Bioprospection of Natural Sources of Polyphenols with Therapeutic Potential for Redox-Related Diseases

 ,
, 
 , , ,
, , ,  and
and
Abstract
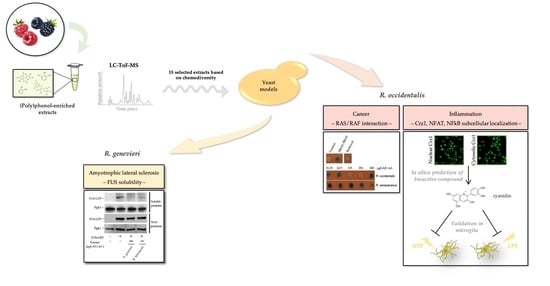
1. Introduction
2. Materials and Methods
2.1. Plant Material and Extraction Procedure
2.2. Total Phenolic Quantification
2.3. Phenolic Profile Determination by LC-ToF-MS
2.4. Component Detection, Peak Alignment, and Integration
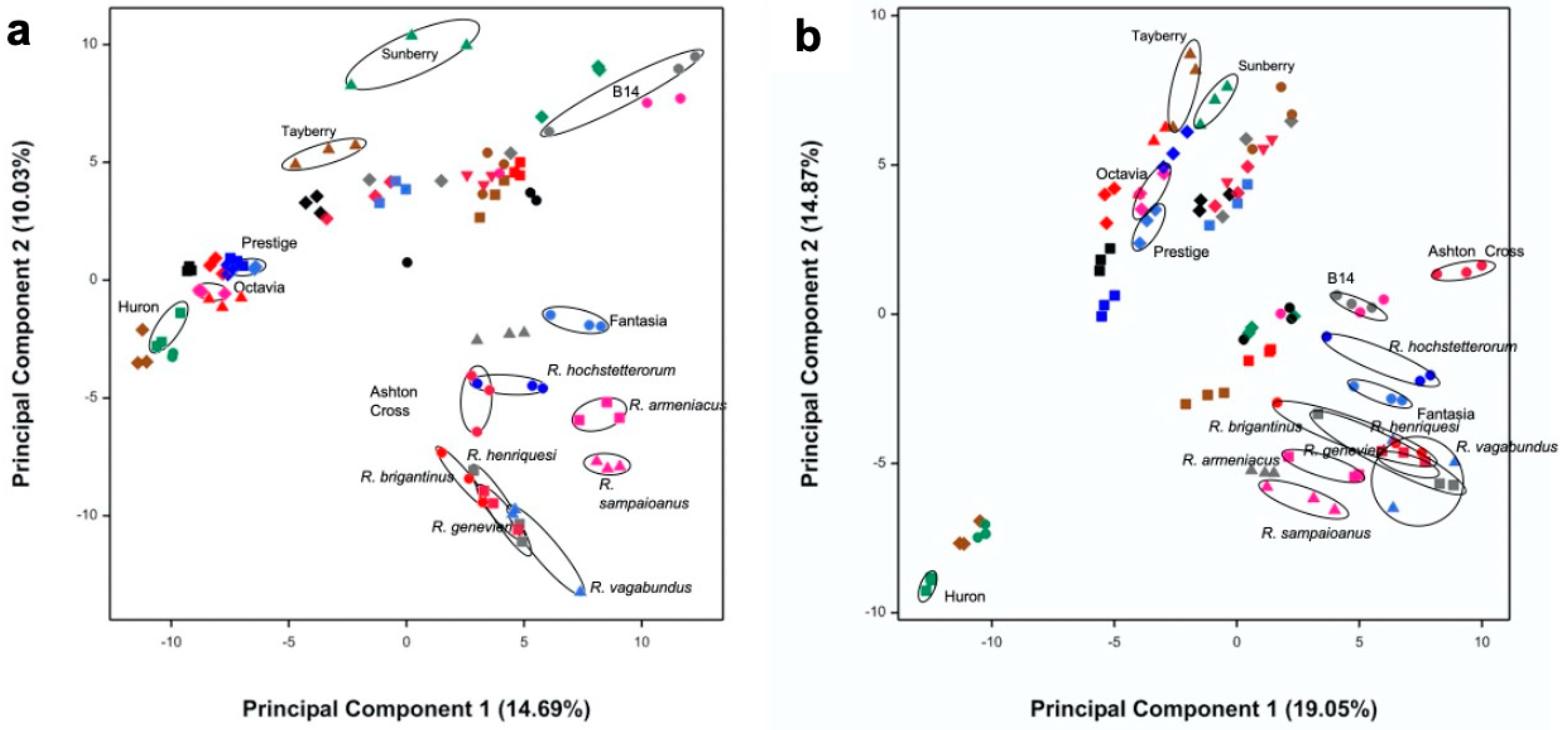
2.5. Multivariate Analysis
2.6. Yeast Strains, Plasmids, and Transformation
2.7. Yeast Growth Conditions
2.8. Growth Assays
2.9. Flow Cytometry
2.10. Fluorescence Microscopy
2.11. Protein Extraction
2.12. Immunoblotting
2.13. β–Galactosidase Assays
2.14. Quantitative Real-Time PCR
2.15. Microglia-Induced Inflammation Model
2.16. Immunofluorescence
2.17. Nitric Oxide (NO) Quantification
2.18. TNF-α Quantification
2.19. Statistical Analysis
3. Results
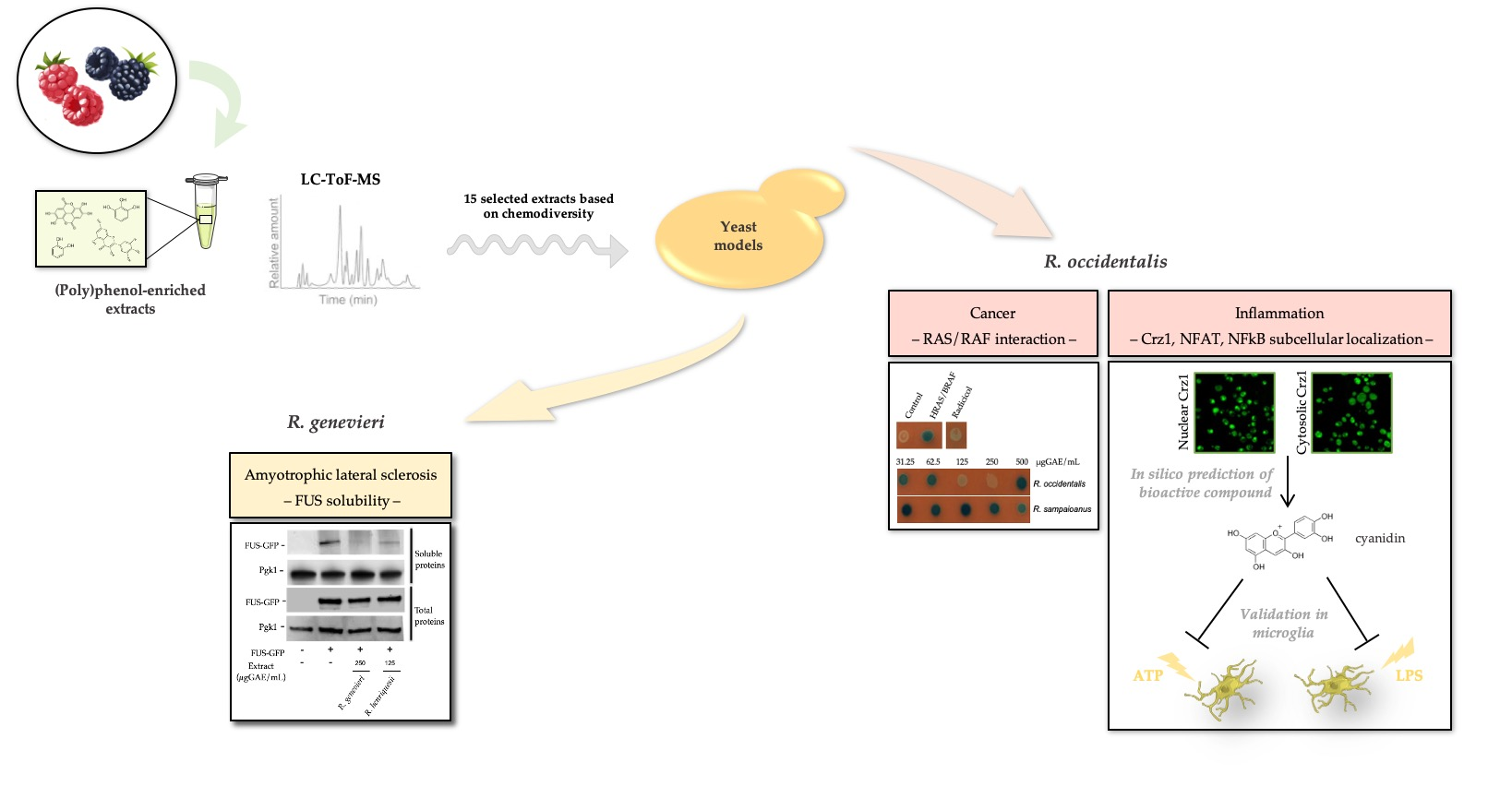
3.1. Selection of Most Chemically Diverse Species
3.2. Identification of Bioactivities Using a SMART Discovery Platform
3.3. Bioactivity Towards the Mitigation of FUS Proteotoxicity
3.4. Bioactives Modulating RAS/RAF Pathological Interactions
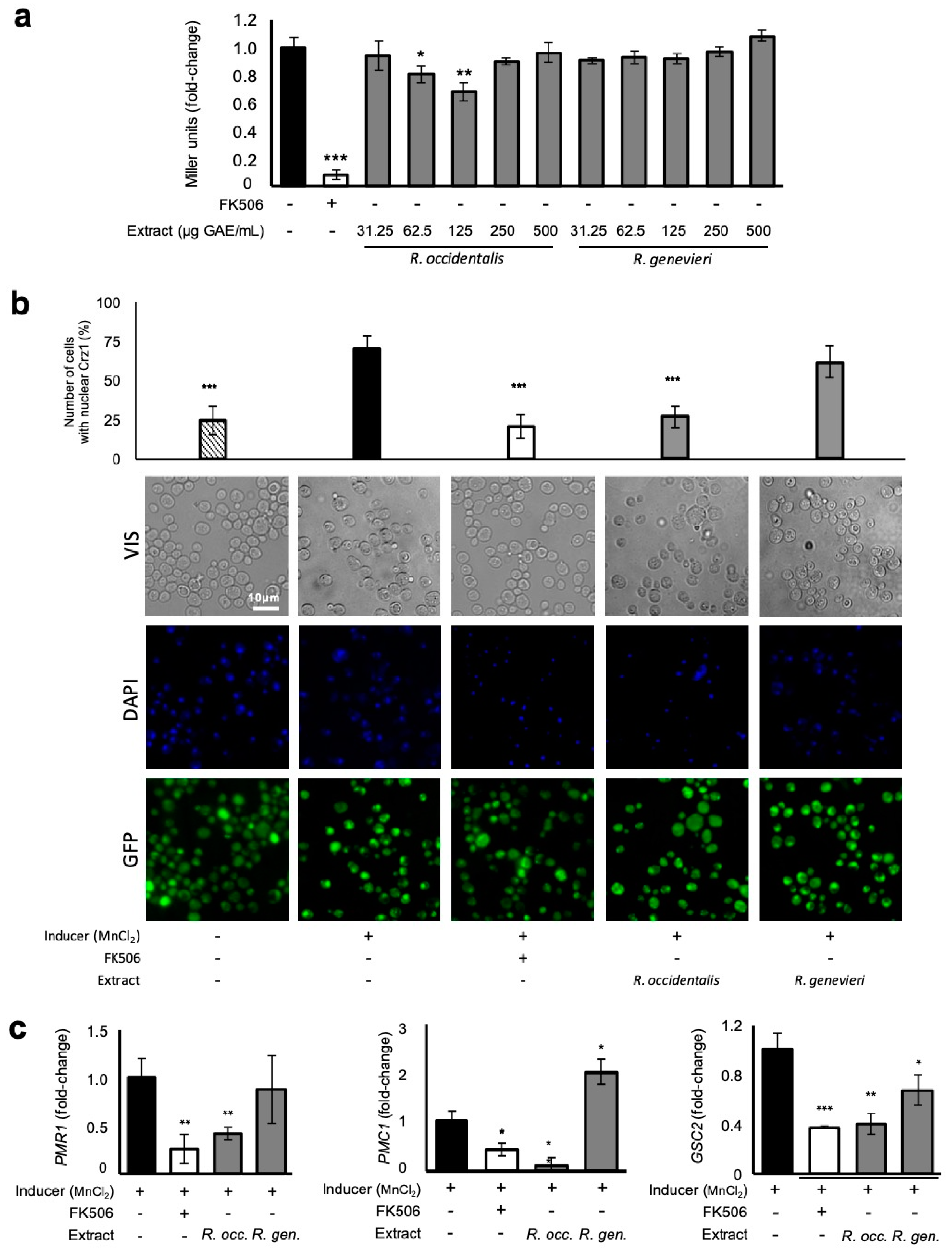
3.5. Attenuation of Crz1 Activation
3.6. Runs Test for the Selection of Potential Components with Bioactivities
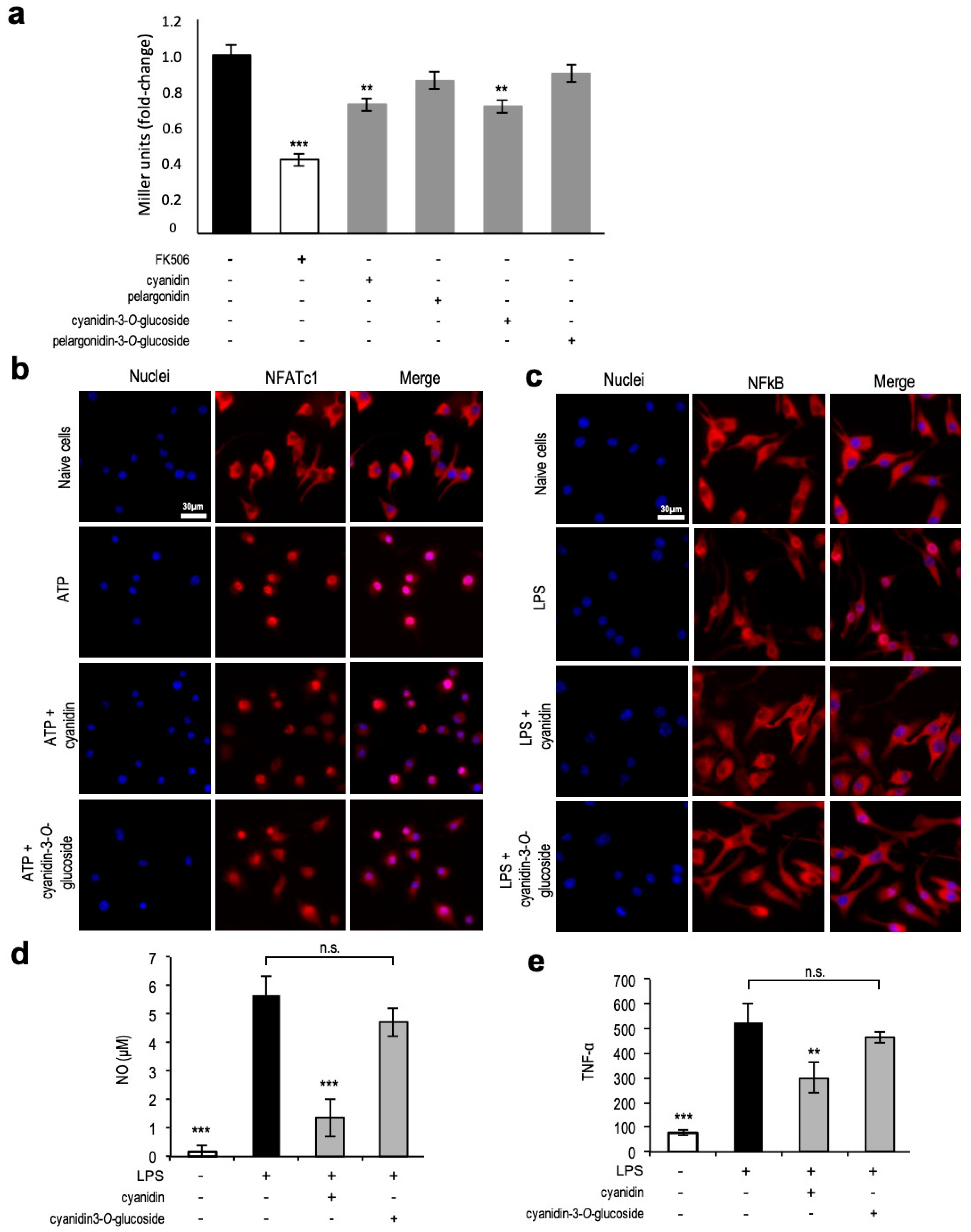
3.7. Unveiling Cyanidin as the Anti-Inflammatory Molecule
4. Discussion
4.1. Cell-Based Assays for Discovery of Health-Promoting Attributes in the Rubus Germplasm
4.2. Exploring Statistical Tools to Expedite Compounds Bioactivity Identification
4.3. Identification of a Single Compound—Cyanidin—with Anti-Inflammatory Properties towards NFAT and NF-kB Transcription Factors
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shen, B. A New Golden Age of Natural Products Drug Discovery. Cell 2015, 163, 1297–1300. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [PubMed]
- Soto, C. Unfolding the role of protein misfolding in neurodegenerative diseases. Nat. Rev. Neurosci. 2003, 4, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Quist, A.; Doudevski, I.; Lin, H.; Azimova, R.; Ng, D.; Frangione, B.; Kagan, B.; Ghiso, J.; Lal, R. Amyloid ion channels: A common structural link for protein-misfolding disease. Proc. Natl. Acad. Sci. USA 2005, 102, 10427–10432. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, R.J.; Wong, P.C. Amyloid precursor protein processing and Alzheimer’s disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef]
- Lee, V.M.; Goedert, M.; Trojanowski, J.Q. Neurodegenerative tauopathies. Annu. Rev. Neurosci. 2001, 24, 1121–1159. [Google Scholar] [CrossRef]
- Nalls, M.A.; Pankratz, N.; Lill, C.M.; Do, C.B.; Hernandez, D.G.; Saad, M.; DeStefano, A.L.; Kara, E.; Bras, J.; Sharma, M.; et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat. Genet. 2014, 46, 989–993. [Google Scholar] [CrossRef]
- Shults, C.W. Lewy bodies. Proc. Natl. Acad. Sci. USA 2006, 103, 1661–1668. [Google Scholar] [CrossRef]
- DiFiglia, M.; Sapp, E.; Chase, K.O.; Davies, S.W.; Bates, G.P.; Vonsattel, J.P.; Aronin, N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 1997, 277, 1990–1993. [Google Scholar] [CrossRef]
- Kwiatkowski, T.J., Jr.; Bosco, D.A.; Leclerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef]
- Vance, C.; Rogelj, B.; Hortobagyi, T.; De Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P.; et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef] [PubMed]
- Dudnik, A.; Almeida, A.F.; Andrade, R.; Avila, B.; Banados, P.; Barbay, D.; Bassard, J.E.; Benkoulouche, M.; Bott, M.; Braga, A.; et al. BacHBerry: BACterial Hosts for production of Bioactive phenolics from bERRY fruits. Phytochem. Rev. 2018, 17, 291–326. [Google Scholar] [CrossRef]
- Kallscheuer, N.; Menezes, R.; Foito, A.; Henriques da Silva, M.D.; Braga, A.; Dekker, W.; Mendez Sevillano, D.; Rosado-Ramos, R.; Jardim, C.; Oliveira, J.; et al. Identification and microbial production of the raspberry phenol salidroside that is active against Huntington’s disease. Plant Physiol. 2018, 179, 969–985. [Google Scholar] [CrossRef] [PubMed]
- Rencus-Lazar, S.; DeRowe, Y.; Adsi, H.; Gazit, E.; Laor, D. Yeast Models for the Study of Amyloid-Associated Disorders and Development of Future Therapy. Front. Mol. Biosci. 2019, 6, 15. [Google Scholar] [CrossRef]
- Bharadwaj, P.; Martins, R.; Macreadie, I. Yeast as a model for studying Alzheimer’s disease. FEMS Yeast Res. 2010, 10, 961–969. [Google Scholar] [CrossRef][Green Version]
- Outeiro, T.F.; Lindquist, S. Yeast cells provide insight into alpha-synuclein biology and pathobiology. Science 2003, 302, 1772–1775. [Google Scholar] [CrossRef]
- Willingham, S.; Outeiro, T.F.; DeVit, M.J.; Lindquist, S.L.; Muchowski, P.J. Yeast genes that enhance the toxicity of a mutant huntingtin fragment or alpha-synuclein. Science 2003, 302, 1769–1772. [Google Scholar] [CrossRef]
- Ju, S.; Tardiff, D.F.; Han, H.; Divya, K.; Zhong, Q.; Maquat, L.E.; Bosco, D.A.; Hayward, L.J.; Brown, R.H., Jr.; Lindquist, S.; et al. A yeast model of FUS/TLS-dependent cytotoxicity. PLoS Biol. 2011, 9, e1001052. [Google Scholar] [CrossRef]
- Khazak, V.; Kato-Stankiewicz, J.; Tamanoi, F.; Golemis, E.A. Yeast screens for inhibitors of Ras-Raf interaction and characterization of MCP inhibitors of Ras-Raf interaction. Methods Enzym. 2006, 407, 612–629. [Google Scholar] [CrossRef]
- Araki, Y.; Wu, H.; Kitagaki, H.; Akao, T.; Takagi, H.; Shimoi, H. Ethanol stress stimulates the Ca2+-mediated calcineurin/Crz1 pathway in Saccharomyces cerevisiae. J. Biosci. Bioeng. 2009, 107, 1–6. [Google Scholar] [CrossRef]
- Menezes, R.; Tenreiro, S.; Macedo, D.; Santos, C.N.; Outeiro, T.F. From the baker to the bedside: Yeast models of Parkinson’s disease. Microb. Cell 2015, 2, 18. [Google Scholar] [CrossRef] [PubMed]
- Tenreiro, S.; Munder, M.C.; Alberti, S.; Outeiro, T.F. Harnessing the power of yeast to unravel the molecular basis of neurodegeneration. J. Neurochem. 2013, 127, 438–452. [Google Scholar] [CrossRef] [PubMed]
- Tardiff, D.F.; Jui, N.T.; Khurana, V.; Tambe, M.A.; Thompson, M.L.; Chung, C.Y.; Kamadurai, H.B.; Kim, H.T.; Lancaster, A.K.; Caldwell, K.A.; et al. Yeast reveal a “druggable” Rsp5/Nedd4 network that ameliorates alpha-synuclein toxicity in neurons. Science 2013, 342, 979–983. [Google Scholar] [CrossRef] [PubMed]
- Tardiff, D.F.; Lindquist, S. Phenotypic screens for compounds that target the cellular pathologies underlying Parkinson’s disease. Drug Discov. Today Technol. 2013, 10, e121–e128. [Google Scholar] [CrossRef] [PubMed]
- Vincent, B.M.; Tardiff, D.F.; Piotrowski, J.S.; Aron, R.; Lucas, M.C.; Chung, C.Y.; Bacherman, H.; Chen, Y.; Pires, M.; Subramaniam, R.; et al. Inhibiting Stearoyl-CoA Desaturase Ameliorates alpha-Synuclein Cytotoxicity. Cell Rep. 2018, 25, 2742–2754. [Google Scholar] [CrossRef] [PubMed]
- Griffioen, G.; Duhamel, H.; Van Damme, N.; Pellens, K.; Zabrocki, P.; Pannecouque, C.; van Leuven, F.; Winderickx, J.; Wera, S. A yeast-based model of alpha-synucleinopathy identifies compounds with therapeutic potential. Biochim. Biophys. Acta 2006, 1762, 312–318. [Google Scholar] [CrossRef]
- Tavares, L.F.S.; Carrilho, C.; McDougall, G.J.; Stewart, D.; Ferreira, R.B.; Santos, C.N. Antioxidant and antiproliferative properties of strawberry tree tissues. J. Berry Res. 2010, 1, 3–12. [Google Scholar] [CrossRef]
- Gietz, R.D.; Schiestl, R.H. Applications of high efficiency lithium acetate transformation of intact yeast cells using single-stranded nucleic acids as carrier. Yeast 1991, 7, 253–263. [Google Scholar] [CrossRef]
- Stathopoulos, A.M.; Cyert, M.S. Calcineurin acts through the CRZ1/TCN1-encoded transcription factor to regulate gene expression in yeast. Genes Dev. 1997, 11, 3432–3444. [Google Scholar] [CrossRef]
- Tenreiro, S.; Rosado-Ramos, R.; Gerhardt, E.; Favretto, F.; Magalhaes, F.; Popova, B.; Becker, S.; Zweckstetter, M.; Braus, G.H.; Outeiro, T.F. Yeast reveals similar molecular mechanisms underlying alpha- and beta-synuclein toxicity. Hum. Mol. Genet. 2016, 25, 275–290. [Google Scholar] [CrossRef]
- Miller-Fleming, L.; Cheong, H.; Antas, P.; Klionsky, D.J. Detection of Saccharomyces cerevisiae Atg13 by western blot. Autophagy 2014, 10, 514–517. [Google Scholar] [CrossRef] [PubMed]
- Garcia, G.; Santos, C.N.; Menezes, R. High-Throughput Yeast-Based Reporter Assay to Identify Compounds with Anti-inflammatory Potential. Methods Mol. Biol. 2016, 1449, 441–452. [Google Scholar] [CrossRef] [PubMed]
- Miller, J. Experiments in Molecular Genetics; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 1972; pp. 352–355. [Google Scholar]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Figueira, I.; Garcia, G.; Pimpao, R.C.; Terrasso, A.P.; Costa, I.; Almeida, A.F.; Tavares, L.; Pais, T.F.; Pinto, P.; Ventura, M.R.; et al. Polyphenols journey through blood-brain barrier towards neuronal protection. Sci. Rep. 2017, 7, 11456. [Google Scholar] [CrossRef] [PubMed]
- Ii, M.; Sunamoto, M.; Ohnishi, K.; Ichimori, Y. beta-Amyloid protein-dependent nitric oxide production from microglial cells and neurotoxicity. Brain Res. 1996, 720, 93–100. [Google Scholar] [CrossRef]
- Ki, S.W.; Kasahara, K.; Kwon, H.J.; Eishima, J.; Takesako, K.; Cooper, J.A.; Yoshida, M.; Horinouchi, S. Identification of radicicol as an inhibitor of in vivo Ras/Raf interaction with the yeast two-hybrid screening system. J. Antibiot. (Tokyo) 1998, 51, 936–944. [Google Scholar] [CrossRef]
- Soga, S.; Kozawa, T.; Narumi, H.; Akinaga, S.; Irie, K.; Matsumoto, K.; Sharma, S.V.; Nakano, H.; Mizukami, T.; Hara, M. Radicicol leads to selective depletion of Raf kinase and disrupts K-Ras-activated aberrant signaling pathway. J. Biol. Chem. 1998, 273, 822–828. [Google Scholar] [CrossRef]
- Lee, S.; Oh, D.G.; Lee, S.; Kim, G.R.; Lee, J.S.; Son, Y.K.; Bae, C.H.; Yeo, J.; Lee, C.H. Chemotaxonomic Metabolite Profiling of 62 Indigenous Plant Species and Its Correlation with Bioactivities. Molecules 2015, 20, 19719–19734. [Google Scholar] [CrossRef]
- Carija, A.; Navarro, S.; de Groot, N.S.; Ventura, S. Protein aggregation into insoluble deposits protects from oxidative stress. Redox Biol. 2017, 12, 699–711. [Google Scholar] [CrossRef]
- Pang, X.; Liu, M. A combination therapy for KRAS-mutant lung cancer by targeting synthetic lethal partners of mutant KRAS. Chin. J. Cancer 2016, 35, 92. [Google Scholar] [CrossRef]
- Pratilas, C.A.; Xing, F.; Solit, D.B. Targeting oncogenic BRAF in human cancer. Curr. Top. Microbiol. Immunol. 2012, 355, 83–98. [Google Scholar] [CrossRef] [PubMed]
- Santarpia, L.; Lippman, S.M.; El-Naggar, A.K. Targeting the MAPK-RAS-RAF signaling pathway in cancer therapy. Expert Opin. Targets 2012, 16, 103–119. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Kummer, M.P.; Latz, E. Innate immune activation in neurodegenerative disease. Nat. Rev. Immunol. 2014, 14, 463–477. [Google Scholar] [CrossRef] [PubMed]
- Foito, A.; Stewart, D. Metabolomics: A high-throughput screen for biochemical and bioactivity diversity in plants and crops. Curr. Pharm. Des. 2018, 24, 2043–2054. [Google Scholar] [CrossRef]
- Suganthy, N.; Devi, K.P.; Nabavi, S.F.; Braidy, N.; Nabavi, S.M. Bioactive effects of quercetin in the central nervous system: Focusing on the mechanisms of actions. Biomed. Pharm. 2016, 84, 892–908. [Google Scholar] [CrossRef]
- Baral, S.; Pariyar, R.; Kim, J.; Lee, H.S.; Seo, J. Quercetin-3-O-glucuronide promotes the proliferation and migration of neural stem cells. Neurobiol. Aging 2017, 52, 39–52. [Google Scholar] [CrossRef]
- Dey, A.; De, J.N. Neuroprotective therapeutics from botanicals and phytochemicals against Huntington’s disease and related neurodegenerative disorders. J. Herb. Med. 2015, 5, 19. [Google Scholar] [CrossRef]
- Testa, D.; Caraceni, T.; Fetoni, V. Branched-chain amino acids in the treatment of amyotrophic lateral sclerosis. J. Neurol. 1989, 236, 445–447. [Google Scholar] [CrossRef]
- Tandan, R.; Bromberg, M.B.; Forshew, D.; Fries, T.J.; Badger, G.J.; Carpenter, J.; Krusinski, P.B.; Betts, E.F.; Arciero, K.; Nau, K. A controlled trial of amino acid therapy in amyotrophic lateral sclerosis: I. Clinical, functional, and maximum isometric torque data. Neurology 1996, 47, 1220–1226. [Google Scholar] [CrossRef]
- The Italian ALS Study Group. Branched-chain amino acids and amyotrophic lateral sclerosis: A treatment failure. Neurology 1993, 43, 2466–2470. [Google Scholar] [CrossRef]
- Plaitakis, A.; Smith, J.; Mandeli, J.; Yahr, M.D. Pilot trial of branched-chain aminoacids in amyotrophic lateral sclerosis. Lancet 1988, 1, 1015–1018. [Google Scholar] [CrossRef]
- Mori, N.; Adachi, Y.; Takeshima, T.; Kashiwaya, Y.; Okada, A.; Nakashima, K. Branched-chain amino acid therapy for spinocerebellar degeneration: A pilot clinical crossover trial. Intern. Med. 1999, 38, 401–406. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Patin, F.; Corcia, P.; Vourc’h, P.; Nadal-Desbarats, L.; Baranek, T.; Goossens, J.F.; Marouillat, S.; Dessein, A.F.; Descat, A.; Madji Hounoum, B.; et al. Omics to Explore Amyotrophic Lateral Sclerosis Evolution: The Central Role of Arginine and Proline Metabolism. Mol. Neurobiol. 2017, 54, 5361–5374. [Google Scholar] [CrossRef] [PubMed]
- Nunez-Sanchez, M.A.; Gonzalez-Sarrias, A.; Romo-Vaquero, M.; Garcia-Villalba, R.; Selma, M.V.; Tomas-Barberan, F.A.; Garcia-Conesa, M.T.; Espin, J.C. Dietary phenolics against colorectal cancer—From promising preclinical results to poor translation into clinical trials: Pitfalls and future needs. Mol. Nutr. Food Res. 2015, 59, 1274–1291. [Google Scholar] [CrossRef] [PubMed]
- McDougall, G.J.; Allwood, J.W.; Pereira-Caro, G.; Brown, E.M.; Verrall, S.; Stewart, D.; Latimer, C.; McMullan, G.; Lawther, R.; O’Connor, G.; et al. Novel colon-available triterpenoids identified in raspberry fruits exhibit antigenotoxic activities in vitro. Mol. Nutr. Food Res. 2017, 61, 1600327. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.H.; Thulasingam, S.; Nagarajan, S. Terpenoids as anti-colon cancer agents—A comprehensive review on its mechanistic perspectives. Eur. J. Pharm. 2017, 795, 169–178. [Google Scholar] [CrossRef]
- Saha, A.; Kuzuhara, T.; Echigo, N.; Suganuma, M.; Fujiki, H. New role of (-)-epicatechin in enhancing the induction of growth inhibition and apoptosis in human lung cancer cells by curcumin. Cancer Prev. Res. (Phila) 2010, 3, 953–962. [Google Scholar] [CrossRef]
- Suganuma, M.; Okabe, S.; Kai, Y.; Sueoka, N.; Sueoka, E.; Fujiki, H. Synergistic effects of (--)-epigallocatechin gallate with (--)-epicatechin, sulindac, or tamoxifen on cancer-preventive activity in the human lung cancer cell line PC-9. Cancer Res. 1999, 59, 44–47. [Google Scholar]
- Arts, I.C.; Jacobs, D.R., Jr.; Gross, M.; Harnack, L.J.; Folsom, A.R. Dietary catechins and cancer incidence among postmenopausal women: The Iowa Women’s Health Study (United States). Cancer Causes Control 2002, 13, 373–382. [Google Scholar] [CrossRef]
- Theodoratou, E.; Kyle, J.; Cetnarskyj, R.; Farrington, S.M.; Tenesa, A.; Barnetson, R.; Porteous, M.; Dunlop, M.; Campbell, H. Dietary flavonoids and the risk of colorectal cancer. Cancer Epidemiol. Biomark. Prev. 2007, 16, 684–693. [Google Scholar] [CrossRef]
- Piszcz, J.; Lemancewicz, D.; Dudzik, D.; Ciborowski, M. Differences and similarities between LC-MS derived serum fingerprints of patients with B-cell malignancies. Electrophoresis 2013, 34, 2857–2864. [Google Scholar] [CrossRef] [PubMed]
- Struck-Lewicka, W.; Kordalewska, M.; Bujak, R.; Yumba Mpanga, A.; Markuszewski, M.; Jacyna, J.; Matuszewski, M.; Kaliszan, R.; Markuszewski, M.J. Urine metabolic fingerprinting using LC-MS and GC-MS reveals metabolite changes in prostate cancer: A pilot study. J. Pharm. Biomed. Anal. 2015, 111, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Morais, C.A.; De Rosso, V.V.; Estadella, D.; Pisani, L.P. Anthocyanins as inflammatory modulators and the role of the gut microbiota. J. Nutr. Biochem. 2016, 33, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, A.; Rogers, G.; Peterson, J.J.; Dwyer, J.T.; Lin, H.; Jacques, P.F. Higher dietary anthocyanin and flavonol intakes are associated with anti-inflammatory effects in a population of US adults. Am. J. Clin. Nutr. 2015, 102, 172–181. [Google Scholar] [CrossRef]
- Jung, H.; Lee, H.J.; Cho, H.; Lee, K.; Kwak, H.K.; Hwang, K.T. Anthocyanins in Rubus fruits and antioxidant and anti-inflammatory activities in RAW 264.7 cells. Food Sci. Biotechnol. 2015, 24, 1879–1886. [Google Scholar] [CrossRef]
- Fratantonio, D.; Speciale, A.; Ferrari, D.; Cristani, M.; Saija, A.; Cimino, F. Palmitate-induced endothelial dysfunction is attenuated by cyanidin-3-O-glucoside through modulation of Nrf2/Bach1 and NF-kappaB pathways. Toxicol. Lett. 2015, 239, 152–160. [Google Scholar] [CrossRef]
- Ferrari, D.; Speciale, A.; Cristani, M.; Fratantonio, D.; Molonia, M.S.; Ranaldi, G.; Saija, A.; Cimino, F. Cyanidin-3-O-glucoside inhibits NF-kB signalling in intestinal epithelial cells exposed to TNF-alpha and exerts protective effects via Nrf2 pathway activation. Toxicol. Lett. 2016, 264, 51–58. [Google Scholar] [CrossRef]
- Ajit, D.; Simonyi, A.; Li, R.; Chen, Z.; Hannink, M.; Fritsche, K.L.; Mossine, V.V.; Smith, R.E.; Dobbs, T.K.; Luo, R.; et al. Phytochemicals and botanical extracts regulate NF-kappaB and Nrf2/ARE reporter activities in DI TNC1 astrocytes. Neurochem. Int. 2016, 97, 49–56. [Google Scholar] [CrossRef]
- Serra, D.; Paixao, J.; Nunes, C.; Dinis, T.C.; Almeida, L.M. Cyanidin-3-glucoside suppresses cytokine-induced inflammatory response in human intestinal cells: Comparison with 5-aminosalicylic acid. PLoS ONE 2013, 8, e73001. [Google Scholar] [CrossRef]
- Cimino, F.; Ambra, R.; Canali, R.; Saija, A.; Virgili, F. Effect of cyanidin-3-O-glucoside on UVB-induced response in human keratinocytes. J. Agric. Food Chem. 2006, 54, 4041–4047. [Google Scholar] [CrossRef]
- Park, K.H.; Gu, D.R.; So, H.S.; Kim, K.J.; Lee, S.H. Dual Role of Cyanidin-3-glucoside on the Differentiation of Bone Cells. J. Dent. Res. 2015, 94, 1676–1683. [Google Scholar] [CrossRef] [PubMed]
- Duarte, L.J.; Chaves, V.C.; Nascimento, M.; Calvete, E.; Li, M.; Ciraolo, E.; Ghigo, A.; Hirsch, E.; Simoes, C.M.O.; Reginatto, F.H.; et al. Molecular mechanism of action of Pelargonidin-3-O-glucoside, the main anthocyanin responsible for the anti-inflammatory effect of strawberry fruits. Food Chem. 2018, 247, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Manach, C.; Williamson, G.; Morand, C.; Scalbert, A.; Remesy, C. Bioavailability and bioefficacy of polyphenols in humans. I. Review of 97 bioavailability studies. Am. J. Clin. Nutr. 2005, 81, 230S–242S. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rubus Samples | Protective Factor in Each Disease Model | |||||
|---|---|---|---|---|---|---|
| αSyn Toxicity a | Aβ42 Toxicity a | HTT Toxicity a | FUS Toxicity a | KRAS/RAF Interaction b,c | Crz1 Activation b,d | |
| R. vagabundus | 0 | 0 | 0 | 0 | 0 | 0 |
| R. brigantinus | 0 | 0 | 10.5 ± 10.1 | 0 | 0 | 35.2 ± 13.1 |
| R. sampaioanus | 0 | 0 | 0 | 0 | 0 | 15.7 ± 9.2 |
| R. genevieri | 0 | 0 | 0 | 39.0 ± 13.9 | 0 | 0 |
| R. hochstetterorum | 0 | 0 | 17.0 ± 10.7 | 0 | 0 | 12.2 ± 8.3 |
| R. henriquesii | 0 | 0 | 16.0 ± 10.1 | 0 | 0 | 10.3 ± 2.9 |
| R. loganobaccus var. Tayberry | 0 | 0 | 0 | 0 | 52.5 ± 1.2 | 0 |
| R. loganobaccus var. Sunberry | 0 | 90.3 ± 6.2 | 87.6 ± 30.7 | 0 | 0 | 6.4 ± 2.6 |
| R. fruticosus var. Fantasia | 0 | 0 | 0 | 0 | 0 | 0 |
| R. fruticosus var. Ashton cross | 0 | 0 | 0 | 0 | 0 | 41.2 ± 5.3 |
| R. idaeus var. Prestige | 0 | 0 | 98.7 ± 28.8 | 49.4 ± 24.0 | 15.4 ± 2.0 | 19.5 ± 0.9 |
| R. idaeus var. Octavia | 12.3 ± 6.6 | 0 | 0 | 22.4 ± 30.0 | 0 | 60.8 ± 8.5 |
| R. occidentalis var. Huron | 0 | 0 | 0 | 0 | 57.7 ± 1.2 | 57 ± 4.9 |
| R. armeniacus var. Himalayan giant | 0 | 0 | 0 | 0 | 27.1 ± 6.2 | 46.4 ± 3.4 |
| Rubus sp. B14 | 0 | 0 | 0 | 0 | 35.9 ± 6.0 | 0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Menezes, R.; Foito, A.; Jardim, C.; Costa, I.; Garcia, G.; Rosado-Ramos, R.; Freitag, S.; Alexander, C.J.; Outeiro, T.F.; Stewart, D.; et al. Bioprospection of Natural Sources of Polyphenols with Therapeutic Potential for Redox-Related Diseases. Antioxidants 2020, 9, 789. https://doi.org/10.3390/antiox9090789
Menezes R, Foito A, Jardim C, Costa I, Garcia G, Rosado-Ramos R, Freitag S, Alexander CJ, Outeiro TF, Stewart D, et al. Bioprospection of Natural Sources of Polyphenols with Therapeutic Potential for Redox-Related Diseases. Antioxidants. 2020; 9(9):789. https://doi.org/10.3390/antiox9090789
Chicago/Turabian StyleMenezes, Regina, Alexandre Foito, Carolina Jardim, Inês Costa, Gonçalo Garcia, Rita Rosado-Ramos, Sabine Freitag, Colin James Alexander, Tiago Fleming Outeiro, Derek Stewart, and et al. 2020. "Bioprospection of Natural Sources of Polyphenols with Therapeutic Potential for Redox-Related Diseases" Antioxidants 9, no. 9: 789. https://doi.org/10.3390/antiox9090789
APA StyleMenezes, R., Foito, A., Jardim, C., Costa, I., Garcia, G., Rosado-Ramos, R., Freitag, S., Alexander, C. J., Outeiro, T. F., Stewart, D., & Santos, C. N. (2020). Bioprospection of Natural Sources of Polyphenols with Therapeutic Potential for Redox-Related Diseases. Antioxidants, 9(9), 789. https://doi.org/10.3390/antiox9090789