J. R. Soc. Med. 2003, 96, 374–378. [Google Scholar] [CrossRef]
- Vabret, N.; Britton, G.J.; Gruber, C.; Hegde, S.; Kim, J.; Kuksin, M.; Levantovsky, R.; Malle, L.; Moreira, A.; Park, M.D.; et al. Immunology of COVID-19: Current State of the Science. Immunity 2020, 52, 910–941. [Google Scholar] [CrossRef]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034. [Google Scholar] [CrossRef]
- Novoa, R.R.; Calderita, G.; Arranz, R.; Fontana, J.; Granzow, H.; Risco, C. Virus factories: Associations of cell organelles for viral replication and morphogenesis. Biol. Cell 2005, 97, 147–172. [Google Scholar] [CrossRef]
- Mercer, J.; Schelhaas, M.; Helenius, A. Virus entry by endocytosis. Annu. Rev. Biochem. 2010, 79, 803–833. [Google Scholar] [CrossRef] [Green Version]
- Jean Beltran, P.M.; Cook, K.C.; Cristea, I.M. Exploring and Exploiting Proteome Organization during Viral Infection. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [Green Version]
- Anand, S.K.; Tikoo, S.K. Viruses as modulators of mitochondrial functions. Adv. Virol. 2013, 2013, 738794. [Google Scholar] [CrossRef]
- Machida, K.; Cheng, K.T.; Lai, C.K.; Jeng, K.S.; Sung, V.M.; Lai, M.M. Hepatitis C virus triggers mitochondrial permeability transition with production of reactive oxygen species, leading to DNA damage and STAT3 activation. J. Virol. 2006, 80, 7199–7207. [Google Scholar] [CrossRef] [Green Version]
- Saffran, H.A.; Pare, J.M.; Corcoran, J.A.; Weller, S.K.; Smiley, J.R. Herpes simplex virus eliminates host mitochondrial DNA. EMBO Rep. 2007, 8, 188–193. [Google Scholar] [CrossRef] [Green Version]
- Halestrap, A.P. A pore way to die: The role of mitochondria in reperfusion injury and cardioprotection. Biochem. Soc. Trans. 2010, 38, 841–860. [Google Scholar] [CrossRef] [Green Version]
- Williamson, C.D.; DeBiasi, R.L.; Colberg-Poley, A.M. Viral product trafficking to mitochondria, mechanisms and roles in pathogenesis. Infect. Disord. Drug Targets 2012, 12, 18–37. [Google Scholar] [CrossRef]
- Everett, H.; Barry, M.; Sun, X.; Lee, S.F.; Frantz, C.; Berthiaume, L.G.; McFadden, G.; Bleackley, R.C. The myxoma poxvirus protein, M11L, prevents apoptosis by direct interaction with the mitochondrial permeability transition pore. J. Exp. Med. 2002, 196, 1127–1139. [Google Scholar] [CrossRef] [Green Version]
- Jacotot, E.; Ravagnan, L.; Loeffler, M.; Ferri, K.F.; Vieira, H.L.; Zamzami, N.; Costantini, P.; Druillennec, S.; Hoebeke, J.; Briand, J.P.; et al. The HIV-1 viral protein R induces apoptosis via a direct effect on the mitochondrial permeability transition pore. J. Exp. Med. 2000, 191, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Claus, C.; Liebert, U.G. A renewed focus on the interplay between viruses and mitochondrial metabolism. Arch. Virol. 2014, 159, 1267–1277. [Google Scholar] [CrossRef]
- Munger, J.; Bajad, S.U.; Coller, H.A.; Shenk, T.; Rabinowitz, J.D. Dynamics of the cellular metabolome during human cytomegalovirus infection. PLoS Pathog. 2006, 2, e132. [Google Scholar] [CrossRef] [Green Version]
- Munger, J.; Bennett, B.D.; Parikh, A.; Feng, X.J.; McArdle, J.; Rabitz, H.A.; Shenk, T.; Rabinowitz, J.D. Systems-level metabolic flux profiling identifies fatty acid synthesis as a target for antiviral therapy. Nat. Biotechnol. 2008, 26, 1179–1186. [Google Scholar] [CrossRef] [Green Version]
- Bardell, D.; Essex, M. Glycolysis during early infection of feline and human cells with feline leukemia virus. Infect. Immun. 1974, 9, 824–827. [Google Scholar] [CrossRef] [Green Version]
- Vastag, L.; Koyuncu, E.; Grady, S.L.; Shenk, T.E.; Rabinowitz, J.D. Divergent effects of human cytomegalovirus and herpes simplex virus-1 on cellular metabolism. PLoS Pathog. 2011, 7, e1002124. [Google Scholar] [CrossRef] [Green Version]
- Sharon-Friling, R.; Goodhouse, J.; Colberg-Poley, A.M.; Shenk, T. Human cytomegalovirus pUL37x1 induces the release of endoplasmic reticulum calcium stores. Proc. Natl. Acad. Sci. USA 2006, 103, 19117–19122. [Google Scholar] [CrossRef] [Green Version]
- Campanella, M.; de Jong, A.S.; Lanke, K.W.; Melchers, W.J.; Willems, P.H.; Pinton, P.; Rizzuto, R.; van Kuppeveld, F.J. The coxsackievirus 2B protein suppresses apoptotic host cell responses by manipulating intracellular Ca2+ homeostasis. J. Biol. Chem. 2004, 279, 18440–18450. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J.; Khan, M.; Quan, J.; Till, A.; Subramani, S.; Siddiqui, A. Hepatitis B virus disrupts mitochondrial dynamics: Induces fission and mitophagy to attenuate apoptosis. PLoS Pathog. 2013, 9, e1003722. [Google Scholar] [CrossRef] [Green Version]
- Meng, G.; Xia, M.; Wang, D.; Chen, A.; Wang, Y.; Wang, H.; Yu, D.; Wei, J. Mitophagy promotes replication of oncolytic Newcastle disease virus by blocking intrinsic apoptosis in lung cancer cells. Oncotarget 2014, 5, 6365–6374. [Google Scholar] [CrossRef] [Green Version]
- Xia, M.; Meng, G.; Li, M.; Wei, J. Mitophagy in viral infections. DNA Cell Biol. 2014, 33, 739–742. [Google Scholar] [CrossRef] [PubMed]
- Ding, B.; Zhang, L.; Li, Z.; Zhong, Y.; Tang, Q.; Qin, Y.; Chen, M. The Matrix Protein of Human Parainfluenza Virus Type 3 Induces Mitophagy that Suppresses Interferon Responses. Cell Host Microbe 2017, 21, 538–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinberg, S.E.; Sena, L.A.; Chandel, N.S. Mitochondria in the regulation of innate and adaptive immunity. Immunity 2015, 42, 406–417. [Google Scholar] [CrossRef] [Green Version]
- Shi, C.S.; Qi, H.Y.; Boularan, C.; Huang, N.N.; Abu-Asab, M.; Shelhamer, J.H.; Kehrl, J.H. SARS-coronavirus open reading frame-9b suppresses innate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6 signalosome. J. Immunol. 2014, 193, 3080–3089. [Google Scholar] [CrossRef] [Green Version]
- Novoa, R.R.; Calderita, G.; Cabezas, P.; Elliott, R.M.; Risco, C. Key Golgi factors for structural and functional maturation of bunyamwera virus. J. Virol. 2005, 79, 10852–10863. [Google Scholar] [CrossRef] [Green Version]
- Roy, J.; Galano, J.M.; Durand, T.; Le Guennec, J.Y.; Lee, J.C. Physiological role of reactive oxygen species as promoters of natural defenses. FASEB J. 2017, 31, 3729–3745. [Google Scholar] [CrossRef] [Green Version]
- Dandekar, A.; Mendez, R.; Zhang, K. Cross talk between ER stress, oxidative stress, and inflammation in health and disease. Methods Mol. Biol. 2015, 1292, 205–214. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxidative Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef]
- Peterhans, E. Sendai virus stimulates chemiluminescence in mouse spleen cells. Biochem. Biophys. Res. Commun. 1979, 91, 383–392. [Google Scholar] [CrossRef]
- Müller, F. Reactive oxygen intermediates and human immunodeficiency virus (HIV) infection. Free Radic. Biol. Med. 1992, 13, 651–657. [Google Scholar] [CrossRef]
- Peterhans, E.; Grob, M.; Bürge, T.; Zanoni, R. Virus-induced formation of reactive oxygen intermediates in phagocytic cells. Free Radic. Res. Commun. 1987, 3, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Reshi, M.L.; Su, Y.C.; Hong, J.R. RNA Viruses: ROS-Mediated Cell Death. Int. J. Cell Biol. 2014, 2014, 467452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dan Dunn, J.; Alvarez, L.A.; Zhang, X.; Soldati, T. Reactive oxygen species and mitochondria: A nexus of cellular homeostasis. Redox Biol. 2015, 6, 472–485. [Google Scholar] [CrossRef]
- Lee, Y.I.; Hwang, J.M.; Im, J.H.; Lee, Y.I.; Kim, N.S.; Kim, D.G.; Yu, D.Y.; Moon, H.B.; Park, S.K. Human hepatitis B virus-X protein alters mitochondrial function and physiology in human liver cells. J. Biol. Chem. 2004, 279, 15460–15471. [Google Scholar] [CrossRef] [Green Version]
- Broz, P.; Monack, D.M. Molecular mechanisms of inflammasome activation during microbial infections. Immunol. Rev. 2011, 243, 174–190. [Google Scholar] [CrossRef]
- Camini, F.C.; da Silva Caetano, C.C.; Almeida, L.T.; de Brito Magalhães, C.L. Implications of oxidative stress on viral pathogenesis. Arch. Virol. 2017, 162, 907–917. [Google Scholar] [CrossRef]
- Leff, J.A.; Oppegard, M.A.; Curiel, T.J.; Brown, K.S.; Schooley, R.T.; Repine, J.E. Progressive increases in serum catalase activity in advancing human immunodeficiency virus infection. Free Radic. Biol. Med. 1992, 13, 143–149. [Google Scholar] [CrossRef]
- Huang, S.H.; Cao, X.J.; Liu, W.; Shi, X.Y.; Wei, W. Inhibitory effect of melatonin on lung oxidative stress induced by respiratory syncytial virus infection in mice. J. Pineal Res. 2010, 48, 109–116. [Google Scholar] [CrossRef]
- Deramaudt, T.B.; Dill, C.; Bonay, M. Regulation of oxidative stress by Nrf2 in the pathophysiology of infectious diseases. Med. Mal. Infect. 2013, 43, 100–107. [Google Scholar] [CrossRef]
- Schachtele, S.J.; Hu, S.; Lokensgard, J.R. Modulation of experimental herpes encephalitis-associated neurotoxicity through sulforaphane treatment. PLoS ONE 2012, 7, e36216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burdon, R.H. Superoxide and hydrogen peroxide in relation to mammalian cell proliferation. Free Radic. Biol. Med. 1995, 18, 775–794. [Google Scholar] [CrossRef]
- Schwarz, K.B. Oxidative stress during viral infection: A review. Free Radic. Biol. Med. 1996, 21, 641–649. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Sinha, K.; Sil, P.C. Cytochrome P450s: Mechanisms and biological implications in drug metabolism and its interaction with oxidative stress. Curr. Drug Metab. 2014, 15, 719–742. [Google Scholar] [CrossRef]
- Babbar, N.; Murray-Stewart, T.; Casero, R.A., Jr. Inflammation and polyamine catabolism: The good, the bad and the ugly. Biochem. Soc. Trans. 2007, 35, 300–304. [Google Scholar] [CrossRef] [Green Version]
- Amarante-Mendes, G.P.; Adjemian, S.; Branco, L.M.; Zanetti, L.C.; Weinlich, R.; Bortoluci, K.R. Pattern Recognition Receptors and the Host Cell Death Molecular Machinery. Front. Immunol. 2018, 9, 2379. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.Y.; Zhen, Z.D.; Fan, D.Y.; Wang, P.G.; An, J. Transcriptomic Analysis Suggests the M1 Polarization and Launch of Diverse Programmed Cell Death Pathways in Japanese Encephalitis Virus-Infected Macrophages. Viruses 2020, 12, 356. [Google Scholar] [CrossRef] [Green Version]
- Futosi, K.; Fodor, S.; Mócsai, A. Neutrophil cell surface receptors and their intracellular signal transduction pathways. Int. Immunopharmacol. 2013, 17, 638–650. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Malo, D.; Hekimi, S. Elevated mitochondrial reactive oxygen species generation affects the immune response via hypoxia-inducible factor-1alpha in long-lived Mclk1+/- mouse mutants. J. Immunol. 2010, 184, 582–590. [Google Scholar] [CrossRef] [Green Version]
- Schreck, R.; Rieber, P.; Baeuerle, P.A. Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-kappa B transcription factor and HIV-1. EMBO J. 1991, 10, 2247–2258. [Google Scholar] [CrossRef]
- Rius, J.; Guma, M.; Schachtrup, C.; Akassoglou, K.; Zinkernagel, A.S.; Nizet, V.; Johnson, R.S.; Haddad, G.G.; Karin, M. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature 2008, 453, 807–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nile, S.H.; Nile, A.; Qiu, J.; Li, L.; Jia, X.; Kai, G. COVID-19: Pathogenesis, cytokine storm and therapeutic potential of interferons. Cytokine Growth Factor Rev. 2020, 53, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Shu, T.; Wu, D.; Mu, J.; Wang, C.; Huang, M.; Han, Y.; Zhang, X.Y.; Zhou, W.; Qiu, Y.; et al. The ORF3a protein of SARS-CoV-2 induces apoptosis in cells. Cell Mol. Immunol. 2020, 17, 881–883. [Google Scholar] [CrossRef] [PubMed]
- van den Brand, J.M.; Haagmans, B.L.; van Riel, D.; Osterhaus, A.D.; Kuiken, T. The pathology and pathogenesis of experimental severe acute respiratory syndrome and influenza in animal models. J. Comp. Pathol. 2014, 151, 83–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.W.; Lin, K.H.; Hsieh, T.H.; Shiu, S.Y.; Li, J.Y. Severe acute respiratory syndrome coronavirus 3C-like protease-induced apoptosis. FEMS Immunol. Med. Microbiol. 2006, 46, 375–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.R.; Cao, Q.D.; Hong, Z.S.; Tan, Y.Y.; Chen, S.D.; Jin, H.J.; Tan, K.S.; Wang, D.Y.; Yan, Y. The origin, transmission and clinical therapies on coronavirus disease 2019 (COVID-19) outbreak—An update on the status. Mil. Med. Res. 2020, 7, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Liu, H.G.; Liu, W.; Liu, J.; Liu, K.; Shang, J.; Deng, Y.; Wei, S. Analysis of clinical features of 29 patients with 2019 novel coronavirus pneumonia. Chin. J. Tuberc. Respir. Dis. 2020, 43, e005. [Google Scholar] [CrossRef]
- Conti, P.; Ronconi, G.; Caraffa, A.; Gallenga, C.E.; Ross, R.; Frydas, I.; Kritas, S.K. Induction of pro-inflammatory cytokines (IL-1 and IL-6) and lung inflammation by Coronavirus-19 (COVI-19 or SARS-CoV-2): Anti-inflammatory strategies. J. Biol. Regul. Homeost. Agents 2020, 34. [Google Scholar] [CrossRef]
- Lee, D.W.; Gardner, R.; Porter, D.L.; Louis, C.U.; Ahmed, N.; Jensen, M.; Grupp, S.A.; Mackall, C.L. Current concepts in the diagnosis and management of cytokine release syndrome. Blood 2014, 124, 188–195. [Google Scholar] [CrossRef] [Green Version]
- Riegler, L.L.; Jones, G.P.; Lee, D.W. Current approaches in the grading and management of cytokine release syndrome after chimeric antigen receptor T-cell therapy. Ther. Clin. Risk Manag. 2019, 15, 323–335. [Google Scholar] [CrossRef] [Green Version]
- Sardar, S.; Chakraborty, A.; Chatterjee, M. Comparative effectiveness of vitamin D3 and dietary vitamin E on peroxidation of lipids and enzymes of the hepatic antioxidant system in Sprague--Dawley rats. Int. J. Vitam. Nutr. Res. 1996, 66, 39–45. [Google Scholar] [PubMed]
- Li, L.; Prabhakaran, K.; Zhang, X.; Zhang, L.; Liu, H.; Borowitz, J.L.; Isom, G.E. 1Alpha,25-dihydroxyvitamin D3 attenuates cyanide-induced neurotoxicity by inhibiting uncoupling protein-2 up-regulation. J. Neurosci. Res. 2008, 86, 1397–1408. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhu, Y.; Wang, X.; Yang, Y.; Cheng, S. Cardioprotective effect of calcitriol on myocardial injury induced by isoproterenol in rats. J. Cardiovasc. Pharmacol. Ther. 2013, 18, 386–391. [Google Scholar] [CrossRef] [PubMed]
- García, I.M.; Altamirano, L.; Mazzei, L.; Fornés, M.; Cuello-Carrión, F.D.; Ferder, L.; Manucha, W. Vitamin D receptor-modulated Hsp70/AT1 expression may protect the kidneys of SHRs at the structural and functional levels. Cell Stress Chaperones 2014, 19, 479–491. [Google Scholar] [CrossRef] [Green Version]
- Uberti, F.; Lattuada, D.; Morsanuto, V.; Nava, U.; Bolis, G.; Vacca, G.; Squarzanti, D.F.; Cisari, C.; Molinari, C. Vitamin D protects human endothelial cells from oxidative stress through the autophagic and survival pathways. J. Clin. Endocrinol. Metab. 2014, 99, 1367–1374. [Google Scholar] [CrossRef] [Green Version]
- Sturza, A.; Văduva, A.; Uțu, D.; Rațiu, C.; Pop, N.; Duicu, O.; Popoiu, C.; Boia, E.; Matusz, P.; Muntean, D.M.; et al. Vitamin D improves vascular function and decreases monoamine oxidase A expression in experimental diabetes. Mol. Cell Biochem. 2019, 453, 33–40. [Google Scholar] [CrossRef]
- Zabul, P.; Wozniak, M.; Slominski, A.T.; Preis, K.; Gorska, M.; Korozan, M.; Wieruszewski, J.; Zmijewski, M.A.; Zabul, E.; Tuckey, R.; et al. A Proposed Molecular Mechanism of High-Dose Vitamin D3 Supplementation in Prevention and Treatment of Preeclampsia. Int. J. Mol. Sci. 2015, 16, 13043–13064. [Google Scholar] [CrossRef] [Green Version]
- Tian, X.; Ma, S.; Wang, Y.; Hou, L.; Shi, Y.; Yao, M.; Wang, X.; Zhang, H.; Jiang, L. Effects of Placental Ischemia Are Attenuated by 1,25-Dihydroxyvitamin D Treatment and Associated with Reduced Apoptosis and Increased Autophagy. DNA Cell Biol. 2016, 35, 59–70. [Google Scholar] [CrossRef]
- Longoni, A.; Kolling, J.; dos Santos, T.M.; dos Santos, J.P.; da Silva, J.S.; Pettenuzzo, L.; Gonçalves, C.A.; de Assis, A.M.; Quincozes-Santos, A.; Wyse, A.T. 1,25-Dihydroxyvitamin D3 exerts neuroprotective effects in an ex vivo model of mild hyperhomocysteinemia. Int. J. Dev. Neurosci. 2016, 48, 71–79. [Google Scholar] [CrossRef]
- Longoni, A.; Kolling, J.; Siebert, C.; Dos Santos, J.P.; da Silva, J.S.; Pettenuzzo, L.F.; Meira-Martins, L.A.; Gonçalves, C.A.; de Assis, A.M.; Wyse, A.T. 1,25-Dihydroxyvitamin D(3) prevents deleterious effects of homocysteine on mitochondrial function and redox status in heart slices. Nutr. Res. 2017, 38, 52–63. [Google Scholar] [CrossRef]
- Molinari, C.; Morsanuto, V.; Ghirlanda, S.; Ruga, S.; Notte, F.; Gaetano, L.; Uberti, F. Role of Combined Lipoic Acid and Vitamin D3 on Astrocytes as a Way to Prevent Brain Ageing by Induced Oxidative Stress and Iron Accumulation. Oxidative Med. Cell. Longev. 2019, 2019, 2843121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamini, P.; Ray, R.S.; Chopra, K. Vitamin D (3) attenuates cognitive deficits and neuroinflammatory responses in ICV-STZ induced sporadic Alzheimer’s disease. Inflammopharmacology 2018, 26, 39–55. [Google Scholar] [CrossRef] [PubMed]
- Gong, Q.; Li, X.; Sun, J.; Ding, G.; Zhou, M.; Zhao, W.; Lu, Y. The effects of calcipotriol on the dendritic morphology of human melanocytes under oxidative stress and a possible mechanism: Is it a mitochondrial protector? J. Dermatol. Sci. 2015, 77, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.; Kang, H.J.; Kim, D.A.; Ryu, E.S.; Yu, M.; Lee, H.; Lee, H.K.; Ryu, H.M.; Park, S.H.; Kim, Y.L.; et al. Paricalcitol attenuates TGF-β1-induced phenotype transition of human peritoneal mesothelial cells (HPMCs) via modulation of oxidative stress and NLRP3 inflammasome. FASEB J. 2019, 33, 3035–3050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussien, N.I.; El-Wakeel, H.S.; Souror, S.M.; Ahmed, I.A. Alleviation of cardiac mitochondrial dysfunction and oxidative stress underlies the protective effect of vitamin D in chronic stress-induced cardiac dysfunction in rats. Gen. Physiol. Biophys. 2019, 38, 51–61. [Google Scholar] [CrossRef]
- Ketsa, O.V.; Marchenko, M.M. Effect of essential lipophilic nutrients on free radical processes in liver mitochondrial fraction of the tumor-bearing rats. Vopr. Pitan. 2019, 88, 32–39. [Google Scholar] [CrossRef]
- Zhang, H.; Xue, L.; Li, B.; Zhang, Z.; Tao, S. Vitamin D Protects Against Alcohol-Induced Liver Cell Injury Within an NRF2-ALDH2 Feedback Loop. Mol. Nutr. Food Res. 2019, 63, e1801014. [Google Scholar] [CrossRef]
- Tong, T.; Liu, Z.; Zhang, H.; Sun, J.; Zhang, D.; Wang, F.; Miao, D.; Shen, Y. Age-dependent expression of the vitamin D receptor and the protective effect of vitamin D receptor activation on H(2)O(2)-induced apoptosis in rat intervertebral disc cells. J. Steroid Biochem. Mol. Biol. 2019, 190, 126–138. [Google Scholar] [CrossRef]
- Zhu, X.; Wu, S.; Guo, H. Active Vitamin D and Vitamin D Receptor Help Prevent High Glucose Induced Oxidative Stress of Renal Tubular Cells via AKT/UCP2 Signaling Pathway. Biomed. Res. Int. 2019, 2019, 9013904. [Google Scholar] [CrossRef]
- Krone, B.; Grange, J.M. Paradigms in multiple sclerosis: Time for a change, time for a unifying concept. Inflammopharmacology 2011, 19, 187–195. [Google Scholar] [CrossRef] [Green Version]
- Mocayar Marón, F.J.; Ferder, L.; Reiter, R.J.; Manucha, W. Daily and seasonal mitochondrial protection: Unraveling common possible mechanisms involving vitamin D and melatonin. J. Steroid Biochem. Mol. Biol. 2020, 199, 105595. [Google Scholar] [CrossRef] [PubMed]
- Keeney, J.T.R.; Förster, S.; Sultana, R.; Brewer, L.D.; Latimer, C.S.; Cai, J.; Klein, J.B.; Porter, N.M.; Butterfield, D.A. Dietary vitamin D deficiency in rats from middle to old age leads to elevated tyrosine nitration and proteomics changes in levels of key proteins in brain: Implications for low vitamin D-dependent age-related cognitive decline. Free Radic. Biol. Med. 2013, 65, 324–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berridge, M.J. Vitamin D deficiency accelerates ageing and age-related diseases: A novel hypothesis. J. Physiol. 2017, 595, 6825–6836. [Google Scholar] [CrossRef] [PubMed]
- Tobore, T.O. On the Etiopathogenesis and Pathophysiology of Alzheimer’s Disease: A Comprehensive Theoretical Review. J. Alzheimers Dis. 2019, 68, 417–437. [Google Scholar] [CrossRef] [PubMed]
- Wimalawansa, S.J. Vitamin D Deficiency: Effects on Oxidative Stress, Epigenetics, Gene Regulation, and Aging. Biology 2019, 8, 30. [Google Scholar] [CrossRef] [Green Version]
- Dzik, K.P.; Kaczor, J.J. Mechanisms of vitamin D on skeletal muscle function: Oxidative stress, energy metabolism and anabolic state. Eur. J. Appl. Physiol. 2019, 119, 825–839. [Google Scholar] [CrossRef] [Green Version]
- Dzik, K.P.; Skrobot, W.; Kaczor, K.B.; Flis, D.J.; Karnia, M.J.; Libionka, W.; Antosiewicz, J.; Kloc, W.; Kaczor, J.J. Vitamin D Deficiency Is Associated with Muscle Atrophy and Reduced Mitochondrial Function in Patients with Chronic Low Back Pain. Oxidative Med. Cell. Longev. 2019, 2019, 6835341. [Google Scholar] [CrossRef]
- Chang, E. 1,25-Dihydroxyvitamin D Decreases Tertiary Butyl-Hydrogen Peroxide-Induced Oxidative Stress and Increases AMPK/SIRT1 Activation in C2C12 Muscle Cells. Molecules 2019, 24, 3903. [Google Scholar] [CrossRef] [Green Version]
- Yao, T.; Ying, X.; Zhao, Y.; Yuan, A.; He, Q.; Tong, H.; Ding, S.; Liu, J.; Peng, X.; Gao, E.; et al. Vitamin D receptor activation protects against myocardial reperfusion injury through inhibition of apoptosis and modulation of autophagy. Antioxid. Redox Signal. 2015, 22, 633–650. [Google Scholar] [CrossRef] [Green Version]
- Consiglio, M.; Destefanis, M.; Morena, D.; Foglizzo, V.; Forneris, M.; Pescarmona, G.; Silvagno, F. The vitamin D receptor inhibits the respiratory chain, contributing to the metabolic switch that is essential for cancer cell proliferation. PLoS ONE 2014, 9, e115816. [Google Scholar] [CrossRef] [Green Version]
- Piotrowska, A.; Wierzbicka, J.; Ślebioda, T.; Woźniak, M.; Tuckey, R.C.; Slominski, A.T.; Żmijewski, M.A. Vitamin D derivatives enhance cytotoxic effects of H2O2 or cisplatin on human keratinocytes. Steroids 2016, 110, 49–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedraza-Chaverri, J.; Sánchez-Lozada, L.G.; Osorio-Alonso, H.; Tapia, E.; Scholze, A. New Pathogenic Concepts and Therapeutic Approaches to Oxidative Stress in Chronic Kidney Disease. Oxidative Med. Cell. Longev. 2016, 2016, 6043601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Favre, G.A.; Esnault, V.L.; Van Obberghen, E. Modulation of glucose metabolism by the renin-angiotensin-aldosterone system. Am. J. Physiol. Endocrinol. Metab. 2015, 308, 435–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Case, A.J.; Yang, R.F.; Schultz, H.D.; Zimmerman, M.C. Over-expressed copper/zinc superoxide dismutase localizes to mitochondria in neurons inhibiting the angiotensin II-mediated increase in mitochondrial superoxide. Redox Biol. 2013, 2, 8–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Indo, H.P.; Yen, H.C.; Nakanishi, I.; Matsumoto, K.; Tamura, M.; Nagano, Y.; Matsui, H.; Gusev, O.; Cornette, R.; Okuda, T.; et al. A mitochondrial superoxide theory for oxidative stress diseases and aging. J. Clin. Biochem. Nutr. 2015, 56, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Somanna, N.K.; Yariswamy, M.; Garagliano, J.M.; Siebenlist, U.; Mummidi, S.; Valente, A.J.; Chandrasekar, B. Aldosterone-induced cardiomyocyte growth, and fibroblast migration and proliferation are mediated by TRAF3IP2. Cell. Signal. 2015, 27, 1928–1938. [Google Scholar] [CrossRef]
- Giam, B.; Kaye, D.M.; Rajapakse, N.W. Role of Renal Oxidative Stress in the Pathogenesis of the Cardiorenal Syndrome. Heart Lung Circ. 2016, 25, 874–880. [Google Scholar] [CrossRef]
- Haas, M.J.; Onstead-Haas, L.; Lee, T.; Torfah, M.; Mooradian, A.D. Angiotensin II receptor one (AT1) mediates dextrose induced endoplasmic reticulum stress and superoxide production in human coronary artery endothelial cells. Int. J. Cardiol. 2016, 220, 842–850. [Google Scholar] [CrossRef]
- Wang, B.; Lin, L.; Wang, H.; Guo, H.; Gu, Y.; Ding, W. Overexpressed cyclophilin B suppresses aldosterone-induced proximal tubular cell injury both in vitro and in vivo. Oncotarget 2016, 7, 69309–69320. [Google Scholar] [CrossRef] [Green Version]
- Ghazi, L.; Drawz, P. Advances in understanding the renin-angiotensin-aldosterone system (RAAS) in blood pressure control and recent pivotal trials of RAAS blockade in heart failure and diabetic nephropathy. F1000Research 2017, 6. [Google Scholar] [CrossRef] [Green Version]
- Nishihara, M.; Takesue, K.; Hirooka, Y. Olmesartan combined with renal denervation reduces blood pressure in association with sympatho-inhibitory and aldosterone-reducing effects in hypertensive mice with chronic kidney disease. Clin. Exp. Hypertens. 2019, 41, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Eid, R.A.; El-Kott, A.F.; Zaki, M.S.A.; Eldeen, M.A.; Al-Hashem, F.H.; Alkhateeb, M.A.; Alassiri, M.; Aldera, H. Acylated ghrelin protects aorta damage post-MI via activation of eNOS and inhibition of angiotensin-converting enzyme induced activation of NAD(P)H-dependent oxidase. Ultrastruct. Pathol 2018, 42, 416–429. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.R.; Fan, X.H.; Chen, G.; Zeng, G.W.; Xue, Y.G.; Liu, X.T.; Wang, C.Y. Irisin attenuates angiotensin II-induced cardiac fibrosis via Nrf2 mediated inhibition of ROS/TGFβ1/Smad2/3 signaling axis. Chem. Biol. Interact. 2019, 302, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, C.; Schulz, I.; Epe, B.; Schupp, N. Angiotensin II-induced hypertension increases the mutant frequency in rat kidney. Arch. Toxicol. 2019, 93, 2045–2055. [Google Scholar] [CrossRef] [PubMed]
- Loffredo, L.; Violi, F. COVID-19 and cardiovascular injury: A role for oxidative stress and antioxidant treatment? Int. J. Cardiol. 2020, 312, 136. [Google Scholar] [CrossRef] [PubMed]
- Alpalhão, M.; Ferreira, J.A.; Filipe, P. Persistent SARS-CoV-2 infection and the risk for cancer. Med. Hypotheses 2020, 143, 109882. [Google Scholar] [CrossRef] [PubMed]
- Iddir, M.; Brito, A.; Dingeo, G.; Fernandez Del Campo, S.S.; Samouda, H.; La Frano, M.R.; Bohn, T. Strengthening the Immune System and Reducing Inflammation and Oxidative Stress through Diet and Nutrition: Considerations during the COVID-19 Crisis. Nutrients 2020, 12, 1562. [Google Scholar] [CrossRef]
- Korakas, E.; Ikonomidis, I.; Kousathana, F.; Balampanis, K.; Kountouri, A.; Raptis, A.; Palaiodimou, L.; Kokkinos, A.; Lambadiari, V. Obesity and COVID-19: Immune and metabolic derangement as a possible link to adverse clinical outcomes. Am. J. Physiol. Endocrinol. Metab. 2020, 319, E105–E109. [Google Scholar] [CrossRef]
- Sawalha, A.H.; Zhao, M.; Coit, P.; Lu, Q. Epigenetic dysregulation of ACE2 and interferon-regulated genes might suggest increased COVID-19 susceptibility and severity in lupus patients. Clin. Immunol. 2020, 215, 108410. [Google Scholar] [CrossRef]
- McCord, J.M.; Hybertson, B.M.; Cota-Gomez, A.; Gao, B. Nrf2 Activator PB125® as a Potential Therapeutic Agent Against COVID-19. Antioxidants 2020. [Google Scholar] [CrossRef]
- Polonikov, A. Endogenous Deficiency of Glutathione as the Most Likely Cause of Serious Manifestations and Death in COVID-19 Patients. ACS Infect. Dis. 2020, 6, 1558–1562. [Google Scholar] [CrossRef] [PubMed]
- Shneider, A.; Kudriavtsev, A.; Vakhrusheva, A. Can melatonin reduce the severity of COVID-19 pandemic? Int. Rev. Immunol. 2020, 39, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Ferder, M.; Inserra, F.; Manucha, W.; Ferder, L. The world pandemic of vitamin D deficiency could possibly be explained by cellular inflammatory response activity induced by the renin-angiotensin system. Am. J. Physiol. Cell Physiol. 2013, 304, C1027–C1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giménez, V.M.M.; Sanz, R.L.; Marón, F.J.M.; Ferder, L.; Manucha, W. Vitamin D-RAAS connection: An Integrative Standpoint into Cardiovascular and Neuroinflammatory Disorders. Curr. Protein Pept. Sci 2020. [Google Scholar] [CrossRef] [PubMed]
- Manucha, W.; Ritchie, B.; Ferder, L. Hypertension and insulin resistance: Implications of mitochondrial dysfunction. Curr. Hypertens. Rep. 2015, 17, 504. [Google Scholar] [CrossRef]
- Li, Y.C.; Qiao, G.; Uskokovic, M.; Xiang, W.; Zheng, W.; Kong, J. Vitamin D: A negative endocrine regulator of the renin-angiotensin system and blood pressure. J. Steroid Biochem. Mol. Biol. 2004, 89–90, 387–392. [Google Scholar] [CrossRef]
- Santoro, D.; Caccamo, D.; Lucisano, S.; Buemi, M.; Sebekova, K.; Teta, D.; De Nicola, L. Interplay of vitamin D, erythropoiesis, and the renin-angiotensin system. Biomed. Res. Int 2015, 2015, 145828. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Yang, J.; Chen, J.; Luo, Q.; Zhang, Q.; Zhang, H. Vitamin D alleviates lipopolysaccharide-induced acute lung injury via regulation of the renin-angiotensin system. Mol. Med. Rep. 2017, 16, 7432–7438. [Google Scholar] [CrossRef] [Green Version]
- Ricca, C.; Aillon, A.; Bergandi, L.; Alotto, D.; Castagnoli, C.; Silvagno, F. Vitamin D Receptor Is Necessary for Mitochondrial Function and Cell Health. Int. J. Mol. Sci. 2018, 19, 1672. [Google Scholar] [CrossRef] [Green Version]
- Silvagno, F.; De Vivo, E.; Attanasio, A.; Gallo, V.; Mazzucco, G.; Pescarmona, G. Mitochondrial localization of vitamin D receptor in human platelets and differentiated megakaryocytes. PLoS ONE 2010, 5, e8670. [Google Scholar] [CrossRef] [Green Version]
- Abadir, P.M.; Foster, D.B.; Crow, M.; Cooke, C.A.; Rucker, J.J.; Jain, A.; Smith, B.J.; Burks, T.N.; Cohn, R.D.; Fedarko, N.S.; et al. Identification and characterization of a functional mitochondrial angiotensin system. Proc. Natl. Acad. Sci. USA 2011, 108, 14849–14854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanz, R.; Mazzei, L.; Santino, N.; Ingrasia, M.; Manucha, W. Vitamin D-mitochondria cross-talk could modulate the signaling pathway involved in hypertension development: A translational integrative overview. Clin. Investig. Arterioscler. 2020, 32, 144–155. [Google Scholar] [CrossRef] [PubMed]
- Carrara, D.; Bruno, R.M.; Bacca, A.; Taddei, S.; Duranti, E.; Ghiadoni, L.; Bernini, G. Cholecalciferol treatment downregulates renin-angiotensin system and improves endothelial function in essential hypertensive patients with hypovitaminosid D. J. Hypertens. 2016, 34, 2199–2205. [Google Scholar] [CrossRef] [PubMed]
- Leung, P.S. The Modulatory Action of Vitamin D on the Renin-Angiotensin System and the Determination of Hepatic Insulin Resistance. Molecules 2019, 24, 2479. [Google Scholar] [CrossRef] [Green Version]
- Kong, J.; Zhu, X.; Shi, Y.; Liu, T.; Chen, Y.; Bhan, I.; Zhao, Q.; Thadhani, R.; Li, Y.C. VDR attenuates acute lung injury by blocking Ang-2-Tie-2 pathway and renin-angiotensin system. Mol. Endocrinol. 2013, 27, 2116–2125. [Google Scholar] [CrossRef]
- Shi, Y.; Liu, T.; Yao, L.; Xing, Y.; Zhao, X.; Fu, J.; Xue, X. Chronic vitamin D deficiency induces lung fibrosis through activation of the renin-angiotensin system. Sci. Rep. 2017, 7, 3312. [Google Scholar] [CrossRef]
- Al-Ishaq, R.K.; Kubatka, P.; Brozmanova, M.; Gazdikova, K.; Caprnda, M.; Büsselberg, D. Health implication of vitamin D on the cardiovascular and the renal system. Arch. Physiol. Biochem. 2019, 1–15. [Google Scholar] [CrossRef]
- He, M.C.; Shi, Z.; Sha, N.N.; Chen, N.; Peng, S.Y.; Liao, D.F.; Wong, M.S.; Dong, X.L.; Wang, Y.J.; Yuan, T.F.; et al. Paricalcitol alleviates lipopolysaccharide-induced depressive-like behavior by suppressing hypothalamic microglia activation and neuroinflammation. Biochem. Pharmacol. 2019, 163, 1–8. [Google Scholar] [CrossRef]
- Turin, A.; Bax, J.J.; Doukas, D.; Joyce, C.; Lopez, J.J.; Mathew, V.; Pontone, G.; Shah, F.; Singh, S.; Wilber, D.J.; et al. Interactions Among Vitamin D, Atrial Fibrillation, and the Renin-Angiotensin-Aldosterone System. Am. J. Cardiol. 2018, 122, 780–784. [Google Scholar] [CrossRef]
- Zhang, W.; Chen, L.; Zhang, L.; Xiao, M.; Ding, J.; Goltzman, D.; Miao, D. Administration of exogenous 1,25(OH)2D3 normalizes overactivation of the central renin-angiotensin system in 1α(OH)ase knockout mice. Neurosci. Lett. 2015, 588, 184–189. [Google Scholar] [CrossRef]
- Li, Y.C.; Kong, J.; Wei, M.; Chen, Z.F.; Liu, S.Q.; Cao, L.P. 1,25-Dihydroxyvitamin D(3) is a negative endocrine regulator of the renin-angiotensin system. J. Clin. Investig. 2002, 110, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.; Pan, W.; Kong, J.; Zheng, W.; Szeto, F.L.; Wong, K.E.; Cohen, R.; Klopot, A.; Zhang, Z.; Li, Y.C. 1,25-dihydroxyvitamin D3 suppresses renin gene transcription by blocking the activity of the cyclic AMP response element in the renin gene promoter. J. Biol. Chem. 2007, 282, 29821–29830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, C.; Lu, F.; Cao, K.; Xu, D.; Goltzman, D.; Miao, D. Calcium-independent and 1,25(OH)2D3-dependent regulation of the renin-angiotensin system in 1alpha-hydroxylase knockout mice. Kidney Int. 2008, 74, 170–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitani, H.; Ishizaka, N.; Aizawa, T.; Ohno, M.; Usui, S.; Suzuki, T.; Amaki, T.; Mori, I.; Nakamura, Y.; Sato, M.; et al. In vivo klotho gene transfer ameliorates angiotensin II-induced renal damage. Hypertension 2002, 39, 838–843. [Google Scholar] [CrossRef] [Green Version]
- Takenaka, T.; Inoue, T.; Miyazaki, T.; Kobori, H.; Nishiyama, A.; Ishii, N.; Hayashi, M.; Suzuki, H. Klotho suppresses the renin-angiotensin system in adriamycin nephropathy. Nephrol. Dial. Transplant. 2017, 32, 791–800. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Mo, H.; Miao, J.; Zhou, D.; Tan, R.J.; Hou, F.F.; Liu, Y. Klotho Ameliorates Kidney Injury and Fibrosis and Normalizes Blood Pressure by Targeting the Renin-Angiotensin System. Am. J. Pathol. 2015, 185, 3211–3223. [Google Scholar] [CrossRef] [Green Version]
- Chandel, N.; Sharma, B.; Husain, M.; Salhan, D.; Singh, T.; Rai, P.; Mathieson, P.W.; Saleem, M.A.; Malhotra, A.; Singhal, P.C. HIV compromises integrity of the podocyte actin cytoskeleton through downregulation of the vitamin D receptor. Am. J. Physiol. Renal. Physiol. 2013, 304, 1347–1357. [Google Scholar] [CrossRef] [Green Version]
- Salhan, D.; Husain, M.; Subrati, A.; Goyal, R.; Singh, T.; Rai, P.; Malhotra, A.; Singhal, P.C. HIV-induced kidney cell injury: Role of ROS-induced downregulated vitamin D receptor. Am. J. Physiol. Renal. Physiol. 2012, 303, F503–F514. [Google Scholar] [CrossRef]
- Shenoy, S. Coronavirus (Covid-19) sepsis: Revisiting mitochondrial dysfunction in pathogenesis, aging, inflammation, and mortality. Inflamm. Res. 2020, 1–9. [Google Scholar] [CrossRef]
- Wang, J.Z.; Zhang, R.Y.; Bai, J. An anti-oxidative therapy for ameliorating cardiac injuries of critically ill COVID-19-infected patients. Int. J. Cardiol. 2020, 312, 137–138. [Google Scholar] [CrossRef]
- Grant, W.B.; Lahore, H.; McDonnell, S.L.; Baggerly, C.A.; French, C.B.; Aliano, J.L.; Bhattoa, H.P. Evidence that Vitamin D Supplementation Could Reduce Risk of Influenza and COVID-19 Infections and Deaths. Nutrients 2020, 12, 988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, S.; Hwang, J.W.; Kirkham, P.A.; Rahman, I. Pharmacological and dietary antioxidant therapies for chronic obstructive pulmonary disease. Curr. Med. Chem. 2013, 20, 1496–1530. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, P.E.; Lu, H.; Mann, E.H.; Chen, Y.H.; Ho, T.R.; Cousins, D.J.; Corrigan, C.; Kelly, F.J.; Mudway, I.S.; Hawrylowicz, C.M. Effects of vitamin D on inflammatory and oxidative stress responses of human bronchial epithelial cells exposed to particulate matter. PLoS ONE 2018, 13, e0200040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Azzawi, M.A.; Ghoneim, A.H.; Elmadbouh, I. Evaluation of Vitamin D, Vitamin D Binding Protein Gene Polymorphism with Oxidant—Antioxidant Profiles in Chronic Obstructive Pulmonary Disease. J. Med. Biochem. 2017, 36, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Mansur, J.L.; Tajer, C.; Mariani, J.; Inserra, F.; Ferder, L.; Manucha, W. Vitamin D high doses supplementation could represent a promising alternative to prevent or treat COVID-19 infection. Clin. Investig. Arterioscler. 2020. [Google Scholar] [CrossRef]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
de las Heras, N.; Martín Giménez, V.M.; Ferder, L.; Manucha, W.; Lahera, V. Implications of Oxidative Stress and Potential Role of Mitochondrial Dysfunction in COVID-19: Therapeutic Effects of Vitamin D. Antioxidants 2020, 9, 897. https://doi.org/10.3390/antiox9090897
de las Heras N, Martín Giménez VM, Ferder L, Manucha W, Lahera V. Implications of Oxidative Stress and Potential Role of Mitochondrial Dysfunction in COVID-19: Therapeutic Effects of Vitamin D. Antioxidants. 2020; 9(9):897. https://doi.org/10.3390/antiox9090897
Chicago/Turabian Stylede las Heras, Natalia, Virna Margarita Martín Giménez, León Ferder, Walter Manucha, and Vicente Lahera. 2020. "Implications of Oxidative Stress and Potential Role of Mitochondrial Dysfunction in COVID-19: Therapeutic Effects of Vitamin D" Antioxidants 9, no. 9: 897. https://doi.org/10.3390/antiox9090897
APA Stylede las Heras, N., Martín Giménez, V. M., Ferder, L., Manucha, W., & Lahera, V. (2020). Implications of Oxidative Stress and Potential Role of Mitochondrial Dysfunction in COVID-19: Therapeutic Effects of Vitamin D. Antioxidants, 9(9), 897. https://doi.org/10.3390/antiox9090897