When Oxidative Stress Meets Epigenetics: Implications in Cancer Development


Abstract
1. Introduction
1.1. Cancer
1.2. Epigenetics
1.2.1. DNA Methylation
1.2.2. Histone Modifications
1.2.3. Non-Coding RNAs
2. Implication of Epigenetics in the Development of Cancer
2.1. microRNAs
2.2. piRNAs and siRNAs
2.3. Long Non-Coding RNAs
3. Oxidative Stress and Its Effect in the Epigenetic Machinery to Promote Cancer
3.1. Effect over DNA Methylation
3.2. Effect over Histone Modifications
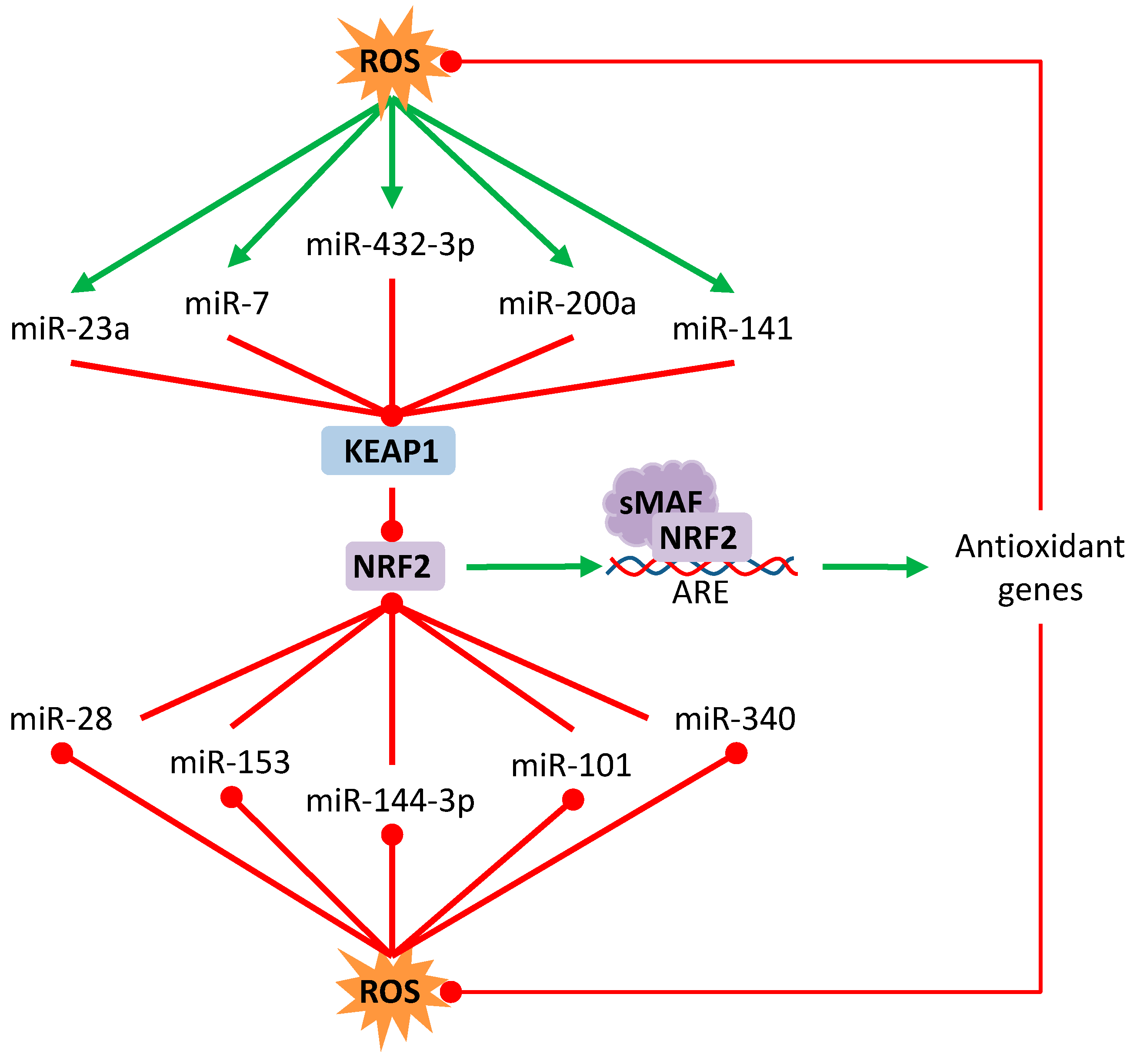
3.3. Effect over Non-Coding RNAs
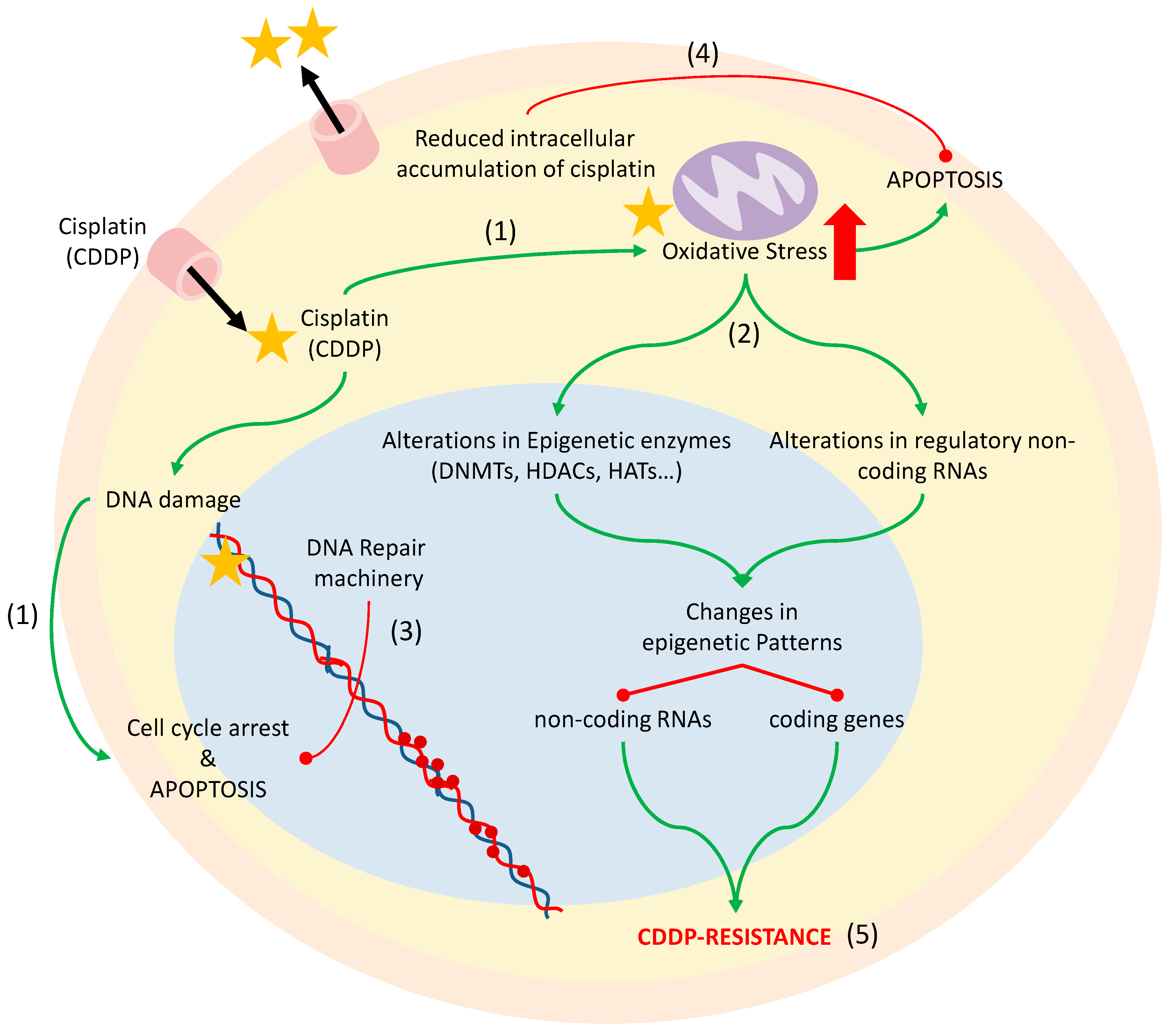
4. Chemotherapy-Induced Oxidative Stress: Epigenetics and Cisplatin Resistance
4.1. Cisplatin Resistance Mechanisms
4.2. DNA Methylation and Histone Modifications
4.3. microRNAs
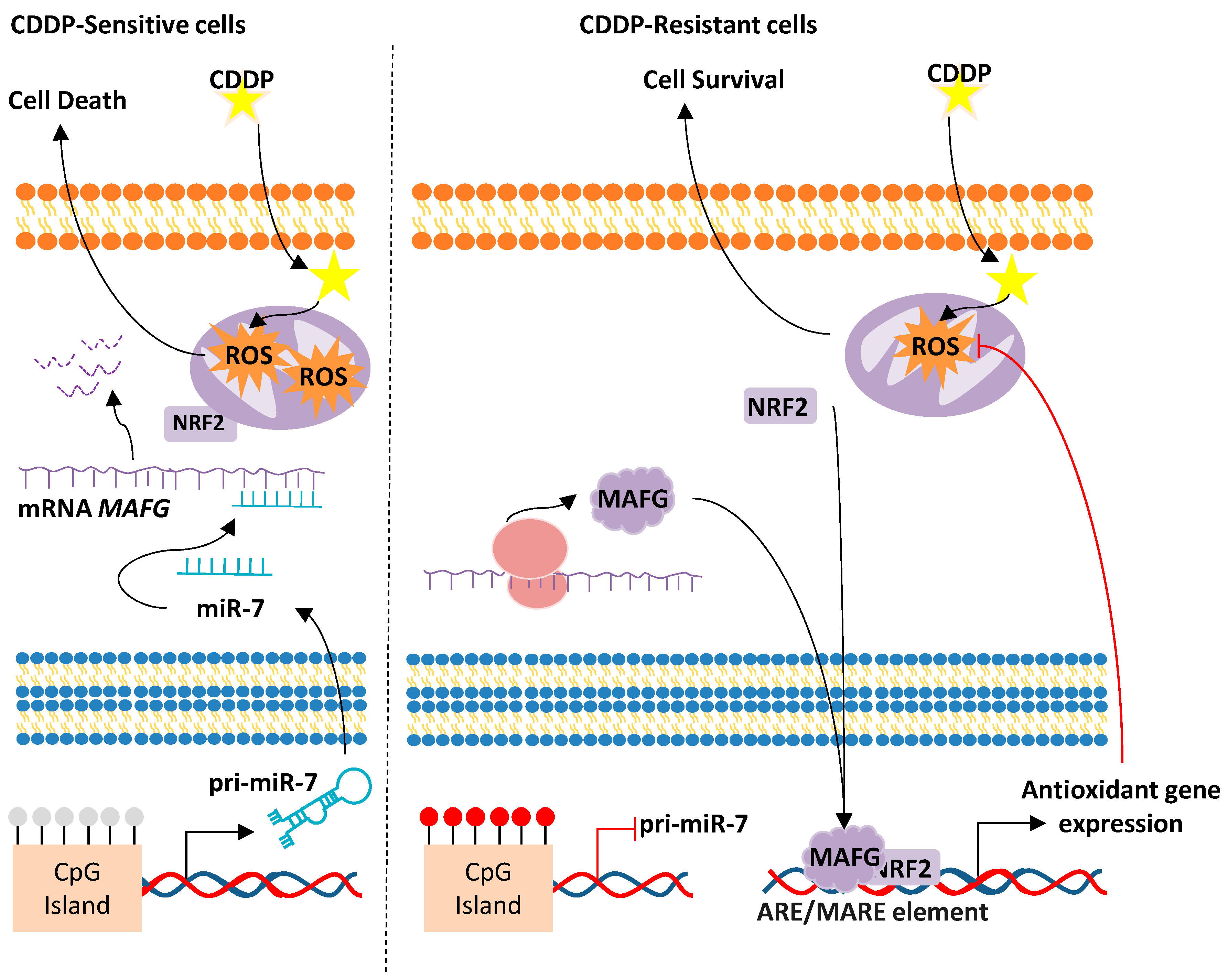
miR-7/MAFG/ROS axis
4.4. Long Non-Coding RNA
5. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- World Health Organ. 2018. Available online: http://www.who.int/cancer/en/ (accessed on 3 May 2020).
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Luo, J.; Solimini, N.L.; Elledge, S.J. Principles of cancer therapy: Oncogene and non-oncogene addiction. Cell 2009, 136, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Bishop, K.S.; Ferguson, L.R. The interaction between epigenetics, nutrition and the development of cancer. Nutrients 2015, 7, 922–947. [Google Scholar] [CrossRef]
- Stirzaker, C.; Taberlay, P.C.; Statham, A.L.; Clark, S.J. Mining cancer methylomes: Prospects and challenges. Trends Genet. TIG 2014, 30, 75–84. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2013, 38, 23–38. [Google Scholar] [CrossRef]
- Zampieri, M.; Ciccarone, F.; Calabrese, R.; Franceschi, C.; Burkle, A.; Caiafa, P. Reconfiguration of DNA methylation in aging. Mech. Ageing Dev. 2015, 151, 60–70. [Google Scholar] [CrossRef]
- Deplus, R.; Brenner, C.; Burgers, W.A.; Putmans, P.; Kouzarides, T.; de Launoit, Y.; Fuks, F. Dnmt3L is a transcriptional repressor that recruits histone deacetylase. Nucleic Acids Res. 2002, 30, 3831–3838. [Google Scholar] [CrossRef]
- Brown, S.D. XIST and the mapping of the X chromosome inactivation centre. Bioessays 1991, 13, 607–612. [Google Scholar] [CrossRef]
- Ng, K.; Pullirsch, D.; Leeb, M.; Wutz, A. Xist and the order of silencing. EMBO Rep. 2007, 8, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Boumil, R.M.; Ogawa, Y.; Sun, B.K.; Huynh, K.D.; Lee, J.T. Differential methylation of Xite and CTCF sites in Tsix mirrors the pattern of X-inactivation choice in mice. Mol. Cell. Biol. 2006, 26, 2109–2117. [Google Scholar] [CrossRef] [PubMed]
- Reik, W.; Walter, J. Genomic imprinting: Parental influence on the genome. Nat. Rev. Genet. 2001, 2, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Portela, A.; Esteller, M. Epigenetic modifications and human disease. Nat. Biotechnol. 2010, 28, 1057–1068. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Xia, Y.; Hawke, D.; Li, X.; Liang, J.; Xing, D.; Aldape, K.; Hunter, T.; Alfred Yung, W.K.; Lu, Z. PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell 2012, 150, 685–696. [Google Scholar] [CrossRef]
- Lau, P.N.; Cheung, P. Histone code pathway involving H3 S28 phosphorylation and K27 acetylation activates transcription and antagonizes polycomb silencing. Proc. Natl. Acad. Sci. USA 2011, 108, 2801–2806. [Google Scholar] [CrossRef]
- Dawson, M.A.; Bannister, A.J.; Gottgens, B.; Foster, S.D.; Bartke, T.; Green, A.R.; Kouzarides, T. JAK2 phosphorylates histone H3Y41 and excludes HP1alpha from chromatin. Nature 2009, 461, 819–822. [Google Scholar] [CrossRef]
- Cohen-Armon, M.; Visochek, L.; Rozensal, D.; Kalal, A.; Geistrikh, I.; Klein, R.; Bendetz-Nezer, S.; Yao, Z.; Seger, R. DNA-independent PARP-1 activation by phosphorylated ERK2 increases Elk1 activity: A link to histone acetylation. Mol. Cell 2007, 25, 297–308. [Google Scholar] [CrossRef]
- Moss, E.; Halkos, M.E. Cost effectiveness of robotic mitral valve surgery. Ann. Cardiothorac. Surg. 2017, 6, 33–37. [Google Scholar] [CrossRef]
- Gill, G. Something about SUMO inhibits transcription. Curr. Opin. Genet. Dev. 2005, 15, 536–541. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.H.; Sharrocks, A.D. SUMO promotes HDAC-mediated transcriptional repression. Mol. Cell 2004, 13, 611–617. [Google Scholar] [CrossRef]
- Meas, R.; Mao, P. Histone ubiquitylation and its roles in transcription and DNA damage response. DNA Repair 2015, 36, 36–42. [Google Scholar] [CrossRef]
- Oya, E.; Nakagawa, R.; Yoshimura, Y.; Tanaka, M.; Nishibuchi, G.; Machida, S.; Shirai, A.; Ekwall, K.; Kurumizaka, H.; Tagami, H.; et al. H3K14 ubiquitylation promotes H3K9 methylation for heterochromatin assembly. EMBO Rep. 2019, 20, e48111. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Guermah, M.; McGinty, R.K.; Lee, J.S.; Tang, Z.; Milne, T.A.; Shilatifard, A.; Muir, T.W.; Roeder, R.G. RAD6-Mediated transcription-coupled H2B ubiquitylation directly stimulates H3K4 methylation in human cells. Cell 2009, 137, 459–471. [Google Scholar] [CrossRef]
- Mattick, J.S.; Makunin, I.V. Non-coding RNA. Hum. Mol. Genet. 2006, 15, R17–R29. [Google Scholar] [CrossRef]
- Gibb, E.A.; Brown, C.J.; Lam, W.L. The functional role of long non-coding RNA in human carcinomas. Mol. Cancer 2011, 10, 38. [Google Scholar] [CrossRef]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef]
- Ponting, C.P.; Oliver, P.L.; Reik, W. Evolution and functions of long noncoding RNAs. Cell 2009, 136, 629–641. [Google Scholar] [CrossRef]
- Sanchez-Perez, I.; Murguia, J.R.; Perona, R. Cisplatin induces a persistent activation of JNK that is related to cell death. Oncogene 1998, 16, 533–540. [Google Scholar] [CrossRef]
- McPherson, S.; McMullin, M.F.; Mills, K. Epigenetics in Myeloproliferative Neoplasms. J. Cell. Mol. Med. 2017, 21, 1660–1667. [Google Scholar] [CrossRef] [PubMed]
- Machova Polakova, K.; Koblihova, J.; Stopka, T. Role of epigenetics in chronic myeloid leukemia. Curr. Hematol. Malig. Rep. 2013, 8, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Amodio, N.; D’Aquila, P.; Passarino, G.; Tassone, P.; Bellizzi, D. Epigenetic modifications in multiple myeloma: Recent advances on the role of DNA and histone methylation. Expert Opin. Ther. Targets 2017, 21, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Rahmani, M.; Talebi, M.; Hagh, M.F.; Feizi, A.A.H.; Solali, S. Aberrant DNA methylation of key genes and Acute Lymphoblastic Leukemia. Biomed. Pharmacother. Biomed. Pharmacother. 2018, 97, 1493–1500. [Google Scholar] [CrossRef]
- Figueroa, M.E.; Lugthart, S.; Li, Y.; Erpelinck-Verschueren, C.; Deng, X.; Christos, P.J.; Schifano, E.; Booth, J.; van Putten, W.; Skrabanek, L.; et al. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell 2010, 17, 13–27. [Google Scholar] [CrossRef]
- Guieze, R.; Wu, C.J. Genomic and epigenomic heterogeneity in chronic lymphocytic leukemia. Blood 2015, 126, 445–453. [Google Scholar] [CrossRef]
- Mansouri, L.; Wierzbinska, J.A.; Plass, C.; Rosenquist, R. Epigenetic deregulation in chronic lymphocytic leukemia: Clinical and biological impact. Semin. Cancer Biol. 2018, 51, 1–11. [Google Scholar] [CrossRef]
- Das, P.M.; Singal, R. DNA methylation and cancer. J. Clin. Oncol. 2004, 22, 4632–4642. [Google Scholar] [CrossRef]
- Esteller, M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat. Rev. Genet. 2007, 8, 286–298. [Google Scholar] [CrossRef]
- Koch, A.; Joosten, S.C.; Feng, Z.; de Ruijter, T.C.; Draht, M.X.; Melotte, V.; Smits, K.M.; Veeck, J.; Herman, J.G.; Van Neste, L.; et al. Analysis of DNA methylation in cancer: Location revisited. Nat. Rev. Clin. Oncol. 2018, 15, 459–466. [Google Scholar] [CrossRef]
- Chi, P.; Allis, C.D.; Wang, G.G. Covalent histone modifications--miswritten, misinterpreted and mis-erased in human cancers. Nat. Rev. Cancer 2010, 10, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Morera, L.; Lubbert, M.; Jung, M. Targeting histone methyltransferases and demethylases in clinical trials for cancer therapy. Clin. Epigenet. 2016, 8, 57. [Google Scholar] [CrossRef] [PubMed]
- Nacev, B.A.; Feng, L.; Bagert, J.D.; Lemiesz, A.E.; Gao, J.; Soshnev, A.A.; Kundra, R.; Schultz, N.; Muir, T.W.; Allis, C.D. The expanding landscape of ‘oncohistone’ mutations in human cancers. Nature 2019, 567, 473–478. [Google Scholar] [CrossRef]
- Gulyaeva, L.F.; Kushlinskiy, N.E. Regulatory mechanisms of microRNA expression. J. Transl. Med. 2016, 14, 143. [Google Scholar] [CrossRef] [PubMed]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef]
- Davis, B.N.; Hata, A. Regulation of MicroRNA Biogenesis: A miRiad of mechanisms. Cell Commun. Signal. CCS 2009, 7, 18. [Google Scholar] [CrossRef]
- Landthaler, M.; Yalcin, A.; Tuschl, T. The human DiGeorge syndrome critical region gene 8 and Its D. melanogaster homolog are required for miRNA biogenesis. Curr. Biol. CB 2004, 14, 2162–2167. [Google Scholar] [CrossRef]
- Calin, G.A.; Dumitru, C.D.; Shimizu, M.; Bichi, R.; Zupo, S.; Noch, E.; Aldler, H.; Rattan, S.; Keating, M.; Rai, K.; et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 15524–15529. [Google Scholar] [CrossRef]
- Mendell, J.T.; Olson, E.N. MicroRNAs in stress signaling and human disease. Cell 2012, 148, 1172–1187. [Google Scholar] [CrossRef]
- Kwon, J.J.; Factora, T.D.; Dey, S.; Kota, J. A Systematic Review of miR-29 in Cancer. Mol. Ther. Oncolytics 2019, 12, 173–194. [Google Scholar] [CrossRef]
- Creighton, C.J.; Hernandez-Herrera, A.; Jacobsen, A.; Levine, D.A.; Mankoo, P.; Schultz, N.; Du, Y.; Zhang, Y.; Larsson, E.; Sheridan, R.; et al. Integrated analyses of microRNAs demonstrate their widespread influence on gene expression in high-grade serous ovarian carcinoma. PLoS ONE 2012, 7, e34546. [Google Scholar] [CrossRef]
- Fabbri, M.; Garzon, R.; Cimmino, A.; Liu, Z.; Zanesi, N.; Callegari, E.; Liu, S.; Alder, H.; Costinean, S.; Fernandez-Cymering, C.; et al. MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc. Natl. Acad. Sci. USA 2007, 104, 15805–15810. [Google Scholar] [CrossRef] [PubMed]
- Parpart, S.; Roessler, S.; Dong, F.; Rao, V.; Takai, A.; Ji, J.; Qin, L.X.; Ye, Q.H.; Jia, H.L.; Tang, Z.Y.; et al. Modulation of miR-29 expression by alpha-fetoprotein is linked to the hepatocellular carcinoma epigenome. Hepatology 2014, 60, 872–883. [Google Scholar] [CrossRef]
- Nguyen, T.; Kuo, C.; Nicholl, M.B.; Sim, M.S.; Turner, R.R.; Morton, D.L.; Hoon, D.S. Downregulation of microRNA-29c is associated with hypermethylation of tumor-related genes and disease outcome in cutaneous melanoma. Epigenetics 2011, 6, 388–394. [Google Scholar] [CrossRef] [PubMed]
- Amodio, N.; Rossi, M.; Raimondi, L.; Pitari, M.R.; Botta, C.; Tagliaferri, P.; Tassone, P. miR-29s: A family of epi-miRNAs with therapeutic implications in hematologic malignancies. Oncotarget 2015, 6, 12837–12861. [Google Scholar] [CrossRef]
- Mazzoccoli, L.; Robaina, M.C.; Apa, A.G.; Bonamino, M.; Pinto, L.W.; Queiroga, E.; Bacchi, C.E.; Klumb, C.E. MiR-29 silencing modulates the expression of target genes related to proliferation, apoptosis and methylation in Burkitt lymphoma cells. J. Cancer Res. Clin. Oncol. 2018, 144, 483–497. [Google Scholar] [CrossRef] [PubMed]
- Bui, T.V.; Mendell, J.T. Myc: Maestro of MicroRNAs. Genes Cancer 2010, 1, 568–575. [Google Scholar] [CrossRef]
- Hermeking, H. MicroRNAs in the p53 network: Micromanagement of tumour suppression. Nat. Rev. Cancer 2012, 12, 613–626. [Google Scholar] [CrossRef]
- Furuta, M.; Kozaki, K.I.; Tanaka, S.; Arii, S.; Imoto, I.; Inazawa, J. miR-124 and miR-203 are epigenetically silenced tumor-suppressive microRNAs in hepatocellular carcinoma. Carcinogenesis 2010, 31, 766–776. [Google Scholar] [CrossRef]
- Balaguer, F.; Link, A.; Lozano, J.J.; Cuatrecasas, M.; Nagasaka, T.; Boland, C.R.; Goel, A. Epigenetic silencing of miR-137 is an early event in colorectal carcinogenesis. Cancer Res. 2010, 70, 6609–6618. [Google Scholar] [CrossRef]
- Iwasaki, Y.W.; Siomi, M.C.; Siomi, H. PIWI-Interacting RNA: Its Biogenesis and Functions. Annu. Rev. Biochem. 2015, 84, 405–433. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Dou, M.; Song, X.; Dong, Y.; Liu, S.; Liu, H.; Tao, J.; Li, W.; Yin, X.; Xu, W. The emerging role of the piRNA/piwi complex in cancer. Mol. Cancer 2019, 18, 123. [Google Scholar] [CrossRef] [PubMed]
- Hall, I.M.; Shankaranarayana, G.D.; Noma, K.; Ayoub, N.; Cohen, A.; Grewal, S.I. Establishment and maintenance of a heterochromatin domain. Science 2002, 297, 2232–2237. [Google Scholar] [CrossRef]
- Martinez, J.; Patkaniowska, A.; Urlaub, H.; Luhrmann, R.; Tuschl, T. Single-stranded antisense siRNAs guide target RNA cleavage in RNAi. Cell 2002, 110, 563–574. [Google Scholar] [CrossRef]
- Aravin, A.A.; Naumova, N.M.; Tulin, A.V.; Vagin, V.V.; Rozovsky, Y.M.; Gvozdev, V.A. Double-stranded RNA-mediated silencing of genomic tandem repeats and transposable elements in the D. melanogaster germline. Curr. Biol. CB 2001, 11, 1017–1027. [Google Scholar] [CrossRef]
- Yao, J.; Wang, Y.W.; Fang, B.B.; Zhang, S.J.; Cheng, B.L. piR-651 and its function in 95-D lung cancer cells. Biomed. Rep. 2016, 4, 546–550. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Luo, Y.; Gao, Y.; Yang, Y.; Wang, Y.; Xu, Y.; Tan, S.; Zhang, Y.; Duan, J.; Yang, Y. piR-651 promotes tumor formation in non-small cell lung carcinoma through the upregulation of cyclin D1 and CDK4. Int. J. Mol. Med. 2016, 38, 927–936. [Google Scholar] [CrossRef]
- Peng, L.; Song, L.; Liu, C.; Lv, X.; Li, X.; Jie, J.; Zhao, D.; Li, D. piR-55490 inhibits the growth of lung carcinoma by suppressing mTOR signaling. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2016, 37, 2749–2756. [Google Scholar] [CrossRef]
- Tan, L.; Mai, D.; Zhang, B.; Jiang, X.; Zhang, J.; Bai, R.; Ye, Y.; Li, M.; Pan, L.; Su, J.; et al. PIWI-interacting RNA-36712 restrains breast cancer progression and chemoresistance by interaction with SEPW1 pseudogene SEPW1P RNA. Mol. Cancer 2019, 18, 9. [Google Scholar] [CrossRef]
- Fu, A.; Jacobs, D.I.; Hoffman, A.E.; Zheng, T.; Zhu, Y. PIWI-interacting RNA 021285 is involved in breast tumorigenesis possibly by remodeling the cancer epigenome. Carcinogenesis 2015, 36, 1094–1102. [Google Scholar] [CrossRef]
- Weng, W.; Liu, N.; Toiyama, Y.; Kusunoki, M.; Nagasaka, T.; Fujiwara, T.; Wei, Q.; Qin, H.; Lin, H.; Ma, Y.; et al. Novel evidence for a PIWI-interacting RNA (piRNA) as an oncogenic mediator of disease progression, and a potential prognostic biomarker in colorectal cancer. Mol. Cancer 2018, 17, 16. [Google Scholar] [CrossRef]
- Singh, G.; Roy, J.; Rout, P.; Mallick, B. Genome-wide profiling of the PIWI-interacting RNA-mRNA regulatory networks in epithelial ovarian cancers. PLoS ONE 2018, 13, e0190485. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, R.A.; Cluster, P.D.; English, J.; Que, Q.; Napoli, C.A. Chalcone synthase cosuppression phenotypes in petunia flowers: Comparison of sense vs. antisense constructs and single-copy vs. complex T-DNA sequences. Plant Mol. Biol. 1996, 31, 957–973. [Google Scholar] [CrossRef]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Elbashir, S.M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001, 411, 494–498. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Trivedi, P.; Jain, N.K. Advances in siRNA delivery in cancer therapy. Artif. Cells Nanomed. Biotechnol. 2018, 46, 274–283. [Google Scholar] [CrossRef]
- Young, S.W.; Stenzel, M.; Yang, J.L. Nanoparticle-siRNA: A potential cancer therapy? Crit. Rev. Oncol. Hematol. 2016, 98, 159–169. [Google Scholar] [CrossRef]
- Dey, B.K.; Mueller, A.C.; Dutta, A. Long non-coding RNAs as emerging regulators of differentiation, development, and disease. Transcription 2014, 5, e944014. [Google Scholar] [CrossRef]
- Wilusz, J.E.; Sunwoo, H.; Spector, D.L. Long noncoding RNAs: Functional surprises from the RNA world. Genes Dev. 2009, 23, 1494–1504. [Google Scholar] [CrossRef]
- Mercer, T.R.; Dinger, M.E.; Mattick, J.S. Long non-coding RNAs: Insights into functions. Nat. Rev. Genet. 2009, 10, 155–159. [Google Scholar] [CrossRef]
- Wang, K.C.; Chang, H.Y. Molecular mechanisms of long noncoding RNAs. Mol. Cell 2011, 43, 904–914. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Bajic, V.B.; Zhang, Z. On the classification of long non-coding RNAs. RNA Biol. 2013, 10, 925–933. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Rinn, J.L. Modular regulatory principles of large non-coding RNAs. Nature 2012, 482, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.L. Linking Long Noncoding RNA Localization and Function. Trends Biochem. Sci. 2016, 41, 761–772. [Google Scholar] [CrossRef]
- Fatica, A.; Bozzoni, I. Long non-coding RNAs: New players in cell differentiation and development. Nat. Rev. Genet. 2014, 15, 7–21. [Google Scholar] [CrossRef]
- Bartolomei, M.S.; Zemel, S.; Tilghman, S.M. Parental imprinting of the mouse H19 gene. Nature 1991, 351, 153–155. [Google Scholar] [CrossRef]
- Brannan, C.I.; Dees, E.C.; Ingram, R.S.; Tilghman, S.M. The product of the H19 gene may function as an RNA. Mol. Cell. Biol. 1990, 10, 28–36. [Google Scholar] [CrossRef]
- Tripathi, V.; Ellis, J.D.; Shen, Z.; Song, D.Y.; Pan, Q.; Watt, A.T.; Freier, S.M.; Bennett, C.F.; Sharma, A.; Bubulya, P.A.; et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol. Cell 2010, 39, 925–938. [Google Scholar] [CrossRef]
- Ji, P.; Diederichs, S.; Wang, W.; Boing, S.; Metzger, R.; Schneider, P.M.; Tidow, N.; Brandt, B.; Buerger, H.; Bulk, E.; et al. MALAT-1, a novel noncoding RNA, and thymosin beta4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene 2003, 22, 8031–8041. [Google Scholar] [CrossRef]
- Zhou, Y.; Xu, X.; Lv, H.; Wen, Q.; Li, J.; Tan, L.; Sheng, X. The Long Noncoding RNA MALAT-1 Is Highly Expressed in Ovarian Cancer and Induces Cell Growth and Migration. PLoS ONE 2016, 11, e0155250. [Google Scholar] [CrossRef]
- Formisano, S.; Brewer, H.B., Jr.; Osborne, J.C., Jr. Effect of pressure and ionic strength on the self-association of Apo-A-I from the human high density lipoprotein complex. J. Biol. Chem. 1978, 253, 354–359. [Google Scholar]
- Hudson, A.M.; McMartin, C. Metabolism of tritiated 1-24 corticotrophin in peripheral tissues immediately after intravenous injection in the rat [proceedings]. J. Endocrinol. 1978, 79, 62P–63P. [Google Scholar] [PubMed]
- Sun, L.; Sun, P.; Zhou, Q.Y.; Gao, X.; Han, Q. Long noncoding RNA MALAT1 promotes uveal melanoma cell growth and invasion by silencing of miR-140. Am. J. Transl. Res. 2016, 8, 3939–3946. [Google Scholar] [PubMed]
- Li, F.; Li, X.; Qiao, L.; Liu, W.; Xu, C.; Wang, X. MALAT1 regulates miR-34a expression in melanoma cells. Cell Death Dis. 2019, 10, 389. [Google Scholar] [CrossRef] [PubMed]
- Amodio, N.; Stamato, M.A.; Juli, G.; Morelli, E.; Fulciniti, M.; Manzoni, M.; Taiana, E.; Agnelli, L.; Cantafio, M.E.G.; Romeo, E.; et al. Drugging the lncRNA MALAT1 via LNA gapmeR ASO inhibits gene expression of proteasome subunits and triggers anti-multiple myeloma activity. Leukemia 2018, 32, 1948–1957. [Google Scholar] [CrossRef]
- Stamato, M.A.; Juli, G.; Romeo, E.; Ronchetti, D.; Arbitrio, M.; Caracciolo, D.; Neri, A.; Tagliaferri, P.; Tassone, P.; Amodio, N. Inhibition of EZH2 triggers the tumor suppressive miR-29b network in multiple myeloma. Oncotarget 2017, 8, 106527–106537. [Google Scholar] [CrossRef]
- Amodio, N.; Raimondi, L.; Juli, G.; Stamato, M.A.; Caracciolo, D.; Tagliaferri, P.; Tassone, P. MALAT1: A druggable long non-coding RNA for targeted anti-cancer approaches. J. Hematol. Oncol. 2018, 11, 63. [Google Scholar] [CrossRef]
- Li, Z.X.; Zhu, Q.N.; Zhang, H.B.; Hu, Y.; Wang, G.; Zhu, Y.S. MALAT1: A potential biomarker in cancer. Cancer Manag. Res. 2018, 10, 6757–6768. [Google Scholar] [CrossRef]
- Huarte, M. The emerging role of lncRNAs in cancer. Nat. Med. 2015, 21, 1253–1261. [Google Scholar] [CrossRef]
- Bhan, A.; Soleimani, M.; Mandal, S.S. Long Noncoding RNA and Cancer: A New Paradigm. Cancer Res. 2017, 77, 3965–3981. [Google Scholar] [CrossRef]
- Peng, W.X.; Koirala, P.; Mo, Y.Y. LncRNA-mediated regulation of cell signaling in cancer. Oncogene 2017, 36, 5661–5667. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, A.M.; Chang, H.Y. Long Noncoding RNAs in Cancer Pathways. Cancer Cell 2016, 29, 452–463. [Google Scholar] [CrossRef] [PubMed]
- Betteridge, D.J. What is oxidative stress? Metab. Clin. Exp. 2000, 49, 3–8. [Google Scholar] [CrossRef]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: A Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef]
- Chen, X.L.; Kunsch, C. Induction of cytoprotective genes through Nrf2/antioxidant response element pathway: A new therapeutic approach for the treatment of inflammatory diseases. Curr. Pharm. Des. 2004, 10, 879–891. [Google Scholar] [CrossRef]
- Katsuoka, F.; Yamamoto, M. Small Maf proteins (MafF, MafG, MafK): History, structure and function. Gene 2016, 586, 197–205. [Google Scholar] [CrossRef]
- Hirotsu, Y.; Katsuoka, F.; Funayama, R.; Nagashima, T.; Nishida, Y.; Nakayama, K.; Engel, J.D.; Yamamoto, M. Nrf2-MafG heterodimers contribute globally to antioxidant and metabolic networks. Nucleic Acids Res. 2012, 40, 10228–10239. [Google Scholar] [CrossRef]
- Katsuoka, F.; Motohashi, H.; Ishii, T.; Aburatani, H.; Engel, J.D.; Yamamoto, M. Genetic evidence that small maf proteins are essential for the activation of antioxidant response element-dependent genes. Mol. Cell. Biol. 2005, 25, 8044–8051. [Google Scholar] [CrossRef]
- Perez, S.; Rius-Perez, S.; Tormos, A.M.; Finamor, I.; Nebreda, A.R.; Talens-Visconti, R.; Sastre, J. Age-dependent regulation of antioxidant genes by p38alpha MAPK in the liver. Redox Biol. 2018, 16, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef]
- Bourgeais, J.; Gouilleux-Gruart, V.; Gouilleux, F. Oxidative metabolism in cancer: A STAT affair? JAK-STAT 2013, 2, e25764. [Google Scholar] [CrossRef]
- Jung, J.E.; Kim, G.S.; Narasimhan, P.; Song, Y.S.; Chan, P.H. Regulation of Mn-superoxide dismutase activity and neuroprotection by STAT3 in mice after cerebral ischemia. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 7003–7014. [Google Scholar] [CrossRef]
- Inoue, I.; Goto, S.; Matsunaga, T.; Nakajima, T.; Awata, T.; Hokari, S.; Komoda, T.; Katayama, S. The ligands/activators for peroxisome proliferator-activated receptor alpha (PPARalpha) and PPARgamma increase Cu2+,Zn2+-superoxide dismutase and decrease p22phox message expressions in primary endothelial cells. Metab. Clin. Exp. 2001, 50, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Schumacker, P.T. Reactive oxygen species in cancer cells: Live by the sword, die by the sword. Cancer Cell 2006, 10, 175–176. [Google Scholar] [CrossRef]
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements. Biomolecules 2019, 9, 735. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Kudo, M. Oxidative stress and epigenetic instability in human hepatocarcinogenesis. Dig. Dis. 2013, 31, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Kreuz, S.; Fischle, W. Oxidative stress signaling to chromatin in health and disease. Epigenomics 2016, 8, 843–862. [Google Scholar] [CrossRef] [PubMed]
- Mahalingaiah, P.K.; Ponnusamy, L.; Singh, K.P. Oxidative stress-induced epigenetic changes associated with malignant transformation of human kidney epithelial cells. Oncotarget 2017, 8, 11127–11143. [Google Scholar] [CrossRef] [PubMed]
- Cyr, A.R.; Domann, F.E. The redox basis of epigenetic modifications: From mechanisms to functional consequences. Antioxid. Redox Signal. 2011, 15, 551–589. [Google Scholar] [CrossRef]
- Garcia-Gimenez, J.L.; Roma-Mateo, C.; Perez-Machado, G.; Peiro-Chova, L.; Pallardo, F.V. Role of glutathione in the regulation of epigenetic mechanisms in disease. Free Radic. Biol. Med. 2017, 112, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.C. Regulation of glutathione synthesis. Mol. Asp. Med. 2009, 30, 42–59. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Schoneveld, O.; Georgakilas, A.G.; Panayiotidis, M.I. Oxidative stress, DNA methylation and carcinogenesis. Cancer Lett. 2008, 266, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Arizumi, T.; Takita, M.; Kitai, S.; Yada, N.; Hagiwara, S.; Inoue, T.; Minami, Y.; Ueshima, K.; Sakurai, T.; et al. Reactive oxygen species induce epigenetic instability through the formation of 8-hydroxydeoxyguanosine in human hepatocarcinogenesis. Dig. Dis. 2013, 31, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Udomsinprasert, W.; Kitkumthorn, N.; Mutirangura, A.; Chongsrisawat, V.; Poovorawan, Y.; Honsawek, S. Global methylation, oxidative stress, and relative telomere length in biliary atresia patients. Sci. Rep. 2016, 6, 26969. [Google Scholar] [CrossRef]
- Ziech, D.; Franco, R.; Pappa, A.; Panayiotidis, M.I. Reactive oxygen species (ROS)--induced genetic and epigenetic alterations in human carcinogenesis. Mutat. Res. 2011, 711, 167–173. [Google Scholar] [CrossRef]
- Arita, A.; Costa, M. Oxidative Stress and the Epigenome in Human Disease. J. Genet. Genome Res. 2014, 1, 5. [Google Scholar] [CrossRef]
- Sundar, I.K.; Yao, H.; Rahman, I. Oxidative stress and chromatin remodeling in chronic obstructive pulmonary disease and smoking-related diseases. Antioxid. Redox Signal. 2013, 18, 1956–1971. [Google Scholar] [CrossRef]
- Yao, H.; Rahman, I. Role of histone deacetylase 2 in epigenetics and cellular senescence: Implications in lung inflammaging and COPD. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 303, L557–L566. [Google Scholar] [CrossRef]
- Sabbagh, Z.; Vatanparast, H. Is calcium supplementation a risk factor for cardiovascular diseases in older women? Nutr. Rev. 2009, 67, 105–108. [Google Scholar] [CrossRef]
- Lawless, M.W.; O’Byrne, K.J.; Gray, S.G. Oxidative stress induced lung cancer and COPD: Opportunities for epigenetic therapy. J. Cell. Mol. Med. 2009, 13, 2800–2821. [Google Scholar] [CrossRef] [PubMed]
- Fuschi, P.; Maimone, B.; Gaetano, C.; Martelli, F. Noncoding RNAs in the Vascular System Response to Oxidative Stress. Antioxid. Redox Signal. 2017. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.; Huang, Z.; Han, J.; Shao, J.; Huang, C. Redox regulation of microRNAs in cancer. Cancer Lett. 2018, 418, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Magenta, A.; Cencioni, C.; Fasanaro, P.; Zaccagnini, G.; Greco, S.; Sarra-Ferraris, G.; Antonini, A.; Martelli, F.; Capogrossi, M.C. miR-200c is upregulated by oxidative stress and induces endothelial cell apoptosis and senescence via ZEB1 inhibition. Cell Death Differ. 2011, 18, 1628–1639. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Li, T.W.; Zhou, Y.; Peng, H.; Liu, T.; Zandi, E.; Martinez-Chantar, M.L.; Mato, J.M.; Lu, S.C. Activation of a novel c-Myc-miR27-prohibitin 1 circuitry in cholestatic liver injury inhibits glutathione synthesis in mice. Antioxid. Redox Signal. 2015, 22, 259–274. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Yan, W.; Lu, L.; Wang, Y.; Lu, W.; Cao, Y.; Cai, W. p38/p53/miR-200a-3p feedback loop promotes oxidative stress-mediated liver cell death. Cell Cycle 2015, 14, 1548–1558. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.; Ren, X.; Zhang, X.; Luo, Y.; Wang, G.; Huang, K.; Feng, S.; Bao, X.; Huang, K.; He, X.; et al. Selective killing of lung cancer cells by miRNA-506 molecule through inhibiting NF-kappaB p65 to evoke reactive oxygen species generation and p53 activation. Oncogene 2015, 34, 691–703. [Google Scholar] [CrossRef] [PubMed]
- Faller, M.; Matsunaga, M.; Yin, S.; Loo, J.A.; Guo, F. Heme is involved in microRNA processing. Nat. Struct. Mol. Biol. 2007, 14, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Barr, I.; Smith, A.T.; Chen, Y.; Senturia, R.; Burstyn, J.N.; Guo, F. Ferric, not ferrous, heme activates RNA-binding protein DGCR8 for primary microRNA processing. Proc. Natl. Acad. Sci. USA 2012, 109, 1919–1924. [Google Scholar] [CrossRef]
- Yi, J.; Huang, W.Z.; Wen, Y.Q.; Yi, Y.C. Effect of miR-101 on proliferation and oxidative stress-induced apoptosis of breast cancer cells via Nrf2 signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 8931–8939. [Google Scholar] [CrossRef]
- Akdemir, B.; Nakajima, Y.; Inazawa, J.; Inoue, J. miR-432 Induces NRF2 Stabilization by Directly Targeting KEAP1. Mol. Cancer Res. 2017, 15, 1570–1578. [Google Scholar] [CrossRef] [PubMed]
- Kabaria, S.; Choi, D.C.; Chaudhuri, A.D.; Jain, M.R.; Li, H.; Junn, E. MicroRNA-7 activates Nrf2 pathway by targeting Keap1 expression. Free Radic. Biol. Med. 2015, 89, 548–556. [Google Scholar] [CrossRef]
- Eades, G.; Yang, M.; Yao, Y.; Zhang, Y.; Zhou, Q. miR-200a regulates Nrf2 activation by targeting Keap1 mRNA in breast cancer cells. J. Biol. Chem. 2011, 286, 40725–40733. [Google Scholar] [CrossRef] [PubMed]
- Petrelli, A.; Perra, A.; Cora, D.; Sulas, P.; Menegon, S.; Manca, C.; Migliore, C.; Kowalik, M.A.; Ledda-Columbano, G.M.; Giordano, S.; et al. MicroRNA/gene profiling unveils early molecular changes and nuclear factor erythroid related factor 2 (NRF2) activation in a rat model recapitulating human hepatocellular carcinoma (HCC). Hepatology 2014, 59, 228–241. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Hu, C.; Xu, Q.; Chen, L.; Ma, K.; Xu, N.; Zhu, H. Methylseleninic acid activates Keap1/Nrf2 pathway via up-regulating miR-200a in human oesophageal squamous cell carcinoma cells. Biosci. Rep. 2015, 35. [Google Scholar] [CrossRef]
- Yang, M.; Yao, Y.; Eades, G.; Zhang, Y.; Zhou, Q. MiR-28 regulates Nrf2 expression through a Keap1-independent mechanism. Breast Cancer Res. Treat. 2011, 129, 983–991. [Google Scholar] [CrossRef]
- Wang, B.; Teng, Y.; Liu, Q. MicroRNA-153 Regulates NRF2 Expression and is Associated with Breast Carcinogenesis. Clin. Lab. 2016, 62, 39–47. [Google Scholar] [CrossRef]
- Khan, A.U.H.; Rathore, M.G.; Allende-Vega, N.; Vo, D.N.; Belkhala, S.; Orecchioni, S.; Talarico, G.; Bertolini, F.; Cartron, G.; Lecellier, C.H.; et al. Human Leukemic Cells performing Oxidative Phosphorylation (OXPHOS) Generate an Antioxidant Response Independently of Reactive Oxygen species (ROS) Production. EBioMedicine 2016, 3, 43–53. [Google Scholar] [CrossRef]
- Vera, O.; Jimenez, J.; Pernia, O.; Rodriguez-Antolin, C.; Rodriguez, C.; Sanchez Cabo, F.; Soto, J.; Rosas, R.; Lopez-Magallon, S.; Esteban Rodriguez, I.; et al. DNA Methylation of miR-7 is a Mechanism Involved in Platinum Response through MAFG Overexpression in Cancer Cells. Theranostics 2017, 7, 4118–4134. [Google Scholar] [CrossRef]
- Qu, J.; Zhang, L.; Li, L.; Su, Y. miR-148b Functions as a Tumor Suppressor by Targeting Endoplasmic Reticulum Metallo Protease 1 in Human Endometrial Cancer Cells. Oncol. Res. 2018, 27, 81–88. [Google Scholar] [CrossRef]
- Degli Esposti, D.; Aushev, V.N.; Lee, E.; Cros, M.P.; Zhu, J.; Herceg, Z.; Chen, J.; Hernandez-Vargas, H. miR-500a-5p regulates oxidative stress response genes in breast cancer and predicts cancer survival. Sci. Rep. 2017, 7, 15966. [Google Scholar] [CrossRef]
- Gomes, S.E.; Pereira, D.M.; Roma-Rodrigues, C.; Fernandes, A.R.; Borralho, P.M.; Rodrigues, C.M.P. Convergence of miR-143 overexpression, oxidative stress and cell death in HCT116 human colon cancer cells. PLoS ONE 2018, 13, e0191607. [Google Scholar] [CrossRef]
- Pajic, M.; Froio, D.; Daly, S.; Doculara, L.; Millar, E.; Graham, P.H.; Drury, A.; Steinmann, A.; de Bock, C.E.; Boulghourjian, A.; et al. miR-139-5p Modulates Radiotherapy Resistance in Breast Cancer by Repressing Multiple Gene Networks of DNA Repair and ROS Defense. Cancer Res. 2018, 78, 501–515. [Google Scholar] [CrossRef] [PubMed]
- Hou, M.; Zuo, X.; Li, C.; Zhang, Y.; Teng, Y. Mir-29b Regulates Oxidative Stress by Targeting SIRT1 in Ovarian Cancer Cells. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2017, 43, 1767–1776. [Google Scholar] [CrossRef] [PubMed]
- Kao, Y.Y.; Chou, C.H.; Yeh, L.Y.; Chen, Y.F.; Chang, K.W.; Liu, C.J.; Fan Chiang, C.Y.; Lin, S.C. MicroRNA miR-31 targets SIRT3 to disrupt mitochondrial activity and increase oxidative stress in oral carcinoma. Cancer Lett. 2019, 456, 40–48. [Google Scholar] [CrossRef]
- Chang, M.; Qiao, L.; Li, B.; Wang, J.; Zhang, G.; Shi, W.; Liu, Z.; Gu, N.; Di, Z.; Wang, X.; et al. Suppression of SIRT6 by miR-33a facilitates tumor growth of glioma through apoptosis and oxidative stress resistance. Oncol. Rep. 2017, 38, 1251–1258. [Google Scholar] [CrossRef]
- Yang, C.; Yan, Z.; Hu, F.; Wei, W.; Sun, Z.; Xu, W. Silencing of microRNA-517a induces oxidative stress injury in melanoma cells via inactivation of the JNK signaling pathway by upregulating CDKN1C. Cancer Cell Int. 2020, 20, 32. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Liu, H.; Xu, J.; Shi, D.; Zhai, L.; Liu, B.; Wang, L.; Liu, G.; Qin, J. miR1443p regulates the resistance of lung cancer to cisplatin by targeting Nrf2. Oncol. Rep. 2018, 40, 3479–3488. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Chen, Z.G.; Wu, L.L.; Zheng, J.J.; Yang, J.R.; Chen, X.F.; Chen, Z.Q.; Liu, C.L.; Chi, S.Y.; Zheng, J.Y.; et al. miR-340 reverses cisplatin resistance of hepatocellular carcinoma cell lines by targeting Nrf2-dependent antioxidant pathway. Asian Pac. J. Cancer Prev. 2014, 15, 10439–10444. [Google Scholar] [CrossRef]
- Van Jaarsveld, M.T.; Helleman, J.; Boersma, A.W.; van Kuijk, P.F.; van Ijcken, W.F.; Despierre, E.; Vergote, I.; Mathijssen, R.H.; Berns, E.M.; Verweij, J.; et al. miR-141 regulates KEAP1 and modulates cisplatin sensitivity in ovarian cancer cells. Oncogene 2013, 32, 4284–4293. [Google Scholar] [CrossRef]
- Moreno Leon, L.; Gautier, M.; Allan, R.; Ilie, M.; Nottet, N.; Pons, N.; Paquet, A.; Lebrigand, K.; Truchi, M.; Fassy, J.; et al. The nuclear hypoxia-regulated NLUCAT1 long non-coding RNA contributes to an aggressive phenotype in lung adenocarcinoma through regulation of oxidative stress. Oncogene 2019, 38, 7146–7165. [Google Scholar] [CrossRef] [PubMed]
- Qi, W.; Li, Z.; Xia, L.; Dai, J.; Zhang, Q.; Wu, C.; Xu, S. LncRNA GABPB1-AS1 and GABPB1 regulate oxidative stress during erastin-induced ferroptosis in HepG2 hepatocellular carcinoma cells. Sci. Rep. 2019, 9, 16185. [Google Scholar] [CrossRef] [PubMed]
- Shao, Q.; Wang, Q.; Wang, J. LncRNA SCAMP1 regulates ZEB1/JUN and autophagy to promote pediatric renal cell carcinoma under oxidative stress via miR-429. Biomed. Pharmacother. Biomed. Pharmacother. 2019, 120, 109460. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.T.; Ye, H.; Wei, P.P.; Han, B.W.; He, B.; Chen, Z.H.; Chen, Y.Q. LncRNAs H19 and HULC, activated by oxidative stress, promote cell migration and invasion in cholangiocarcinoma through a ceRNA manner. J. Hematol. Oncol. 2016, 9, 117. [Google Scholar] [CrossRef]
- Kim, K.H.; Roberts, C.W. Targeting EZH2 in cancer. Nat. Med. 2016, 22, 128–134. [Google Scholar] [CrossRef]
- Zhou, L.; Xu, D.Y.; Sha, W.G.; Shen, L.; Lu, G.Y.; Yin, X.; Wang, M.J. High glucose induces renal tubular epithelial injury via Sirt1/NF-kappaB/microR-29/Keap1 signal pathway. J. Transl. Med. 2015, 13, 352. [Google Scholar] [CrossRef]
- Sun, L.; Zhang, J.; Li, Y. Chronic central miR-29b antagonism alleviates angiotensin II-induced hypertension and vascular endothelial dysfunction. Life Sci. 2019, 235, 116862. [Google Scholar] [CrossRef]
- Li, C.; Fan, K.; Qu, Y.; Zhai, W.; Huang, A.; Sun, X.; Xing, S. Deregulation of UCA1 expression may be involved in the development of chemoresistance to cisplatin in the treatment of non-small-cell lung cancer via regulating the signaling pathway of microRNA-495/NRF2. J. Cell. Physiol. 2020, 235, 3721–3730. [Google Scholar] [CrossRef]
- Gao, M.; Li, C.; Xu, M.; Liu, Y.; Liu, S. LncRNA UCA1 attenuates autophagy-dependent cell death through blocking autophagic flux under arsenic stress. Toxicol. Lett. 2018, 284, 195–204. [Google Scholar] [CrossRef]
- Siddik, Z.H. Cisplatin: Mode of cytotoxic action and molecular basis of resistance. Oncogene 2003, 22, 7265–7279. [Google Scholar] [CrossRef]
- Rabik, C.A.; Dolan, M.E. Molecular mechanisms of resistance and toxicity associated with platinating agents. Cancer Treat. Rev. 2007, 33, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Lippard, S.J. Cellular processing of platinum anticancer drugs. Nat. Rev. Drug Discov. 2005, 4, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [PubMed]
- Marullo, R.; Werner, E.; Degtyareva, N.; Moore, B.; Altavilla, G.; Ramalingam, S.S.; Doetsch, P.W. Cisplatin induces a mitochondrial-ROS response that contributes to cytotoxicity depending on mitochondrial redox status and bioenergetic functions. PLoS ONE 2013, 8, e81162. [Google Scholar] [CrossRef] [PubMed]
- Martins, N.M.; Santos, N.A.; Curti, C.; Bianchi, M.L.; Santos, A.C. Cisplatin induces mitochondrial oxidative stress with resultant energetic metabolism impairment, membrane rigidification and apoptosis in rat liver. J. Appl. Toxicol. JAT 2008, 28, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Schaaf, G.J.; Maas, R.F.; de Groene, E.M.; Fink-Gremmels, J. Management of oxidative stress by heme oxygenase-1 in cisplatin-induced toxicity in renal tubular cells. Free Radic. Res. 2002, 36, 835–843. [Google Scholar] [CrossRef]
- Kartalou, M.; Essigmann, J.M. Mechanisms of resistance to cisplatin. Mutat. Res. 2001, 478, 23–43. [Google Scholar] [CrossRef]
- Holzer, A.K.; Samimi, G.; Katano, K.; Naerdemann, W.; Lin, X.; Safaei, R.; Howell, S.B. The copper influx transporter human copper transport protein 1 regulates the uptake of cisplatin in human ovarian carcinoma cells. Mol. Pharmacol. 2004, 66, 817–823. [Google Scholar] [CrossRef]
- Wu, C.; Wangpaichitr, M.; Feun, L.; Kuo, M.T.; Robles, C.; Lampidis, T.; Savaraj, N. Overcoming cisplatin resistance by mTOR inhibitor in lung cancer. Mol. Cancer 2005, 4, 25. [Google Scholar] [CrossRef]
- Nicholson, L.J.; Smith, P.R.; Hiller, L.; Szlosarek, P.W.; Kimberley, C.; Sehouli, J.; Koensgen, D.; Mustea, A.; Schmid, P.; Crook, T. Epigenetic silencing of argininosuccinate synthetase confers resistance to platinum-induced cell death but collateral sensitivity to arginine auxotrophy in ovarian cancer. Int. J. Cancer 2009, 125, 1454–1463. [Google Scholar] [CrossRef]
- Ibragimova, I.; Ibanez de Caceres, I.; Hoffman, A.M.; Potapova, A.; Dulaimi, E.; Al-Saleem, T.; Hudes, G.R.; Ochs, M.F.; Cairns, P. Global reactivation of epigenetically silenced genes in prostate cancer. Cancer Prev. Res. (Phila.) 2010, 3, 1084–1092. [Google Scholar] [CrossRef] [PubMed]
- Cortes-Sempere, M.; de Miguel, M.P.; Pernia, O.; Rodriguez, C.; de Castro Carpeno, J.; Nistal, M.; Conde, E.; Lopez-Rios, F.; Belda-Iniesta, C.; Perona, R.; et al. IGFBP-3 methylation-derived deficiency mediates the resistance to cisplatin through the activation of the IGFIR/Akt pathway in non-small cell lung cancer. Oncogene 2013, 32, 1274–1283. [Google Scholar] [CrossRef] [PubMed]
- Chang, X.; Monitto, C.L.; Demokan, S.; Kim, M.S.; Chang, S.S.; Zhong, X.; Califano, J.A.; Sidransky, D. Identification of hypermethylated genes associated with cisplatin resistance in human cancers. Cancer Res. 2010, 70, 2870–2879. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xiang, S.; Williams, K.A.; Dong, H.; Bai, W.; Nicosia, S.V.; Khochbin, S.; Bepler, G.; Zhang, X. Depletion of HDAC6 enhances cisplatin-induced DNA damage and apoptosis in non-small cell lung cancer cells. PLoS ONE 2012, 7, e44265. [Google Scholar] [CrossRef]
- Gruosso, T.; Mieulet, V.; Cardon, M.; Bourachot, B.; Kieffer, Y.; Devun, F.; Dubois, T.; Dutreix, M.; Vincent-Salomon, A.; Miller, K.M.; et al. Chronic oxidative stress promotes H2AX protein degradation and enhances chemosensitivity in breast cancer patients. Embo Mol. Med. 2016, 8, 527–549. [Google Scholar] [CrossRef]
- Kim, N.H.; Choi, S.H.; Kim, C.H.; Lee, C.H.; Lee, T.R.; Lee, A.Y. Reduced MiR-675 in exosome in H19 RNA-related melanogenesis via MITF as a direct target. J. Investig. Dermatol. 2014, 134, 1075–1082. [Google Scholar] [CrossRef]
- Ceppi, P.; Mudduluru, G.; Kumarswamy, R.; Rapa, I.; Scagliotti, G.V.; Papotti, M.; Allgayer, H. Loss of miR-200c expression induces an aggressive, invasive, and chemoresistant phenotype in non-small cell lung cancer. Mol. Cancer Res. 2010, 8, 1207–1216. [Google Scholar] [CrossRef]
- Gu, Y.; Zhang, Z.; Yin, J.; Ye, J.; Song, Y.; Liu, H.; Xiong, Y.; Lu, M.; Zheng, G.; He, Z. Epigenetic silencing of miR-493 increases the resistance to cisplatin in lung cancer by targeting tongue cancer resistance-related protein 1(TCRP1). J. Exp. Clin. Cancer Res. 2017, 36, 114. [Google Scholar] [CrossRef]
- Liu, X.; Feng, M.; Zheng, G.; Gu, Y.; Wang, C.; He, Z. TCRP1 expression is associated with platinum sensitivity in human lung and ovarian cancer cells. Oncol. Lett. 2017, 13, 1398–1405. [Google Scholar] [CrossRef]
- Jia, X.; Zhang, Z.; Luo, K.; Zheng, G.; Lu, M.; Song, Y.; Liu, H.; Qiu, H.; He, Z. TCRP1 transcriptionally regulated by c-Myc confers cancer chemoresistance in tongue and lung cancer. Sci. Rep. 2017, 7, 3744. [Google Scholar] [CrossRef]
- Ibanez de Caceres, I.; Cortes-Sempere, M.; Moratilla, C.; Machado-Pinilla, R.; Rodriguez-Fanjul, V.; Manguan-Garcia, C.; Cejas, P.; Lopez-Rios, F.; Paz-Ares, L.; de CastroCarpeno, J.; et al. IGFBP-3 hypermethylation-derived deficiency mediates cisplatin resistance in non-small-cell lung cancer. Oncogene 2010, 29, 1681–1690. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Antolin, C.; Felguera-Selas, L.; Pernia, O.; Vera, O.; Esteban, I.; Losantos Garcia, I.; de Castro, J.; Rosas-Alonso, R.; Ibanez de Caceres, I. miR-7 methylation as a biomarker to predict poor survival in early-stage non-small cell lung cancer patients. Cell Biosci. 2019, 9, 63. [Google Scholar] [CrossRef] [PubMed]
- Young, R.P.; Hopkins, R.J.; Christmas, T.; Black, P.N.; Metcalf, P.; Gamble, G.D. COPD prevalence is increased in lung cancer, independent of age, sex and smoking history. Eur. Respir. J. 2009, 34, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Rosas-Alonso, R.; Galera, R.; Sanchez-Pascuala, J.J.; Casitas, R.; Burdiel, M.; Martinez-Ceron, E.; Vera, O.; Rodriguez-Antolin, C.; Pernia, O.; De Castro, J.; et al. Hypermethylation of Anti-oncogenic MicroRNA 7 is Increased in Emphysema Patients. Arch. Bronconeumol. 2019. [Google Scholar] [CrossRef]
- Vera-Puente, O.; Rodriguez-Antolin, C.; Salgado-Figueroa, A.; Michalska, P.; Pernia, O.; Reid, B.M.; Rosas, R.; Garcia-Guede, A.; SacristAn, S.; Jimenez, J.; et al. MAFG is a potential therapeutic target to restore chemosensitivity in cisplatin-resistant cancer cells by increasing reactive oxygen species. Transl. Res. 2018, 200, 1–17. [Google Scholar] [CrossRef]
- Shavit, J.A.; Motohashi, H.; Onodera, K.; Akasaka, J.; Yamamoto, M.; Engel, J.D. Impaired megakaryopoiesis and behavioral defects in mafG-null mutant mice. Genes Dev. 1998, 12, 2164–2174. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Liu, T.; Wang, J.; Li, T.W.; Fan, W.; Peng, H.; Krishnan, A.; Gores, G.J.; Mato, J.M.; Lu, S.C. Deregulated methionine adenosyltransferase alpha1, c-Myc, and Maf proteins together promote cholangiocarcinoma growth in mice and humans(double dagger). Hepatology 2016, 64, 439–455. [Google Scholar] [CrossRef]
- Fan, W.; Yang, H.; Liu, T.; Wang, J.; Li, T.W.; Mavila, N.; Tang, Y.; Yang, J.; Peng, H.; Tu, J.; et al. Prohibitin 1 suppresses liver cancer tumorigenesis in mice and human hepatocellular and cholangiocarcinoma cells. Hepatology 2017, 65, 1249–1266. [Google Scholar] [CrossRef]
- Liu, T.; Yang, H.; Fan, W.; Tu, J.; Li, T.W.H.; Wang, J.; Shen, H.; Yang, J.; Xiong, T.; Steggerda, J.; et al. Mechanisms of MAFG Dysregulation in Cholestatic Liver Injury and Development of Liver Cancer. Gastroenterology 2018, 155, 557–571. [Google Scholar] [CrossRef]
- Wang, J.P.; Leng, J.Y.; Zhang, R.K.; Zhang, L.; Zhang, B.; Jiang, W.Y.; Tong, L. Functional analysis of gene expression profiling-based prediction in bladder cancer. Oncol. Lett. 2018, 15, 8417–8423. [Google Scholar] [CrossRef]
- Yang, H.; Li, T.W.; Peng, J.; Mato, J.M.; Lu, S.C. Insulin-like growth factor 1 activates methionine adenosyltransferase 2A transcription by multiple pathways in human colon cancer cells. Biochem. J. 2011, 436, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Torres, S.; Garcia-Palmero, I.; Marin-Vicente, C.; Bartolome, R.A.; Calvino, E.; Fernandez-Acenero, M.J.; Casal, J.I. Proteomic Characterization of Transcription and Splicing Factors Associated with a Metastatic Phenotype in Colorectal Cancer. J. Proteome Res. 2018, 17, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.; Hutchinson, L.; Deng, A.; Green, M.R. Common BRAF(V600E)-directed pathway mediates widespread epigenetic silencing in colorectal cancer and melanoma. Proc. Natl. Acad. Sci. USA 2016, 113, 1250–1255. [Google Scholar] [CrossRef]
- Fang, M.; Ou, J.; Hutchinson, L.; Green, M.R. The BRAF oncoprotein functions through the transcriptional repressor MAFG to mediate the CpG Island Methylator phenotype. Mol. Cell 2014, 55, 904–915. [Google Scholar] [CrossRef]
- Katsuoka, F.; Motohashi, H.; Engel, J.D.; Yamamoto, M. Nrf2 transcriptionally activates the mafG gene through an antioxidant response element. J. Biol. Chem. 2005, 280, 4483–4490. [Google Scholar] [CrossRef] [PubMed]
- Schembri, F.; Sridhar, S.; Perdomo, C.; Gustafson, A.M.; Zhang, X.; Ergun, A.; Lu, J.; Liu, G.; Bowers, J.; Vaziri, C.; et al. MicroRNAs as modulators of smoking-induced gene expression changes in human airway epithelium. Proc. Natl. Acad. Sci. USA 2009, 106, 2319–2324. [Google Scholar] [CrossRef]
- Kim, T.; Mehta, S.L.; Morris-Blanco, K.C.; Chokkalla, A.K.; Chelluboina, B.; Lopez, M.; Sullivan, R.; Kim, H.T.; Cook, T.D.; Kim, J.Y.; et al. The microRNA miR-7a-5p ameliorates ischemic brain damage by repressing alpha-synuclein. Sci. Signal. 2018, 11. [Google Scholar] [CrossRef]
- Junn, E.; Lee, K.W.; Jeong, B.S.; Chan, T.W.; Im, J.Y.; Mouradian, M.M. Repression of alpha-synuclein expression and toxicity by microRNA-7. Proc. Natl. Acad. Sci. USA 2009, 106, 13052–13057. [Google Scholar] [CrossRef]
- Titze-de-Almeida, R.; Titze-de-Almeida, S.S. miR-7 Replacement Therapy in Parkinson’s Disease. Curr. Gene Ther. 2018, 18, 143–153. [Google Scholar] [CrossRef]
- Fields, C.R.; Bengoa-Vergniory, N.; Wade-Martins, R. Targeting Alpha-Synuclein as a Therapy for Parkinson’s Disease. Front. Mol. Neurosci. 2019, 12, 299. [Google Scholar] [CrossRef]
- Perfeito, R.; Ribeiro, M.; Rego, A.C. Alpha-synuclein-induced oxidative stress correlates with altered superoxide dismutase and glutathione synthesis in human neuroblastoma SH-SY5Y cells. Arch. Toxicol. 2017, 91, 1245–1259. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.B.; Kjems, J.; Damgaard, C.K. Circular RNA and miR-7 in cancer. Cancer Res. 2013, 73, 5609–5612. [Google Scholar] [CrossRef] [PubMed]
- Akar, R.O.; Selvi, S.; Ulukaya, E.; Aztopal, N. Key actors in cancer therapy: Epigenetic modifiers. Turk. J. Biol. Turk. Biyol. Derg. 2019, 43, 155–170. [Google Scholar] [CrossRef]
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting epigenetic regulators for cancer therapy: Mechanisms and advances in clinical trials. Signal Transduct. Target. Ther. 2019, 4, 62. [Google Scholar] [CrossRef] [PubMed]
- Hanna, J.; Hossain, G.S.; Kocerha, J. The Potential for microRNA Therapeutics and Clinical Research. Front. Genet. 2019, 10, 478. [Google Scholar] [CrossRef]
- Bonneau, E.; Neveu, B.; Kostantin, E.; Tsongalis, G.J.; De Guire, V. How close are miRNAs from clinical practice? A perspective on the diagnostic and therapeutic market. Ejifcc 2019, 30, 114–127. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Spot of Epigenetic Modification | Potentially Active Chromatin | Potentially Inactive Chromatin |
|---|---|---|
| DNA | DNA methylation outside CpG islands | DNA methylation in CpG islands of regulatory regions |
| Histones | Acetylated Unmethylated H3K4 Methylation | Deacetylated Methylated H3K4 unmethylation |
| Chromatin conformation | Open | Condensed Constitutive Heterochromatin |
| Name | Cancer TYPE | Target | Effect over Antioxidant Response | Reference |
|---|---|---|---|---|
| MIR-101 | Breast | NRF2 | downregulated | [141] |
| MIR-28 | Breast | NRF2 | downregulated | [147] |
| MIR-153 | Breast | NRF2 | downregulated | [148] |
| MIR-432-3P | Esophageal Squamous | KEAP1 | upregulated | [142] |
| MIR-200A | Breast, Hepatocelullar, Esophageal squamous | KEAP1 | upregulated | [144,145,146] |
| MIR-23A | Leukemic | KEAP1 | upregulated | [149] |
| MIR-7 | Neuroblastoma | KEAP1 | upregulated | [143] |
| MIR-7 | Non small cell lung | MAFG | downregulated | [150] |
| MIR-148B | Endometrial | ERMP1 | downregulated | [151] |
| MIR-500A-5P | Breast | TXNRD1 and NFE2L2 | downregulated | [152] |
| MIR-143 | Colon | SOD1 | downregulated | [153] |
| MIR-139-5P | Breast | MAT2A | downregulated | [154] |
| MIR-29B | Ovary | SIRT1 | upregulated | [155] |
| MIR-31 | Oral squamous | SIRT3 | downregulated | [156] |
| MIR-33A | Glioma | SIRT6 | downregulated | [157] |
| MIR-517A | Melanoma | JNK sig. path. | downregulated | [158] |
| MIR-144-3P | Lung | NRF2 | downregulated | [159] |
| MIR-340 | Hepatocelullar | NRF2 | downregulated | [160] |
| MIR-141 | Ovary | KEAP1 | upregulated | [161] |
| Name | Cancer TYPE | Intermediates/Effectors | Effect over Antioxidant Response | References |
|---|---|---|---|---|
| NLUCAT1 | Lung cancer | HIF2A, NF-KB, NRF2 | Upregulated | [162] |
| GABPB1-AS1 | Hepatocellular carcinoma | GABPB1 | Upregulated | [163] |
| SCAMP1 | Renal cell carcinoma | miR-429/ZEB, JUN | Downregulated | [164] |
| HULC | Cholangiocarcinoma | Let-7a, Let-7b/IL-6 | Upregulated | [165] |
| H19 | Cholangiocarcinoma | miR-372, miR-373/CXCR4 | Upregulated | [165] |
| UCA1 | Non-small cell lung cancer | miR-495/NRF2 | Upregulated | [169] |
| UCA1 | Hepatocellular carcinoma | miR-184/OSGIN1 | Upregulated | [170] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Guede, Á.; Vera, O.; Ibáñez-de-Caceres, I. When Oxidative Stress Meets Epigenetics: Implications in Cancer Development. Antioxidants 2020, 9, 468. https://doi.org/10.3390/antiox9060468
García-Guede Á, Vera O, Ibáñez-de-Caceres I. When Oxidative Stress Meets Epigenetics: Implications in Cancer Development. Antioxidants. 2020; 9(6):468. https://doi.org/10.3390/antiox9060468
Chicago/Turabian StyleGarcía-Guede, Álvaro, Olga Vera, and Inmaculada Ibáñez-de-Caceres. 2020. "When Oxidative Stress Meets Epigenetics: Implications in Cancer Development" Antioxidants 9, no. 6: 468. https://doi.org/10.3390/antiox9060468
APA StyleGarcía-Guede, Á., Vera, O., & Ibáñez-de-Caceres, I. (2020). When Oxidative Stress Meets Epigenetics: Implications in Cancer Development. Antioxidants, 9(6), 468. https://doi.org/10.3390/antiox9060468