Oxidative Stress, Proton Fluxes, and Chloroquine/Hydroxychloroquine Treatment for COVID-19



Abstract
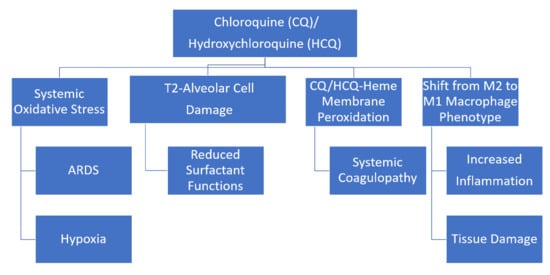
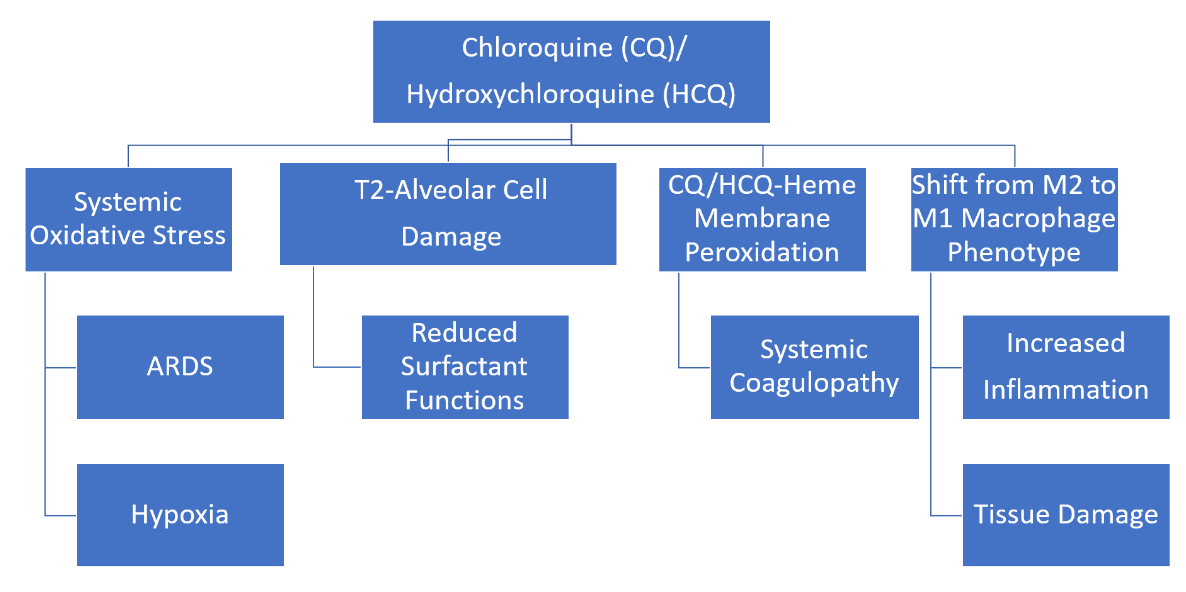
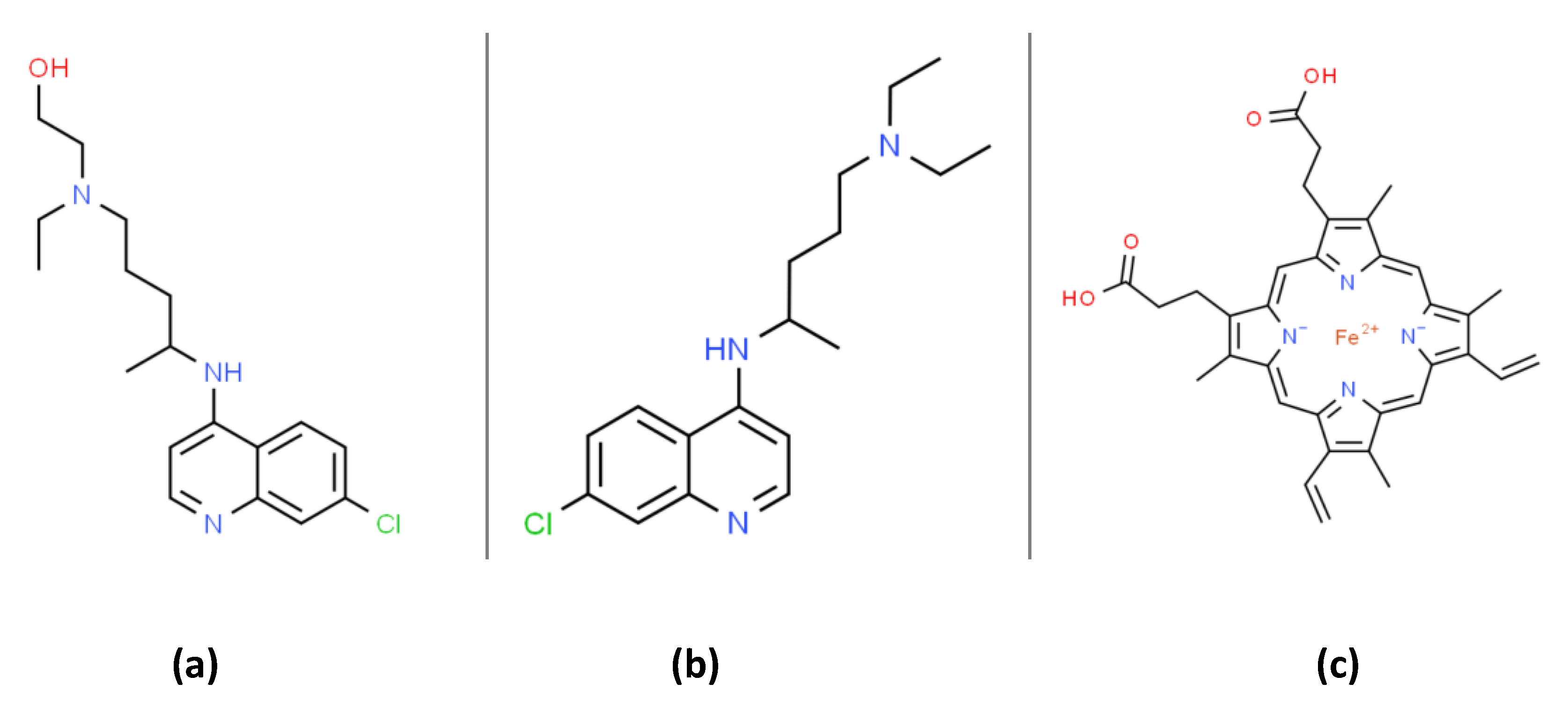
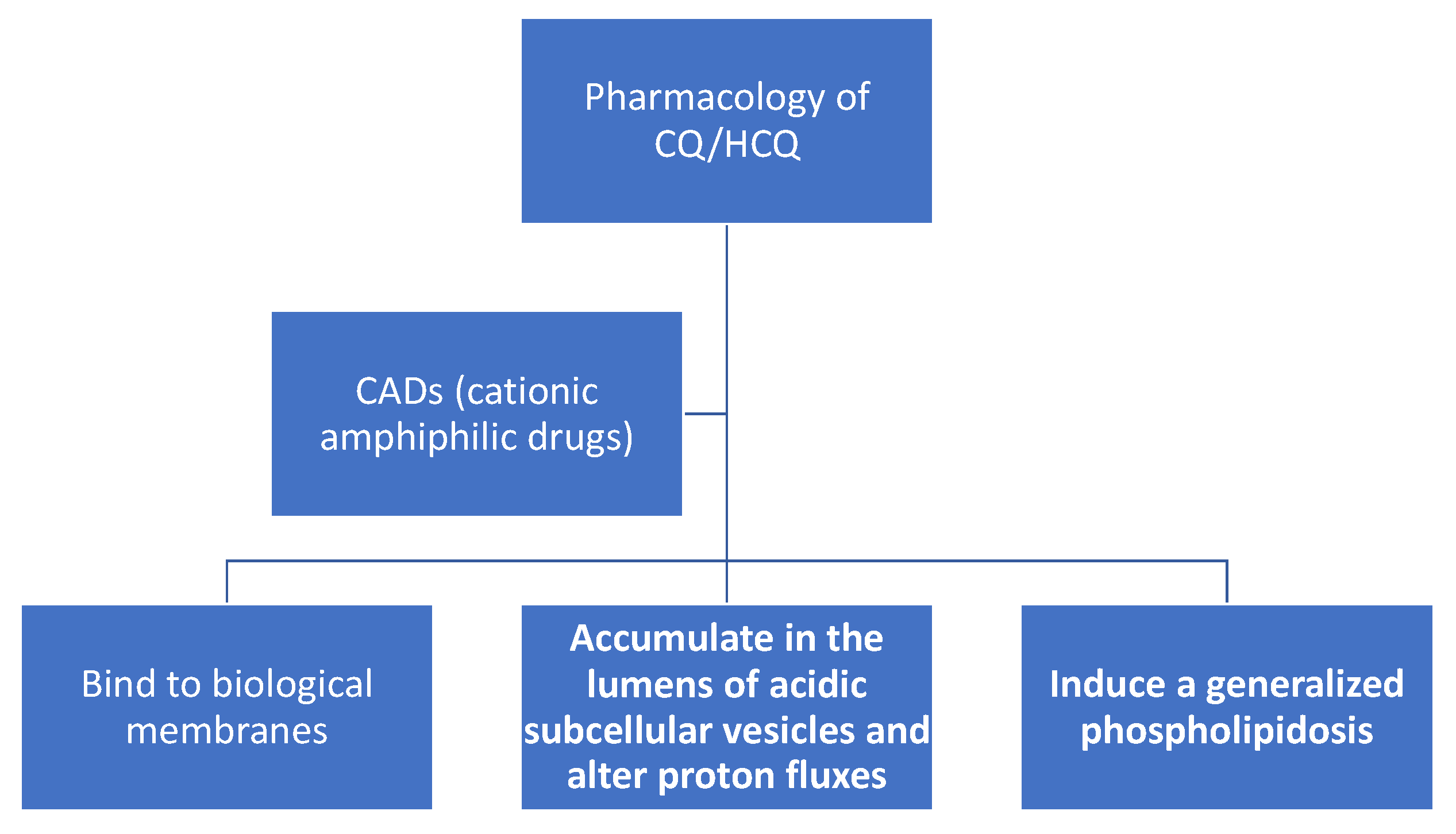
:
{kind=link}
{kind=link}
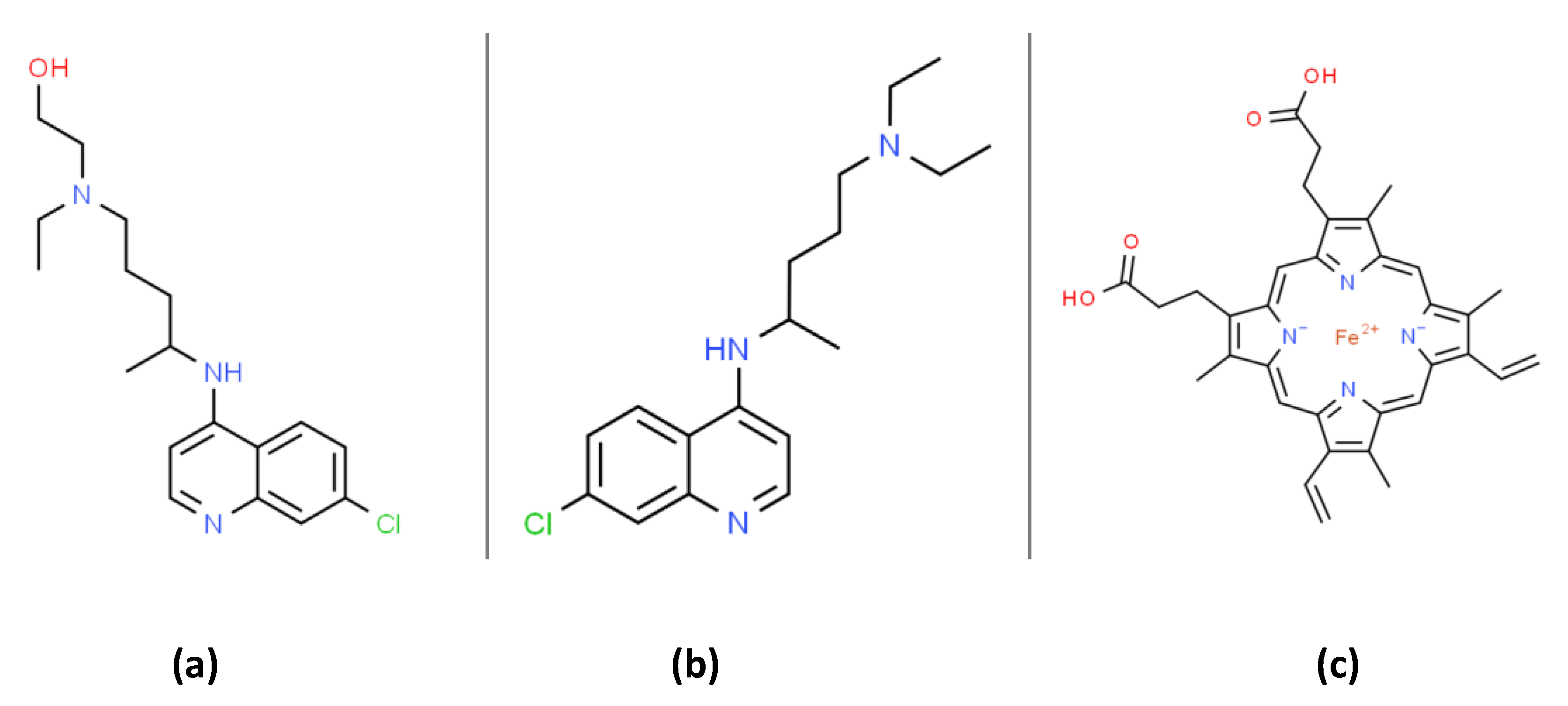
{kind=link}
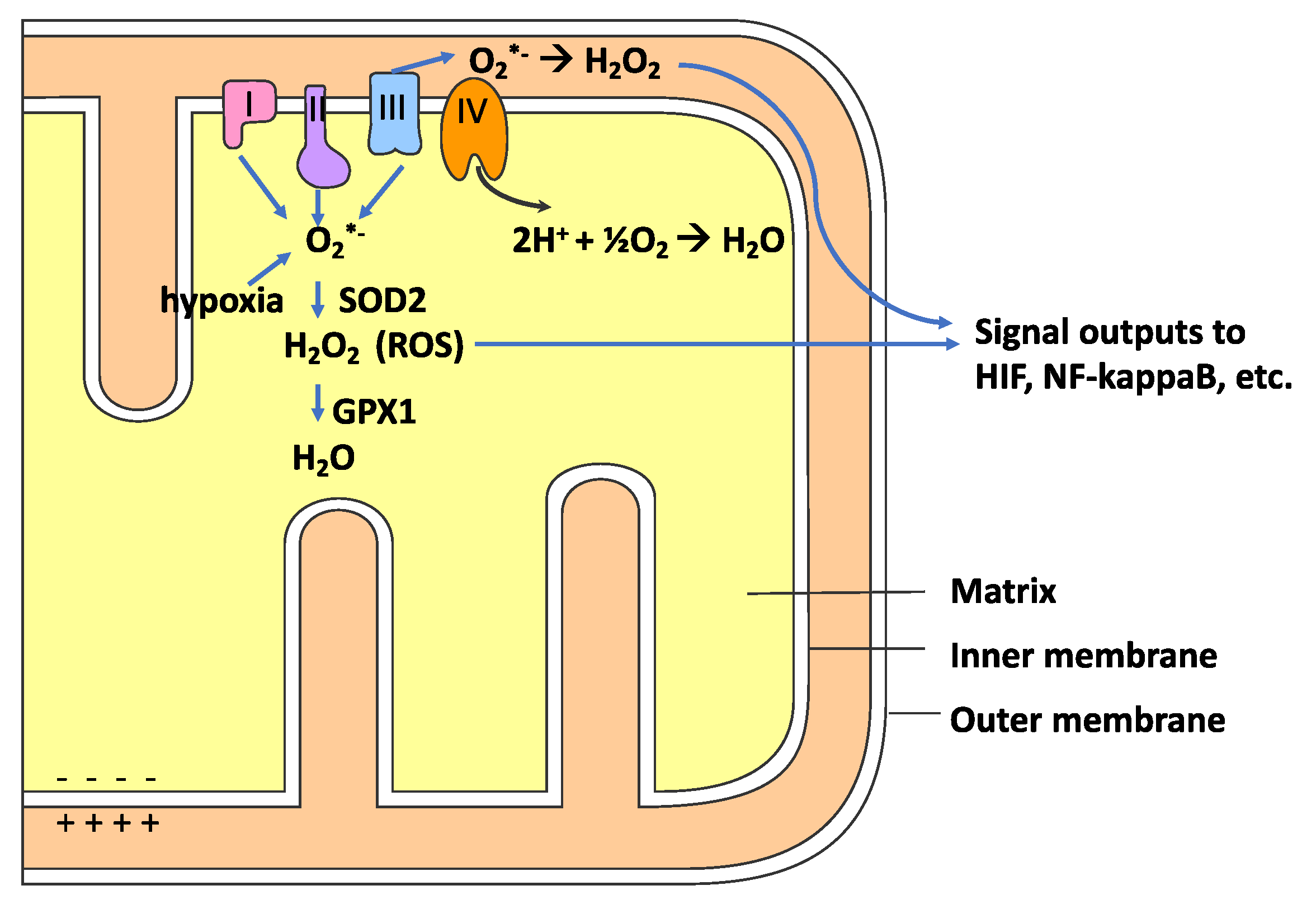{kind=link}
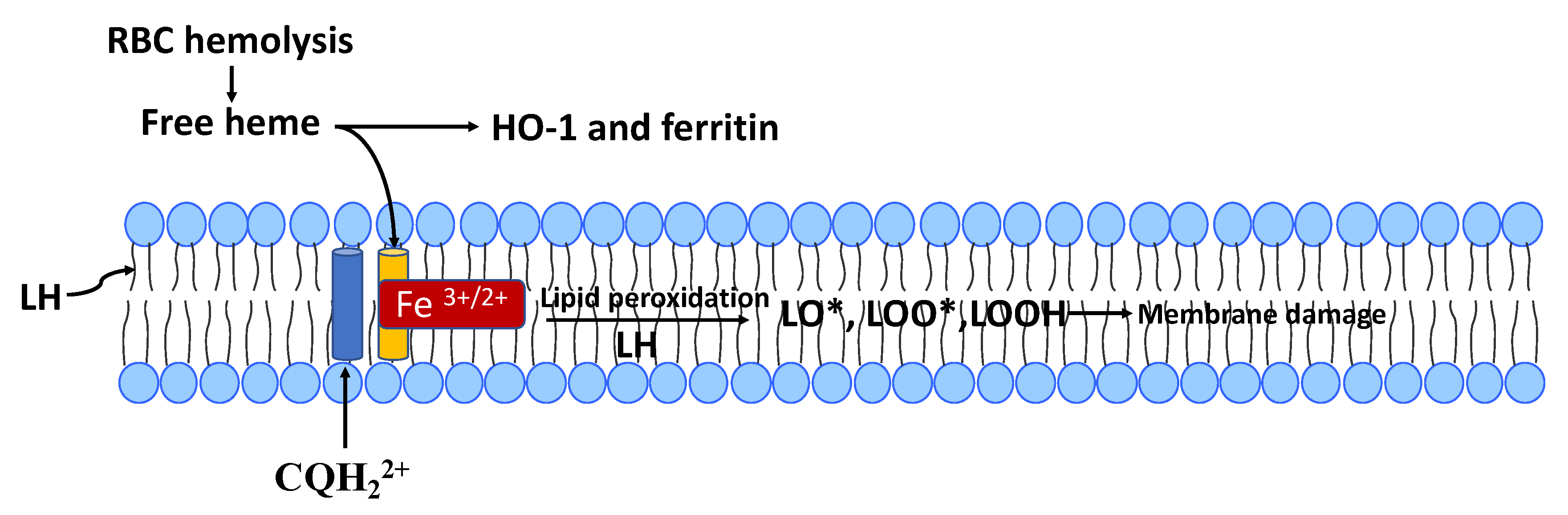{kind=link}
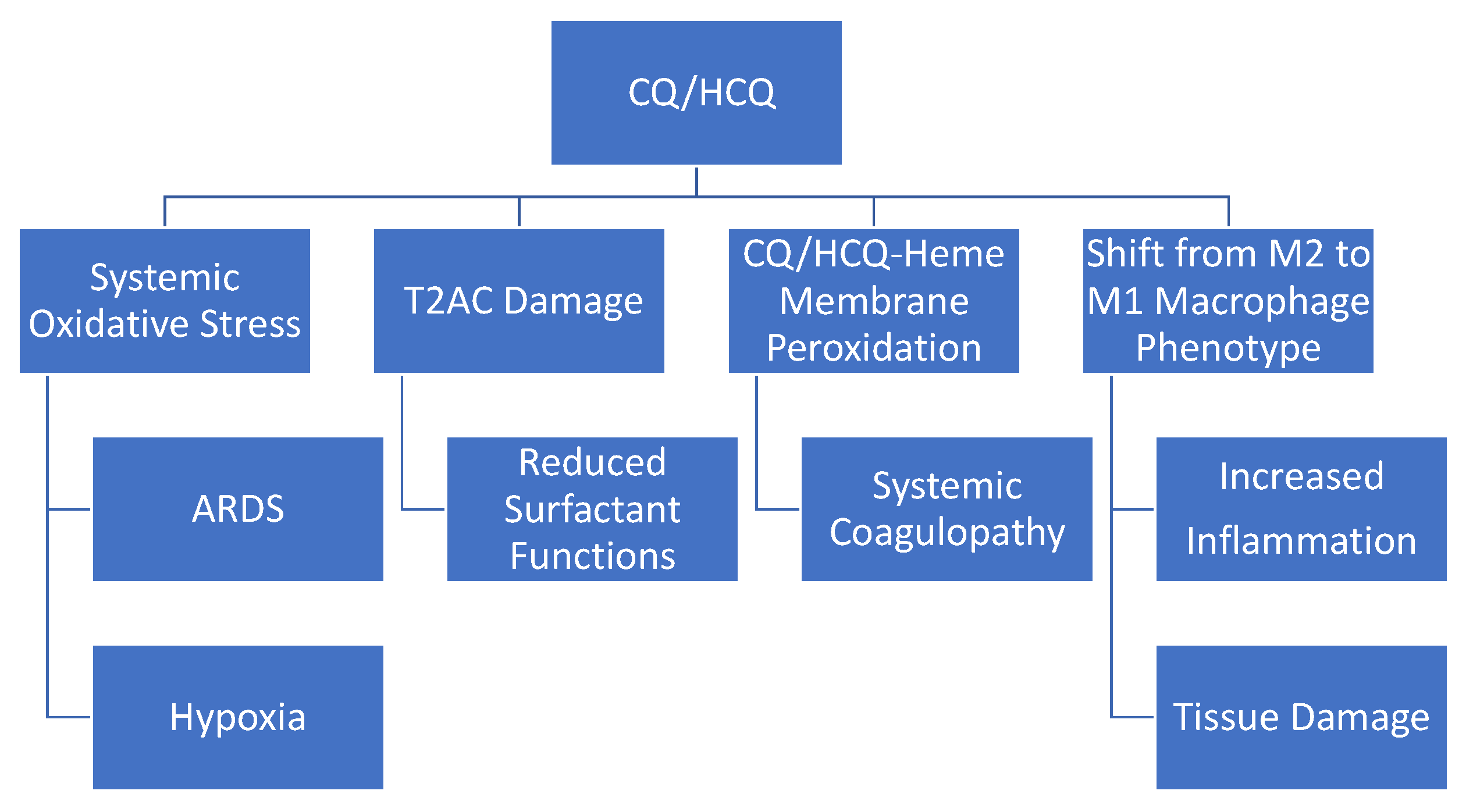{kind=link}
1. Introduction
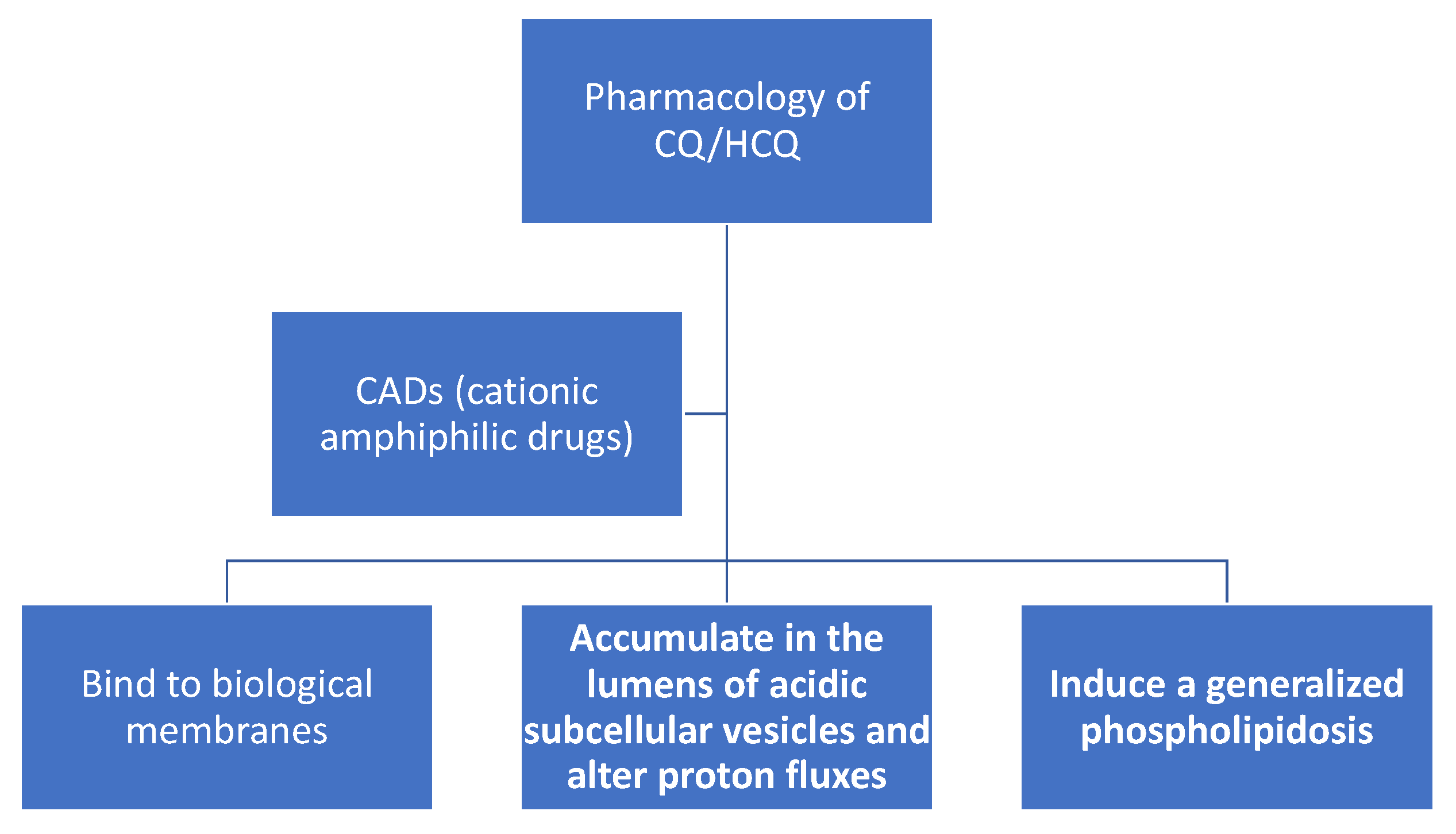
2. CQ/HCQ Pharmacology and Alterations in Subcellular Organelles Proton Fluxes
3. CQ/HCQ Bind to Biological Membranes and Alters their Structure/Function
4. CQ/HCQ Accumulate in the Lumens of Acidic Subcellular Vesicles and Alter Proton Fluxes
5. Endosomal-Lysosomal Proton Fluxes, CQ/HCQ and SARS-CoV-2
6. Redox Physiology, Reactive Oxygen Species (ROS) and Oxidative Stress
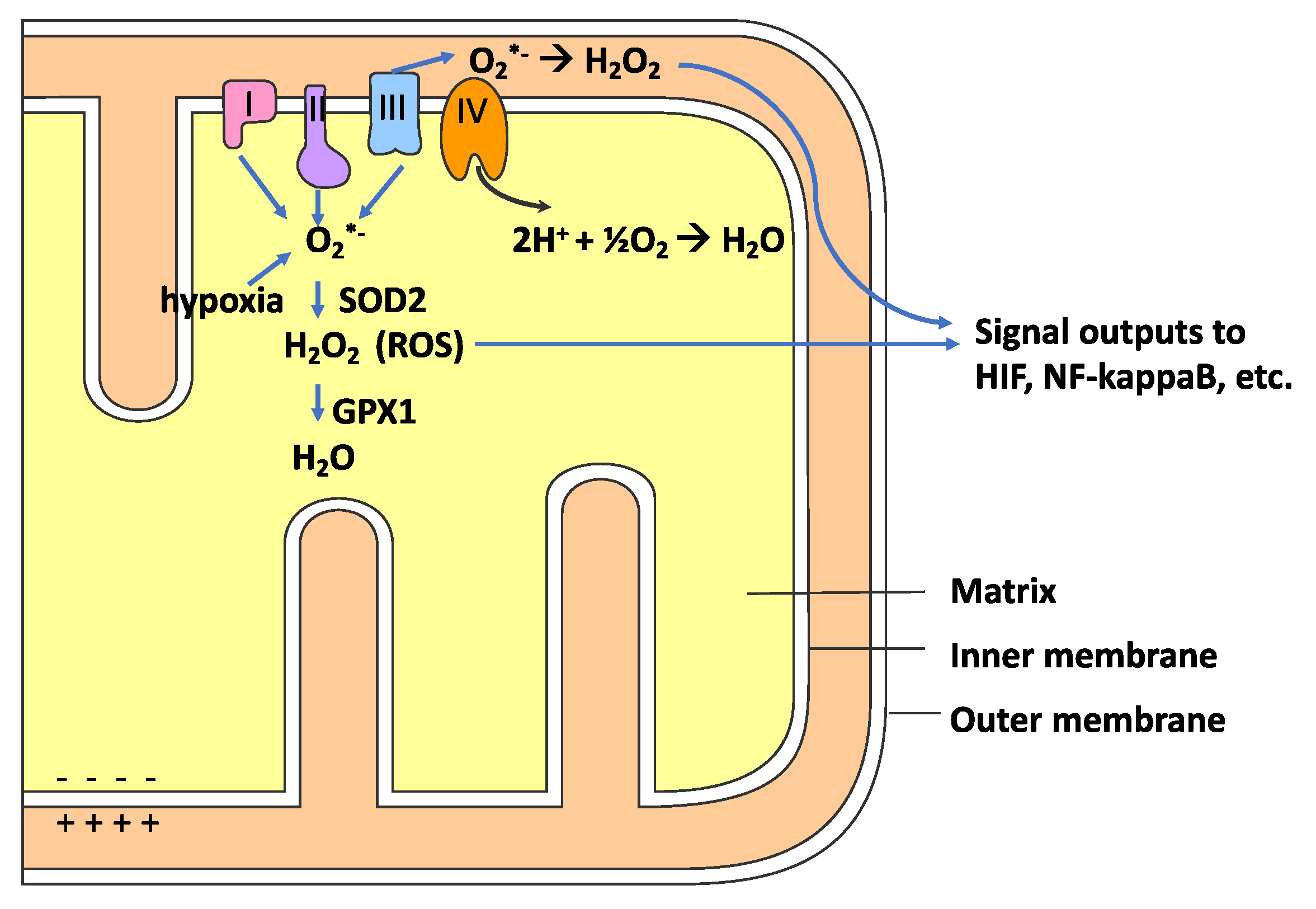
7. Mitochondria, ROS, Hypoxia, and CQ/HCQ
8. Phagocytes, Proton Flux, ROS and CQ/HCQ
9. Evidence for Oxidative Stress Induced by CQ/HCQ
10. CQ/HCQ Influence on Oxidative Stress In Vivo and Ex Vivo
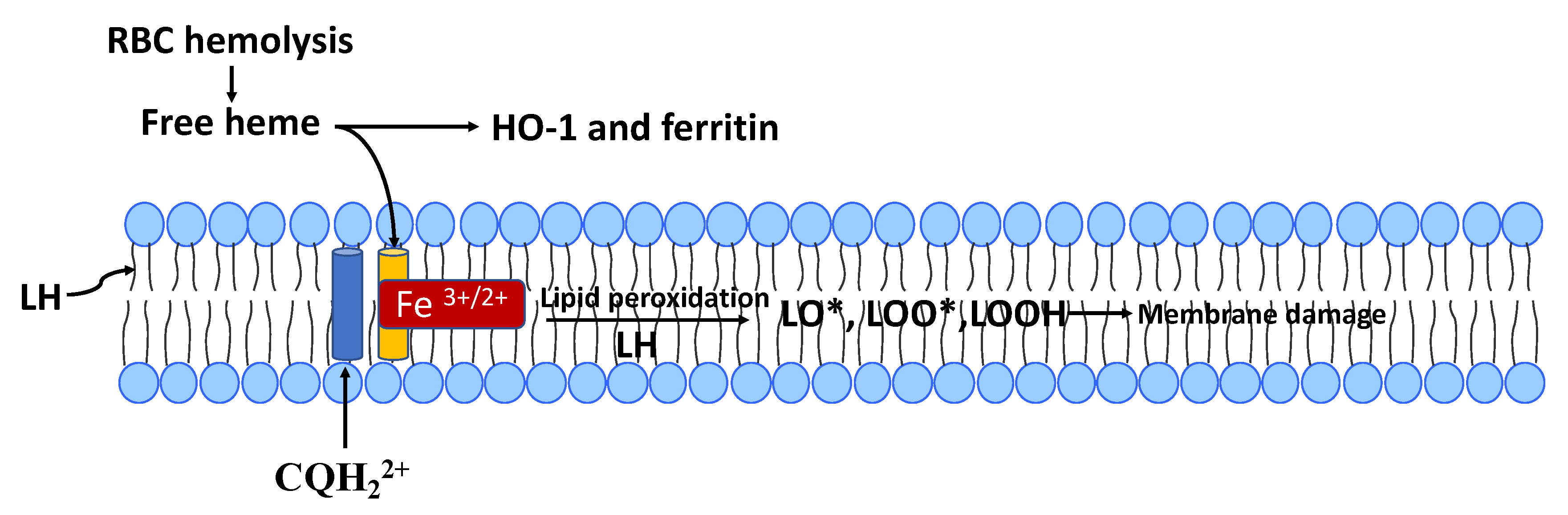
11. CQ/HCQ Oxidative Stress In Vitro and the Role of Free-Heme
12. Zinc and CQ/HCQ Treatment for COVID-19
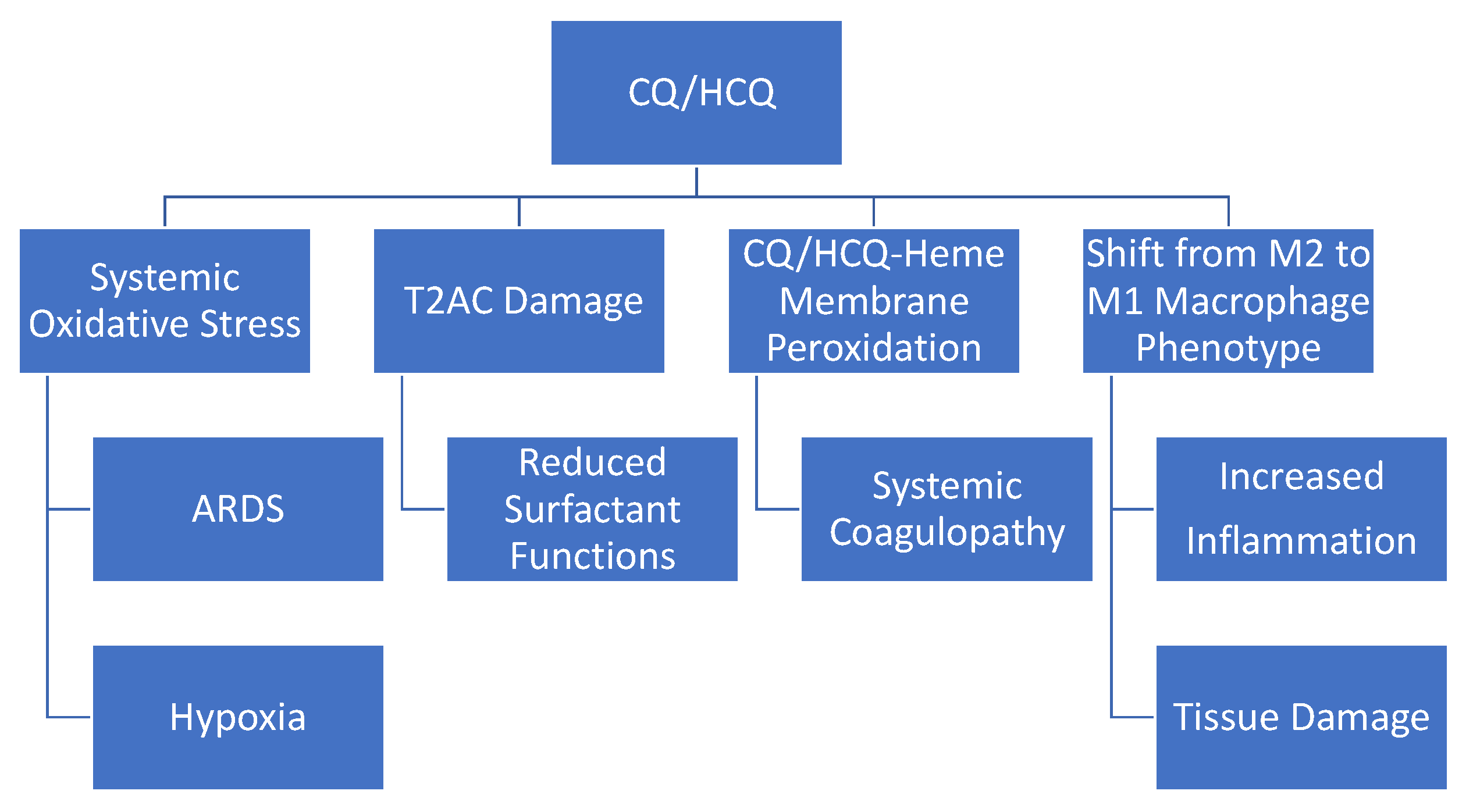
13. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Duan, Y.J.; Liu, Q.; Zhao, S.Q.; Huang, F.; Ren, L.; Liu, L.; Zhou, Y.W. The Trial of Chloroquine in the Treatment of Corona Virus Disease 2019 (COVID-19) and Its Research Progress in Forensic Toxicology. Fa Yi Xue Za Zhi 2020, 36. [Google Scholar] [CrossRef]
- Gautret, P.; Lagier, J.C.; Parola, P.; Hoang, V.T.; Meddeb, L.; Mailhe, M.; Doudier, B.; Courjon, J.; Giordanengo, V.; Vieira, V.E.; et al. Hydroxychloroquine and azithromycin as a treatment of COVID-19: Results of an open-label non-randomized clinical trial. Int. J. Antimicrob. Agents 2020, 105949. [Google Scholar] [CrossRef]
- Zhou, D.; Dai, S.M.; Tong, Q. COVID-19: A recommendation to examine the effect of hydroxychloroquine in preventing infection and progression. J. Antimicrob. Chemother. 2020. [Google Scholar] [CrossRef]
- Cortegiani, A.; Ingoglia, G.; Ippolito, M.; Giarratano, A.; Einav, S. A systematic review on the efficacy and safety of chloroquine for the treatment of COVID-19. J. Crit. Care 2020. [Google Scholar] [CrossRef]
- Yazdany, J.; Kim, A.H.J. Use of Hydroxychloroquine and Chloroquine During the COVID-19 Pandemic: What Every Clinician Should Know. Ann. Intern. Med. 2020. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Cao, R.; Xu, M.; Wang, X.; Zhang, H.; Hu, H.; Li, Y.; Hu, Z.; Zhong, W.; Wang, M. Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARS-CoV-2 infection in vitro. Cell Discov. 2020, 6, 16. [Google Scholar] [CrossRef] [Green Version]
- Shippey, E.A.; Wagler, V.D.; Collamer, A.N. Hydroxychloroquine: An old drug with new relevance. Cleve Clin. J. Med. 2018, 85, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Willis, R.; Seif, A.M.; McGwin, G.; Martinez-Martinez, L.A.; González, E.B.; Dang, N.; Papalardo, E.; Liu, J.; Vilá, L.M.; Reveille, J.D.; et al. Effect of hydroxychloroquine treatment on pro-inflammatory cytokines and disease activity in SLE patients: Data from LUMINA (LXXV), a multiethnic US cohort. Lupus 2012, 21, 830–835. [Google Scholar] [CrossRef] [PubMed]
- Meyerowitz, E.A.; Vannier, A.G.L.; Friesen, M.G.N.; Schoenfeld, S.; Gelfand, J.A.; Callahan, M.V.; Kim, A.Y.; Reeves, P.M.; Poznansky, M.C. Rethinking the role of hydroxychloroquine in the treatment of COVID-19. FASEB J. 2020, 34, 6027–6037. [Google Scholar] [CrossRef]
- Keyaerts, E.; Li, S.; Vijgen, L.; Rysman, E.; Verbeeck, J.; Van Ranst, M.; Maes, P. Antiviral activity of chloroquine against human coronavirus OC43 infection in newborn mice. Antimicrob. Agents Chemother. 2009, 53, 3416–3421. [Google Scholar] [CrossRef] [Green Version]
- Yao, X.; Ye, F.; Zhang, M.; Cui, C.; Huang, B.; Niu, P.; Liu, X.; Zhao, L.; Dong, E.; Song, C.; et al. In Vitro Antiviral Activity and Projection of Optimized Dosing Design of Hydroxychloroquine for the Treatment of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2). Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef] [Green Version]
- Vincent, M.J.; Bergeron, E.; Benjannet, S.; Erickson, B.R.; Rollin, P.E.; Ksiazek, T.G.; Seidah, N.G.; Nichol, S.T. Chloroquine is a potent inhibitor of SARS coronavirus infection and spread. Virol. J. 2005, 2, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- FDA. Available online: https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-revokes-emergency-use-authorization-chloroquine-and (accessed on 17 September 2020).
- Canton, J.; Khezri, R.; Glogauer, M.; Grinstein, S. Contrasting phagosome pH regulation and maturation in human M1 and M2 macrophages. Mol. Biol. Cell 2014, 25, 3330–3341. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.; Lowther, S.; Stambas, J. Inhibition of reactive oxygen species production ameliorates inflammation induced by influenza A viruses via upregulation of SOCS1 and SOCS3. J. Virol. 2015, 89, 2672–2683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, Q.; Wang, B.; Mao, J. The pathogenesis and treatment of the ‘Cytokine Storm’ in COVID-19. J. Infect. 2020, 80, 607–613. [Google Scholar] [CrossRef]
- Herraiz, T.; Guillén, H.; González-Peña, D.; Arán, V.J. Antimalarial Quinoline Drugs Inhibit β-Hematin and Increase Free Hemin Catalyzing Peroxidative Reactions and Inhibition of Cysteine Proteases. Sci. Rep. 2019, 9, 15398. [Google Scholar] [CrossRef] [Green Version]
- Toler, S.M.; Noe, D.; Sharma, A. Selective enhancement of cellular oxidative stress by chloroquine: Implications for the treatment of glioblastoma multiforme. Neurosurg. Focus 2006, 21, E10. [Google Scholar] [CrossRef] [Green Version]
- Farombi, E.O.; Shyntum, Y.Y.; Emerole, G.O. Influence of chloroquine treatment and Plasmodium falciparum malaria infection on some enzymatic and non-enzymatic antioxidant defense indices in humans. Drug Chem. Toxicol. 2003, 26, 59–71. [Google Scholar] [CrossRef]
- Farombi, E.O. Genotoxicity of chloroquine in rat liver cells: Protective role of free radical scavengers. Cell Biol. Toxicol. 2006, 22, 159–167. [Google Scholar] [CrossRef]
- Lehane, A.M.; McDevitt, C.A.; Kirk, K.; Fidock, D.A. Degrees of chloroquine resistance in Plasmodium—Is the redox system involved? Int. J. Parasitol. Drugs Drug Resist. 2012, 2, 47–57. [Google Scholar] [CrossRef] [Green Version]
- Camini, F.C.; da Silva Caetano, C.C.; Almeida, L.T.; de Brito Magalhães, C.L. Implications of oxidative stress on viral pathogenesis. Arch. Virol. 2017, 162, 907–917. [Google Scholar] [CrossRef]
- Wu, C.; Chen, X.; Cai, Y.; Xia, J.; Zhou, X.; Xu, S.; Huang, H.; Zhang, L.; Du, C.; Zhang, Y.; et al. Risk Factors Associated with Acute Respiratory Distress Syndrome and Death in Patients with Coronavirus Disease 2019 Pneumonia in Wuhan, China. JAMA Intern. Med. 2020. [Google Scholar] [CrossRef] [Green Version]
- Chow, C.W.; Herrera Abreu, M.T.; Suzuki, T.; Downey, G.P. Oxidative stress and acute lung injury. Am. J. Respir. Cell Mol. Biol. 2003, 29, 427–431. [Google Scholar] [CrossRef]
- Rocksén, D.; Ekstrand-Hammarström, B.; Johansson, L.; Bucht, A. Vitamin E reduces transendothelial migration of neutrophils and prevents lung injury in endotoxin-induced airway inflammation. Am. J. Respir. Cell Mol. Biol. 2003, 28, 199–207. [Google Scholar] [CrossRef]
- Stone, W.L.; Mukherjee, S.; Smith, M.; Das, S.K. Therapeutic uses of antioxidant liposomes. Methods Mol. Biol. (Clifton N. J.) 2002, 199, 145–161. [Google Scholar] [CrossRef]
- Stone, W.L.; Smith, M. Therapeutic uses of antioxidant liposomes. Mol. Biotechnol. 2004, 27, 217–230. [Google Scholar] [CrossRef]
- Al-Bari, M.A. Chloroquine analogues in drug discovery: New directions of uses, mechanisms of actions and toxic manifestations from malaria to multifarious diseases. J. Antimicrob. Chemother. 2015, 70, 1608–1621. [Google Scholar] [CrossRef] [Green Version]
- Schrezenmeier, E.; Dörner, T. Mechanisms of action of hydroxychloroquine and chloroquine: Implications for rheumatology. Nat. Rev. Rheumatol. 2020, 16, 155–166. [Google Scholar] [CrossRef]
- Breiden, B.; Sandhoff, K. Emerging mechanisms of drug-induced phospholipidosis. Biol. Chem. 2019, 401, 31–46. [Google Scholar] [CrossRef]
- Lüllmann, H.; Wehling, M. The binding of drugs to different polar lipids in vitro. Biochem. Pharmacol. 1979, 28, 3409–3415. [Google Scholar] [CrossRef]
- Seydel, J.K.; Wassermann, O. NMR-studies on the molecular basis of drug-induced phospholipidosis--II. Interaction between several amphiphilic drugs and phospholipids. Biochem. Pharmacol. 1976, 25, 2357–2364. [Google Scholar] [CrossRef]
- Halliwell, W.H. Cationic amphiphilic drug-induced phospholipidosis. Toxicol. Pathol. 1997, 25, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Forgac, M. Vacuolar ATPases: Rotary proton pumps in physiology and pathophysiology. Nat. Rev. Mol. Cell Biol. 2007, 8, 917–929. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Sung, T.; Lin, N.; Abraham, R.T.; Jessen, B.A. Lysosomal adaptation: How cells respond to lysosomotropic compounds. PLoS ONE 2017, 12, e0173771. [Google Scholar] [CrossRef] [Green Version]
- Homewood, C.A.; Warhurst, D.C.; Peters, W.; Baggaley, V.C. Lysosomes, pH and the anti-malarial action of chloroquine. Nature 1972, 235, 50–52. [Google Scholar] [CrossRef]
- Ohkuma, S.; Poole, B. Fluorescence probe measurement of the intralysosomal pH in living cells and the perturbation of pH by various agents. Proc. Natl. Acad. Sci. USA 1978, 75, 3327–3331. [Google Scholar] [CrossRef] [Green Version]
- Daniel, W.A.; Bickel, M.H.; Honegger, U.E. The contribution of lysosomal trapping in the uptake of desipramine and chloroquine by different tissues. Pharmacol. Toxicol. 1995, 77, 402–406. [Google Scholar] [CrossRef]
- Ndiaye, N.; Petrognani, R.; Diatta, B.; Seck, M.; Theobald, X.; Adnet, P. Chloroquine poisoning with respiratory distress and fatal outcome. Ann. Fr. Anesth. Reanim. 1999, 18, 683–685. [Google Scholar] [CrossRef]
- Fois, G.; Hobi, N.; Felder, E.; Ziegler, A.; Miklavc, P.; Walther, P.; Radermacher, P.; Haller, T.; Dietl, P. A new role for an old drug: Ambroxol triggers lysosomal exocytosis via pH-dependent Ca2+ release from acidic Ca2+ stores. Cell Calcium 2015, 58, 628–637. [Google Scholar] [CrossRef]
- Schmitz, G.; Müller, G. Structure and function of lamellar bodies, lipid-protein complexes involved in storage and secretion of cellular lipids. J. Lipid Res. 1991, 32, 1539–1570. [Google Scholar]
- Akella, A.; Deshpande, S.B. Pulmonary surfactants and their role in pathophysiology of lung disorders. Indian J. Exp. Biol 2013, 51, 5–22. [Google Scholar] [PubMed]
- Mason, R.J. Pathogenesis of COVID-19 from a cell biology perspective. Eur. Respir. J. 2020, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.N.; Sun, J.P.; Xue, X.Y.; Wang, J.X. Exogenous pulmonary surfactant for acute respiratory distress syndrome in adults: A systematic review and meta-analysis. Exp. Ther. Med. 2013, 5, 237–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, S.S.; Chang, W.; Lu, Z.H.; Xie, J.F.; Qiu, H.B.; Yang, Y.; Guo, F.M. Effect of surfactant administration on outcomes of adult patients in acute respiratory distress syndrome: A meta-analysis of randomized controlled trials. BMC Pulm. Med. 2019, 19, 9. [Google Scholar] [CrossRef] [PubMed]
- Beers, M.F. Inhibition of cellular processing of surfactant protein C by drugs affecting intracellular pH gradients. J. Biol. Chem. 1996, 271, 14361–14370. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Capote, K.; Manzanares, D.; Haines, T.; Possmayer, F. Reactive oxygen species inactivation of surfactant involves structural and functional alterations to surfactant proteins SP-B and SP-C. Biophys. J. 2006, 90, 2808–2821. [Google Scholar] [CrossRef] [Green Version]
- Hamaguchi, R.; Haginaka, J.; Tanimoto, T.; Kuroda, Y. Maintenance of luminal pH and protease activity in lysosomes/late endosomes by vacuolar ATPase in chlorpromazine-treated RAW264 cells accumulating phospholipids. Cell Biol. Toxicol. 2014, 30, 67–77. [Google Scholar] [CrossRef]
- Tietz, P.S.; Yamazaki, K.; LaRusso, N.F. Time-dependent effects of chloroquine on pH of hepatocyte lysosomes. Biochem. Pharmacol. 1990, 40, 1419–1421. [Google Scholar] [CrossRef]
- Shayman, J.A.; Abe, A. Drug induced phospholipidosis: An acquired lysosomal storage disorder. Biochim. Biophys. Acta 2013, 1831, 602–611. [Google Scholar] [CrossRef] [Green Version]
- Robison, R.L.; Visscher, G.E.; Roberts, S.A.; Engstrom, R.G.; Hartman, H.A.; Ballard, F.H. Generalized phospholipidosis induced by an amphiphilic cationic psychotropic drug. Toxicol. Pathol. 1985, 13, 335–348. [Google Scholar] [CrossRef] [Green Version]
- Jankowski, A.; Scott, C.C.; Grinstein, S. Determinants of the phagosomal pH in neutrophils. J. Biol. Chem. 2002, 277, 6059–6066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jankowski, A.; Grinstein, S. Modulation of the cytosolic and phagosomal pH by the NADPH oxidase. Antioxid. Redox Signal. 2002, 4, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Nunes, P.; Demaurex, N.; Dinauer, M.C. Regulation of the NADPH oxidase and associated ion fluxes during phagocytosis. Traffic 2013, 14, 1118–1131. [Google Scholar] [CrossRef]
- Luzio, J.P.; Pryor, P.R.; Bright, N.A. Lysosomes: Fusion and function. Nat. Rev. Mol. Cell Biol. 2007, 8, 622–632. [Google Scholar] [CrossRef]
- Burkard, C.; Verheije, M.H.; Wicht, O.; van Kasteren, S.I.; van Kuppeveld, F.J.; Haagmans, B.L.; Pelkmans, L.; Rottier, P.J.; Bosch, B.J.; de Haan, C.A. Coronavirus cell entry occurs through the endo-/lysosomal pathway in a proteolysis-dependent manner. PLoS Pathog. 2014, 10, e1004502. [Google Scholar] [CrossRef] [Green Version]
- Yang, N.; Shen, H.M. Targeting the Endocytic Pathway and Autophagy Process as a Novel Therapeutic Strategy in COVID-19. Int. J. Biol. Sci. 2020, 16, 1724–1731. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e278. [Google Scholar] [CrossRef] [PubMed]
- Inoue, Y.; Tanaka, N.; Tanaka, Y.; Inoue, S.; Morita, K.; Zhuang, M.; Hattori, T.; Sugamura, K. Clathrin-dependent entry of severe acute respiratory syndrome coronavirus into target cells expressing ACE2 with the cytoplasmic tail deleted. J. Virol. 2007, 81, 8722–8729. [Google Scholar] [CrossRef] [Green Version]
- Al-Bari, M.A.A. Targeting endosomal acidification by chloroquine analogs as a promising strategy for the treatment of emerging viral diseases. Pharmacol. Res. Perspect. 2017, 5, e00293. [Google Scholar] [CrossRef]
- Singh, A.K.; Singh, A.; Shaikh, A.; Singh, R.; Misra, A. Chloroquine and hydroxychloroquine in the treatment of COVID-19 with or without diabetes: A systematic search and a narrative review with a special reference to India and other developing countries. Diabetes Metab. Syndr. 2020, 14, 241–246. [Google Scholar] [CrossRef]
- Demine, S.; Renard, P.; Arnould, T. Mitochondrial Uncoupling: A Key Controller of Biological Processes in Physiology and Diseases. Cells 2019, 8, 795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsujimoto, Y. Apoptosis and necrosis: Intracellular ATP level as a determinant for cell death modes. Cell Death Differ. 1997, 4, 429–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kvansakul, M. Viral Infection and Apoptosis. Viruses 2017, 9, 356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinon, F. Signaling by ROS drives inflammasome activation. Eur. J. Immunol. 2010, 40, 616–619. [Google Scholar] [CrossRef] [PubMed]
- Collin, F. Chemical Basis of Reactive Oxygen Species Reactivity and Involvement in Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 2407. [Google Scholar] [CrossRef] [Green Version]
- Stone, W.L.; Basit, H.; Mohiuddin, S.S. Biochemistry, Antioxidants. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2019. [Google Scholar]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Onukwufor, J.O.; Berry, B.J.; Wojtovich, A.P. Physiologic Implications of Reactive Oxygen Species Production by Mitochondrial Complex I Reverse Electron Transport. Antioxidants 2019, 8, 285. [Google Scholar] [CrossRef] [Green Version]
- Hamanaka, R.B.; Chandel, N.S. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem. Sci. 2010, 35, 505–513. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal. Transduct. Target. Ther. 2017, 2. [Google Scholar] [CrossRef] [Green Version]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Kwon, D.; Choi, C.; Oh, J.W.; Benveniste, E.N. Chloroquine induces activation of nuclear factor-kappaB and subsequent expression of pro-inflammatory cytokines by human astroglial cells. J. Neurochem. 2003, 84, 1266–1274. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Qiang, L.; Sample, A.; Shah, P.; He, Y.Y. NF-κB Signaling Activation Induced by Chloroquine Requires Autophagosome, p62 Protein, and c-Jun N-terminal Kinase (JNK) Signaling and Promotes Tumor Cell Resistance. J. Biol. Chem. 2017, 292, 3379–3388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, M.P.; Smith, R.A. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 629–656. [Google Scholar] [CrossRef] [PubMed]
- Anderson, E.J.; Katunga, L.A.; Willis, M.S. Mitochondria as a source and target of lipid peroxidation products in healthy and diseased heart. Clin. Exp. Pharmacol. Physiol. 2012, 39, 179–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkerson, R.G.; Adler, J.D.; Shah, N.G.; Brown, R. Silent hypoxia: A harbinger of clinical deterioration in patients with COVID-19. Am. J. Emerg. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxidative Med. Cell Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef]
- Handy, D.E.; Lubos, E.; Yang, Y.; Galbraith, J.D.; Kelly, N.; Zhang, Y.Y.; Leopold, J.A.; Loscalzo, J. Glutathione peroxidase-1 regulates mitochondrial function to modulate redox-dependent cellular responses. J. Biol. Chem. 2009, 284, 11913–11921. [Google Scholar] [CrossRef] [Green Version]
- Deepalakshmi, P.D.; Parasakthy, K.; Shanthi, S.; Devaraj, N.S. Effect of chloroquine on rat liver mitochondria. Indian J. Exp. Biol. 1994, 32, 797–799. [Google Scholar]
- Katewa, S.D.; Katyare, S.S. Treatment with antimalarials adversely affects the oxidative energy metabolism in rat liver mitochondria. Drug Chem. Toxicol. 2004, 27, 41–53. [Google Scholar] [CrossRef]
- Berry, B.J.; Trewin, A.J.; Amitrano, A.M.; Kim, M.; Wojtovich, A.P. Use the Protonmotive Force: Mitochondrial Uncoupling and Reactive Oxygen Species. J. Mol. Biol. 2018, 430, 3873–3891. [Google Scholar] [CrossRef]
- Nainu, F.; Shiratsuchi, A.; Nakanishi, Y. Induction of Apoptosis and Subsequent Phagocytosis of Virus-Infected Cells As an Antiviral Mechanism. Front. Immunol. 2017, 8, 1220. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.L.; Debets-Ossenkopp, Y.; Verbrugh, H.A.; Verhoef, J. Initiation of the respiratory burst of human neutrophils by influenza virus. Infect. Immun. 1981, 32, 1200–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coperchini, F.; Chiovato, L.; Croce, L.; Magri, F.; Rotondi, M. The cytokine storm in COVID-19: An overview of the involvement of the chemokine/chemokine-receptor system. Cytokine Growth Factor Rev. 2020, 53, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020. [Google Scholar] [CrossRef]
- Westlin, W.F.; Gimbrone, M.A. Neutrophil-mediated damage to human vascular endothelium. Role of cytokine activation. Am. J. Pathol. 1993, 142, 117–128. [Google Scholar]
- Szmitko, P.E.; Wang, C.H.; Weisel, R.D.; de Almeida, J.R.; Anderson, T.J.; Verma, S. New markers of inflammation and endothelial cell activation: Part I. Circulation 2003, 108, 1917–1923. [Google Scholar] [CrossRef]
- Winterbourn, C.C.; Kettle, A.J. Redox reactions and microbial killing in the neutrophil phagosome. Antioxid. Redox Signal. 2013, 18, 642–660. [Google Scholar] [CrossRef]
- Singel, K.L.; Segal, B.H. NOX2-dependent regulation of inflammation. Clin. Sci. 2016, 130, 479–490. [Google Scholar] [CrossRef] [Green Version]
- Zemans, R.L.; Matthay, M.A. What drives neutrophils to the alveoli in ARDS? Thorax 2017, 72, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Xiu, H.; Zhang, S.; Zhang, G. The Role of Macrophages in the Pathogenesis of ALI/ARDS. Med. Inflamm. 2018, 2018, 1264913. [Google Scholar] [CrossRef]
- Minari, J.B.; Oloyede, O.B. Immunosupressive effect of chloroquine through the inhibition of myeloperoxidase. In Proceedings of the 2nd International Conference on Clinical & Cellular Immunology, Hampton Inn Tropicana, Las Vegas, NV, USA, 15–17 October 2013. [Google Scholar]
- Labro, M.T.; Babin-Chevaye, C. Effects of amodiaquine, chloroquine, and mefloquine on human polymorphonuclear neutrophil function in vitro. Antimicrob. Agents Chemother. 1988, 32, 1124–1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Xie, J.; Fiskesund, R.; Dong, W.; Liang, X.; Lv, J.; Jin, X.; Liu, J.; Mo, S.; Zhang, T.; et al. Publisher Correction: Chloroquine modulates antitumor immune response by resetting tumor-associated macrophages toward M1 phenotype. Nat. Commun. 2018, 9, 1808. [Google Scholar] [CrossRef] [PubMed]
- Ley, K. M1 Means Kill; M2 Means Heal. J. Immunol. 2017, 199, 2191–2193. [Google Scholar] [CrossRef] [PubMed]
- Kourtzelis, I.; Hajishengallis, G.; Chavakis, T. Phagocytosis of Apoptotic Cells in Resolution of Inflammation. Front. Immunol. 2020, 11, 553. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Roche, L.; Mesta, F. Oxidative Stress as Key Player in Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV) Infection. Arch. Med. Res. 2020, 51, 384–387. [Google Scholar] [CrossRef]
- Sugioka, Y.; Suzuki, M.; Sugioka, K.; Nakano, M. A ferriprotoporphyrin IX-chloroquine complex promotes membrane phospholipid peroxidation. A possible mechanism for antimalarial action. FEBS Lett. 1987, 223, 251–254. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharyya, B.; Chatterjee, T.K.; Ghosh, J.J. Effects of chloroquine on lysosomal enzymes, NADPH-induced lipid peroxidation, and antioxidant enzymes of rat retina. Biochem. Pharmacol. 1983, 32, 2965–2968. [Google Scholar] [CrossRef]
- Yusuf, I.H.; Sharma, S.; Luqmani, R.; Downes, S.M. Hydroxychloroquine retinopathy. Eye 2017, 31, 828–845. [Google Scholar] [CrossRef]
- Ogunbayo, O.A.; Adisa, R.A.; Ademowo, O.G.; Olorunsogo, O. Incidence of Chloroquine Induced Oxidative Stress in the Blood of Rabbit. Int. J. Pharmacol. 2006, 2, 121–125. [Google Scholar] [CrossRef]
- Giovanella, F.; Ferreira, G.K.; de Prá, S.D.; Carvalho-Silva, M.; Gomes, L.M.; Scaini, G.; Gonçalves, R.C.; Michels, M.; Galant, L.S.; Longaretti, L.M.; et al. Effects of primaquine and chloroquine on oxidative stress parameters in rats. An. Acad. Bras. Cienc. 2015, 87, 1487–1496. [Google Scholar] [CrossRef] [Green Version]
- Fang, L.; Neutzner, A.; Turtschi, S.; Flammer, J.; Mozaffarieh, M. Comet assay as an indirect measure of systemic oxidative stress. J. Vis. Exp. 2015, e52763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, W.L.; Farnsworth, C.C.; Dratz, E.A. A reinvestigation of the fatty acid content of bovine, rat and frog retinal rod outer segments. Exp. Eye Res. 1979, 28, 387–397. [Google Scholar] [CrossRef]
- Song, J.H.; Fujimoto, K.; Miyazawa, T. Polyunsaturated (n-3) Fatty Acids Susceptible to Peroxidation Are Increased in Plasma and Tissue Lipids of Rats Fed Docosahexaenoic Acid–Containing Oils. J. Nutr. 2000, 130, 3028–3033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jančinová, V.; Pažoureková, S.; Lucová, M.; Perečko, T.; Mihalová, D.; Bauerová, K.; Nosáľ, R.; Drábiková, K. Selective inhibition of extracellular oxidants liberated from human neutrophils—A new mechanism potentially involved in the anti-inflammatory activity of hydroxychloroquine. Int. Immunopharmacol. 2015, 28, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Larsen, R.; Gozzelino, R.; Jeney, V.; Tokaji, L.; Bozza, F.A.; Japiassú, A.M.; Bonaparte, D.; Cavalcante, M.M.; Chora, A.; Ferreira, A.; et al. A central role for free heme in the pathogenesis of severe sepsis. Sci. Transl. Med. 2010, 2, 51ra71. [Google Scholar] [CrossRef] [Green Version]
- Chiabrando, D.; Vinchi, F.; Fiorito, V.; Mercurio, S.; Tolosano, E. Heme in pathophysiology: A matter of scavenging, metabolism and trafficking across cell membranes. Front. Pharmacol. 2014, 5, 61. [Google Scholar] [CrossRef] [Green Version]
- de Dios, A.C.; Tycko, R.; Ursos, L.M.B.; Roepe, P.D. NMR Studies of Chloroquine−Ferriprotoporphyrin IX Complex. J. Phys. Chem. A 2003, 107, 5821–5825. [Google Scholar] [CrossRef]
- Atamna, H. Heme, iron, and the mitochondrial decay of ageing. Ageing Res. Rev. 2004, 3, 303–318. [Google Scholar] [CrossRef]
- Belcher, J.D.; Beckman, J.D.; Balla, G.; Balla, J.; Vercellotti, G. Heme degradation and vascular injury. Antioxid. Redox Signal. 2010, 12, 233–248. [Google Scholar] [CrossRef] [Green Version]
- Roumenina, L.T.; Rayes, J.; Lacroix-Desmazes, S.; Dimitrov, J.D. Heme: Modulator of Plasma Systems in Hemolytic Diseases. Trends Mol. Med. 2016, 22, 200–213. [Google Scholar] [CrossRef]
- Sparkenbaugh, E.M.; Chantrathammachart, P.; Wang, S.; Jonas, W.; Kirchhofer, D.; Gailani, D.; Gruber, A.; Kasthuri, R.; Key, N.S.; Mackman, N.; et al. Excess of heme induces tissue factor-dependent activation of coagulation in mice. Haematologica 2015, 100, 308–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connors, J.M.; Levy, J.H. COVID-19 and its implications for thrombosis and anticoagulation. Blood 2020, 135, 2033–2040. [Google Scholar] [CrossRef] [PubMed]
- Kander, T. Coagulation disorder in COVID-19. Lancet Haematol. 2020. [Google Scholar] [CrossRef]
- Arosio, P.; Levi, S. Ferritin, iron homeostasis, and oxidative damage. Free Radic. Biol. Med. 2002, 33, 457–463. [Google Scholar] [CrossRef]
- Hooper, P.L. COVID-19 and heme oxygenase: Novel insight into the disease and potential therapies. Cell Stress Chaperones 2020. [Google Scholar] [CrossRef]
- Kwon, K.J.; Kim, J.N.; Kim, K.M.; Lee, J.; Ignarro, L.J.; Kim, H.-J.; Shin, C.Y.; Han, S.H. Melatonin synergistically increases resveratrol-induced heme oxygenase-1 expression through the inhibition of ubiquitin-dependent proteasome pathway: A possible role in neuroprotection. J. Pineal Res. 2011, 50, 110–123. [Google Scholar] [CrossRef]
- Wessels, I.; Rolles, B.; Rink, L. The Potential Impact of Zinc Supplementation on COVID-19 Pathogenesis. Front. Immunol. 2020, 11, 1712. [Google Scholar] [CrossRef]
- Velthius, A.J.W.T.; van den Worm, S.H.E.; Sims, A.C.; Baric, R.S.; Snijder, E.J.; van Hemert, M.J. Zn2+ Inhibits Coronavirus and Arterivirus RNA Polymerase Activity In Vitro and Zinc Ionophores Block the Replication of These Viruses in Cell Culture. PLoS Pathog. 2010, 6, e1001176. [Google Scholar] [CrossRef]
- Xue, J.; Moyer, A.; Peng, B.; Wu, J.; Hannafon, B.N.; Ding, W.-Q. Chloroquine Is a Zinc Ionophore. PLoS ONE 2014, 9, e109180. [Google Scholar] [CrossRef] [Green Version]
- Wong-ekkabut, J.; Xu, Z.; Triampo, W.; Tang, I.-M.; Tieleman, D.P.; Monticelli, L. Effect of Lipid Peroxidation on the Properties of Lipid Bilayers: A Molecular Dynamics Study. Biophys. J. 2007, 93, 4225–4236. [Google Scholar] [CrossRef] [Green Version]
- Dabbagh-Bazarbachi, H.; Clergeaud, G.; Quesada, I.; Ortiz, M.; O’Sullivan, C.; Fernandez-Larrea, J. Zinc Ionophore Activity of Quercetin and Epigallocatechin-gallate: From Hepa 1-6 Cells to a Liposome Model. J. Agric. Food Chem. 2014, 62, 8085–8093. [Google Scholar] [CrossRef] [PubMed]
- NIH. Available online: https://clinicaltrials.gov/ct2/show/NCT04370782 (accessed on 17 September 2020).
- Yarosz, E.L.; Chang, C.H. The Role of Reactive Oxygen Species in Regulating T Cell-mediated Immunity and Disease. Immune Netw. 2018, 18, e14. [Google Scholar] [CrossRef]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Mösbauer, K.; Hofmann-Winkler, H.; Kaul, A.; Kleine-Weber, H.; Krüger, N.; Gassen, N.C.; Müller, M.A.; Drosten, C.; Pöhlmann, S. Chloroquine does not inhibit infection of human lung cells with SARS-CoV-2. Nature 2020. [Google Scholar] [CrossRef] [PubMed]
- Skipper, C.P.; Pastick, K.A.; Engen, N.W.; Bangdiwala, A.S.; Abassi, M.; Lofgren, S.M.; Williams, D.A.; Okafor, E.C.; Pullen, M.F.; Nicol, M.R.; et al. Hydroxychloroquine in Nonhospitalized Adults with Early COVID-19: A Randomized Trial. Ann. Intern. Med. 2020. [Google Scholar] [CrossRef]
- Boulware, D.R.; Pullen, M.F.; Bangdiwala, A.S.; Pastick, K.A.; Lofgren, S.M.; Okafor, E.C.; Skipper, C.P.; Nascene, A.A.; Nicol, M.R.; Abassi, M.; et al. A Randomized Trial of Hydroxychloroquine as Postexposure Prophylaxis for Covid-19. N. Eng. J. Med. 2020. [Google Scholar] [CrossRef]
- Cavalcanti, A.B.; Zampieri, F.G.; Rosa, R.G.; Azevedo, L.C.P.; Veiga, V.C.; Avezum, A.; Damiani, L.P.; Marcadenti, A.; Kawano-Dourado, L.; Lisboa, T.; et al. Hydroxychloroquine with or without Azithromycin in Mild-to-Moderate Covid-19. N. Eng. J. Med. 2020. [Google Scholar] [CrossRef]
- Stone, W.L.; LeClair, I.; Ponder, T.; Baggs, G.; Reis, B.B. Infants discriminate between natural and synthetic vitamin E. Am. J. Clin. Nutr. 2003, 77, 899–906. [Google Scholar] [CrossRef] [Green Version]
- Marcello, A.; Civra, A.; Milan Bonotto, R.; Nascimento Alves, L.; Rajasekharan, S.; Giacobone, C.; Caccia, C.; Cavalli, R.; Adami, M.; Brambilla, P.; et al. The cholesterol metabolite 27-hydroxycholesterol inhibits SARS-CoV-2 and is markedly decreased in COVID-19 patients. Redox Biol. 2020, 36, 101682. [Google Scholar] [CrossRef]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klouda, C.B.; Stone, W.L. Oxidative Stress, Proton Fluxes, and Chloroquine/Hydroxychloroquine Treatment for COVID-19. Antioxidants 2020, 9, 894. https://doi.org/10.3390/antiox9090894
Klouda CB, Stone WL. Oxidative Stress, Proton Fluxes, and Chloroquine/Hydroxychloroquine Treatment for COVID-19. Antioxidants. 2020; 9(9):894. https://doi.org/10.3390/antiox9090894
Chicago/Turabian StyleKlouda, Christina B., and William L. Stone. 2020. "Oxidative Stress, Proton Fluxes, and Chloroquine/Hydroxychloroquine Treatment for COVID-19" Antioxidants 9, no. 9: 894. https://doi.org/10.3390/antiox9090894
APA StyleKlouda, C. B., & Stone, W. L. (2020). Oxidative Stress, Proton Fluxes, and Chloroquine/Hydroxychloroquine Treatment for COVID-19. Antioxidants, 9(9), 894. https://doi.org/10.3390/antiox9090894