Lindera obtusiloba Attenuates Oxidative Stress and Airway Inflammation in a Murine Model of Ovalbumin-Challenged Asthma

 and
and
Abstract
1. Introduction
2. Materials and Methods
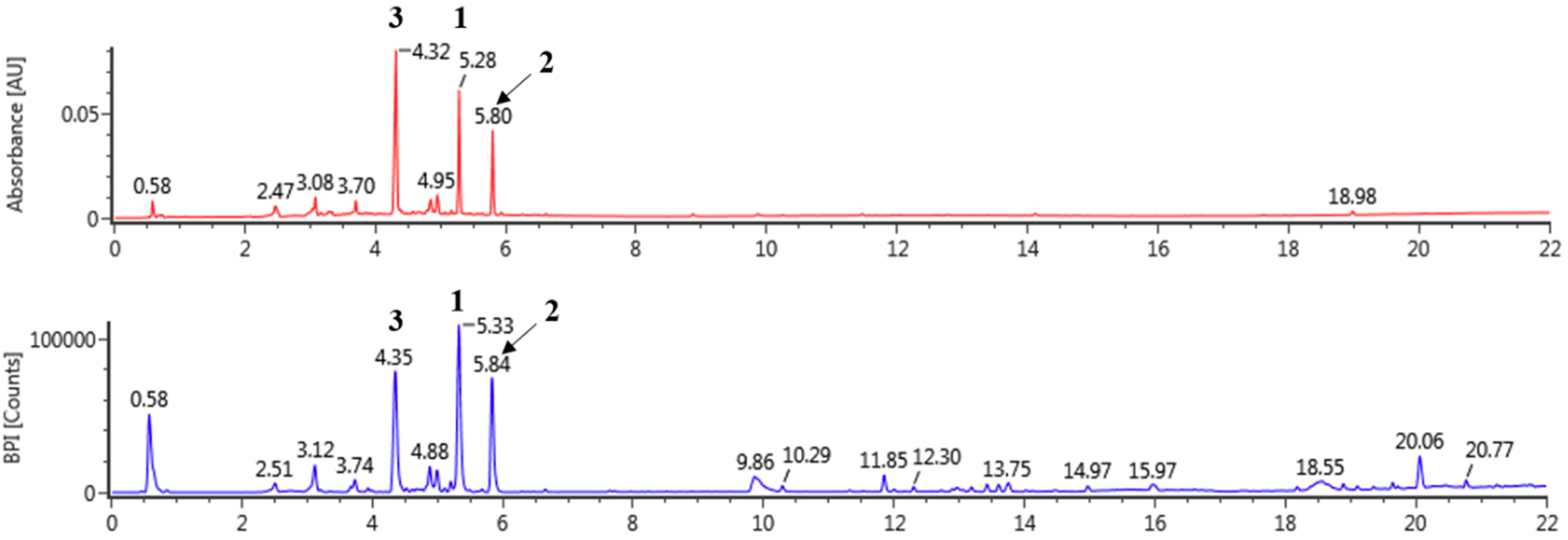
2.1. Ultra-Performance Liquid Chromatography-Quadrupole-Time of Flight Mass Spectrometry (UPLC Q-ToF/MS) Analysis
2.2. Test Compound and Reagents
2.3. Experimental Procedure
- Normal control (NC) group: treated with vehicle (2% DMSO) from day 21 to day 25 and PBS sensitization/challenge
- OVA group: treated with vehicle (2% DMSO) from day 21 to day 25 and OVA sensitization/challenge
- DEX group: treated with 3 mg/kg from day 21 to day 25 and OVA sensitization/challenge
- LOL50 group: treated with LOL 50 mg/kg and OVA sensitization/challenge
- LOL100 group: treated with LOL 100 mg/kg and OVA sensitization/challenge
2.4. Measurement of Airway Hyper-Responsiveness
2.5. Bronchoalveolar Lavage Fluid (BALF) Collection and Inflammatory Cell Count
2.6. Measurement of Th2 Cytokines, Eotaxin in BALF, and OVA-Specific IgE in Serum
2.7. Histopathological Examination of Lungs
2.8. Immunoblotting in Lung Tissues
2.9. Measurement of Nitric Oxide (NO) and ROS Level
2.10. Oxidative Stress Markers Analysis
2.11. Cell Culture and Cell Viability Assay
2.12. Measurement of 2,2-Diphenyl-1-Picryl Hydrazyl (DPPH) Radical Scavenging Activity
2.13. Measurement of Levels of Pro-inflammatory Cytokine Production and ROS, and Oxidative Stress Marker in TNF-α-Stimulated NCI-H292 Cells
2.14. Immunoblotting in TNF-α-Stimulated NCI-H292 Cells
2.15. Quantitative Real-Time Polymerase Chain Reaction (PCR) Analysis of Cytokines
2.16. Statistical Analysis
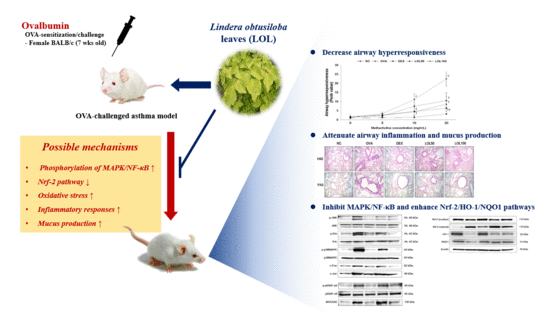
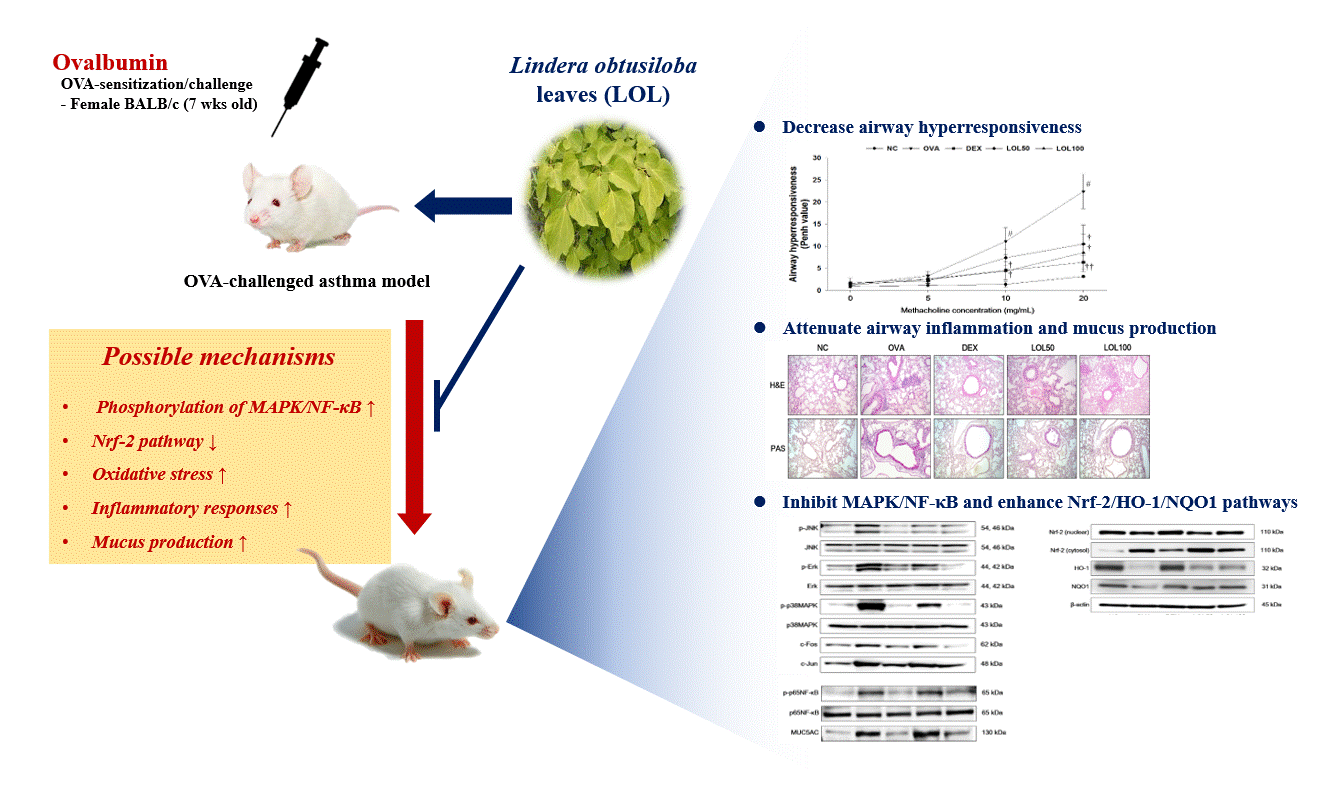
3. Results
3.1. Tentative Characterization of LOL Extract
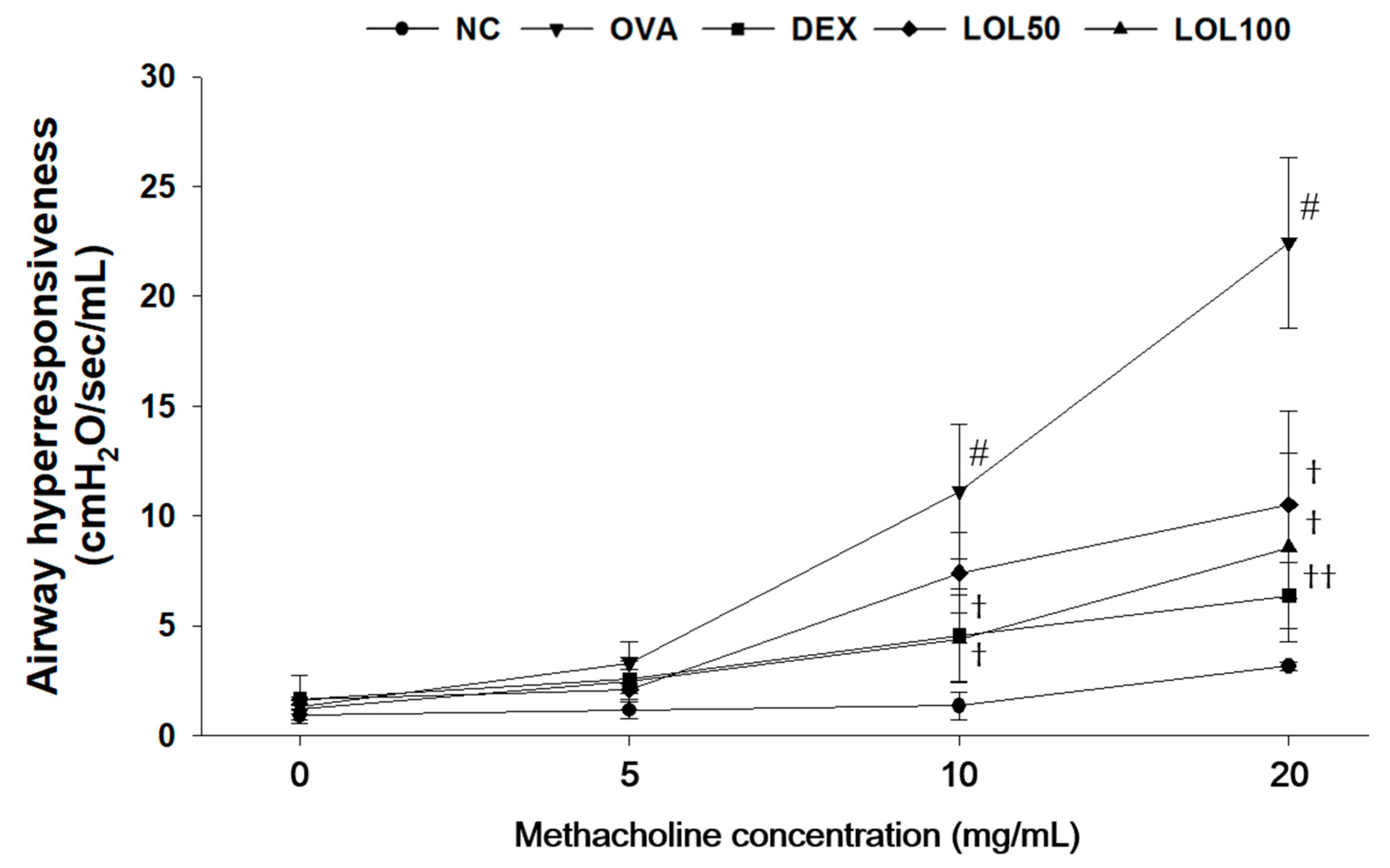
3.2. Effects of LOL on Airway Hyper-Responsiveness in OVA-Challenged Asthma Model
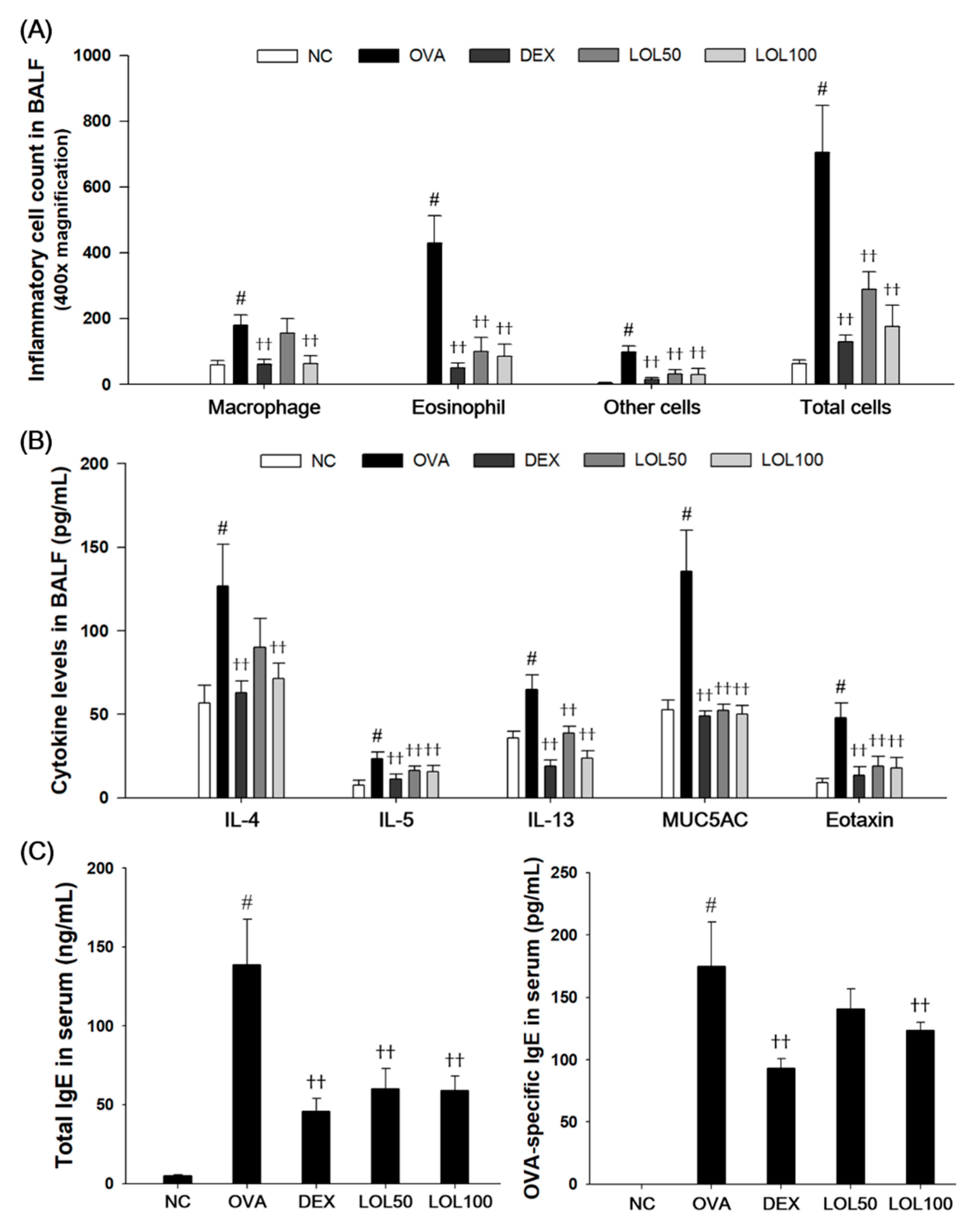
3.3. Effects of LOL on Inflammatory Cell Counts, Th2 Cytokines, Eotaxin, MUC5AC of BALF, and OVA-Specific IgE of Serum in OVA-Challenged Asthma Model
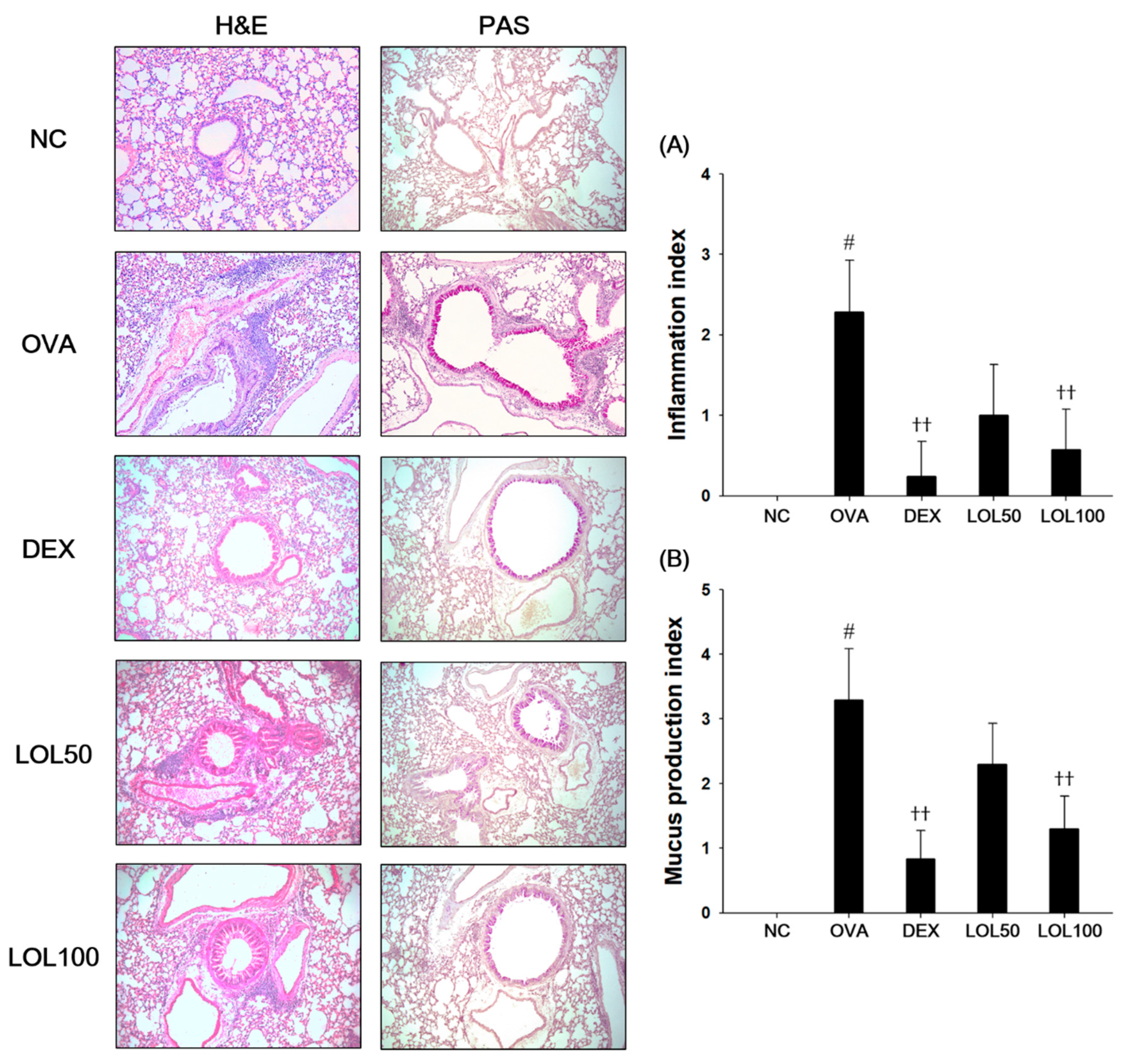
3.4. Effects of LOL on Inflammatory Response and Mucus Production in the Lung Tissue from the OVA-Challenged Asthma Model
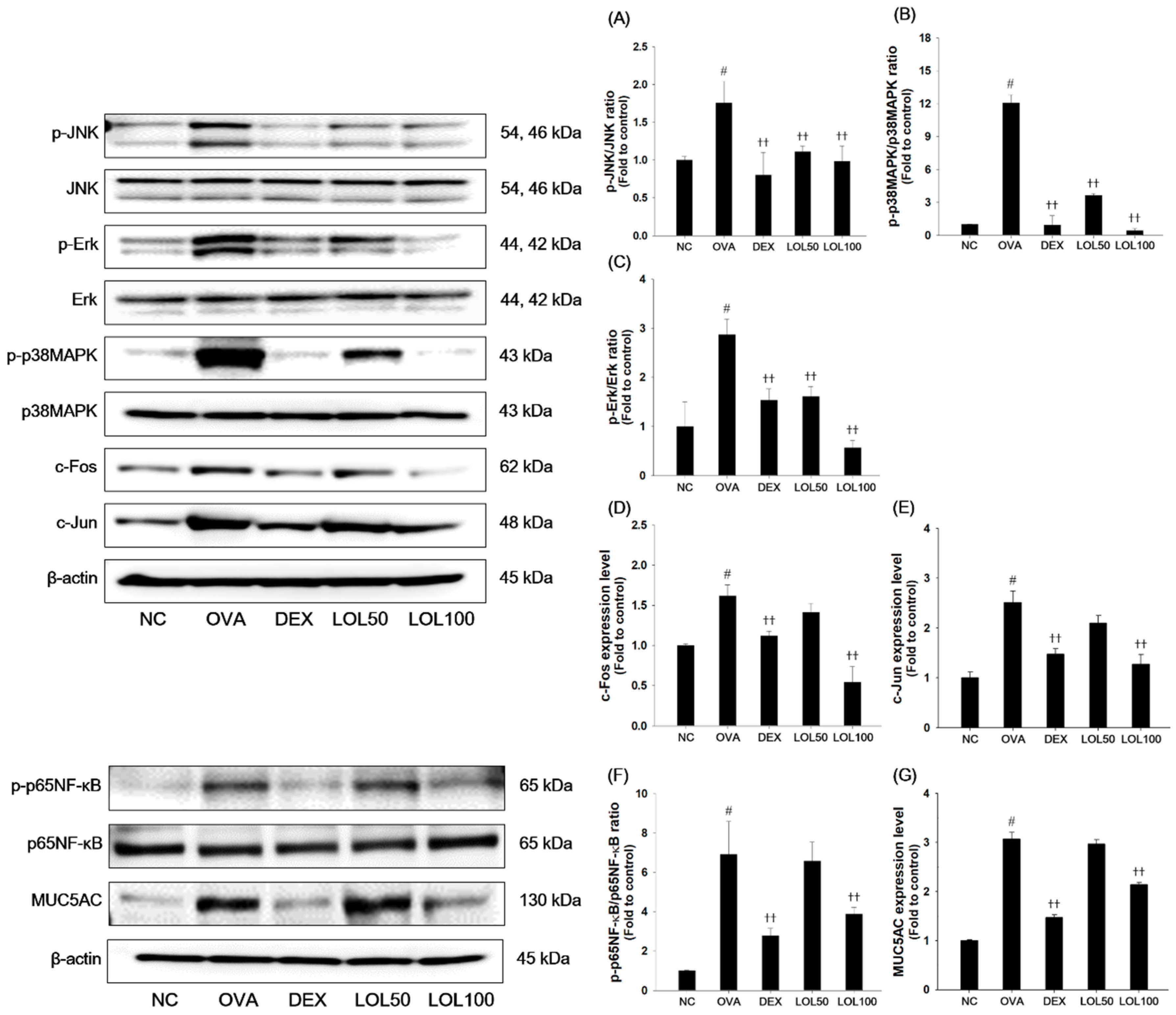
3.5. Effects of LOL on MAPKs/AP-1, p65NF-κB and MUC5AC in the Lung Tissue of the OVA-Challenged Asthma Model
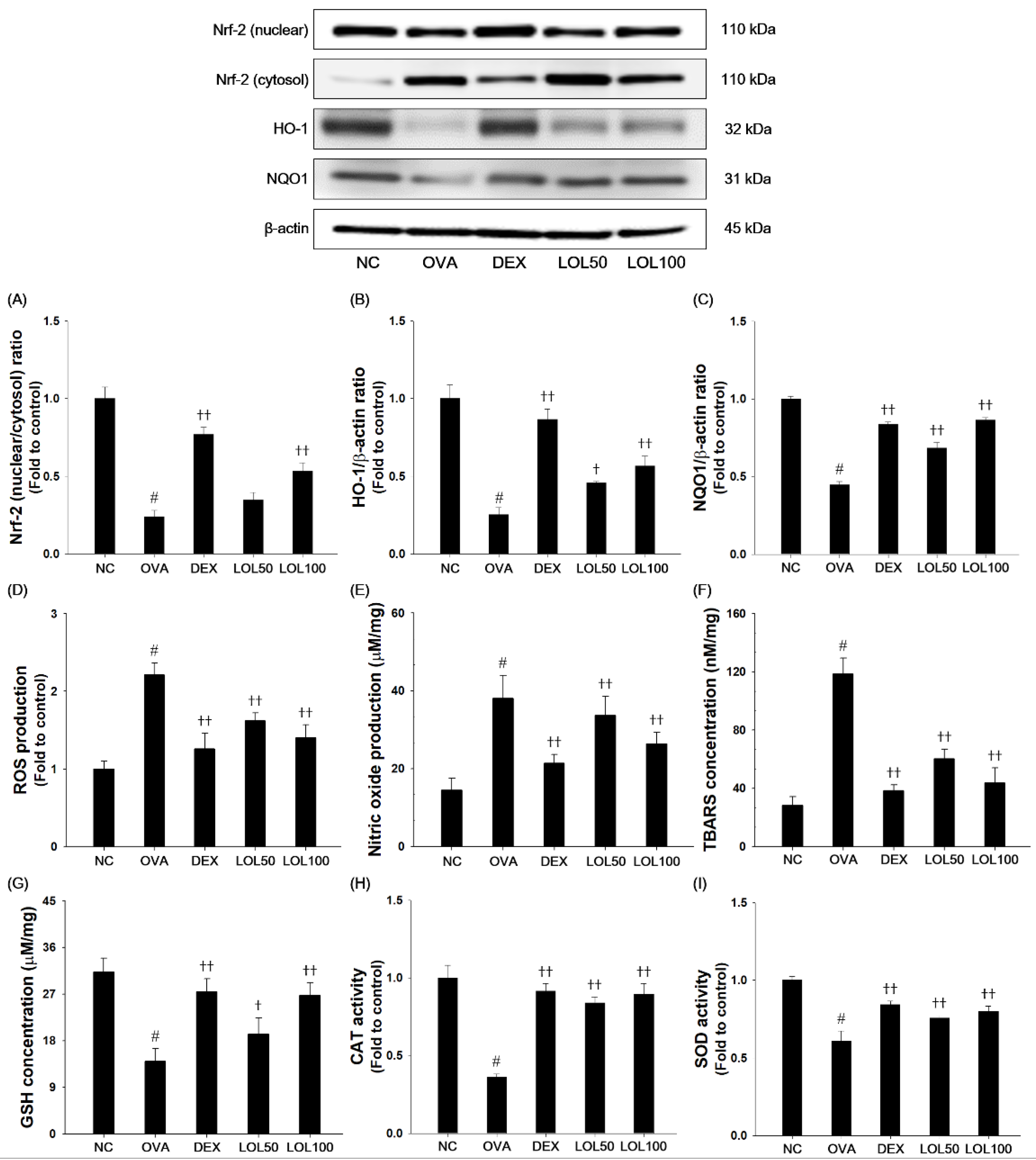
3.6. Effects of LOL on Nrf-2 Pathway, Productions of ROS and NO, and Oxidative Stress Markers in the Lung Tissue of the OVA-Challenged Asthma Model
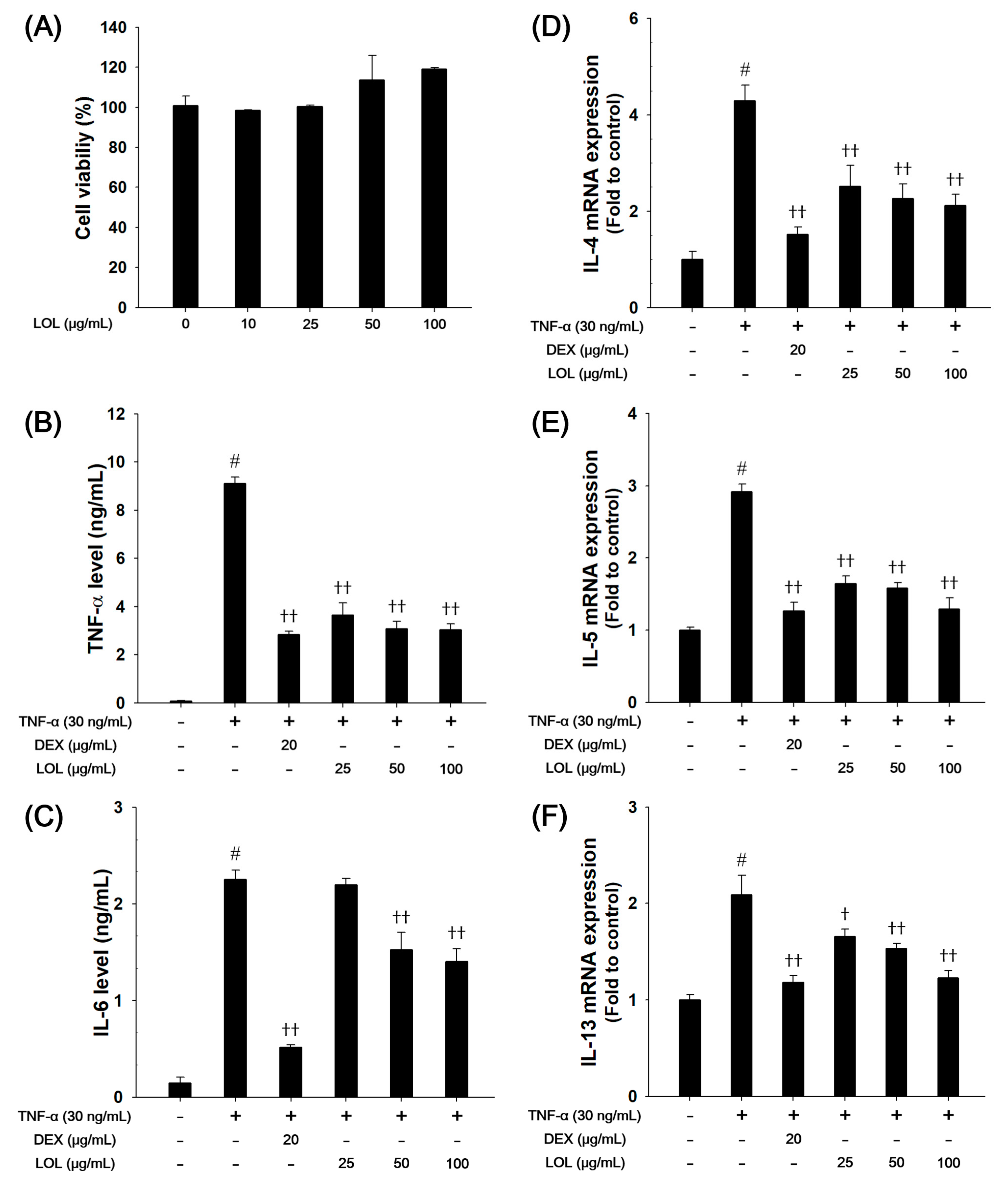
3.7. Effects of LOL on Pro-inflammatory Cytokines and Th2 Cytokines Production in TNF-α-Stimulated NCI-H292 Cells
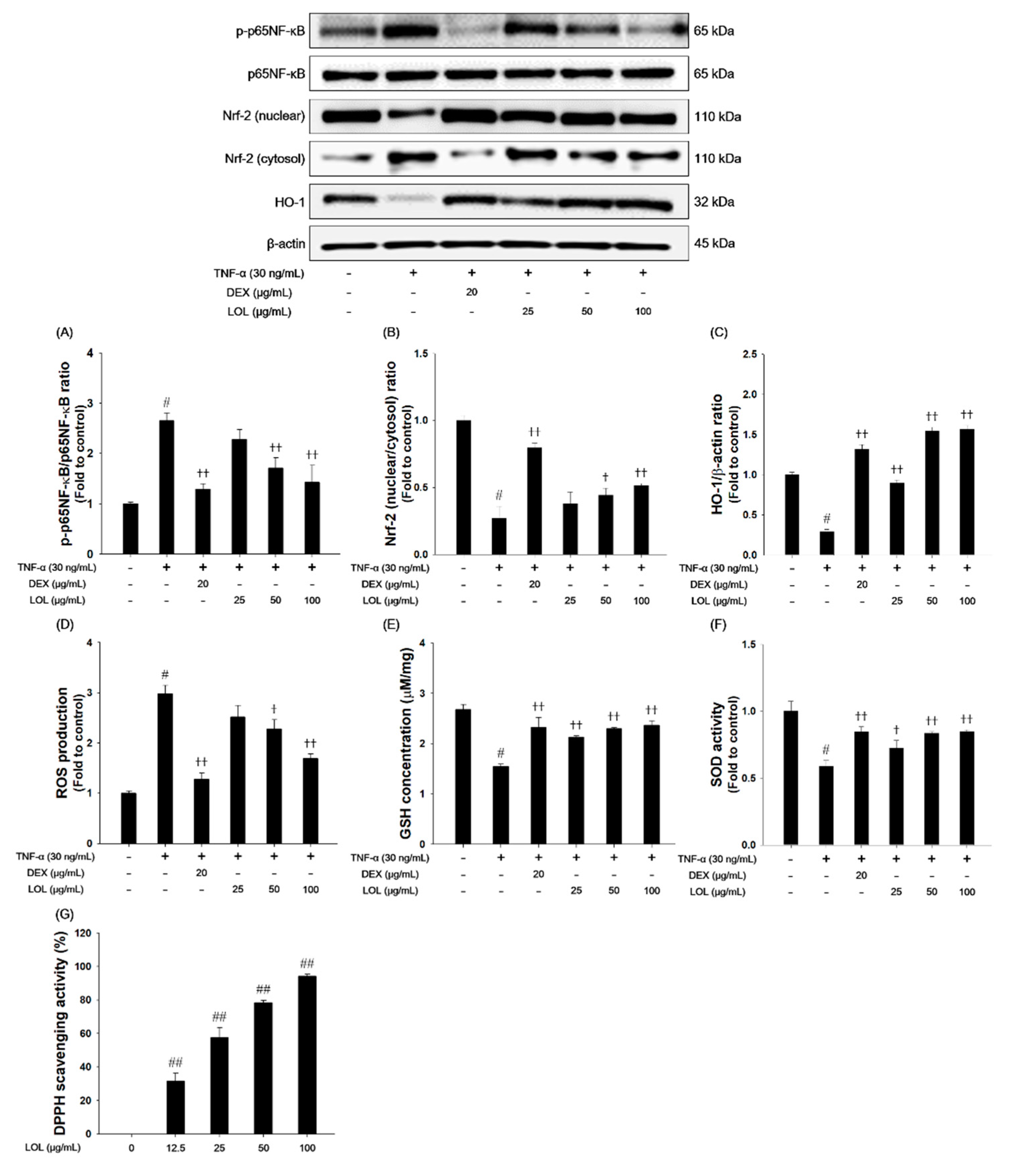
3.8. Effects of LOL on P65NF-κB and Nrf-2 Pathway, ROS Production, Oxidaive Stress Markers and DPPH Radical Scavenging Activity in TNF-α-Stimulated NCI-H292 Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Welte, T.; Groneberg, D.A. Asthma and COPD. Exp. Toxicol. Pathol. 2006, 2, 35–40. [Google Scholar] [CrossRef]
- Shin, I.S.; Hong, J.; Jeon, C.M.; Shin, N.R.; Kwon, O.K.; Kim, H.S.; Kim, J.C.; Oh, S.R.; Ahn, K.S. Diallyl-disulfide, an organosulfur compound of garlic, attenuates airway inflammation via activation of the Nrf-2/HO-1 pathway and NF-kappaB suppression. Food Chem. Toxicol. 2013, 62, 506–513. [Google Scholar] [CrossRef]
- Mehta, A.K.; Arora, N.; Gaur, S.N.; Singh, B.P. Choline supplementation reduces oxidative stress in mouse model of allergic airway disease. Eur. J. Clin. Investig. 2009, 39, 934–941. [Google Scholar] [CrossRef]
- Won, H.Y.; Sohn, J.H.; Min, H.J.; Lee, K.; Woo, H.A.; Ho, Y.S.; Park, J.W.; Rhee, S.G.; Hwang, E.S. Glutathione peroxidase 1 deficiency attenuates allergen induced airway inflammation by suppressing Th2 and Th17 cell development. Antioxid. Redox Signal. 2010, 13, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.Y.; Seo, C.S.; Lee, J.A.; Lee, N.H.; Kim, J.H.; Ha, H.; Zheng, M.S.; Son, J.K.; Shin, H.K. Anti-asthmatic effects of Angelica dahurica against ovalbumin-induced airway inflammation via upregulation of heme oxygenase-1. Food Chem. Toxicol. 2011, 49, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Hamelmann, E.; Tadeda, K.; Oshiba, A.; Gelfand, E.W. Role of IgE in the development of allergic airway inflammation and airway hyperresponsiveness—A murine model. Allergy 1999, 54, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Leigh, R.; Ellis, I.R.; Wattie, J.N.; Hirota, J.A.; Matthaei, K.I.; Foster, P.S.; O’Byrne, P.M.; Inman, M.D. Type 2 cytokines in the pathogenesis of sustained airway dysfunction and airway remodeling in mice. Am. J. Respir. Crit. Care Med. 2004, 169, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Nadeem, A.; Masood, A.; Siddiqui, N. Oxidant-antioxidant imbalance in asthma, scientific evidence, epidemiological data and possible therapeutic options. Ther. Adv. Respir. Dis. 2008, 2, 215–235. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Johnson, J.A. An important role of Nrf2-ARE pathway in the cellular defense mechanism. J. Biochem. Mol. Biol. 2004, 37, 139–143. [Google Scholar] [CrossRef]
- Nguyen, T.; Sherratt, P.J.; Pickett, C.B. Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu. Rev. Pharmacol. Toxicol. 2003, 43, 233–260. [Google Scholar] [CrossRef]
- Choi, Y.H. The cytoprotective effects of ethanol extract of Ecklonia cava against oxidative stress are associated with upregulation of Nrf2-mediated HO-1 and NQO-1 expression through activation of the MAPK pathway. Gen. Physiol. Biophys. 2016, 35, 45–53. [Google Scholar] [PubMed]
- Wang, C.; Choi, Y.H.; Xian, Z.; Zheng, M.; Piao, H.; Yan, G. Aloperine suppresses allergic airway inflammation through NF-κB, MAPK, and Nrf2/HO-1 signaling pathways in mice. Int. Immunopharmacol. 2018, 65, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, H.; Ichinose, M. Oxidative and nitrative stress in bronchial asthma. Antioxid. Redox Signal. 2008, 10, 785–797. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Ge, A.; Zhu, W.; Liu, Y.N.; Ji, N.F.; Zha, W.J.; Zhang, J.X.; Zeng, X.N.; Huang, M. Morin attenuates ovalbumin-induced airway inflammation by modulating oxidative stress-responsive MAPK signaling. Oxidative Med. Cell. Longev. 2016, 2016, 5843672. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Ko, J.W.; Shin, N.R.; Shin, D.H.; Cho, Y.K.; Seo, C.S.; Kim, J.C.; Kim, J.S.; Shin, I.S. 4-Hydroxycinnamic acid protects mice from cigarette smoke-induced pulmonary inflammation via MAPK pathways. Food Chem. Toxicol. 2017, 110, 151–155. [Google Scholar] [CrossRef]
- Wegmann, M. Th2 cells as targets for therapeutic intervention in allergic bronchial asthma. Expert Rev. Mol. Diagn. 2014, 9, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.; Seong, K.J.; Bae, S.W.; Chun, C.; Kim, W.J.; Jung, J.Y. A synthetic diosgenin primary amine derivative attenuates LPS-stimulated inflammation via inhibition of NF-κB and JNK MAPK signaling in microglial BV2 cells. Int. Immunopharmacol. 2018, 61, 204–214. [Google Scholar] [CrossRef]
- Tian, C.; Zhang, P.; Yang, J.; Zhang, Z.; Wang, H.; Guo, Y.; Liu, M. The protective effect of the flavonoid fraction of Abutilon theophrasti Medic. Leaves on LPS-induced acute lung injury in mice via the NF-κB and MAPK signaling pathways. Biomed. Pharmacother. 2019, 109, 1024–1031. [Google Scholar] [CrossRef]
- Xu, X.; Yin, P.; Wan, C.; Chong, X.; Liu, M.; Cheng, P.; Chen, J.; Liu, F.; Xu, J. Punicalagin inhibits inflammation in LPS-Induced RAW264.7 macrophages via the suppression of TLR4-mediated MAPKs and NF-κB activation. Inflammation 2014, 37, 956–965. [Google Scholar] [CrossRef]
- Gao, Y.; Jiang, W.; Dong, C.; Li, C.; Fu, X.; Min, L.; Tian, J.; Jin, H.; Shen, J. Anti-inflammatory effects of sophocarpine in LPS-induced Raw 264.7 cells via NF-κB and MAPKs signaling pathways. Toxicol. In Vitro 2012, 26, 1–6. [Google Scholar] [CrossRef]
- Zhang, W.; Li, X.; Zhang, Y. Rho-kinase inhibitor attenuates airway mucus hypersecretion and inflammation partly by downregulation of IL-13 and the JNK1/2-AP1 signaling pathway. Biochem. Biophys. Res. Commun. 2019, 516, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Minden, A.; Karin, M. Regulation and function of the JNK subgroup of MAP kinases. Biochim. Biophys. Acta Rev. Cancer 1997, 1333, F85–F104. [Google Scholar] [CrossRef]
- Lee, W.; Mitchell, P.; Tjian, R. Purified transcription factor AP-1 interacts with TPA-inducible enhancer elements. Cell 1987, 49, 741–752. [Google Scholar] [CrossRef]
- Gensch, E.; Gallup, M.; Sucher, A.; Li, D.; Gebremichael, A.; Lemjabbar, H.; Mengistab, A.; Dasari, V.; Hotchkiss, J.; Harkema, J.; et al. Tobacco smoke control of mucin production in lung cells requires oxygen radicals AP-1 and JNK. J. Biol. Chem. 2004, 279, 39085–39093. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Li, H.; Yu, L.; Wang, N.; Li, X.; Chen, W. IL-1β upregulates Muc5ac expression via NF-κB-induced HIF-1α in asthma. Immunol. Lett. 2017, 192, 20–26. [Google Scholar] [CrossRef]
- Kim, D.Y.; Yang, W.M. Panax ginseng ameliorates airway inflammation in an ovalbumin-sensitized mouse allergic asthma model. J. Ethnopharmacol. 2011, 136, 230–235. [Google Scholar] [CrossRef]
- Kuo, C.Y.; Wang, T.; Dai, T.Y.; Wang, C.H.; Chen, K.N.; Chen, Y.P.; Chen, M.J. Effect of the velvet antler of formosan sambar deer (Cervus unicolor swinhoei) on the prevention of an allergic airway response in mice. Evid. Based Complement. Altern. Med. 2012, 2012, 10. [Google Scholar] [CrossRef]
- Sun, J.; Li, L.; Wu, J.; Liu, B.; Gong, W.; Lv, Y.; Luo, Q.; Duan, X.; Dong, J. Effects of baicalin on airway remodeling in asthmatic mice. Planta Med. 2013, 79, 199–206. [Google Scholar] [CrossRef]
- Santini, A.; Tenore, G.C.; Novellino, E. Nutraceuticals, a paradigm of proactive medicine. Eur. J. Pharm. Sci. 2017, 1, 53–61. [Google Scholar] [CrossRef]
- Nanni, V.; Canuti, L.; Gismondi, A.; Canini, A. Hydroalcoholic extract of Spartium junceum L. flowers inhibits growth and melanogenesis in B16-F10 cells by inducing senescence. Pytomedicine 2018, 15, 1–10. [Google Scholar] [CrossRef]
- Nardi, G.M.; Januario, A.G.F.; Freire, C.G.; Megiolaro, F.; Schneider, K.; Perazzoli, M.R.A.; Nascimento, S.R.D.; Gon, A.C.; Mariano, L.N.B.; Wagner, G.; et al. Anti-inflammatory activity of Berry fruits in mice model of inflammation is based on oxidative stress modulation. Pharm. Res. 2016, 8, S42–S49. [Google Scholar]
- Gutierrez, R.; Alvarado, J.L.; Presno, M.; Perez-Veyna, O.; Serrano, C.J.; Yahuaca, P. Oxidative stress modulation by Rosmarinus officinalis in CCI4-induced liver cirrhosis. Phytother. Res. 2010, 24, 595–601. [Google Scholar] [CrossRef] [PubMed]
- Yook, C. Medical Plants of Korea; Academy Publishing Co.: Seoul, Korea, 1989; p. 184. [Google Scholar]
- Choi, E.J.; Lee, S.; Kim, H.H.; Singh, T.S.; Choi, J.K.; Choi, H.G.; Suh, W.M.; Lee, S.H.; Kim, S.H. Suppression of dust mite extract and 2,4-dinitro-chloro-benzene-induced atopic dermatitis by the water extract of Lindera obtusiloba. J. Ethnopharmacol. 2011, 137, 802–807. [Google Scholar] [CrossRef] [PubMed]
- Won, D.H.; Han, S.B.; Hwang, J.P.; Kim, S.J.; Park, J.; Park, S.N. Antioxidative effect and tyrosinase inhibitory activity of Lindera obtusiloba blume extracts. J. Soc. Cosmet. Sci. Korea 2012, 38, 297–304. [Google Scholar]
- Suh, W.M.; Park, S.B.; Lee, S.; Kim, H.H.; Suk, K.; Son, J.H.; Kwon, T.K.; Choi, H.G.; Lee, S.H.; Kim, S.H. Suppression of mast-cell-mediated allergic inflammation by Lindera obtusiloba. Exp. Biol. Med. 2011, 236, 204–246. [Google Scholar] [CrossRef] [PubMed]
- Melgert, B.N.; Postma, D.S.; Kuipers, I.; Geerlings, M.; Luinge, M.A.; Van Der Strate, B.W.; Kerstjens, H.A.; Timens, W.; Hylkema, M.N. Female mice are more susceptible to the development of allergic airway inflammation than male mice. Clin. Exp. Allergy 2005, 35, 1496–1503. [Google Scholar] [CrossRef]
- Shin, I.S.; Park, J.W.; Shin, N.R.; Jeon, C.M.; Kwon, O.K.; Kim, J.S.; Kim, J.C.; Oh, S.R.; Ahn, K.S. Melatonin reduces airway inflammation in ovalbumin-induced asthma. Immunobiology 2014, 219, 901–908. [Google Scholar] [CrossRef]
- Shalaby, K.H.; Gold, L.G.; Schuessler, T.F.; Martin, J.G.; Robichaud, A. Combined forced oscillation and forced expiration measurements in mice for the assessment of airway hyperresponsiveness. Respir. Res. 2010, 11, 82. [Google Scholar] [CrossRef]
- Shin, N.R.; Shin, I.S.; Song, H.H.; Hong, J.M.; Kwon, O.K.; Jeon, C.M.; Kim, J.H.; Lee, S.W.; Lee, J.K.; Jin, H.; et al. Callicarpa japonica Thunb. reduces inflammatory responses: A mouse model of lipopolysaccharide-induced acute lung injury. Int. Immunopharmacol. 2015, 26, 174–180. [Google Scholar] [CrossRef]
- Abderrahim, F.; Arribas, S.M.; Gonzalez, M.C.; Condezo-Hoyos, L. Rapid high-throughput assay to assess scavenging capacity index using DPPH. Food Chem. 2013, 15, 788–794. [Google Scholar] [CrossRef] [PubMed]
- Kawamoto, Y.; Morinaga, Y.; Kimura, Y.; Kaku, N.; Kosai, K.; Uno, N.; Hasegawa, H.; Yanagihara, K. TNF-alpha inhibits the growth of Legionella pneumophila in airway epithelial cells by inducing apoptosis. J. Infect. Chemother. 2017, 23, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Ryu, E.K.; Kim, T.H.; Jang, E.J.; Choi, Y.S.; Kim, S.T.; Hahm, K.B. Wogonin, a plant flavone from Scutellariae radix, attenuated ovalbumin-induced airway inflammation in mouse model of asthma via the suppression of IL-4/STAT6 signaling. J. Clin. Biochem. Nutr. 2015, 57, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Kurakula, K.; Vos, M.; Logiantara, A.; Roelofs, J.J.; Nieuwenhum, M.A.; Koppelman, G.H.; Postma, D.S.; Van Rijt, L.S.; De Vries, C.J. Nuclear receptor Nur 77 attenuates airway inflammation in mice by suppressing NF-κB activity in lung epithelial cells. J. Immunol. 2015, 195, 1388–1398. [Google Scholar] [CrossRef]
- Hvattum, E. Determination of phenolic compounds in rose hip (Rosa canina) using liquid chromatography coupled to electrospray ionisation tandem mass spectrometry and diode-array detection. Rapid Commun. Mass Spectrom. 2002, 16, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, T.; Nebehaj, E.; Albert, L. The high-performance liquid chromatography/multistage electrospray mass spectrometric investigation and extraction optimization of beech (Fagus sylvatica L.) bark polyphenols. J. Chromatogr. A 2015, 1393, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Abu-Reidah, I.M.; Del Mar, C.M.; Arraez-Roman, D.; Fernandez-Gutierrez, A.; Segura-Carretero, A. UHPLC-ESI-QTOF-MS-based metabolic profiling of Vicia faba L. (Fabaceae) seeds as a key strategy for characterization in foodomics. Electrophoresis 2014, 35, 1571–1581. [Google Scholar] [CrossRef]
- Hong, C.O.; Rhee, C.H.; Won, N.H.; Choi, H.D.; Lee, K.W. Protective effect of 70% ethanolic extract of Lindera obtusiloba Blume on tert-butyl hydroperoxide-induced oxidative hepatotoxicity in rats. Food Chem. Toxicol. 2013, 53, 214–220. [Google Scholar] [CrossRef]
- Gauvreau, G.M.; Ellis, A.K.; Denburg, J.A. Haemopoietic processes in allergic disease, eosinophil/basophil development. Clin. Exp. Allergy 2009, 39, 1297–1306. [Google Scholar] [CrossRef]
- Uhm, T.G.; Kim, B.S.; Chung, I.Y. Eosinophil development, regulation of eosinophil-specific genes, and role of eosinophils in the pathogenesis of asthma. Allergy Asthma Immunol. Res. 2012, 4, 68–79. [Google Scholar] [CrossRef]
- Yuk, J.E.; Lee, M.Y.; Kwon, O.K.; Cai, X.F.; Jang, H.Y.; Oh, S.R.; Lee, H.K.; Ahn, K.S. Effects of astilbic acid on airway hyperresponsiveness and inflammation in a mouse model of allergic asthma. Int. Immunopharmacol. 2011, 11, 266–273. [Google Scholar] [CrossRef]
- Inam, A.; Shahzad, M.; Shabbir, A.; Shahid, H.; Shahid, K.; Javeed, A. Carica papaya ameliorates allergic asthma via down regulation of IL-4, IL-5, eotaxin, TNF-α, NF-κB, and iNOS levels. Phytomedicine 2017, 32, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Rothenberg, M.E.; Hogan, S.P. The eosinophil. Annu. Rev. Immunol. 2006, 24, 147–174. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Dai, J.; Liu, H.; Li, R.R.; Sun, P.L.; Du, Q.; Pang, L.L.; Chen, Z.; Yin, K.S. Naringenin inhibits allergen-induced airway inflammation and airway responsiveness and inhibits NF-κB activity in a murine model of asthma. Can. J. Physiol. Pharmacol. 2009, 87, 729–735. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Luo, F.; Lu, Q.; Liu, J.; Li, P.; Wang, X.; Fu, Y.; Hao, K.; Yan, T.; Ding, X. The protective effect of Trillin LPS-induced acute lung injury by the regulations of inflammation and oxidative state. Chem. -Bioloqical Interact. 2016, 243, 127–134. [Google Scholar] [CrossRef]
- Yao, W.; Luo, G.; Zhu, G.; Chi, X.; Zhang, A.; Xia, Z.; Hei, Z. Propofol activation of the Nrf2 pathway is associated with amelioration of acute lung injury in a rat liver transplantation model. Oxidative Med. Cell. Longev. 2014, 2014, 258567. [Google Scholar] [CrossRef]
- Ganesh, Y.V.; Negi, G.; Sharma, S.S.; Kumar, A. Potential therapeutic effects of the simultaneous targeting of the Nrf2 and NF-κB pathways in diabetic neuropathy. Redox Biol. 2013, 1, 394–397. [Google Scholar] [CrossRef] [PubMed]
- Park, K.R.; Nam, D.; Yun, H.M.; Lee, S.G.; Jang, H.J.; Sethi, G.; Cho, S.K.; Ahn, K.S. β-caryophyllene oxide inhibits growth and induces apoptosis through the suppression of PI3K/AKT/mTOR/S6K1 pathways and ROS-mediated MAPKs activation. Cancer Lett. 2011, 312, 178–188. [Google Scholar] [CrossRef]
- Alam, R.; Gorska, M.M. Mitogen-activated protein kinase signaling and ERK1/2 bistability in asthma. Clin. Exp. Allergy 2010, 41, 149–159. [Google Scholar] [CrossRef]
- Henderson, W.R.; Chi, E.Y.; Teo, J.L.; Nguyen, C.; Kahn, M. A small molecule inhibitor of redox-regulated NF-kappa B and activator protein-1 transcription blocks allergic airway inflammation in a mouse asthma model. J. Immunol. 2002, 169, 5294–5299. [Google Scholar] [CrossRef]
- Su, Y.W.; Chiou, W.F.; Chao, S.H.; Lee, M.H.; Chen, C.C.; Tsai, Y.C. Ligustilide prevents LPS-induced iNOS expression in RAW 264.7 macrophages by preventing ROS production and down-regulating the MAPK, NF-kappaB and AP-1 signaling pathways. Int. Immunopharmacol. 2011, 11, 1166–1172. [Google Scholar] [CrossRef]
- Chen, R.; Lim, J.H.; Jono, H.; Gu, X.X.; Kim, Y.S.; Basbaum, C.B.; Murphy, T.F.; Li, J.D. Nontypeable Haemophilus influenzae lipoprotein P6 induces MUC5AC mucin transcription via TLR2-TAK1-dependent p38 MAPK-AP1 and IKKbeta-Ikappa-Balpha-NF-kappaB signaling pathways. Biochem. Biophys. Res. Commun. 2004, 324, 1087–1094. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.C.; Choi, S.U.; Lee, J.O.; Bae, K.H.; Zee, O.P.; Lee, K.R. Two new lignans from Lindera obtusiloba blume. Arch. Pharmacal Res. 1999, 22, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Freise, C.; Ruehl, M.; Erben, U.; Neumann, U.; Seehofer, D.; Kim, K.Y.; Trowitzsch, K.W.; Stroh, T.; Zeitz, M.; Somasundaram, R. A hepatoprotective Lindera obtusiloba extract suppresses growth and attenuates insulin like growth factor-1 receptor signaling and NF-kappaB activity in human liver cancer cell lines. BMC Complement. Altern. Med. 2011, 11, 39. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, M.Z.; Guzel, A.; Torun, A.C.; Okuyucu, A.; Salis, O.; Karli, R.; Gacar, A.; Guvenc, T.; Paksu, S.; Urey, V.; et al. The therapeutic effects of anti-oxidant and anti-inflammatory drug quercetin on aspiration-induced lung injury in rats. J. Mol. Histol. 2014, 45, 195–203. [Google Scholar] [CrossRef]
- Chung, M.J.; Pandey, R.P.; Choi, J.W.; Sohng, J.K.; Choi, D.J.; Park, Y.I. Inhibitory effects of kaempferol-3-O-rhamnoside on ovalbumin-induced lung inflammation in a mouse model of allergic asthma. Int. Immunopharmacol. 2015, 25, 302–310. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NO. | tR (min) | Formula | Detected m/z | Exacted m/z | Error (ppm) | Fragments | Identification | Content (mg/g) |
|---|---|---|---|---|---|---|---|---|
| 1 | 5.33 | C21H20O11 | 447.09323 | 447.09329 | 0.12 | 447, 301, 300, 271,255, 243 | Quercetin rhamnoside | 26.04 ± 0.05 |
| 2 | 5.84 | C21H20O10 | 431.09840 | 431.09837 | 0.07 | 431, 285 | Kaempferol rhamnoside | 15.82 ± 0.02 |
| 3 | 4.35 | C20H20O11 | 435.09433 | 435.09329 | 1.26 | 435, 303, 285, 151 | Unknown | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, B.-W.; Ha, J.-H.; Shin, H.-G.; Jeong, S.-H.; Kim, J.-H.; Lee, J.; Park, J.-Y.; Kwon, H.-J.; Jung, K.; Lee, W.-S.; et al. Lindera obtusiloba Attenuates Oxidative Stress and Airway Inflammation in a Murine Model of Ovalbumin-Challenged Asthma. Antioxidants 2020, 9, 563. https://doi.org/10.3390/antiox9070563
Lee B-W, Ha J-H, Shin H-G, Jeong S-H, Kim J-H, Lee J, Park J-Y, Kwon H-J, Jung K, Lee W-S, et al. Lindera obtusiloba Attenuates Oxidative Stress and Airway Inflammation in a Murine Model of Ovalbumin-Challenged Asthma. Antioxidants. 2020; 9(7):563. https://doi.org/10.3390/antiox9070563
Chicago/Turabian StyleLee, Ba-Wool, Ji-Hye Ha, Han-Gyo Shin, Seong-Hun Jeong, Ju-Hong Kim, Jihye Lee, Ji-Young Park, Hyung-Jun Kwon, Kyungsook Jung, Woo-Song Lee, and et al. 2020. "Lindera obtusiloba Attenuates Oxidative Stress and Airway Inflammation in a Murine Model of Ovalbumin-Challenged Asthma" Antioxidants 9, no. 7: 563. https://doi.org/10.3390/antiox9070563
APA StyleLee, B.-W., Ha, J.-H., Shin, H.-G., Jeong, S.-H., Kim, J.-H., Lee, J., Park, J.-Y., Kwon, H.-J., Jung, K., Lee, W.-S., Ryu, Y.-B., Jeong, J.-H., & Lee, I.-C. (2020). Lindera obtusiloba Attenuates Oxidative Stress and Airway Inflammation in a Murine Model of Ovalbumin-Challenged Asthma. Antioxidants, 9(7), 563. https://doi.org/10.3390/antiox9070563