Cardamonin Inhibits Oxazolone-Induced Atopic Dermatitis by the Induction of NRF2 and the Inhibition of Th2 Cytokine Production



Abstract
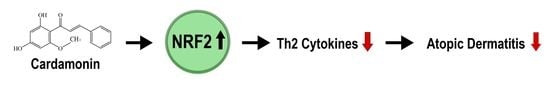
:
1. Introduction
2. Materials and Methods
2.1. Cell Culture, Chemicals, and Reagents
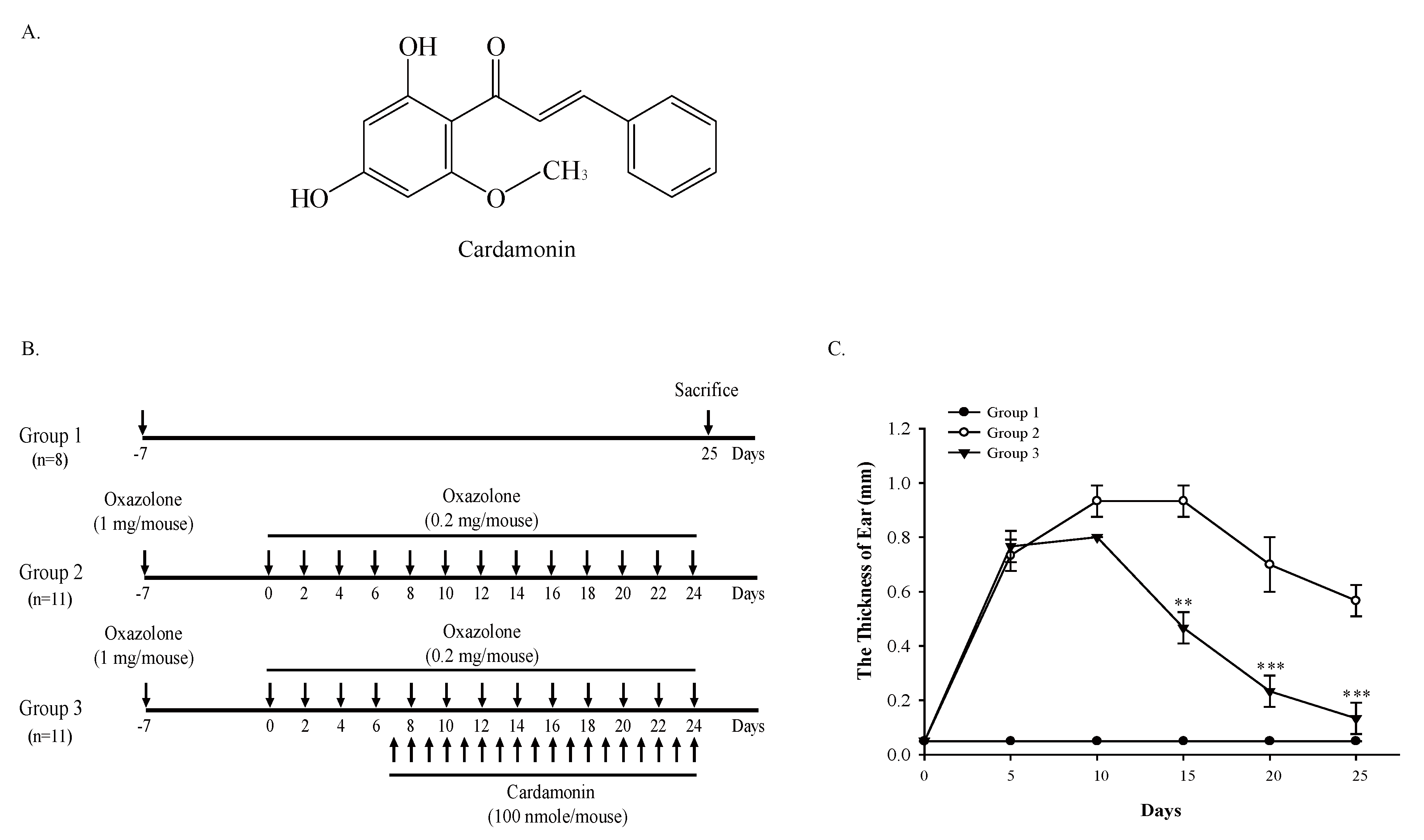
2.2. Examination of the Effect of Cardamonin on Oxazolone-Induced Atopic Dermatitis
2.3. Tissue Dehydration and Paraffin Embedding
2.4. Hematoxylin & Eosin (H&E) Staining
2.5. Preparation of Primary Mouse Embryonic Fibroblasts (MEFs)
2.6. Masson’s Trichrome Staining
2.7. Immunohistochemistry Staining with DAB
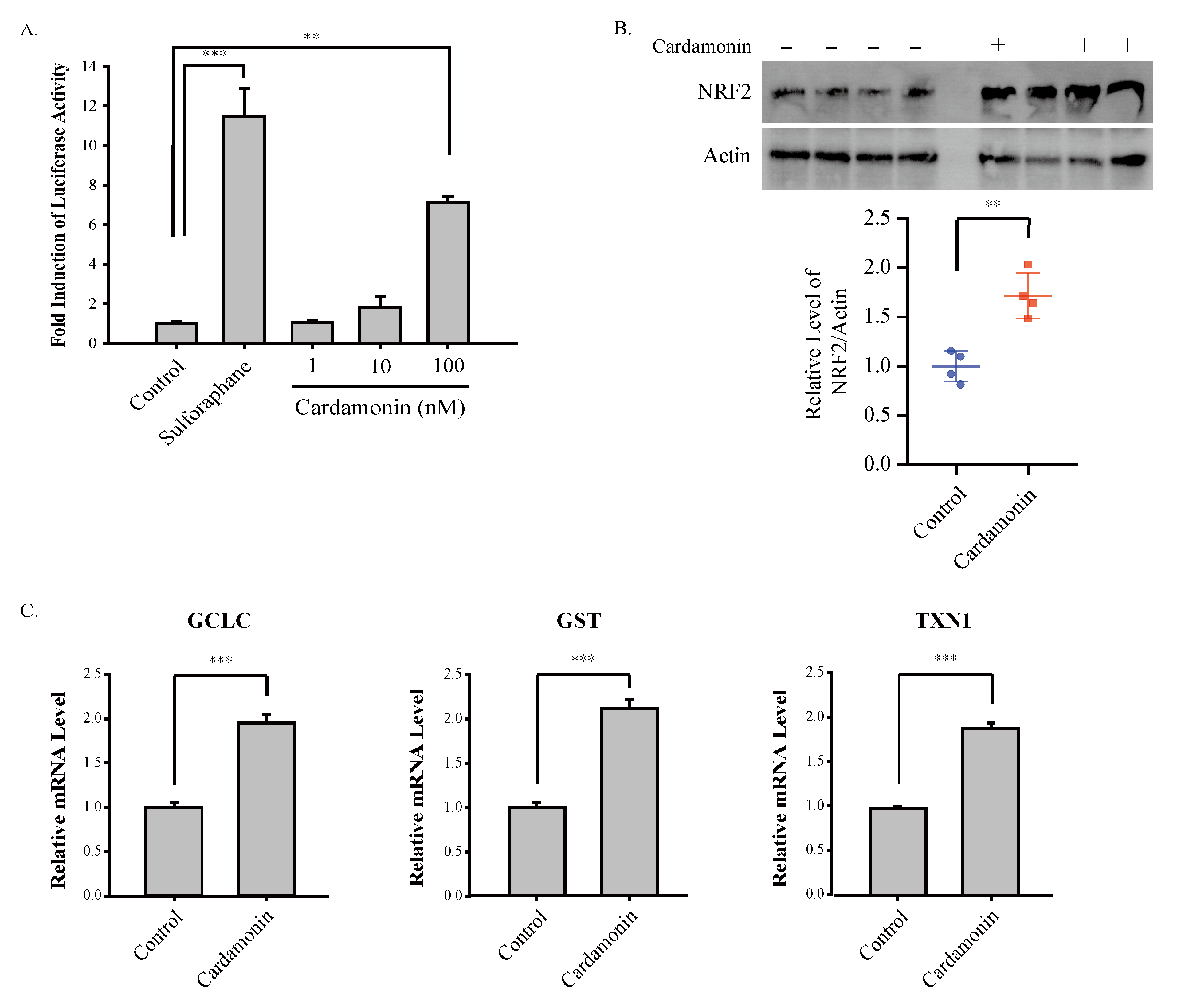
2.8. Measurement of ARE-Luciferase Activity in HaCaT-ARE-Luciferase Cells
2.9. Western Blot Analysis
2.10. Real-Time RT-PCR Assay
2.11. Statistical Analysis
3. Results
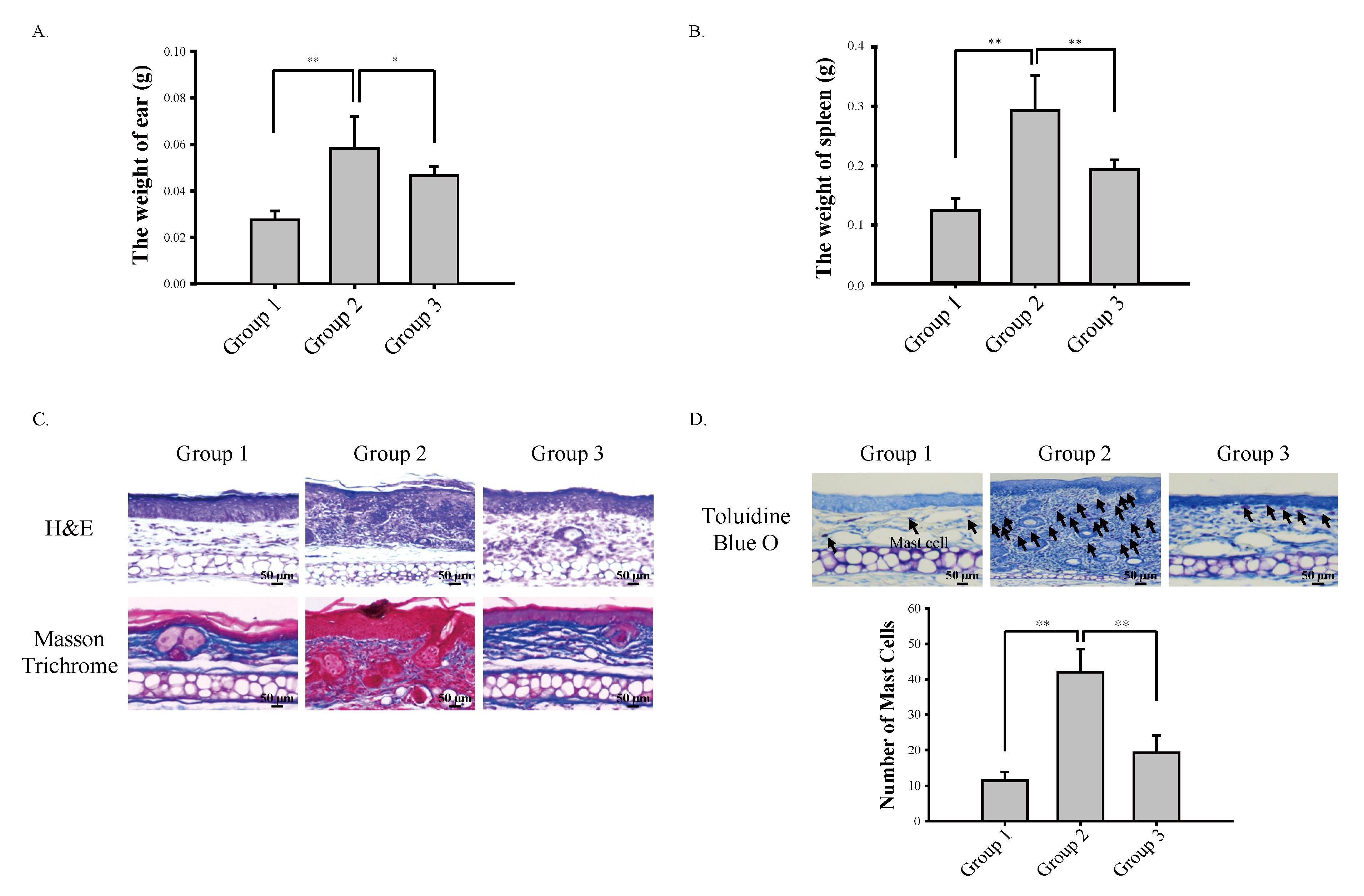
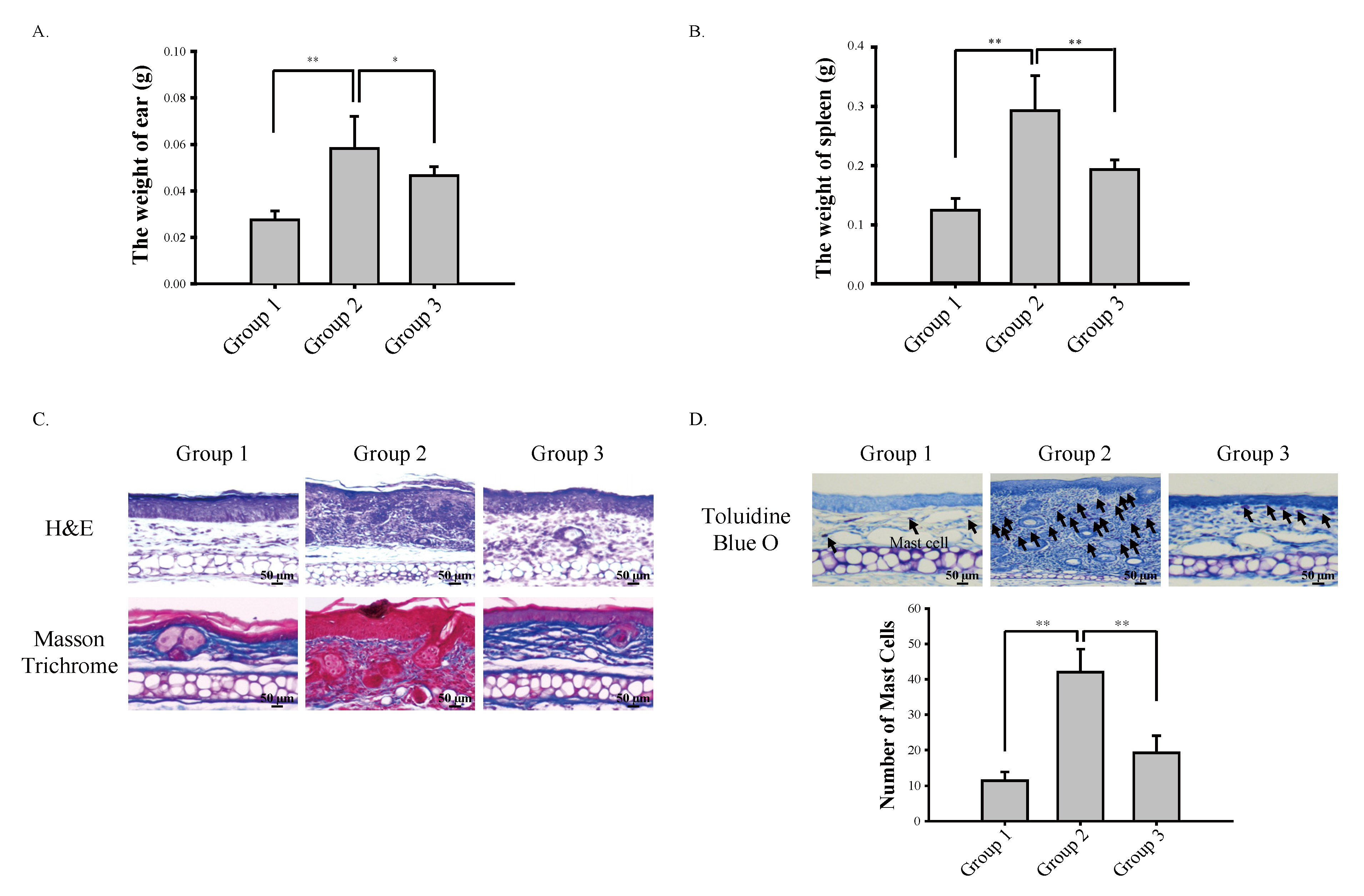
3.1. Cardamonin Suppresses Oxazolone-Induced Atopic Dermatitis In Vivo
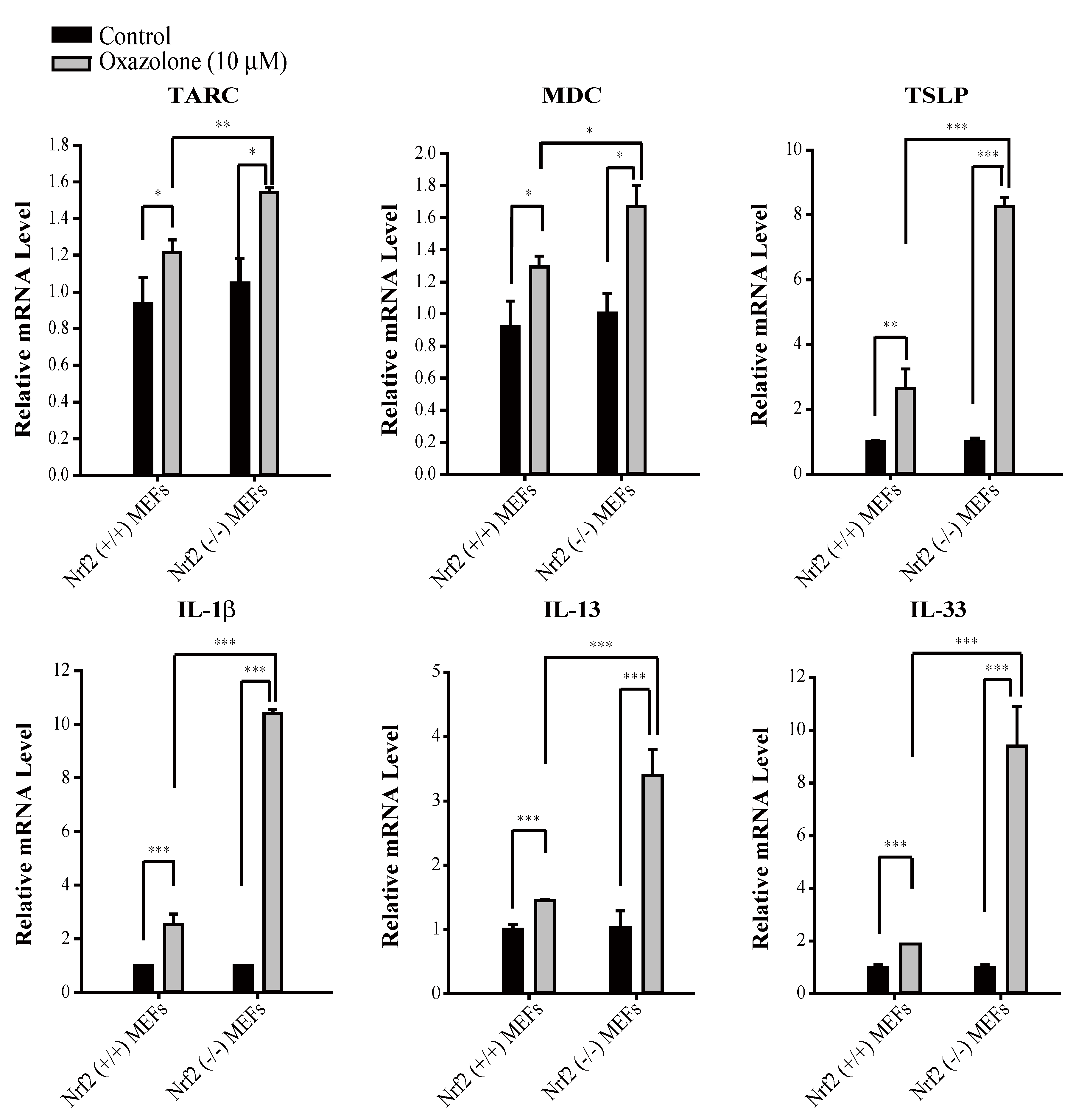
3.2. Cardamonin Suppresses Oxazolone-Induced Production of Th2 Cytokines In Vivo
3.3. NRF2 Is Responsible for Suppressing Oxazolone-Induced Production of Th2 Cytokines
3.4. Cardamonin Induces NRF2 and Attenuates Oxazolone-Induced Oxidative Damages In Vivo
4. Discussion
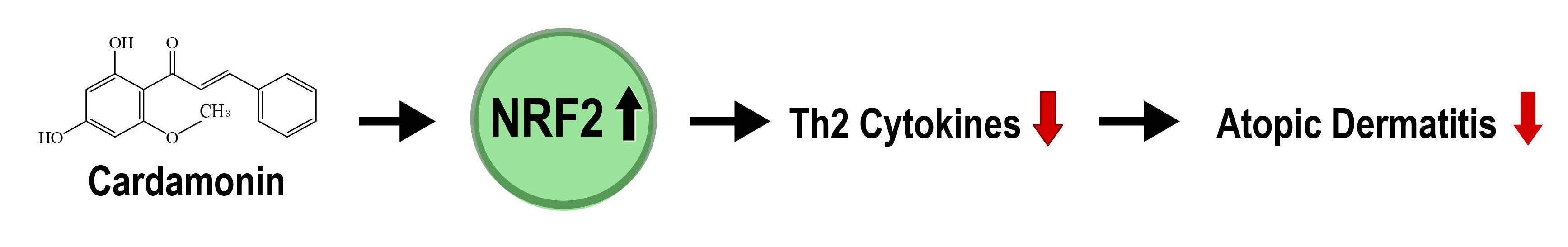
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Yang, E.J.; Sekhon, S.; Sanchez, I.M.; Beck, K.M.; Bhutani, T. Recent Developments in Atopic Dermatitis. Pediatrics 2018, 142. [Google Scholar] [CrossRef] [Green Version]
- Weidinger, S.; Novak, N. Atopic dermatitis. Lancet 2016, 387, 1109–1122. [Google Scholar] [CrossRef]
- Walker, J.A.; McKenzie, A.N.J. TH2 cell development and function. Nat. Rev. Immunol. 2018, 18, 121–133. [Google Scholar] [CrossRef]
- Mack, M.R.; Kim, B.S. The Itch-Scratch Cycle: A Neuroimmune Perspective. Trends Immunol. 2018, 39, 980–991. [Google Scholar] [CrossRef]
- Lin, Y.T.; Wang, C.T.; Chiang, B.L. Role of bacterial pathogens in atopic dermatitis. Clin. Rev. Allergy Immunol. 2007, 33, 167–177. [Google Scholar] [CrossRef]
- Weidinger, S.; Beck, L.A.; Bieber, T.; Kabashima, K.; Irvine, A.D. Atopic dermatitis. Nat. Rev. Dis. Primers 2018, 4, 1. [Google Scholar] [CrossRef]
- Jin, H.; He, R.; Oyoshi, M.; Geha, R.S. Animal models of atopic dermatitis. J. Investig. Dermatol. 2009, 129, 31–40. [Google Scholar] [CrossRef] [Green Version]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef]
- Ji, H.; Li, X.K. Oxidative Stress in Atopic Dermatitis. Oxid. Med. Cell Longev. 2016, 2016, 2721469. [Google Scholar] [CrossRef]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: A Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef] [Green Version]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef] [Green Version]
- Orlikova, B.; Tasdemir, D.; Golais, F.; Dicato, M.; Diederich, M. Dietary chalcones with chemopreventive and chemotherapeutic potential. Genes Nutr. 2011, 6, 125–147. [Google Scholar] [CrossRef] [Green Version]
- Goncalves, L.M.; Valente, I.M.; Rodrigues, J.A. An overview on cardamonin. J. Med. Food 2014, 17, 633–640. [Google Scholar] [CrossRef] [Green Version]
- Nawaz, J.; Rasul, A.; Shah, M.A.; Hussain, G.; Riaz, A.; Sarfraz, I.; Zafar, S.; Adnan, M.; Khan, A.H.; Selamoglu, Z. Cardamonin: A new player to fight cancer via multiple cancer signaling pathways. Life Sci. 2020, 250, 117591. [Google Scholar] [CrossRef]
- Lee, J.; Mailar, K.; Yoo, O.K.; Choi, W.J.; Keum, Y.S. Marliolide inhibits skin carcinogenesis by activating NRF2/ARE to induce heme oxygenase-1. Eur. J. Med. Chem. 2018, 150, 113–126. [Google Scholar] [CrossRef]
- Martin, S.F.; Esser, P.R.; Weber, F.C.; Jakob, T.; Freudenberg, M.A.; Schmidt, M.; Goebeler, M. Mechanisms of chemical-induced innate immunity in allergic contact dermatitis. Allergy 2011, 66, 1152–1163. [Google Scholar] [CrossRef]
- Erkes, D.A.; Selvan, S.R. Hapten-induced contact hypersensitivity, autoimmune reactions, and tumor regression: Plausibility of mediating antitumor immunity. J. Immunol. Res. 2014, 2014, 175265. [Google Scholar] [CrossRef]
- Honda, T.; Egawa, G.; Grabbe, S.; Kabashima, K. Update of immune events in the murine contact hypersensitivity model: Toward the understanding of allergic contact dermatitis. J. Investig. Dermatol. 2013, 133, 303–315. [Google Scholar] [CrossRef] [Green Version]
- Chaplin, D.D. Overview of the immune response. J. Allergy Clin. Immunol. 2010, 125, S3–S23. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int. Immunol. 2009, 21, 317–337. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [Green Version]
- Bonilla, F.A.; Oettgen, H.C. Adaptive immunity. J. Allergy Clin. Immunol. 2010, 125, S33–S40. [Google Scholar] [CrossRef] [PubMed]
- Krangel, M.S. Mechanics of T cell receptor gene rearrangement. Curr. Opin. Immunol. 2009, 21, 133–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taniuchi, I. CD4 Helper and CD8 Cytotoxic T Cell Differentiation. Annu. Rev. Immunol. 2018, 36, 579–601. [Google Scholar] [CrossRef]
- Oliphant, C.J.; Barlow, J.L.; McKenzie, A.N. Insights into the initiation of type 2 immune responses. Immunology 2011, 134, 378–385. [Google Scholar] [CrossRef]
- Biedermann, T.; Skabytska, Y.; Kaesler, S.; Volz, T. Regulation of T Cell Immunity in Atopic Dermatitis by Microbes: The Yin and Yang of Cutaneous Inflammation. Front. Immunol. 2015, 6, 353. [Google Scholar] [CrossRef]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef]
- Nakayama, T.; Hirahara, K.; Onodera, A.; Endo, Y.; Hosokawa, H.; Shinoda, K.; Tumes, D.J.; Okamoto, Y. Th2 Cells in Health and Disease. Annu. Rev. Immunol. 2017, 35, 53–84. [Google Scholar] [CrossRef]
- Lee, J.M.; Chan, K.; Kan, Y.W.; Johnson, J.A. Targeted disruption of Nrf2 causes regenerative immune-mediated hemolytic anemia. Proc. Natl. Acad. Sci. USA 2004, 101, 9751–9756. [Google Scholar] [CrossRef] [Green Version]
- Keum, Y.S.; Choi, B.Y. Molecular and chemical regulation of the Keap1-Nrf2 signaling pathway. Molecules 2014, 19, 10074–10089. [Google Scholar] [CrossRef] [Green Version]
- Nam, L.B.; Keum, Y.S. Binding partners of NRF2: Functions and regulatory mechanisms. Arch. Biochem. Biophys. 2019, 678, 108184. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.S.; Nam, L.B.; Yoo, O.K.; Keum, Y.S. Molecular mechanisms and systemic targeting of NRF2 dysregulation in cancer. Biochem. Pharmacol. 2020, 177, 114002. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Accession Number | Primer Sequence | |
|---|---|---|---|
| TARC | NM_011332 | Forward Reverse | 5′-CTG CTC GAG CCA CCA ATG TA-3′ 5′-TGC CCT GGA CAG TCA GAA AC-3′ |
| MDC | NM_009137 | Forward Reverse | 5′-GCT GTG GCA ATT CAG ACC TC-3′ 5′-TGA CGG ATG TAG TCC TGG CA-3′ |
| IL-1β | NM_008361 | Forward Reverse | 5′-GAA ATG CCA CCT TTT GAC AGT-3′ 5′-GAA GGT CCA CGG GAA AGA CA-3′ |
| IL-13 | NM_008355 | Forward Reverse | 5′-CTG TGT AGC CCT GGA TTC CC-3′ 5′-AGG CCA TGC AAT ATC CTC TGG-3′ |
| IL-33 | NM_001164724 | Forward Reverse | 5′-TGG TCC CGC CTT GCA AAA TA-3′ 5′-GAC GCA GCA AAT GCT TGG AT-3′ |
| TSLP | NM_021367 | Forward Reverse | 5′-ACT GCA ACT TCA CGT CAA TTA CG-3′ 5′-TTG CTC GAA CTT AGC CCC TTT-3′ |
| GCLC | NM_010295 | Forward Reverse | 5′-CTA CCA CGC AGT CAA GGA CC-3′ 5′-CCT TCC GGC GTT TCC TCA TA-3′ |
| GST | NM_013541 | Forward Reverse | 5′-CGG CAA ATA TGT CAC CCT C-3′ 5′-CCT TCC GGC GTT TCC TCA TA-3′ |
| TXN1 | NM_011660 | Forward Reverse | 5′-GCT TGT CGT GGT GGA CTT CT-3′ 5′-AAC TCC CCC ACC TTT TGA CC-3′ |
| GAPDH | NM_001289726 | Forward Reverse | 5′-GGA GAG TGT TTC CTC GTC CC-3′ 5′-ACT GTG CCG TTG AAT TTG CC-3′ |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoo, O.-K.; Choi, W.J.; Keum, Y.-S. Cardamonin Inhibits Oxazolone-Induced Atopic Dermatitis by the Induction of NRF2 and the Inhibition of Th2 Cytokine Production. Antioxidants 2020, 9, 834. https://doi.org/10.3390/antiox9090834
Yoo O-K, Choi WJ, Keum Y-S. Cardamonin Inhibits Oxazolone-Induced Atopic Dermatitis by the Induction of NRF2 and the Inhibition of Th2 Cytokine Production. Antioxidants. 2020; 9(9):834. https://doi.org/10.3390/antiox9090834
Chicago/Turabian StyleYoo, Ok-Kyung, Won Jun Choi, and Young-Sam Keum. 2020. "Cardamonin Inhibits Oxazolone-Induced Atopic Dermatitis by the Induction of NRF2 and the Inhibition of Th2 Cytokine Production" Antioxidants 9, no. 9: 834. https://doi.org/10.3390/antiox9090834
APA StyleYoo, O.-K., Choi, W. J., & Keum, Y.-S. (2020). Cardamonin Inhibits Oxazolone-Induced Atopic Dermatitis by the Induction of NRF2 and the Inhibition of Th2 Cytokine Production. Antioxidants, 9(9), 834. https://doi.org/10.3390/antiox9090834