BVR-A Deficiency Leads to Autophagy Impairment through the Dysregulation of AMPK/mTOR Axis in the Brain—Implications for Neurodegeneration


 ,
,  ,
,  and
and 
Abstract
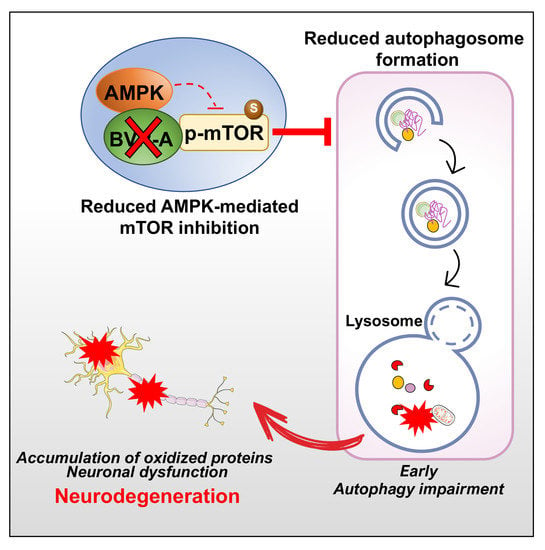
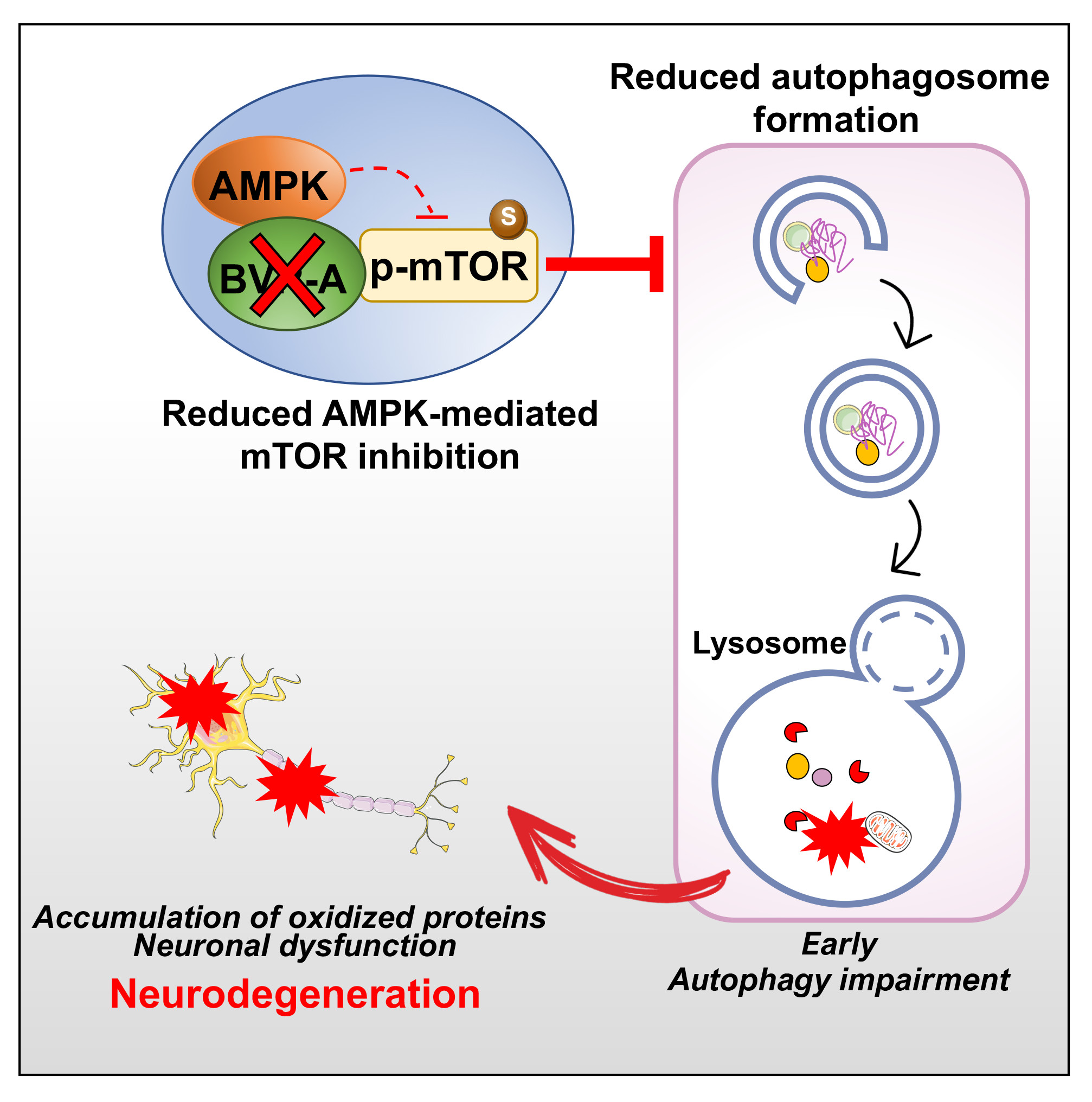
1. Introduction
2. Material and Methods
2.1. Animals
2.2. Samples Preparation
2.3. Slot Blot Analysis
2.4. Western Blot Analysis
2.5. Statistical Analysis
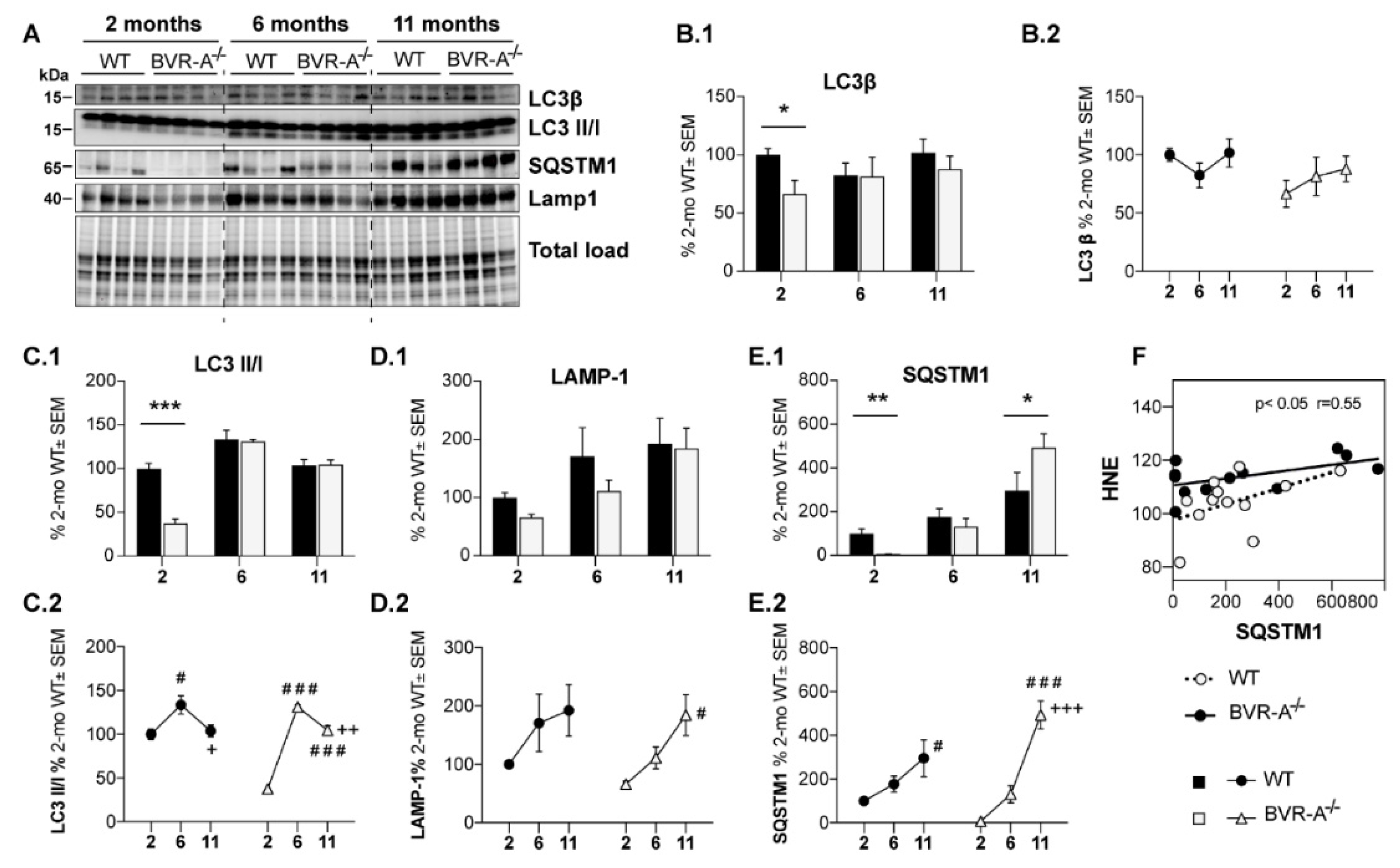
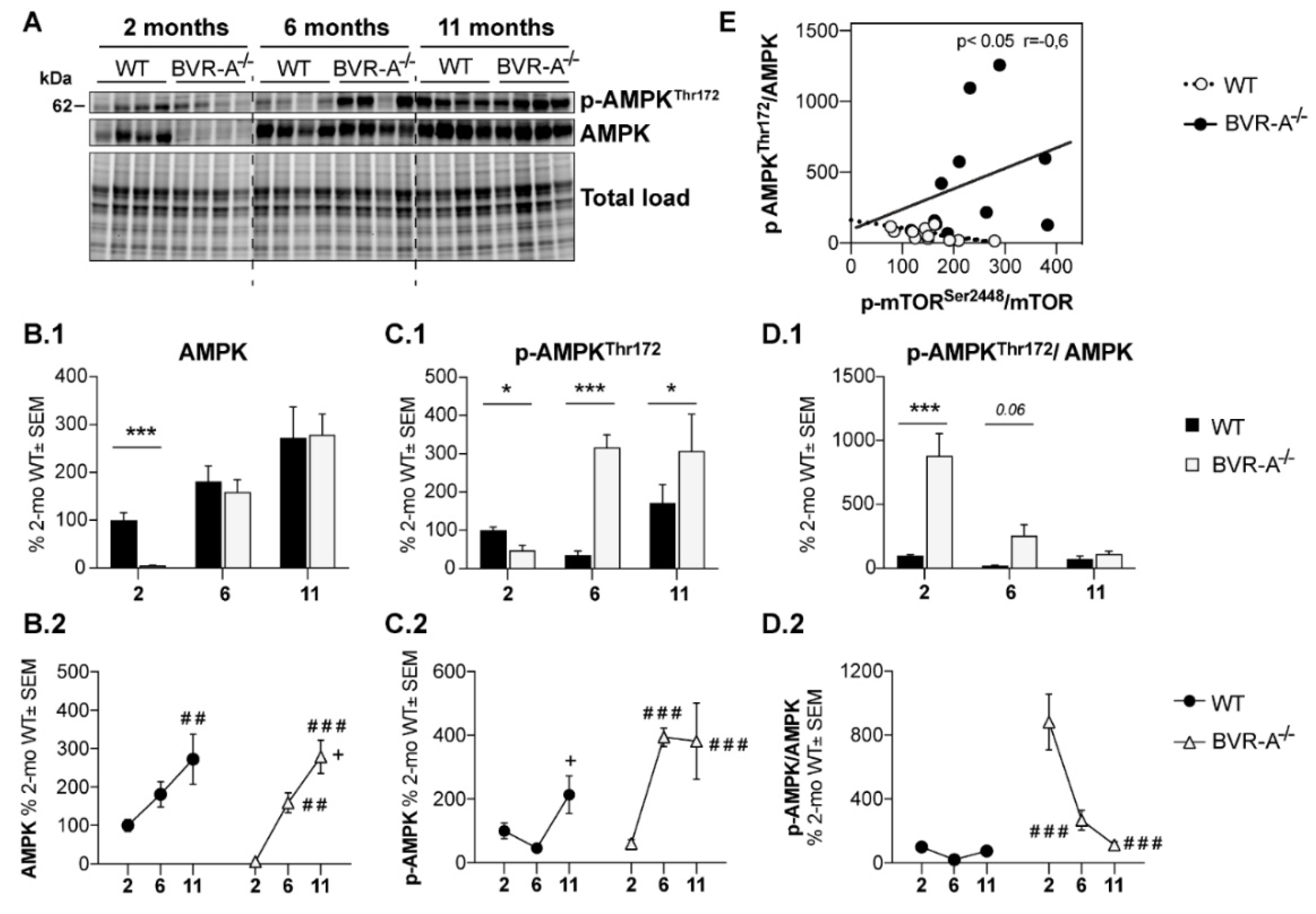
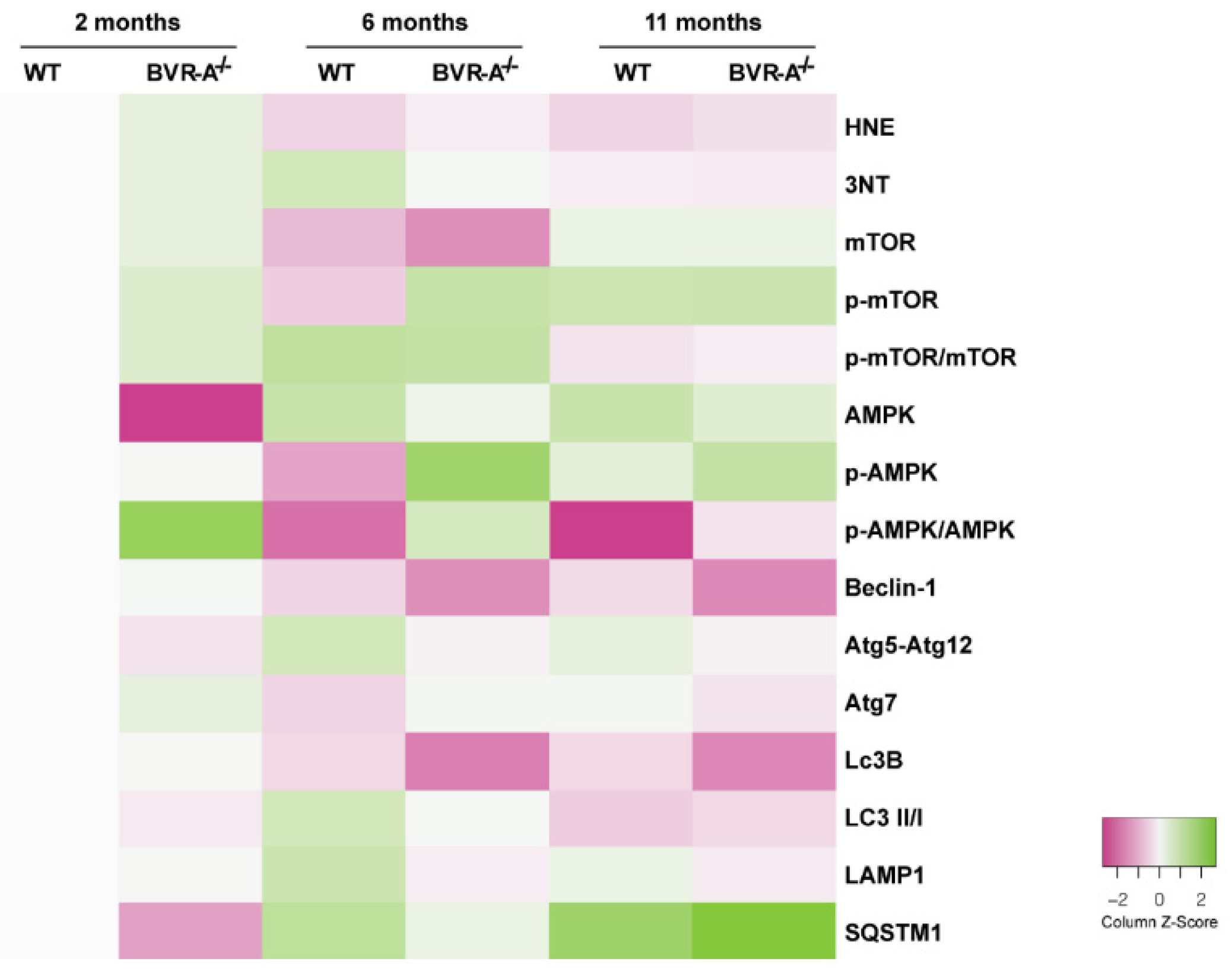
3. Results
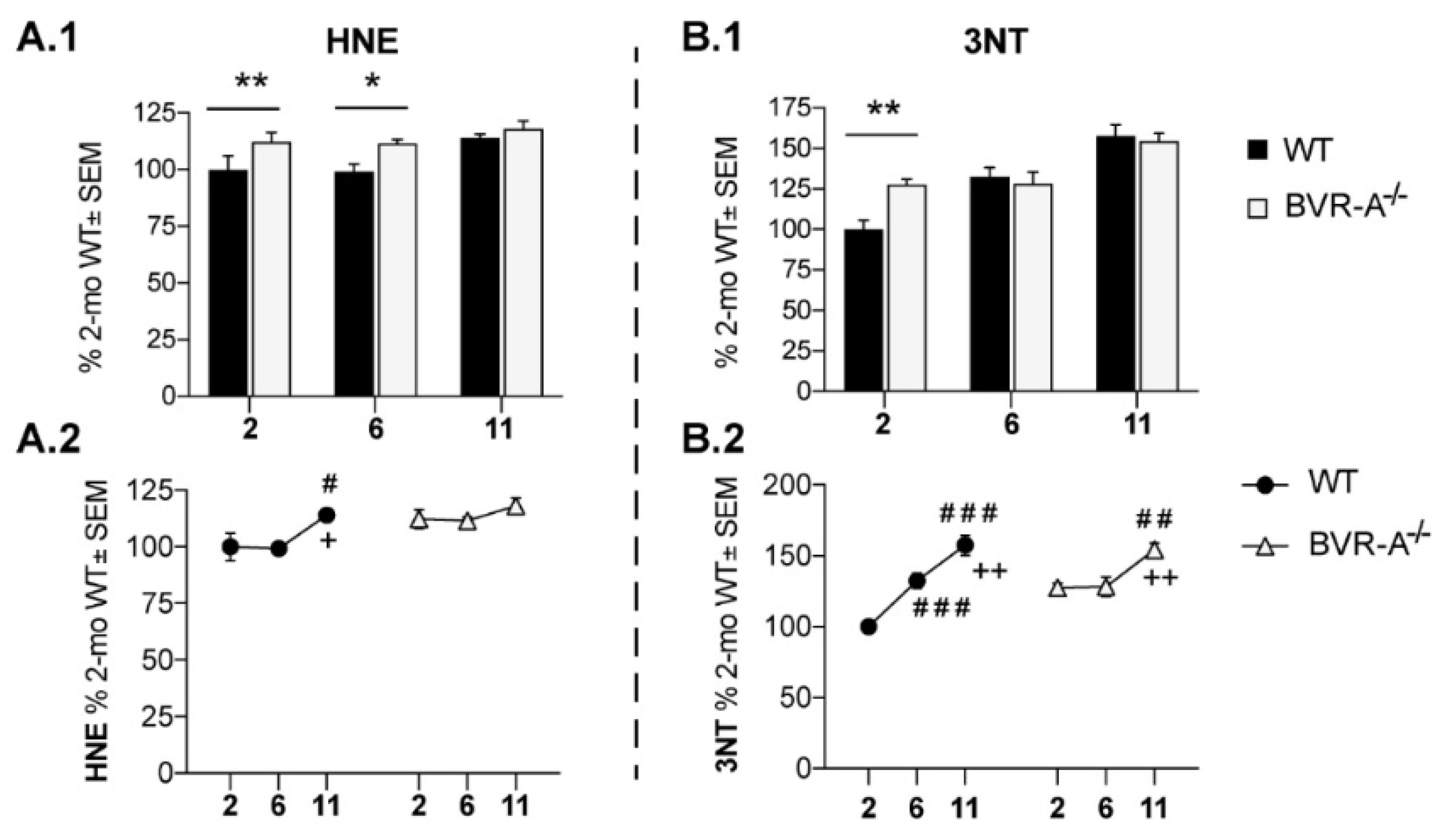
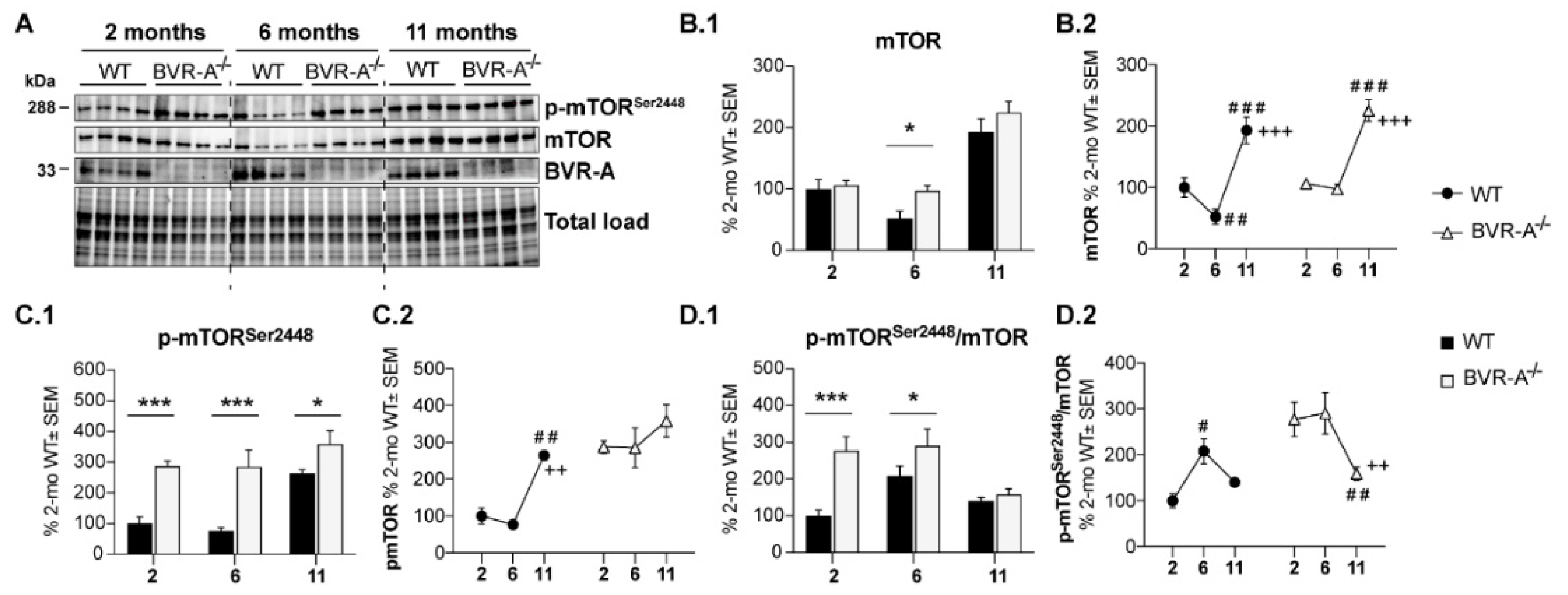
3.1. Oxidatively-Damaged Proteins Accumulation along with mTOR Hyper-Activation Occur in BVR-A−/− Mice
3.2. mTOR Hyper-Active Leads to Impaired Autophagy in BVR-A−/− Mice
3.3. Reduced AMPK Protein Levels and Activation Lead to mTOR Hyper-Activation in BVR-A−/− Mice
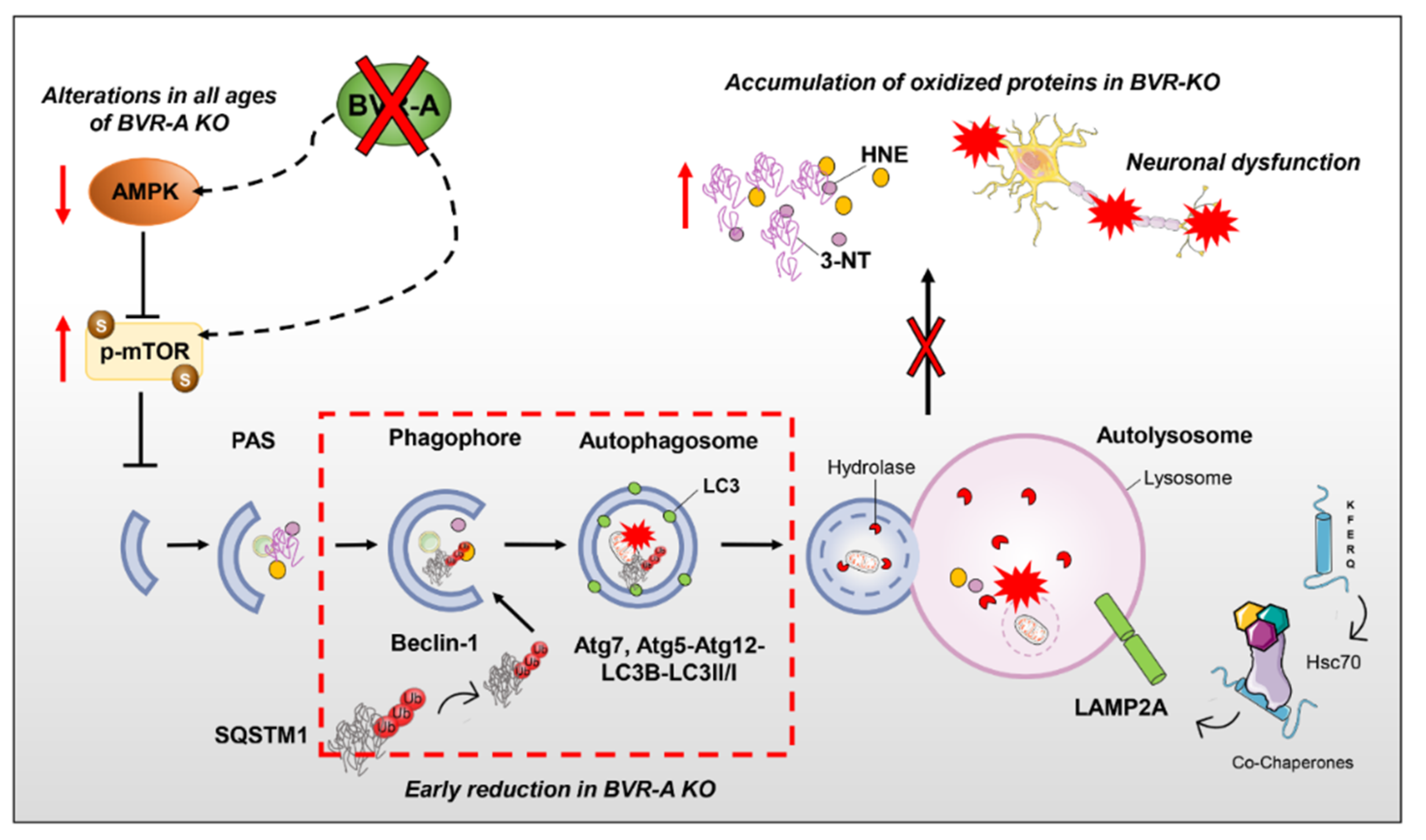
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| ATG | autophagy-related proteins |
| AMPK | 5’ adenosine monophosphate-activated protein kinase |
| BIR | brain insulin resistance |
| IRS-1 | insulin receptor substrate-1 |
| BVR | Biliverdin reductase |
| HO | heme oxygenase |
| IGF-1 | insulin-like growth factor-1 |
| LAMP | lysosome-associated membrane protein |
| LC3 | microtubule-associated protein 1A/1B-light chain 3 |
| KO | knock-out |
| MAPK | mitogen-activated protein kinase |
| MCI | mild cognitive impairment |
| mTORC1 | mammalian target of rapamycin complex 1 |
| mTOR | mammalian target of rapamycin |
| OS | oxidative stress |
| PBMC | peripheral blood mononuclear cells |
| PI3K | phosphatidylinositol-3-kinase |
| SQSTM1 | sequestosome-1 |
| 3-NT | 3-nitro-tyrosine |
| HNE | 4-hydroxy-2-nonenals |
| TLR4 | Toll like receptor 4 |
References
- Kapitulnik, J.; Maines, M.D. Pleiotropic functions of biliverdin reductase: Cellular signaling and generation of cytoprotective and cytotoxic bilirubin. Trends Pharmacol. Sci. 2009, 30, 129–137. [Google Scholar] [CrossRef]
- Barone, E.; Di Domenico, F.; Mancuso, C.; Butterfield, D.A. The Janus face of the heme oxygenase/biliverdin reductase system in Alzheimer disease: It’s time for reconciliation. Neurobiol. Dis. 2014, 62, 144–159. [Google Scholar] [CrossRef] [PubMed]
- Canesin, G.; Hejazi, S.M.; Swanson, K.D.; Wegiel, B. Heme-Derived Metabolic Signals Dictate Immune Responses. Front. Immunol. 2020, 11, 66. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, L.; Hosick, P.A.; John, K.; Stec, D.E.; Hinds, T.D., Jr. Biliverdin reductase isozymes in metabolism. Trends Endocrinol. Metab. 2015, 26, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Maghzal, G.J.; Ayer, A.; Suarna, C.; Dunn, L.L.; Stocker, R. Absence of the biliverdin reductase-a gene is associated with increased endogenous oxidative stress. Free Radic. Biol. Med. 2018, 115, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Kravets, A.; Hu, Z.; Miralem, T.; Torno, M.D.; Maines, M.D. Biliverdin reductase, a novel regulator for induction of activating transcription factor-2 and heme oxygenase-1. J. Biol. Chem. 2004, 279, 19916–19923. [Google Scholar] [CrossRef] [PubMed]
- Maines, M.D.; Ewing, J.F.; Huang, T.J.; Panahian, N. Nuclear localization of biliverdin reductase in the rat kidney: Response to nephrotoxins that induce heme oxygenase-1. J. Pharmacol. Exp. Ther. 2001, 296, 1091–1097. [Google Scholar]
- Gibbs, P.E.; Miralem, T.; Lerner-Marmarosh, N.; Tudor, C.; Maines, M.D. Formation of ternary complex of human biliverdin reductase-protein kinase Cdelta-ERK2 protein is essential for ERK2-mediated activation of Elk1 protein, nuclear factor-kappaB, and inducible nitric-oxidase synthase (iNOS). J. Biol. Chem. 2012, 287, 1066–1079. [Google Scholar] [CrossRef]
- Lerner-Marmarosh, N.; Miralem, T.; Gibbs, P.E.; Maines, M.D. Human biliverdin reductase is an ERK activator; hBVR is an ERK nuclear transporter and is required for MAPK signaling. Proc. Natl. Acad. Sci. USA 2008, 105, 6870–6875. [Google Scholar] [CrossRef] [PubMed]
- Wegiel, B.; Gallo, D.; Csizmadia, E.; Roger, T.; Kaczmarek, E.; Harris, C.; Zuckerbraun, B.S.; Otterbein, L.E. Biliverdin inhibits Toll-like receptor-4 (TLR4) expression through nitric oxide-dependent nuclear translocation of biliverdin reductase. Proc. Natl. Acad. Sci. USA 2011, 108, 18849–18854. [Google Scholar] [CrossRef]
- Miralem, T.; Lerner-Marmarosh, N.; Gibbs, P.E.; Tudor, C.; Hagen, F.K.; Maines, M.D. The human biliverdin reductase-based peptide fragments and biliverdin regulate protein kinase Cdelta activity: The peptides are inhibitors or substrate for the protein kinase C. J. Biol. Chem. 2012, 287, 24698–24712. [Google Scholar] [CrossRef]
- Barone, E.; Di Domenico, F.; Cassano, T.; Arena, A.; Tramutola, A.; Lavecchia, M.A.; Coccia, R.; Butterfield, D.A.; Perluigi, M. Impairment of biliverdin reductase-A promotes brain insulin resistance in Alzheimer disease: A new paradigm. Free Radic. Biol. Med. 2016, 91, 127–142. [Google Scholar] [CrossRef]
- Lerner-Marmarosh, N.; Shen, J.; Torno, M.D.; Kravets, A.; Hu, Z.; Maines, M.D. Human biliverdin reductase: A member of the insulin receptor substrate family with serine/threonine/tyrosine kinase activity. Proc. Natl. Acad. Sci. USA 2005, 102, 7109–7114. [Google Scholar] [CrossRef]
- Gibbs, P.E.; Lerner-Marmarosh, N.; Poulin, A.; Farah, E.; Maines, M.D. Human biliverdin reductase-based peptides activate and inhibit glucose uptake through direct interaction with the kinase domain of insulin receptor. FASEB J. 2014, 28, 2478–2491. [Google Scholar] [CrossRef]
- Miralem, T.; Lerner-Marmarosh, N.; Gibbs, P.E.; Jenkins, J.L.; Heimiller, C.; Maines, M.D. Interaction of human biliverdin reductase with Akt/protein kinase B and phosphatidylinositol-dependent kinase 1 regulates glycogen synthase kinase 3 activity: A novel mechanism of Akt activation. FASEB J. 2016, 30, 2926–2944. [Google Scholar] [CrossRef]
- Hinds, T.D., Jr.; Burns, K.A.; Hosick, P.A.; McBeth, L.; Nestor-Kalinoski, A.; Drummond, H.A.; AlAmodi, A.A.; Hankins, M.W.; Vanden Heuvel, J.P.; Stec, D.E. Biliverdin Reductase A Attenuates Hepatic Steatosis by Inhibition of Glycogen Synthase Kinase (GSK) 3beta Phosphorylation of Serine 73 of Peroxisome Proliferator-activated Receptor (PPAR) alpha. J. Biol. Chem. 2016, 291, 25179–25191. [Google Scholar] [CrossRef]
- Stec, D.E.; Gordon, D.M.; Nestor-Kalinoski, A.L.; Donald, M.C.; Mitchell, Z.L.; Creeden, J.F.; Hinds, T.D., Jr. Biliverdin Reductase A (BVRA) Knockout in Adipocytes Induces Hypertrophy and Reduces Mitochondria in White Fat of Obese Mice. Biomolecules 2020, 10, 387. [Google Scholar] [CrossRef]
- Cimini, F.A.; Arena, A.; Barchetta, I.; Tramutola, A.; Ceccarelli, V.; Lanzillotta, C.; Fontana, M.; Bertoccini, L.; Leonetti, F.; Capoccia, D.; et al. Reduced biliverdin reductase-A levels are associated with early alterations of insulin signaling in obesity. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1490–1501. [Google Scholar] [CrossRef]
- Barone, E.; Tramutola, A.; Triani, F.; Calcagnini, S.; Di Domenico, F.; Ripoli, C.; Gaetani, S.; Grassi, C.; Butterfield, D.A.; Cassano, T.; et al. Biliverdin Reductase-A Mediates the Beneficial Effects of Intranasal Insulin in Alzheimer Disease. Mol. Neurobiol. 2019, 56, 2922–2943. [Google Scholar] [CrossRef]
- Vasavda, C.; Kothari, R.; Malla, A.P.; Tokhunts, R.; Lin, A.; Ji, M.; Ricco, C.; Xu, R.; Saavedra, H.G.; Sbodio, J.I.; et al. Bilirubin Links Heme Metabolism to Neuroprotection by Scavenging Superoxide. Cell Chem. Biol. 2019, 26, 1450–1460. [Google Scholar] [CrossRef]
- Sharma, N.; Tramutola, A.; Lanzillotta, C.; Arena, A.; Blarzino, C.; Cassano, T.; Butterfield, D.A.; Di Domenico, F.; Perluigi, M.; Barone, E. Loss of biliverdin reductase-A favors Tau hyper-phosphorylation in Alzheimer’s disease. Neurobiol. Dis. 2019, 125, 176–189. [Google Scholar] [CrossRef]
- Kim, S.J.; Shin, M.J.; Kim, D.W.; Yeo, H.J.; Yeo, E.J.; Choi, Y.J.; Sohn, E.J.; Han, K.H.; Park, J.; Lee, K.W.; et al. Tat-Biliverdin Reductase A Exerts a Protective Role in Oxidative Stress-Induced Hippocampal Neuronal Cell Damage by Regulating the Apoptosis and MAPK Signaling. Int. J. Mol. Sci. 2020, 21, 2672. [Google Scholar] [CrossRef]
- Triani, F.; Tramutola, A.; Di Domenico, F.; Sharma, N.; Butterfield, D.A.; Head, E.; Perluigi, M.; Barone, E. Biliverdin reductase-A impairment links brain insulin resistance with increased Abeta production in an animal model of aging: Implications for Alzheimer disease. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3181–3194. [Google Scholar] [CrossRef]
- Di Domenico, F.; Perluigi, M.; Barone, E. Biliverdin Reductase-A correlates with inducible nitric oxide synthasein in atorvastatin treated aged canine brain. Neural Regen. Res. 2013, 8, 1925–1937. [Google Scholar]
- Barone, E.; Mancuso, C.; Di Domenico, F.; Sultana, R.; Murphy, M.P.; Head, E.; Butterfield, D.A. Biliverdin reductase-A: A novel drug target for atorvastatin in a dog pre-clinical model of Alzheimer disease. J. Neurochem. 2012, 120, 135–146. [Google Scholar] [CrossRef]
- Barone, E.; Di Domenico, F.; Cenini, G.; Sultana, R.; Coccia, R.; Preziosi, P.; Perluigi, M.; Mancuso, C.; Butterfield, D.A. Oxidative and nitrosative modifications of biliverdin reductase-A in the brain of subjects with Alzheimer’s disease and amnestic mild cognitive impairment. J. Alzheimers Dis. 2011, 25, 623–633. [Google Scholar] [CrossRef]
- Barone, E.; Di Domenico, F.; Cenini, G.; Sultana, R.; Cini, C.; Preziosi, P.; Perluigi, M.; Mancuso, C.; Butterfield, D.A. Biliverdin reductase—A protein levels and activity in the brains of subjects with Alzheimer disease and mild cognitive impairment. Biochim. Biophys. Acta 2011, 1812, 480–487. [Google Scholar] [CrossRef]
- Di Domenico, F.; Pupo, G.; Mancuso, C.; Barone, E.; Paolini, F.; Arena, A.; Blarzino, C.; Schmitt, F.A.; Head, E.; Butterfield, D.A.; et al. Bach1 overexpression in Down syndrome correlates with the alteration of the HO-1/BVR-a system: Insights for transition to Alzheimer’s disease. J. Alzheimers Dis. 2015, 44, 1107–1120. [Google Scholar] [CrossRef]
- Gonzalez, C.D.; Resnik, R.; Vaccaro, M.I. Secretory Autophagy and Its Relevance in Metabolic and Degenerative Disease. Front. Endocrinol. 2020, 11, 266. [Google Scholar] [CrossRef]
- Garcia-Arencibia, M.; Hochfeld, W.E.; Toh, P.P.; Rubinsztein, D.C. Autophagy, a guardian against neurodegeneration. Semin. Cell Dev. Biol. 2010, 21, 691–698. [Google Scholar] [CrossRef]
- Cuervo, A.M. Autophagy and aging: Keeping that old broom working. Trends Genet. 2008, 24, 604–612. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Kang, J.H.; Lee, S. Autophagy in Neurodegenerative Diseases: A Hunter for Aggregates. Int. J. Mol. Sci. 2020, 21, 3369. [Google Scholar] [CrossRef] [PubMed]
- Di Domenico, F.; Zuliani, I.; Tramutola, A. Shining a light on defective autophagy by proteomics approaches: Implications for neurodegenerative illnesses. Expert Rev. Proteom. 2019, 16, 951–964. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. mTOR Signaling. Cold Spring Harb. Perspect. Biol. 2012, 4, a011593. [Google Scholar] [CrossRef]
- Perluigi, M.; Di Domenico, F.; Butterfield, D.A. mTOR signaling in aging and neurodegeneration: At. the crossroad between metabolism dysfunction and impairment of autophagy. Neurobiol. Dis. 2015, 84, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Tramutola, A.; Triplett, J.C.; Di Domenico, F.; Niedowicz, D.M.; Murphy, M.P.; Coccia, R.; Perluigi, M.; Butterfield, D.A. Alteration of mTOR signaling occurs early in the progression of Alzheimer disease (AD): Analysis of brain from subjects with pre-clinical AD, amnestic mild cognitive impairment and late-stage AD. J. Neurochem. 2015, 133, 739–749. [Google Scholar] [CrossRef]
- O’Neill, C. PI3-kinase/Akt/mTOR signaling: Impaired on/off switches in aging, cognitive decline and Alzheimer’s disease. Exp. Gerontol. 2013, 48, 647–653. [Google Scholar] [CrossRef]
- Caccamo, A.; Majumder, S.; Richardson, A.; Strong, R.; Oddo, S. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and Tau: Effects on cognitive impairments. J. Biol. Chem. 2010, 285, 13107–13120. [Google Scholar] [CrossRef]
- Vartak, R.S.; Rodin, A.; Oddo, S. Differential activation of the mTOR/autophagy pathway predicts cognitive performance in APP/PS1 mice. Neurobiol. Aging 2019, 83, 105–113. [Google Scholar] [CrossRef]
- Di Domenico, F.; Tramutola, A.; Barone, E.; Lanzillotta, C.; Defever, O.; Arena, A.; Zuliani, I.; Foppoli, C.; Iavarone, F.; Vincenzoni, F.; et al. Restoration of aberrant mTOR signaling by intranasal rapamycin reduces oxidative damage: Focus on HNE-modified proteins in a mouse model of down syndrome. Redox Biol. 2019, 23, 101162. [Google Scholar] [CrossRef]
- Perluigi, M.; Pupo, G.; Tramutola, A.; Cini, C.; Coccia, R.; Barone, E.; Head, E.; Butterfield, D.A.; Di Domenico, F. Neuropathological role of PI3K/Akt/mTOR axis in Down syndrome brain. Biochim. Biophys. Acta 2014, 1842, 1144–1153. [Google Scholar] [CrossRef]
- Tramutola, A.; Lanzillotta, C.; Arena, A.; Barone, E.; Perluigi, M.; Di Domenico, F. Increased Mammalian Target. of Rapamycin Signaling Contributes to the Accumulation of Protein Oxidative Damage in a Mouse Model. of Down’s Syndrome. Neurodegener. Dis. 2016, 16, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Tramutola, A.; Lanzillotta, C.; Di Domenico, F. Targeting mTOR to reduce Alzheimer-related cognitive decline: From current hits to future therapies. Expert Rev. Neurother. 2017, 17, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Di Domenico, F.; Pupo, G.; Tramutola, A.; Giorgi, A.; Schinina, M.E.; Coccia, R.; Head, E.; Butterfield, D.A.; Perluigi, M. Redox proteomics analysis of HNE-modified proteins in Down syndrome brain: Clues for understanding the development of Alzheimer disease. Free Radic. Biol. Med. 2014, 71, 270–280. [Google Scholar] [CrossRef] [PubMed]
- Di Domenico, F.; Barone, E.; Perluigi, M.; Butterfield, D.A. The Triangle of Death in Alzheimer’s Disease Brain: The Aberrant Cross-Talk Among Energy Metabolism, Mammalian Target. of Rapamycin Signaling, and Protein Homeostasis Revealed by Redox Proteomics. Antioxid. Redox Signal 2017, 26, 364–387. [Google Scholar] [CrossRef]
- Majumder, S.; Richardson, A.; Strong, R.; Oddo, S. Inducing autophagy by rapamycin before, but not after, the formation of plaques and tangles ameliorates cognitive deficits. PLoS ONE 2011, 6, e25416. [Google Scholar] [CrossRef]
- Tramutola, A.; Lanzillotta, C.; Barone, E.; Arena, A.; Zuliani, I.; Mosca, L.; Blarzino, C.; Butterfield, D.A.; Perluigi, M.; Di Domenico, F. Intranasal rapamycin ameliorates Alzheimer-like cognitive decline in a mouse model of Down syndrome. Transl. Neurodegener. 2018, 7, 28. [Google Scholar] [CrossRef]
- Switon, K.; Kotulska, K.; Janusz-Kaminska, A.; Zmorzynska, J.; Jaworski, J. Molecular neurobiology of mTOR. Neuroscience 2017, 341, 112–153. [Google Scholar] [CrossRef]
- Arroyo, D.S.; Gaviglio, E.A.; Peralta Ramos, J.M.; Bussi, C.; Rodriguez-Galan, M.C.; Iribarren, P. Autophagy in inflammation, infection, neurodegeneration and cancer. Int. Immunopharmacol. 2014, 18, 55–65. [Google Scholar] [CrossRef]
- Saha, S.; Panigrahi, D.P.; Patil, S.; Bhutia, S.K. Autophagy in health and disease: A comprehensive review. Biomed. Pharmacother. 2018, 104, 485–495. [Google Scholar] [CrossRef]
- Metcalf, D.J.; Garcia-Arencibia, M.; Hochfeld, W.E.; Rubinsztein, D.C. Autophagy and misfolded proteins in neurodegeneration. Exp. Neurol. 2012, 238, 22–28. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T.; Levine, B. Methods in mammalian autophagy research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Cuervo, A.M.; Ravikumar, B.; Sarkar, S.; Korolchuk, V.; Kaushik, S.; Klionsky, D.J. In search of an “autophagomometer”. Autophagy 2009, 5, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Eskelinen, E.L. Roles of LAMP-1 and LAMP-2 in lysosome biogenesis and autophagy. Mol. Asp. Med. 2006, 27, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Ji, J.; Yan, X.H. Cross-talk between AMPK and mTOR in regulating energy balance. Crit. Rev. Food Sci. Nutr. 2012, 52, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Ghavami, S.; Shojaei, S.; Yeganeh, B.; Ande, S.R.; Jangamreddy, J.R.; Mehrpour, M.; Christoffersson, J.; Chaabane, W.; Moghadam, A.R.; Kashani, H.H.; et al. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog. Neurobiol. 2014, 112, 24–49. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Gu, L.; Di Domenico, F.; Robinson, R.A. Mass spectrometry and redox proteomics: Applications in disease. Mass Spectrom. Rev. 2014, 33, 277–301. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Halliwell, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef]
- Di Domenico, F.; Tramutola, A.; Butterfield, D.A. Role of 4-hydroxy-2-nonenal (HNE) in the pathogenesis of alzheimer disease and other selected age-related neurodegenerative disorders. Free Radic. Biol. Med. 2017, 111, 253–261. [Google Scholar] [CrossRef]
- Jansen, T.; Daiber, A. Direct Antioxidant Properties of Bilirubin and Biliverdin. Is there a Role for Biliverdin Reductase? Front. Pharmacol. 2012, 3, 30. [Google Scholar] [CrossRef] [PubMed]
- Stocker, R.; Yamamoto, Y.; McDonagh, A.F.; Glazer, A.N.; Ames, B.N. Bilirubin is an antioxidant of possible physiological importance. Science 1987, 235, 1043–1046. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, C.; Barone, E.; Guido, P.; Miceli, F.; Di Domenico, F.; Perluigi, M.; Santangelo, R.; Preziosi, P. Inhibition of lipid peroxidation and protein oxidation by endogenous and exogenous antioxidants in rat brain microsomes in vitro. Neurosci. Lett. 2012, 518, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, V.; Cornelius, C.; Mancuso, C.; Barone, E.; Calafato, S.; Bates, T.; Rizzarelli, E.; Kostova, A.T. Vitagenes, dietary antioxidants and neuroprotection in neurodegenerative diseases. Front. Biosci. 2009, 14, 376–397. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, C.; Scapagini, G.; Curro, D.; Giuffrida Stella, A.M.; De Marco, C.; Butterfield, D.A.; Calabrese, V. Mitochondrial dysfunction, free radical generation and cellular stress response in neurodegenerative disorders. Front. Biosci. 2007, 12, 1107–1123. [Google Scholar] [CrossRef] [PubMed]
- Maines, M.D. The heme oxygenase system: A regulator of second messenger gases. Annu. Rev. Pharmacol. Toxicol. 1997, 37, 517–554. [Google Scholar] [CrossRef]
- Maines, M.D. The heme oxygenase system and its functions in the brain. Cell. Mol. Biol. 2000, 46, 573–585. [Google Scholar]
- Mancuso, C. Heme oxygenase and its products in the nervous system. Antioxid. Redox Signal 2004, 6, 878–887. [Google Scholar]
- Barone, E.; Di Domenico, F.; Sultana, R.; Coccia, R.; Mancuso, C.; Perluigi, M.; Butterfield, D.A. Heme oxygenase-1 posttranslational modifications in the brain of subjects with Alzheimer disease and mild cognitive impairment. Free Radic. Biol. Med. 2012, 52, 2292–2301. [Google Scholar] [CrossRef]
- Schipper, H.M.; Song, W.; Tavitian, A.; Cressatti, M. The sinister face of heme oxygenase-1 in brain aging and disease. Prog. Neurobiol. 2019, 172, 40–70. [Google Scholar] [CrossRef]
- Tudor, C.; Lerner-Marmarosh, N.; Engelborghs, Y.; Gibbs, P.E.; Maines, M.D. Biliverdin reductase is a transporter of haem into the nucleus and is essential for regulation of HO-1 gene expression by haematin. Biochem. J. 2008, 413, 405–416. [Google Scholar] [CrossRef]
- Bisht, K.; Wegiel, B.; Tampe, J.; Neubauer, O.; Wagner, K.H.; Otterbein, L.E.; Bulmer, A.C. Biliverdin modulates the expression of C5aR in response to endotoxin in part via mTOR signaling. Biochem. Biophys. Res. Commun. 2014, 449, 94–99. [Google Scholar] [CrossRef][Green Version]
- Gibbs, P.E.; Miralem, T.; Maines, M.D. Characterization of the human biliverdin reductase gene structure and regulatory elements: Promoter activity is enhanced by hypoxia and suppressed by TNF-alpha-activated NF-kappaB. FASEB J. 2010, 24, 3239–3354. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.M. Regulation and function of AMPK in physiology and diseases. Exp. Mol. Med. 2016, 48, e245. [Google Scholar] [CrossRef] [PubMed]
- Carling, D. AMPK signalling in health and disease. Curr. Opin. Cell Biol. 2017, 45, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Peixoto, C.A.; Oliveira, W.H.; Araujo, S.; Nunes, A.K.S. AMPK activation: Role in the signaling pathways of neuroinflammation and neurodegeneration. Exp. Neurol. 2017, 298, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Leprivier, G.; Rotblat, B. How does mTOR sense glucose starvation? AMPK is the usual suspect. Cell Death Discov. 2020, 6, 27. [Google Scholar] [CrossRef] [PubMed]
- Davie, E.; Forte, G.M.; Petersen, J. Nitrogen regulates AMPK to control TORC1 signaling. Curr. Biol. 2015, 25, 445–454. [Google Scholar] [CrossRef]
- Hindupur, S.K.; Gonzalez, A.; Hall, M.N. The opposing actions of target of rapamycin and AMP-activated protein kinase in cell growth control. Cold Spring Harb. Perspect. Biol. 2015, 7, a019141. [Google Scholar] [CrossRef]
- Ciechanover, A. The unravelling of the ubiquitin system. Nat. Rev. Mol. Cell Biol. 2015, 16, 322–324. [Google Scholar] [CrossRef]
- Seibenhener, M.L.; Babu, J.R.; Geetha, T.; Wong, H.C.; Krishna, N.R.; Wooten, M.W. Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol. Cell. Biol. 2004, 24, 8055–8068. [Google Scholar] [CrossRef]
- Korolchuk, V.I.; Mansilla, A.; Menzies, F.M.; Rubinsztein, D.C. Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol. Cell 2009, 33, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Katsuragi, Y.; Ichimura, Y.; Komatsu, M. p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J. 2015, 282, 4672–4678. [Google Scholar] [CrossRef] [PubMed]
- Bitto, A.; Sell, C.; Crowe, E.; Lorenzini, A.; Malaguti, M.; Hrelia, S.; Torres, C. Stress-induced senescence in human and rodent astrocytes. Exp. Cell Res. 2010, 316, 2961–2968. [Google Scholar] [CrossRef]
- Bhat, R.; Crowe, E.P.; Bitto, A.; Moh, M.; Katsetos, C.D.; Garcia, F.U.; Johnson, F.B.; Trojanowski, J.Q.; Sell, C.; Torres, C. Astrocyte senescence as a component of Alzheimer’s disease. PLoS ONE 2012, 7, e45069. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Ojala, J.; Kaarniranta, K.; Haapasalo, A.; Hiltunen, M.; Soininen, H. Astrocytes in the aging brain express characteristics of senescence-associated secretory phenotype. Eur. J. Neurosci. 2011, 34, 3–11. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target of Analysis | 2-WAY ANOVA | |||||
|---|---|---|---|---|---|---|
| Age | Genotype (WT-BVR-A−/−) | Interaction | ||||
| F (DFn, DFd) | p | F (DFn, DFd) | p | F (DFn, DFd) | p | |
| HNE | F (2, 18) = 5.18 | p < 0.05 | n.s. | n.s | n.s. | n.s. |
| 3-NT | F (2, 18) = 28,70 | p < 0,0001 | n.s. | n.s. | F (2, 18) = 5,141 | p < 0,05 |
| p-mTOR | F (2, 18) = 10.53 | p < 0.001 | F (1, 18) = 40.78 | p < 0.0001 | n.s. | n.s. |
| mTOR | F (2, 18) = 45.89 | p < 0.0001 | F (1, 18) = 5.33 | p < 0.05 | n.s. | n.s. |
| p-mTOR/mTOR | F (2, 18) = 6.43 | p < 0.01 | F (1, 18) = 16.40 | p < 0.001 | F (2, 18) = 4.02 | p < 0.05 |
| Beclin-1 | F (2, 18) = 5.08 | p < 0.05 | F (1, 18) = 7.52 | p < 0.05 | F (2, 18) = 5.59 | p < 0.05 |
| Atg12-Atg5 | F (2, 18) = 20.30 | p < 0.0001 | F (1, 18) = 9.27 | p < 0.01 | n.s. | n.s. |
| Atg7 | F (2, 18) = 8.26 | p < 0.01 | n.s. | n.s. | F (2, 18) = 7.34 | p < 0.01 |
| LC3β | n.s. | n.s. | n.s. | n.s. | n.s. | n.s. |
| LC3 II/I | F (2, 18) = 49.28 | p < 0.0001 | F (1, 18) = 16.58 | p < 0.001 | F (2, 18) = 15.50 | p < 0.0001 |
| SQSTM1 | F (2, 18) = 25.27 | p < 0.0001 | n.s. | n.s. | F (2, 18) = 5.02 | p < 0.05 |
| LAMP1 | F (2, 18) = 5.54 | p < 0.05 | n.s. | n.s. | n.s. | n.s. |
| AMPK | F (2, 18) = 18.45 | p < 0.0001 | n.s. | n.s. | n.s. | n.s. |
| p-AMPK | F (2, 18) = 7.42 | p < 0.01 | F (1, 18) = 11.55 | p < 0.01 | F (2, 18) = 5.74 | p < 0.05 |
| p-AMPK/AMPK | F (2, 18) = 19.88 | p < 0.0001 | F (1, 18) = 36.12 | p < 0.0001 | F (2, 18) = 17.40 | p < 0.0001 |
| Target of Analysis | WT | BVR-A−/− | ||
|---|---|---|---|---|
| r | p | r | p | |
| p-AMPK | 0.445 | n.s. | 0.086 | n.s. |
| AMPK | 0.584 | <0.05 | 0.468 | n.s. |
| p-AMPK/AMPK | 0.114 | n.s. | −0.339 | n.s. |
| Beclin-1 | 0.688 | <0.01 | −0.240 | n.s. |
| Lamp1 | 0.622 | <0.05 | 0.476 | n.s. |
| SQSTM-1 | 0.579 | <0.05 | 0.480 | n.s. |
| Atg5-12 | 0.676 | <0.05 | 0.467 | n.s. |
| Atg7 | 0.653 | <0.05 | 0.307 | n.s. |
| Lc3β | 0.725 | <0.01 | 0.420 | n.s. |
| LC3 II/I | −0.231 | n.s. | 0.021 | n.s. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lanzillotta, C.; Zuliani, I.; Vasavda, C.; Snyder, S.H.; Paul, B.D.; Perluigi, M.; Di Domenico, F.; Barone, E. BVR-A Deficiency Leads to Autophagy Impairment through the Dysregulation of AMPK/mTOR Axis in the Brain—Implications for Neurodegeneration. Antioxidants 2020, 9, 671. https://doi.org/10.3390/antiox9080671
Lanzillotta C, Zuliani I, Vasavda C, Snyder SH, Paul BD, Perluigi M, Di Domenico F, Barone E. BVR-A Deficiency Leads to Autophagy Impairment through the Dysregulation of AMPK/mTOR Axis in the Brain—Implications for Neurodegeneration. Antioxidants. 2020; 9(8):671. https://doi.org/10.3390/antiox9080671
Chicago/Turabian StyleLanzillotta, Chiara, Ilaria Zuliani, Chirag Vasavda, Solomon H. Snyder, Bindu D. Paul, Marzia Perluigi, Fabio Di Domenico, and Eugenio Barone. 2020. "BVR-A Deficiency Leads to Autophagy Impairment through the Dysregulation of AMPK/mTOR Axis in the Brain—Implications for Neurodegeneration" Antioxidants 9, no. 8: 671. https://doi.org/10.3390/antiox9080671
APA StyleLanzillotta, C., Zuliani, I., Vasavda, C., Snyder, S. H., Paul, B. D., Perluigi, M., Di Domenico, F., & Barone, E. (2020). BVR-A Deficiency Leads to Autophagy Impairment through the Dysregulation of AMPK/mTOR Axis in the Brain—Implications for Neurodegeneration. Antioxidants, 9(8), 671. https://doi.org/10.3390/antiox9080671