The Structural Integrity of the Model Lipid Membrane during Induced Lipid Peroxidation: The Role of Flavonols in the Inhibition of Lipid Peroxidation

 , ,
, ,  , , ,
, , , 

Abstract
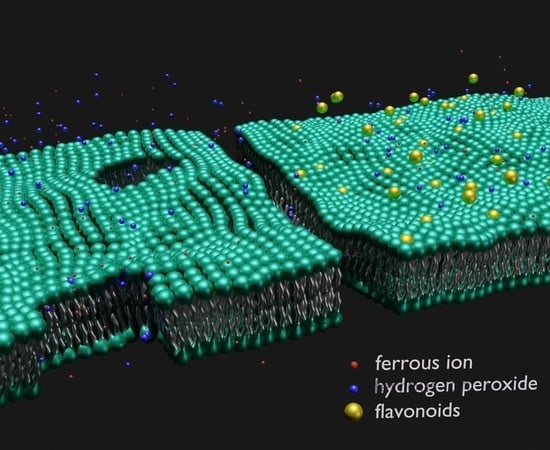
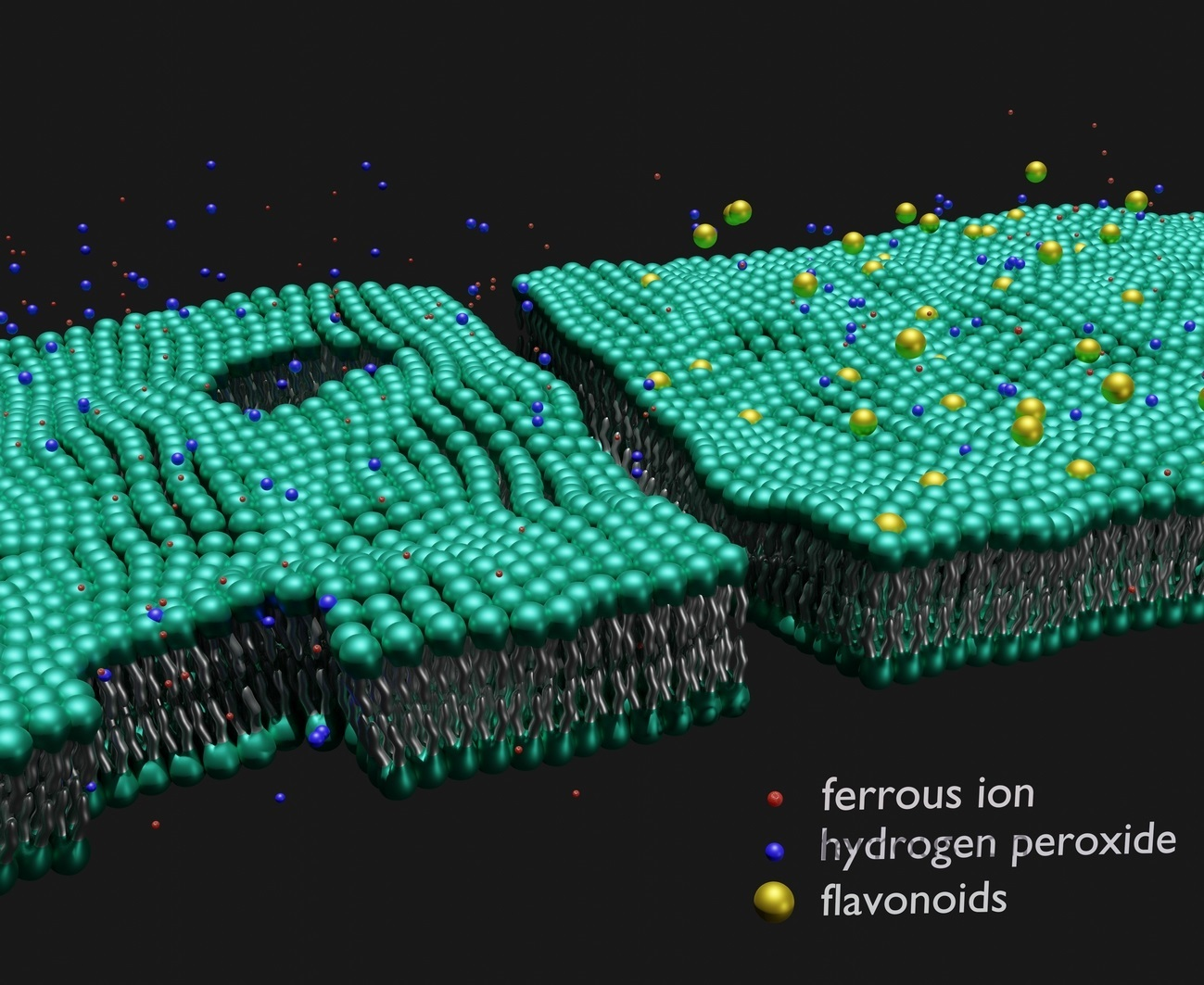
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Preparation and Oxidation of Liposomes with and without Inserted Flavonols
2.3. EPR Spectroscopy
2.3.1. Antioxidant Activity
2.3.2. Fluidity Change during Lipid Peroxidation
2.4. High Resolution Mass Spectrometry
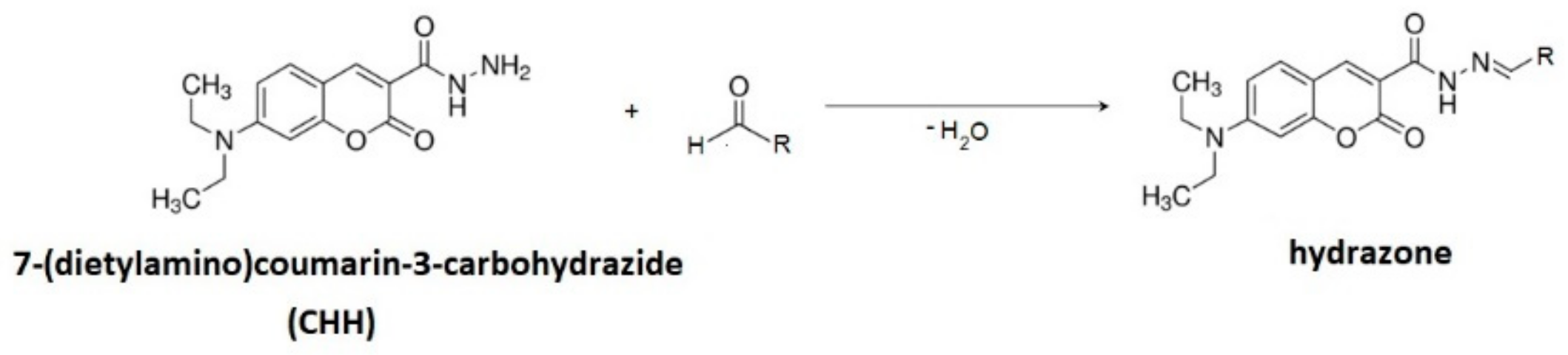
2.4.1. Derivatization with 7-(Diethylamino)coumarin-3-carbohydrazide (CHH)
2.4.2. Measurement Parameters
2.5. FTIR-ATR Spectroscopy
Data Analysis—The Extent of Lipid Peroxidation
2.6. Dynamic Light Scattering (DLS) and Electrophoretic (ELS) Measurement
2.7. Small Angle X-ray Scattering (SAXS)
2.8. Atomic Force Microscopy (AFM) and Force Spectroscopy (FS)
2.8.1. Preparation of the Supported Lipid Bilayers (SLBs)
2.8.2. AFM Imaging in Fluid and FS Measurements before and after Lipid Peroxidation
2.9. Statistical Data Analysis
3. Results and Discussion
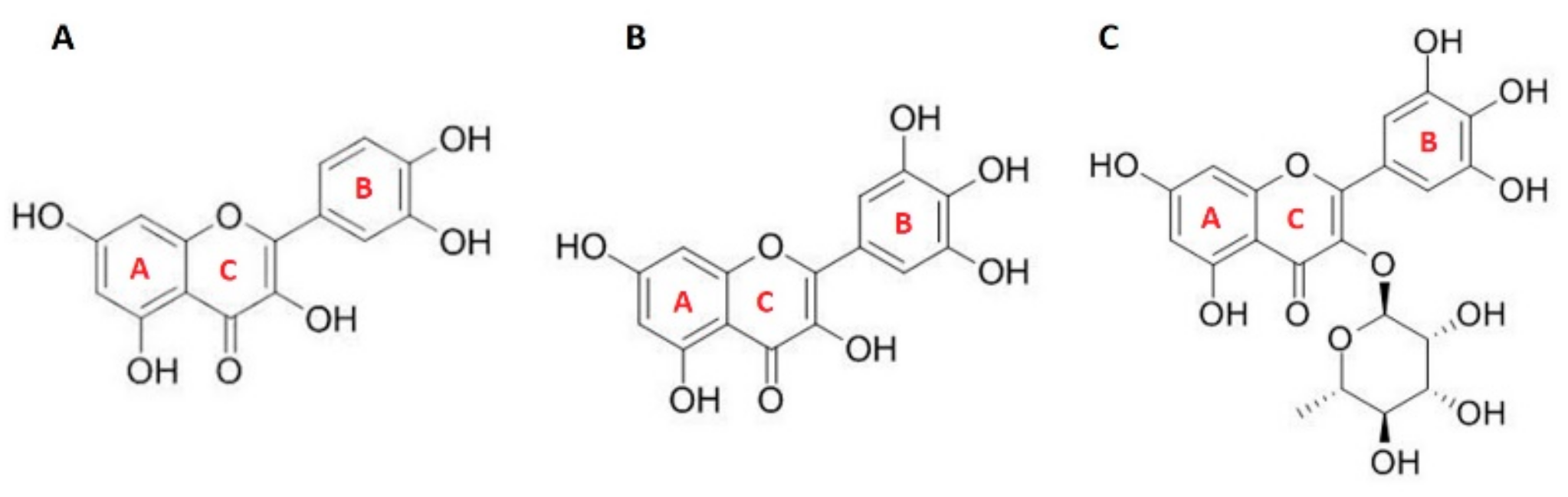
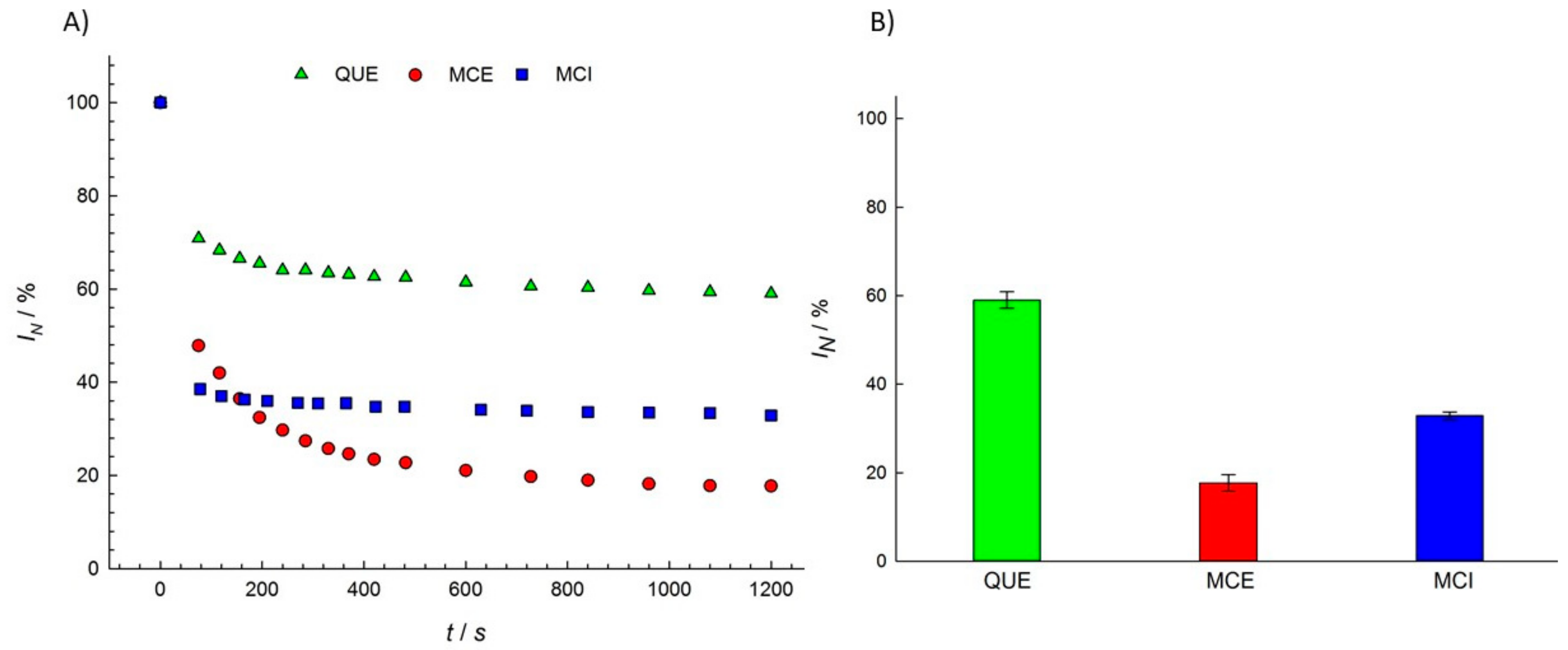
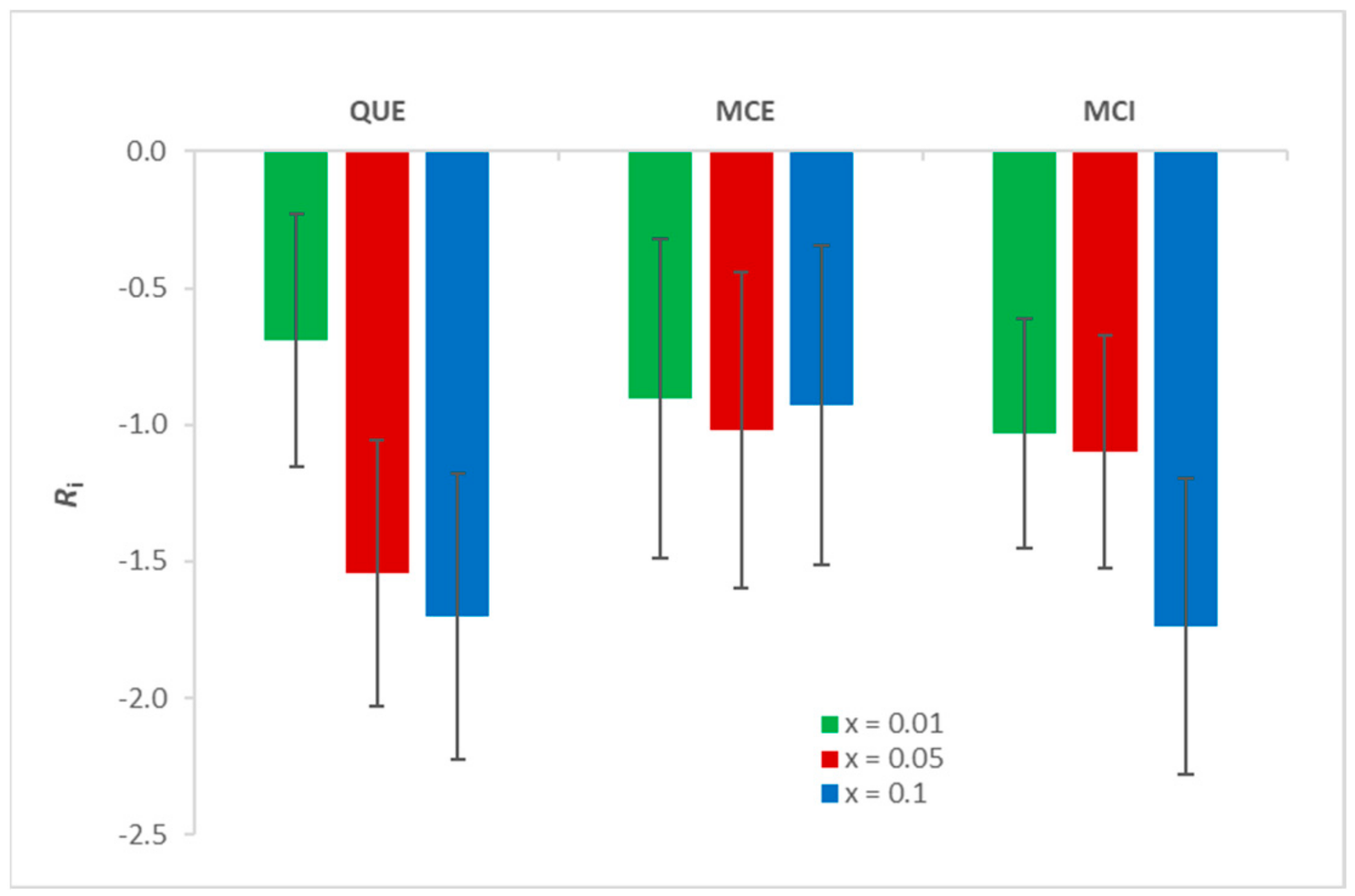
3.1. Antioxidant Activity of Flavonols
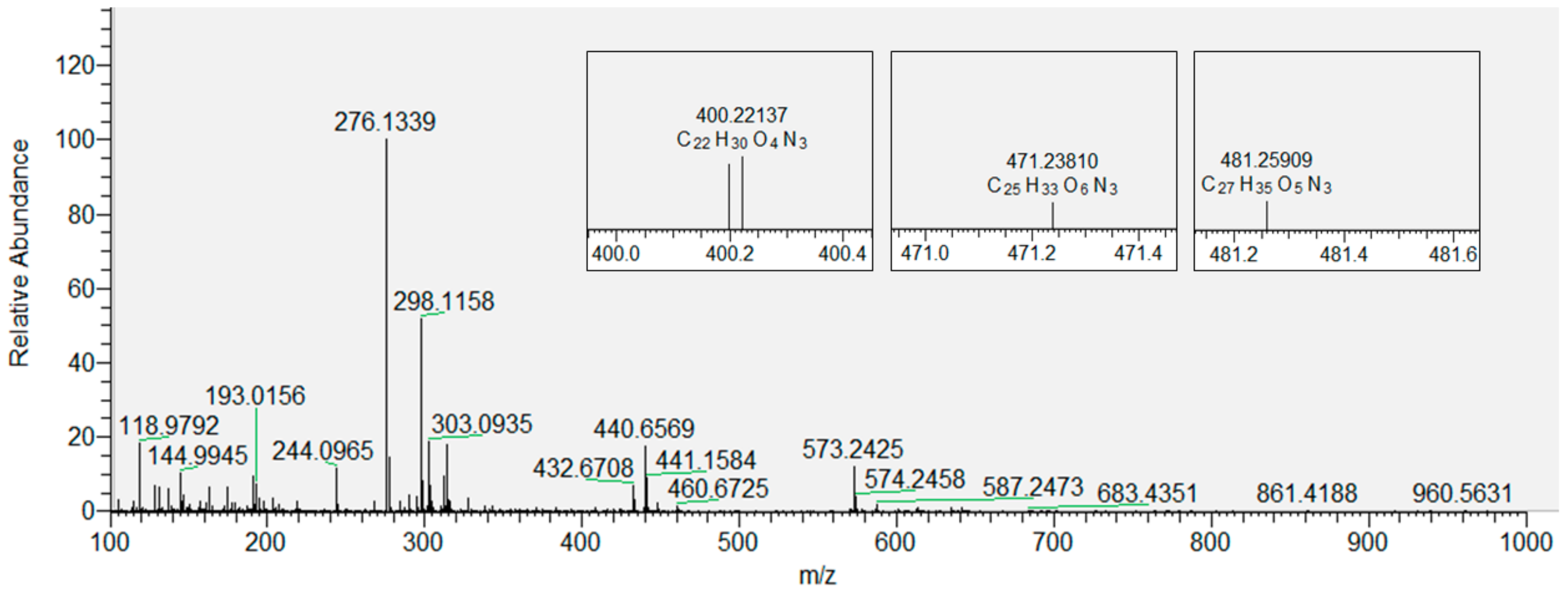
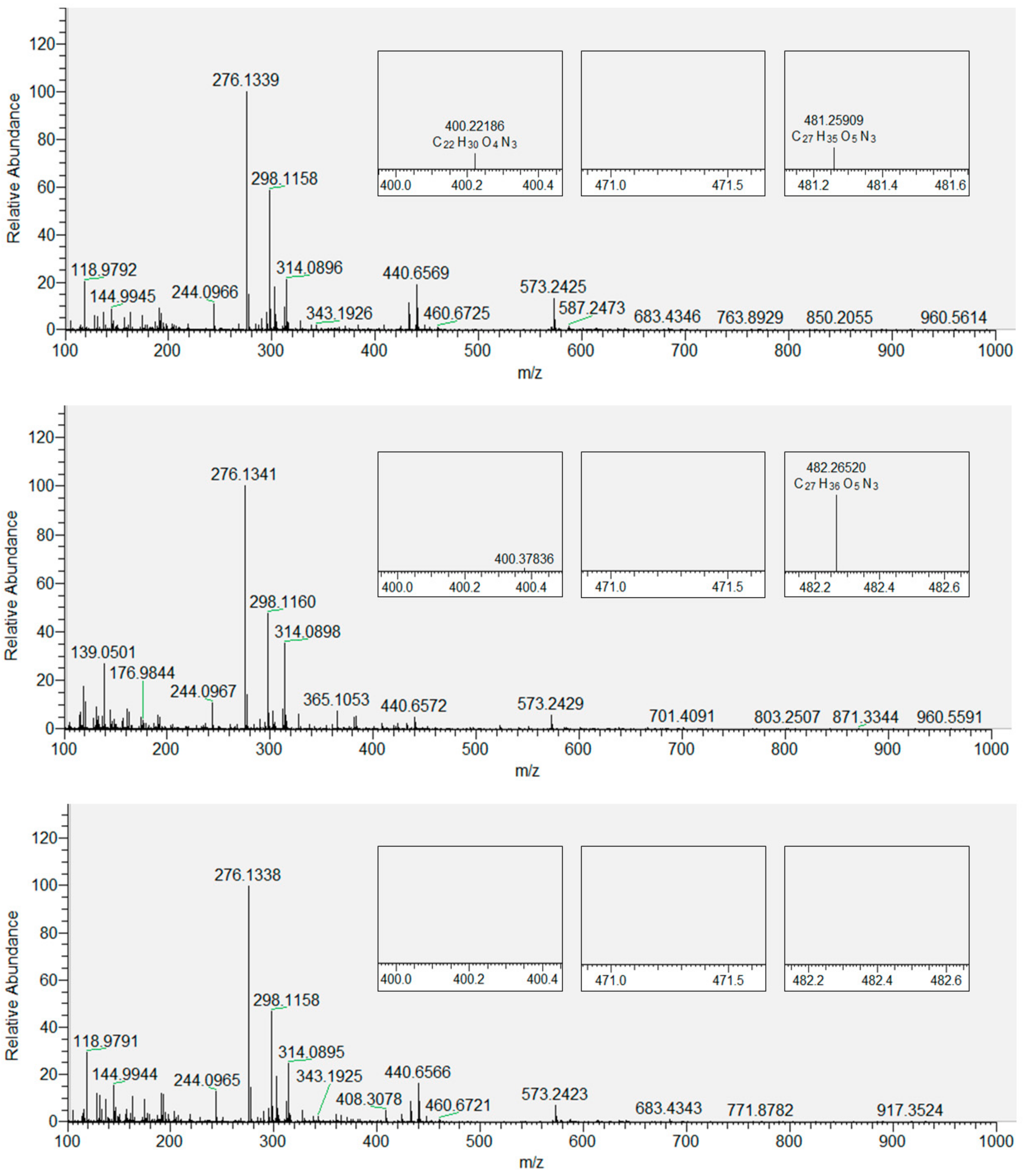
3.2. Products of Lipid Peroxidation with and without Inserted Flavonols from Mass Spectrometry (MS)
3.3. FTIR-ATR Spectroscopy
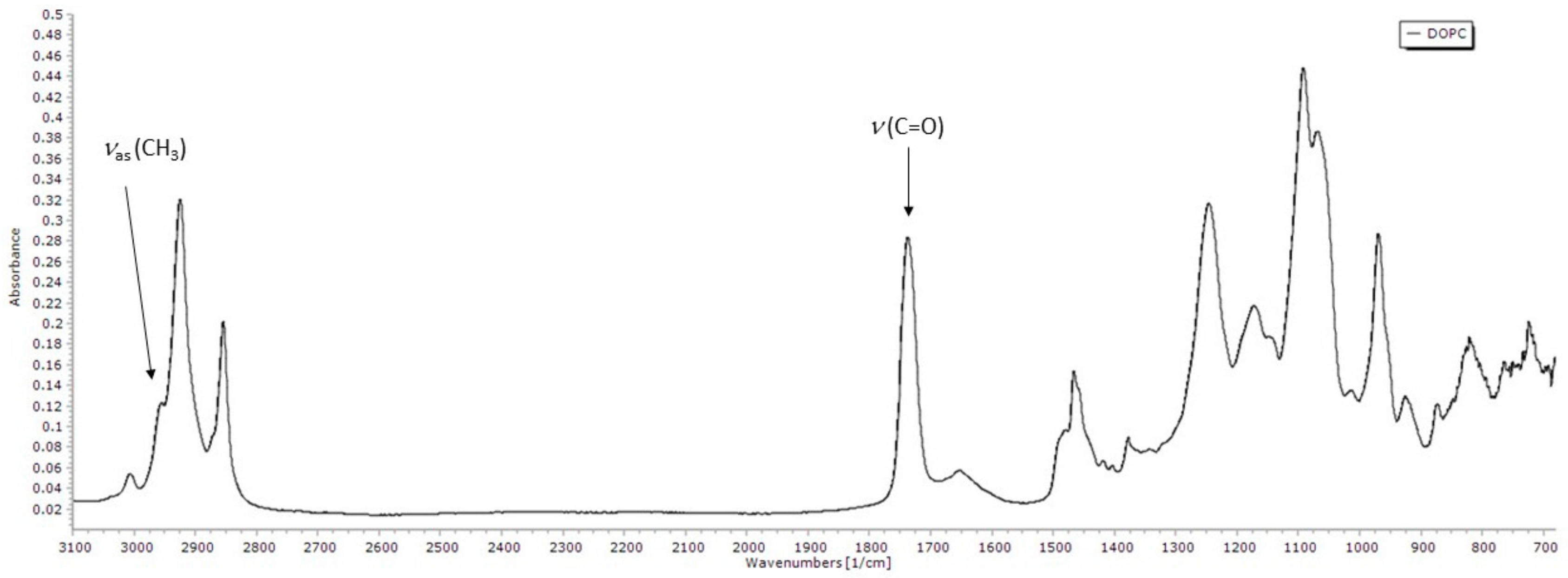
3.3.1. FTIR Spectrum of DOPC Lipid Film
3.3.2. The Extent of Lipid Peroxidation
3.4. Characterization of the Structural Changes of DOPC and Flavonol-Inserted DOPC Liposomes Resulting from Induced Lipid Peroxidation
3.4.1. Dynamic Light Scattering (DLS) and Electrophoretic (ELS) Measurements
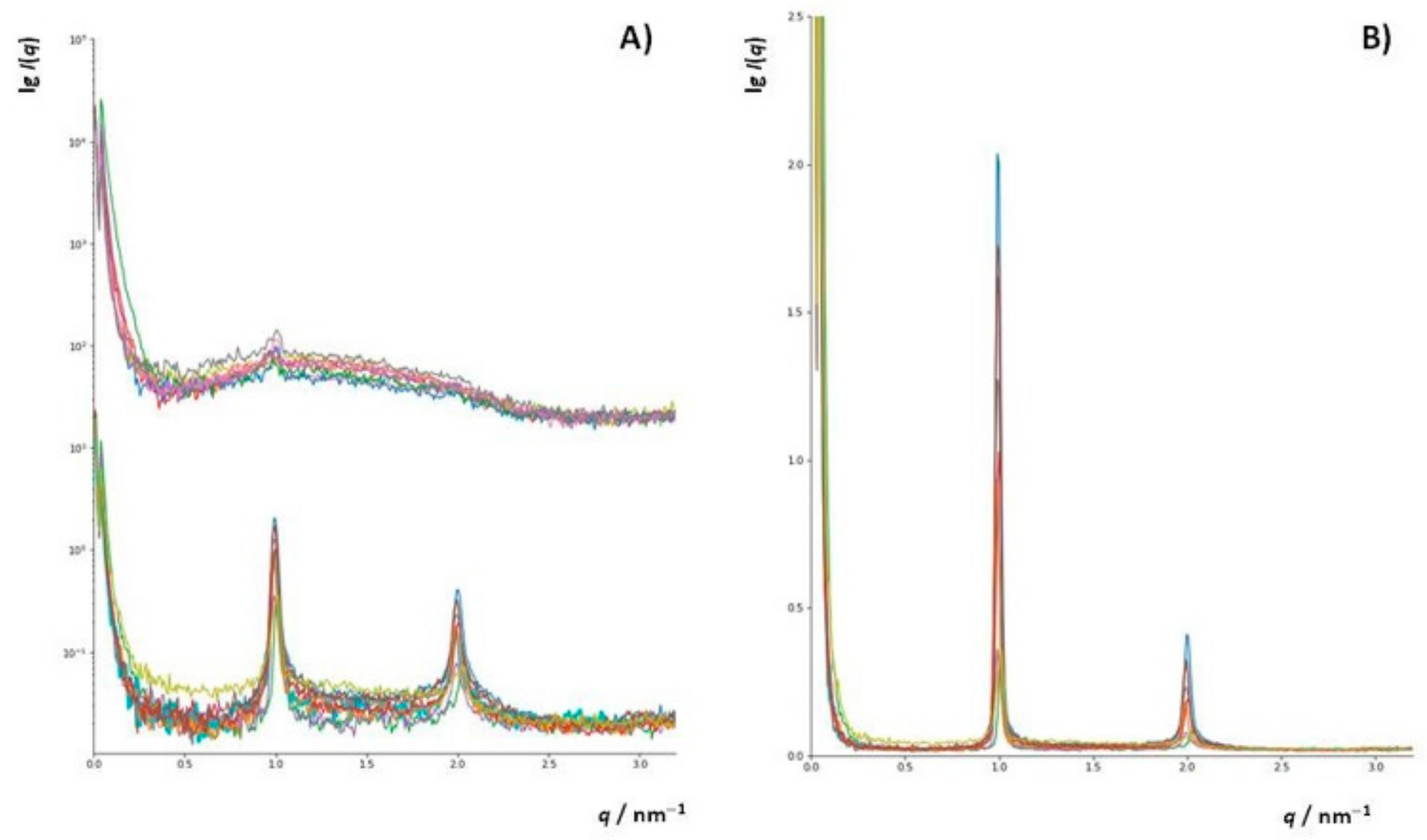
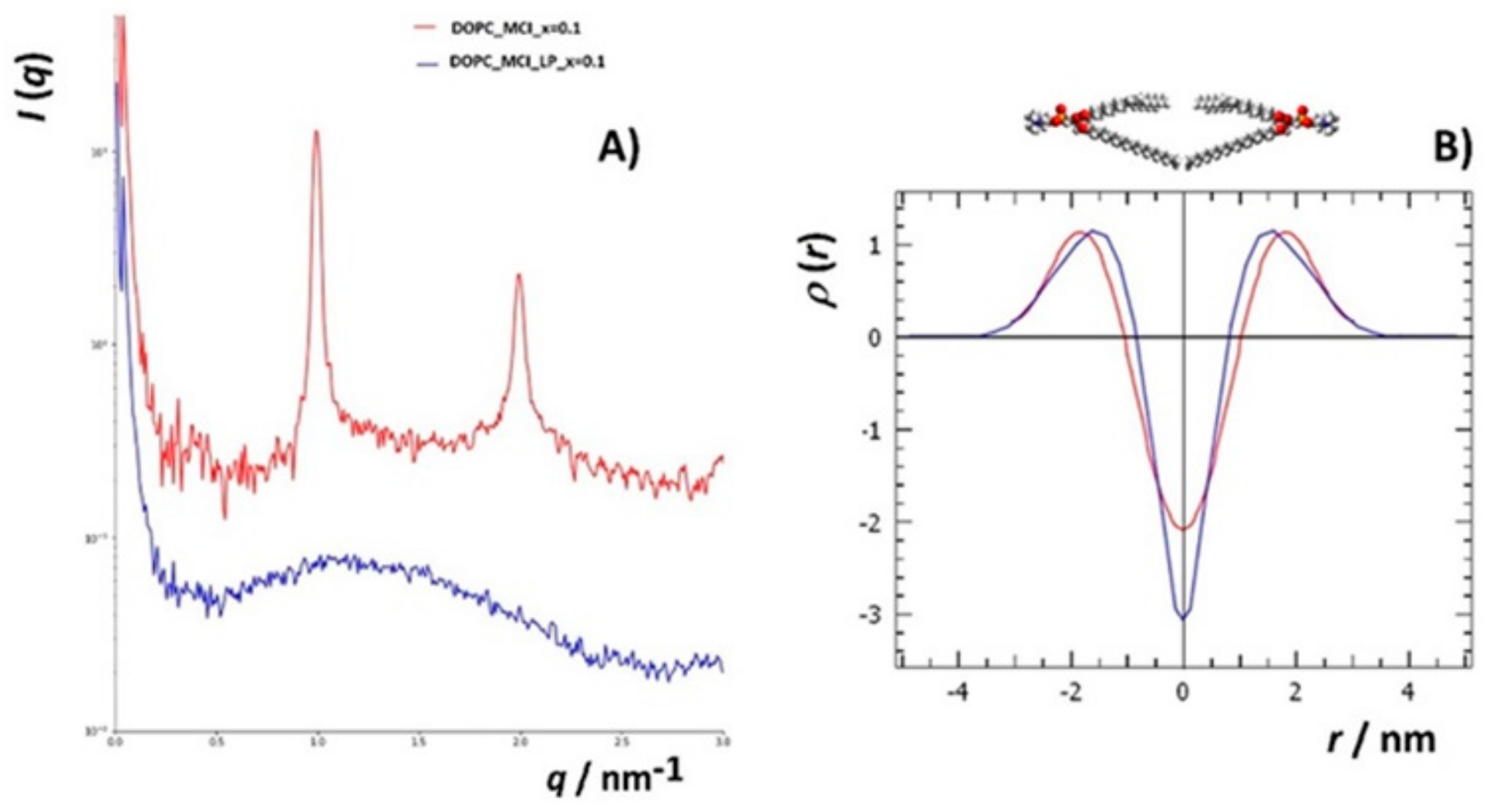
3.4.2. Small Angle X-ray Scattering (SAXS)
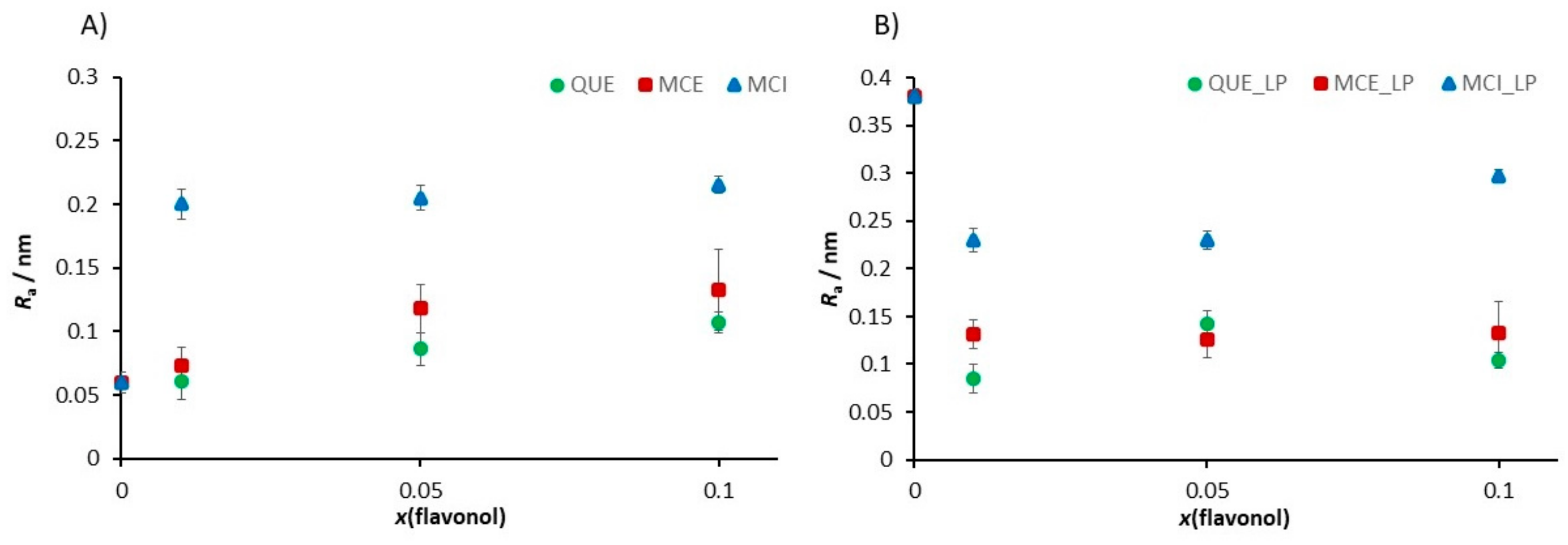
3.4.3. Nanomechanical Properties of Supported Lipid Bilayers (SLBs) before and after Induced Lipid Peroxidation
3.5. Fluidity Change upon Initiated Lipid Peroxidation by EPR
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
References
- Repetto, M.; Semprine, J.; Boveris, A. Lipid Peroxidation: Chemical Mechanism, Biological Implications and Analytical Determination. In Lipid Peroxidation; InTechOpen Limited: London, UK, 2012. [Google Scholar]
- Devasagayam, T.P.A.; Boloor, K.K.; Ramasarma, T. Methods for estimating lipid peroxidation: An analysis of merits and demerits. Indian J. Biochem. Biophys. 2003, 40, 300–308. [Google Scholar] [PubMed]
- Shahidi, F.; Zhong, Y. Measurement of antioxidant activity. J. Funct. Foods 2015, 18, 757–781. [Google Scholar] [CrossRef]
- Aruoma, O.I. Free radicals, oxidative stress, and antioxidants in human health and disease. J. Am. Oil Chem. Soc. 1998, 75, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Parola, M.; Bellomo, G.; Robino, G.; Barrera, G.; Dianzani, M.U. 4-Hydroxynonenal As a Biological Signal: Molecular Basis and Pathophysiological Implications. Antioxid. Redox Signal. 1999, 1, 255–284. [Google Scholar] [CrossRef]
- Cajone, F.; Bernelli-Zazzera, A. The Action of 4-Hydroxynonenal on Heat Shock Gene Expression in Cultured Hepatoma Cells. Free Radic. Res. Commun. 1989, 7, 189–194. [Google Scholar] [CrossRef]
- Cajone, F.; Crescente, M. In vitro activation of heat shock transcription factor by 4-hydroxynonenal. Chem. Biol. Interact. 1992, 84, 97–112. [Google Scholar] [CrossRef]
- Sies, H. Oxidative stress: From basic research to clinical application. Am. J. Med. 1991, 91, S31–S38. [Google Scholar] [CrossRef]
- Sies, H. Biochemistry of oxidative stress. Eur. J. Cancer Clin. Oncol. 1987, 23, 1798. [Google Scholar] [CrossRef]
- Kähkönen, M.P.; Hopia, A.I.; Vuorela, H.J.; Rauha, J.P.; Pihlaja, K.; Kujala, T.S.; Heinonen, M. Antioxidant activity of plant extracts containing phenolic compounds. J. Agric. Food Chem. 1999, 47, 3954–3962. [Google Scholar] [CrossRef]
- Tsao, R.; Yang, R. Optimization of a new mobile phase to know the complex and real polyphenolic composition: Towards a total phenolic index using high-performance liquid chromatography. J. Chromatogr. A 2003, 1018, 29–40. [Google Scholar] [CrossRef]
- Heim, K.E.; Tagliaferro, A.R.; Bobilya, D.J. Flavonoid antioxidants: Chemistry, metabolism and structure-activity relationships. J. Nutr. Biochem. 2002, 13, 572–584. [Google Scholar] [CrossRef]
- Ignat, I.; Volf, I.; Popa, V.I. A critical review of methods for characterisation of polyphenolic compounds in fruits and vegetables. Food Chem. 2011, 126, 1821–1835. [Google Scholar] [CrossRef] [PubMed]
- Hollman, P.C.H.; Katan, M.B. Dietary flavonoids: Intake, health effects and bioavailability. Food Chem. Toxicol. 1999, 37, 937–942. [Google Scholar] [CrossRef]
- Panche, A.N.; Diwan, A.D.; Chandra, S.R. Flavonoids: An overview. J. Nutr. Sci. 2016, 5, 1–15. [Google Scholar]
- Rice-Evans, C.A.; Miller, N.J.; Paganga, G. Structure-antioxidant activity relationships of flavonoids and phenolic acids. Free Radic. Biol. Med. 1996, 20, 933–956. [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J.M.C. Oxygen toxicity, oxygen radicals, transition metals and disease. Biochem. J. 1984, 219, 1–14. [Google Scholar] [CrossRef]
- Repetto, M.G.; Ferrarotti, N.F.; Boveris, A. The involvement of transition metal ions on iron-dependent lipid peroxidation. Arch. Toxicol. 2010, 84, 255–262. [Google Scholar] [CrossRef]
- Ohyashiki, T.; Suzuki, S.; Satoh, E.; Uemori, Y. A marked stimulation of Fe2+-initiated lipid peroxidation in phospholipid liposomes by a lipophilic aluminum complex, aluminum acetylacetonate. Biochim. Biophys. Acta Lipids Lipid Metab. 1998, 1389, 141–149. [Google Scholar]
- Tadolini, B.; Hakim, G. The mechanism of iron (III) stimulation of lipid peroxidation. Free Radic. Res. 1996, 25, 221–227. [Google Scholar] [CrossRef]
- Ohyashiki, T.; Kadoya, A.; Kushida, K. The role of Fe3+ on Fe2+-dependent lipid peroxidation in phospholipid liposomes. Chem. Pharm. Bull. 2002, 50, 203–207. [Google Scholar] [CrossRef]
- Chance, B.; Sies, H.; Boveris, A. Hydroperoxide metabolism in mammalian organs. Physiol. Rev. 1979, 59, 527–605. [Google Scholar] [CrossRef] [PubMed]
- Oteiza, P.I.; Erlejman, A.G.; Verstraeten, S.V.; Keen, C.L.; Fraga, C.G. Flavonoid-membrane interactions: A protective role of flavonoids at the membrane surface? Clin. Dev. Immunol. 2005, 12, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, C.; Driessen, A.J.M.; Recourt, K. The uncoupling efficiency and affinity of flavonoids for vesicles. Biochem. Pharmacol. 2000, 60, 1593–1600. [Google Scholar] [CrossRef]
- Verstraeten, S.V.; Nogueira, L.V.; Schreier, S.; Oteiza, P.I. Effect of trivalent metal ions on phase separation and membrane lipid packing: Role in lipid peroxidation. Arch. Biochem. Biophys. 1997, 338, 121–127. [Google Scholar] [CrossRef]
- Semwal, D.; Semwal, R.; Combrinck, S.; Viljoen, A. Myricetin: A Dietary Molecule with Diverse Biological Activities. Nutrients 2016, 8, 90. [Google Scholar] [CrossRef]
- Pereira, M.; Siba, I.P.; Chioca, L.R.; Correia, D.; Vital, M.A.B.F.; Pizzolatti, M.G.; Santos, A.R.S.; Andreatini, R. Myricitrin, a nitric oxide and protein kinase C inhibitor, exerts antipsychotic-like effects in animal models. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2011, 35, 1636–1644. [Google Scholar] [CrossRef]
- Choi, S.-M.; Kim, B.C.; Cho, Y.-H.; Choi, K.-H.; Chang, J.; Park, M.-S.; Kim, M.-K.; Cho, K.-H.; Kim, J.-K. Effects of Flavonoid Compounds on β-amyloid-peptide-induced Neuronal Death in Cultured Mouse Cortical Neurons. Chonnam Med. J. 2014, 50, 45. [Google Scholar] [CrossRef]
- Boots, A.W.; Haenen, G.R.M.M.; Bast, A. Health effects of quercetin: From antioxidant to nutraceutical. Eur. J. Pharmacol. 2008, 585, 325–337. [Google Scholar] [CrossRef]
- Lee, E.-S.; Lee, H.-E.; Shin, J.-Y.; Yoon, S.; Moon, J.-O. The flavonoid quercetin inhibits dimethylnitrosamine-induced liver damage in rats. J. Pharm. Pharmacol. 2003, 55, 1169–1174. [Google Scholar] [CrossRef]
- Bucki, R.; Pastore, T.J.J.; Giraud, F.; Sulpicejand, J.C.; Janmey, P.A. Flavonoid inhibition of platelet procoagulant activity and phosphoinositide synthesis. J. Thromb. Haemost. 2003, 1, 1820–1828. [Google Scholar] [CrossRef]
- Cushnie, T.P.T.; Lamb, A.J. Antimicrobial activity of flavonoids. Int. J. Antimicrob. Agents 2005, 26, 343–356. [Google Scholar] [CrossRef] [PubMed]
- Shimosaki, S.; Tsurunaga, Y.; Itamura, H.; Nakamura, M. Anti-allergic effect of the flavonoid myricitrin from Myrica rubra leaf extracts invitro and invivo. Nat. Prod. Res. 2011, 25, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Domitrović, R.; Rashed, K.; Cvijanović, O.; Vladimir-Knežević, S.; Škoda, M.; Višnić, A. Myricitrin exhibits antioxidant, anti-inflammatory and antifibrotic activity in carbon tetrachloride-intoxicated mice. Chem. Biol. Interact. 2015, 230, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Meotti, F.C.; Luiz, A.P.; Pizzolatti, M.G.; Santos, A.R.S. Analysis of the Antinociceptive Effect of the Flavonoid Myricitrin: Evidence for a Role of the L-arginine-nitric oxide and protein kinase C pathways. J. Pharmacol. Exp. Ther. 2006, 316, 789–796. [Google Scholar] [CrossRef]
- Reis, A.; Spickett, C.M. Chemistry of phospholipid oxidation. Biochim. Biophys. Acta Biomembr. 2012, 1818, 2374–2387. [Google Scholar] [CrossRef]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef]
- Buettner, G.R. The Pecking Order of Free Radicals and Antioxidants: Lipid Peroxidation, α-Tocopherol, and Ascorbate. Arch. Biochem. Biophys. 1993, 300, 535–543. [Google Scholar] [CrossRef]
- Rehncrona, S.; Smith, D.S.; Åkesson, B.; Westerberg, E.; Siesjö, B.K. Peroxidative Changes in Brain Cortical Fatty Acids and Phospholipids, as Characterized During Fe2+ and Ascorbic Acid-Stimulated Lipid Peroxidation in vitro. J. Neurochem. 1980, 34, 1630–1638. [Google Scholar] [CrossRef]
- Carr, A.; Frei, B. Does vitamin C act as a pro-oxidant under physiological conditions? FASEB J. 1999, 13, 1007–1024. [Google Scholar] [CrossRef]
- Arsov, Z.; Rappolt, M.; Grdadolnik, J. Weakened Hydrogen Bonds in Water Confined between Lipid Bilayers: The Existence of a Long-Range Attractive Hydration Force. ChemPhysChem 2009, 10, 1438–1441. [Google Scholar] [CrossRef]
- Oleszko, A.; Olsztyńska-Janus, S.; Walski, T.; Grzeszczuk-Kuć, K.; Bujok, J.; Gałecka, K.; Czerski, A.; Witkiewicz, W.; Komorowska, M. Application of FTIR-ATR spectroscopy to determine the extent of lipid peroxidation in plasma during haemodialysis. BioMed Res. Int. 2015, 2015, 245607. [Google Scholar] [CrossRef] [PubMed]
- Bradley, M.S.; Krech, J.H. High-pressure Raman spectra of the acetone carbonyl stretch in acetone-methanol mixtures. J. Phys. Chem. 1993, 97, 575–580. [Google Scholar] [CrossRef]
- Bergmann, A.; Fritz, G.; Glatter, O. Solving the generalized indirect Fourier transformation (GIFT) by Boltzmann simplex simulated annealing (BSSA). J. Appl. Crystallogr. 2000, 33, 1212–1216. [Google Scholar] [CrossRef]
- Glatter, O. Convolution square root of band-limited symmetrical functions and its application to small-angle scattering data. J. Appl. Crystallogr. 1981, 14, 101–108. [Google Scholar] [CrossRef]
- Kriechbaum, M. Electron Density Calculator for 1D-Lamellar Lattices. Available online: https://sas.neocities.org/xitami/java/lamdens.html (accessed on 10 April 2020).
- Mandić, L.; Sadžak, A.; Strasser, V.; Baranović, G.; Jurašin, D.D.; Sikirić, M.D.; Šegota, S. Enhanced protection of biological membranes during lipid peroxidation: Study of the interactions between flavonoid loaded mesoporous silica nanoparticles and model cell membranes. Int. J. Mol. Sci. 2019, 20, 2709. [Google Scholar] [CrossRef]
- Šegota, S.; Vojta, D.; Pletikapić, G.; Baranović, G. Ionic strength and composition govern the elasticity of biological membranes. A study of model DMPC bilayers by force- and transmission IR spectroscopy. Chem. Phys. Lipids 2015, 186, 17–29. [Google Scholar] [CrossRef]
- Jazvinšćak Jembrek, M.; Vlainić, J.; Čadež, V.; Šegota, S. Atomic force microscopy reveals new biophysical markers for monitoring subcellular changes in oxidative injury: Neuroprotective effects of quercetin at the nanoscale. PLoS ONE 2018, 13, e0200119. [Google Scholar] [CrossRef]
- Shahidi, F.; Wanasundara, U. Effect of Natural Antioxidants on the Stability of Canola Oil; Elsevier: Amsterdam, The Netherlands, 1995; pp. 469–479. [Google Scholar]
- Taga, M.S.; Miller, E.E.; Pratt, D.E. Chia seeds as a source of natural lipid antioxidants. J. Am. Oil Chem. Soc. 1984, 61, 928–931. [Google Scholar] [CrossRef]
- Das, N.P.; Pereira, T.A. Effects of flavonoids on thermal autoxidation of palm oil: Structure-activity relationships. J. Am. Oil Chem. Soc. 1990, 67, 255–258. [Google Scholar] [CrossRef]
- Chen, Z.Y.; Chan, P.T.; Ho, K.Y.; Fung, K.P.; Wang, J. Antioxidant activity of natural flavonoids is governed by number and location of their aromatic hydroxyl groups. Chem. Phys. Lipids 1996, 79, 157–163. [Google Scholar] [CrossRef]
- Frankel, E.N.; Waterhouse, A.L.; Teissedre, P.L. Principal Phenolic Phytochemicals in Selected California Wines and Their Antioxidant Activity in Inhibiting Oxidation of Human Low-Density Lipoproteins. J. Agric. Food Chem. 1995, 43, 890–894. [Google Scholar] [CrossRef]
- Teissedre, P.L.; Frankel, E.N.; Waterhouse, A.L.; Peleg, H.; German, J.B. Inhibition of in vitro Human LDL Oxidation by Phenolic Antioxidants from Grapes and Wines. J. Sci. Food Agric. 1996, 70, 55–61. [Google Scholar] [CrossRef]
- Vinson, J.A.; Dabbagh, Y.A.; Serry, M.M.; Jang, J. Plant Flavonoids, Especially Tea Flavonols, Are Powerful Antioxidants Using an in Vitro Oxidation Model for Heart Disease. J. Agric. Food Chem. 1995, 43, 2800–2802. [Google Scholar] [CrossRef]
- Mehta, A.; Seshadri, T.R. Flavonoids as antioxidants. J. Sci. Ind. Res. 1959, 18B, 24–28. [Google Scholar]
- Ramanathan, L.; Das, N.P.; Li, Q.-T. Studies on lipid oxidation in fish phospholipid liposomes. Biol. Trace Elem. Res. 1994, 40, 59–70. [Google Scholar] [CrossRef]
- Gordon, M.H.; Roedig-Penman, A. Antioxidant activity of quercetin and myricetin in liposomes. Chem. Phys. Lipids 1998, 97, 79–85. [Google Scholar] [CrossRef]
- Tai, W.-Y.; Yang, Y.-C.; Lin, H.-J.; Huang, C.-P.; Cheng, Y.-L.; Chen, M.-F.; Yen, H.-L.; Liau, I. Interplay between Structure and Fluidity of Model Lipid Membranes under Oxidative Attack. J. Phys. Chem. B 2010, 114, 15642–15649. [Google Scholar] [CrossRef]
- Wratten, M.L.; Van Ginkel, G.; Van’t Veld, A.A.; Bekker, A.; Van Faassen, E.E.; Sevanian, A. Structural and dynamic effects of oxidatively modified phospholipids in unsaturated lipid membranes. Biochemistry 1992, 31, 10901–10907. [Google Scholar] [CrossRef]
- Beranova, L.; Cwiklik, L.; Jurkiewicz, P.; Hof, M.; Jungwirth, P. Oxidation Changes Physical Properties of Phospholipid Bilayers: Fluorescence Spectroscopy and Molecular Simulations. Langmuir 2010, 26, 6140–6144. [Google Scholar] [CrossRef]
- Richter, C. Biophysical consequences of lipid peroxidation in membranes. Chem. Phys. Lipids 1987, 44, 175–189. [Google Scholar] [CrossRef]
- Niki, E.; Yoshida, Y.; Saito, Y.; Noguchi, N. Lipid peroxidation: Mechanisms, inhibition, and biological effects. Biochem. Biophys. Res. Commun. 2005, 338, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Kelsey, N.A.; Wilkins, H.M.; Linseman, D.A. Nutraceutical Antioxidants as Novel Neuroprotective Agents. Molecules 2010, 15, 7792–7814. [Google Scholar] [CrossRef] [PubMed]
- Suh, J.H.; Niu, Y.S.; Hung, W.-L.; Ho, C.-T.; Wang, Y. Lipidomic analysis for carbonyl species derived from fish oil using liquid chromatography–tandem mass spectrometry. Talanta 2017, 168, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Milić, I.; Hoffmann, R.; Fedorova, M. Simultaneous detection of low and high molecular weight carbonylated compounds derived from lipid peroxidation by electrospray ionization-tandem mass spectrometry. Anal. Chem. 2013, 85, 156–162. [Google Scholar] [CrossRef]
- Hollebrands, B.; Varvaki, E.; Kaal, S.; Janssen, H.-G. Selective labeling for the identification and semi-quantification of lipid aldehydes in food products. Anal. Bioanal. Chem. 2018, 410, 5421–5429. [Google Scholar] [CrossRef]
- Bernardes, C.P.; Santos, N.A.G.; Costa, T.R.; Sisti, F.; Amaral, L.; Menaldo, D.L.; Amstalden, M.K.; Ribeiro, D.L.; Antunes, L.M.G.; Sampaio, S.V.; et al. A Synthetic Snake-Venom-Based Tripeptide Protects PC12 Cells from the Neurotoxicity of Acrolein by Improving Axonal Plasticity and Bioenergetics. Neurotox. Res. 2020, 37, 227–237. [Google Scholar] [CrossRef]
- Park, J.H.; Choi, J.-Y.; Jo, C.; Koh, Y.H. Involvement of ADAM10 in acrolein-induced astrocytic inflammation. Toxicol. Lett. 2020, 318, 44–49. [Google Scholar] [CrossRef]
- Mano, J.I.; Biswas, M.; Sugimoto, K. Reactive Carbonyl Species: A Missing Link in ROS Signaling. Plants 2019, 8, 391. [Google Scholar] [CrossRef]
- PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/ (accessed on 10 April 2020).
- Toro-Uribe, S.; López-Giraldo, L.J.; Decker, E.A. Relationship between the Physiochemical Properties of Cocoa Procyanidins and Their Ability to Inhibit Lipid Oxidation in Liposomes. J. Agric. Food Chem. 2018, 66, 4490–4502. [Google Scholar] [CrossRef]
- Sánchez-Alonso, I.; Carmona, P.; Careche, M. Vibrational spectroscopic analysis of hake (Merluccius merluccius L.) lipids during frozen storage. Food Chem. 2012, 132, 160–167. [Google Scholar] [CrossRef]
- Blume, A. Properties of lipid vesicles: FT-IR spectroscopy and fluorescence probe studies. Curr. Opin. Colloid Interface Sci. 1996, 1, 64–77. [Google Scholar] [CrossRef]
- Arrondo, J.L.R.; Goñi, F.M. Infrared studies of protein-induced perturbation of lipids in lipoproteins and membranes. Chem. Phys. Lipids 1998, 96, 53–68. [Google Scholar] [CrossRef]
- Rudolphi-Skórska, E.; Filek, M.; Zembala, M. The Effects of the Structure and Composition of the Hydrophobic Parts of Phosphatidylcholine-Containing Systems on Phosphatidylcholine Oxidation by Ozone. J. Membr. Biol. 2017, 250, 493–505. [Google Scholar] [CrossRef]
- Maity, P.; Saha, B.; Kumar, G.S.; Karmakar, S. Binding of monovalent alkali metal ions with negatively charged phospholipid membranes. Biochim. Biophys. Acta Biomembr. 2016, 1858, 706–714. [Google Scholar] [CrossRef] [PubMed]
- Harris, C.S.; Mo, F.; Migahed, L.; Chepelev, L.; Haddad, P.S.; Wright, J.S.; Willmore, W.G.; Arnason, J.T.; Bennett, S.A.L. Plant phenolics regulate neoplastic cell growth and survival: A quantitative structure–activity and biochemical analysisThis article is one of a selection of papers published in this special issue (part 2 of 2) on the Safety and Efficacy of Natural Health. Can. J. Physiol. Pharmacol. 2007, 85, 1124–1138. [Google Scholar] [CrossRef] [PubMed]
- Álvarez-Diduk, R.; Ramírez-Silva, M.T.; Galano, A.; Merkoçi, A. Deprotonation Mechanism and Acidity Constants in Aqueous Solution of Flavonols: A Combined Experimental and Theoretical Study. J. Phys. Chem. B 2013, 117, 12347–12359. [Google Scholar] [CrossRef]
- Bi, S.; Wang, T.; Zhao, T.; Wang, Y.; Pang, B. Study of the interaction of salmon sperm DNA with myricitrin–CPB based on the enhanced resonance light scattering signal and its potential application. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2013, 112, 397–402. [Google Scholar] [CrossRef]
- Mosca, M.; Ceglie, A.; Ambrosone, L. Effect of membrane composition on lipid oxidation in liposomes. Chem. Phys. Lipids 2011, 164, 158–165. [Google Scholar] [CrossRef]
- Makky, A.; Tanaka, M. Impact of Lipid Oxidization on Biophysical Properties of Model Cell Membranes. J. Phys. Chem. B 2015, 119, 5857–5863. [Google Scholar] [CrossRef]
- Albertini, R.; Rindi, S.; Passi, A.; Pallavicini, G.; De Luca, G. Heparin protection against Fe2+ -and Cu2+ -mediated oxidation of liposomes. FEBS Lett. 1996, 383, 155–158. [Google Scholar] [CrossRef]
- Wong-ekkabut, J.; Xu, Z.; Triampo, W.; Tang, I.-M.; Peter Tieleman, D.; Monticelli, L. Effect of Lipid Peroxidation on the Properties of Lipid Bilayers: A Molecular Dynamics Study. Biophys. J. 2007, 93, 4225–4236. [Google Scholar] [CrossRef] [PubMed]
- Attwood, S.; Choi, Y.; Leonenko, Z. Preparation of DOPC and DPPC Supported Planar Lipid Bilayers for Atomic Force Microscopy and Atomic Force Spectroscopy. Int. J. Mol. Sci. 2013, 14, 3514–3539. [Google Scholar] [CrossRef] [PubMed]
- Engel, A.; Schoenenberger, C.A.; Müller, D.J. High resolution imaging of native biological sample surfaces using scanning probe microscopy. Curr. Opin. Struct. Biol. 1997, 7, 279–284. [Google Scholar] [CrossRef]
- Picas, L.; Rico, F.; Scheuring, S. Direct Measurement of the Mechanical Properties of Lipid Phases in Supported Bilayers. Biophys. J. 2012, 102, L01–L03. [Google Scholar] [CrossRef] [PubMed]
- Ohki, K.; Takamura, T.; Nozawai, Y. Effect of.ALPHA.-tocopherol on lipid peroxidation and acyl chain mobility of liver microsomes from vitamin E-difficient rat. J. Nutr. Sci. Vitaminol. 1984, 30, 221–234. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | dH/nm | ζ/mV |
|---|---|---|
| DOPC | 1049 ± 140 | −3.4 ± 0.6 |
| DOPC/H2O2 + Fe2+ | 932 ± 105 | −2.8 ± 0.9 |
| DOPC/QUE (x = 0.01) | 148 ± 48 | −3.9 ± 0.3 |
| DOPC/QUE (x = 0.01)/H2O2 + Fe2+ | 1319 ± 57 | −2.9 ± 0.6 |
| DOPC / QUE (x = 0.05) | 1258 ± 117 | −3.8 ± 0.4 |
| DOPC/QUE (x = 0.05)/H2O2 + Fe2+ | 1351 ± 205 | −4.9 ± 0.3 |
| DOPC/QUE (x = 0.1) | 202 ± 79 | −5.7 ± 0.5 |
| DOPC/QUE (x = 0.1)/H2O2 + Fe2+ | 487 ± 64 | −7.1 ± 0.3 |
| DOPC/MCE (x = 0.01) | 107 ± 16 | −2.4 ± 0.2 |
| DOPC/MCE (x = 0.01)/ H2O2 + Fe2+ | 577 ± 159 | −3.6 ± 0.4 |
| DOPC/MCE (x = 0.05) | 72 ± 6 | −2.5 ± 0.5 |
| DOPC/MCE (x = 0.05)/H2O2 + Fe2+ | 1031 ± 152 | −3.6 ± 0.2 |
| DOPC/MCE (x = 0.1) | 203 ± 119 | −3.7 ± 0.5 |
| DOPC/MCE (x = 0.1)/H2O2 + Fe2+ | 406 ± 105 | −9.8 ± 2.2 |
| DOPC/MCI (x = 0.01) | 163 ± 87 | −3.5 ± 0.3 |
| DOPC/MCI (x = 0.01)/H2O2 + Fe2+ | 180 ± 42 | −4.0 ± 0.6 |
| DOPC/MCI (x = 0.05) | 222 ± 42 | −2.8 ± 0.2 |
| DOPC/MCI (x = 0.05)/H2O2 + Fe2+ | 266 ± 44 | −4.3 ± 0.2 |
| DOPC/MCI (x = 0.1) | 155 ± 30 | −3.9 ± 0.5 |
| DOPC/MCI (x = 0.1)/H2O2 + Fe2+ | 1746 ± 654 | −8.7 ± 1.2 |
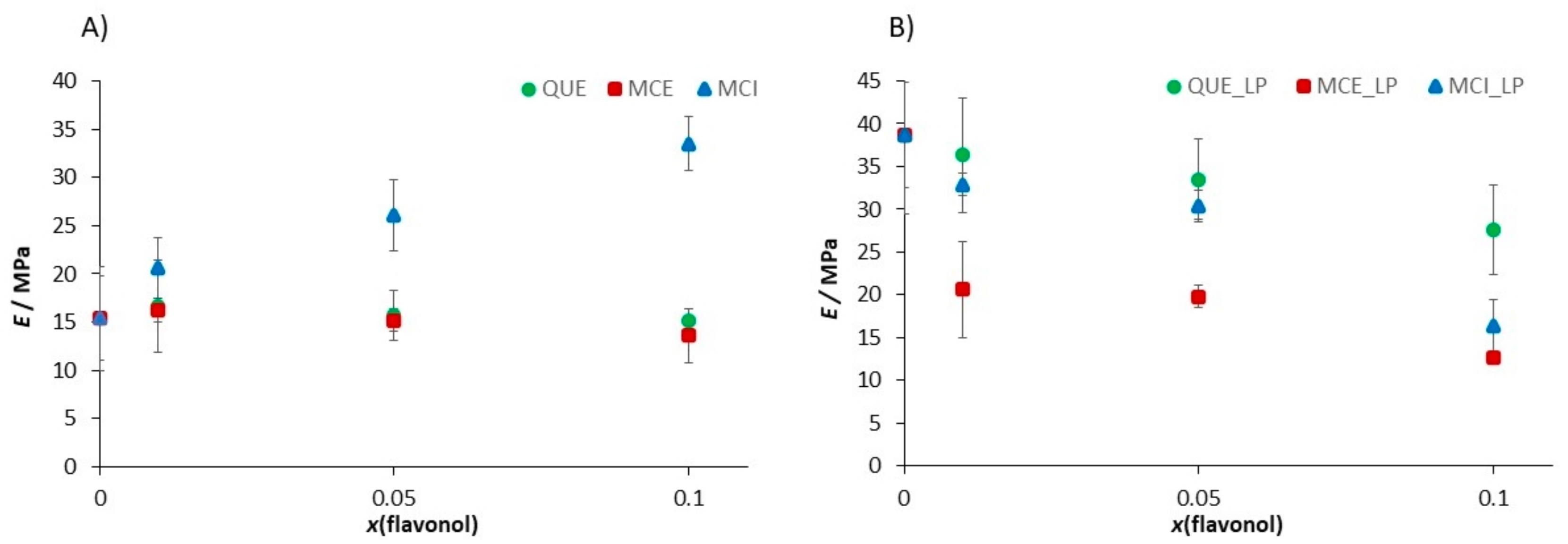
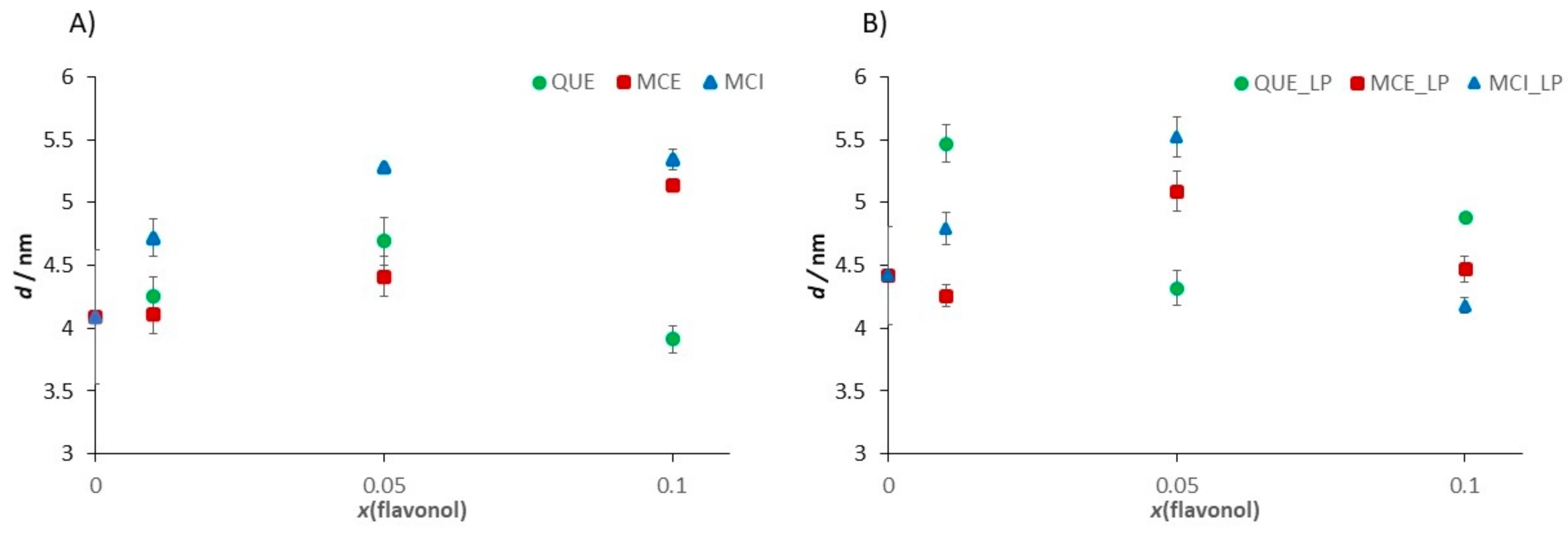
| Sample | Ra/nm | E/MPa | d/nm |
|---|---|---|---|
| DOPC | 0.060 ± 0.008 | 15.4 ± 5.4 | 4.09 ± 0.53 |
| DOPC/H2O2 + Fe2+ | 0.381 ± 0.018 | 38.7 ± 6.2 | 4.42 ± 0.39 |
| DOPC/QUE (x = 0.01) | 0.061 ± 0.015 | 16.6 ± 4.8 | 4.25 ± 0.16 |
| DOPC/QUE (x = 0.01)/H2O2 + Fe2+ | 0.085 ± 0.026 | 36.3 ± 6.7 | 5.47 ± 015 |
| DOPC/QUE (x = 0.05) | 0.086 ± 0.013 | 15.7 ± 2.6 | 4.69 ± 0.19 |
| DOPC/QUE (x = 0.05)/H2O2 + Fe2+ | 0.143 ± 0.018 | 33.5 ± 4.7 | 4.32 ± 0.14 |
| DOPC/QUE (x = 0.1) | 0.107 ± 0.008 | 15.2 ± 1.1 | 3.91 ± 0.11 |
| DOPC/QUE (x = 0.1)/H2O2 + Fe2+ | 0.104 ± 0.024 | 27.6 ± 5.3 | 4.88 ± 0.03 |
| DOPC/MCE (x = 0.01) | 0.073 ± 0.015 | 16.2 ± 1.2 | 4.11 ± 0.15 |
| DOPC/MCE (x = 0.01)/H2O2 + Fe2+ | 0.131 ± 0.034 | 20.6 ± 5.6 | 4.26 ± 0.09 |
| DOPC/MCE (x = 0.05) | 0.118 ± 0.019 | 15.2 ± 1.1 | 4.41 ± 0.16 |
| DOPC/MCE (x = 0.05)/H2O2 + Fe2+ | 0.126 ± 0.019 | 19.8 ± 1.3 | 5.09 ± 0.16 |
| DOPC/MCE (x = 0.1) | 0.133 ± 0.032 | 13.6 ± 2.8 | 5.14 ± 0.02 |
| DOPC/MCE (x = 0.1)/H2O2 + Fe2+ | 0.133 ± 0.030 | 12.6 ± 0.1 | 4.78 ± 0.10 |
| DOPC/MCI (x = 0.01) | 0.200 ± 0.012 | 20.6 ± 3.2 | 4.72 ± 0.15 |
| DOPC/MCI (x = 0.01)/H2O2 + Fe2+ | 0.230 ± 0.023 | 32.9 ± 1.3 | 4.79 ± 0.13 |
| DOPC/MCI (x = 0.05) | 0.205 ± 0.094 | 26.1 ± 3.7 | 5.28 ± 0.02 |
| DOPC/MCI (x = 0.05)/H2O2 + Fe2+ | 0.230 ± 0.003 | 30.4 ± 1.8 | 5.52 ± 0.16 |
| DOPC/MCI (x = 0.1) | 0.215 ± 0.006 | 33.5 ± 2.8 | 5.34 ± 0.08 |
| DOPC/MCI (x = 0.1)/H2O2 + Fe2+ | 0.297 ± 0.005 | 16.3 ± 3.1 | 4.18 ± 0.06 |
| Sample | 2AZZ/G | aoN/G | w(S)/% | w(F)/% | S |
|---|---|---|---|---|---|
| DOPC | 58.32 | - | 100 | - | 1 |
| DOPC + H2O2 + Fe2+ | 58.20 | 15.99 | 32.94 | 67.06 | 0.33 |
| DOPC + QUE (x = 0.01) | 58.97 | 14.89 | 91.34 | 8.64 | 0.91 |
| DOPC + QUE (x = 0.01) + H2O2 + Fe2+ | 60.39 | 14.62 | 94.30 | 5.70 | 0.94 |
| DOPC + QUE (x = 0.05) | 59.68 | 15.00 | 89.43 | 10.57 | 0.89 |
| DOPC + QUE (x = 0.05) + H2O2 + Fe2+ | 60.01 | 14.29 | 96.23 | 3.77 | 0.96 |
| DOPC + QUE (x = 0.1) | 66.40 | 13.98 | 93.78 | 6.22 | 0.94 |
| DOPC + QUE (x = 0.1) + H2O2 + Fe2+ | 64.72 | 14.15 | 92.39 | 7.61 | 0.92 |
| DOPC + MCE (x = 0.01) | 58.37 | - | 100 | - | 1 |
| DOPC + MCE (x = 0.01) + H2O2 + Fe2+ | 60.91 | 15.79 | 75.01 | 24.99 | 0.75 |
| DOPC + MCE (x = 0.05) | 60.18 | 15.08 | 95.52 | 4.48 | 0.96 |
| DOPC + MCE (x = 0.05) + H2O2 + Fe2+ | 61.45 | 14.92 | 92.80 | 7.20 | 0.93 |
| DOPC + MCE (x = 0.1) | 60.65 | 14.97 | 92.64 | 7.36 | 0.93 |
| DOPC + MCE (x = 0.1) + H2O2 + Fe2+ | 62.06 | 15.05 | 91.50 | 8.50 | 0.92 |
| DOPC + MCI (x = 0.01) | 59.56 | 13.87 | 94.28 | 5.72 | 0.94 |
| DOPC + MCI (x = 0.01) + H2O2 + Fe2+ | 62.09 | 13.89 | 93.81 | 6.19 | 0.94 |
| DOPC + MCI (x = 0.05) | 60.03 | 14.02 | 93.52 | 6.48 | 0.94 |
| DOPC + MCI (x = 0.05) + H2O2 + Fe2+ | 61.57 | 13.91 | 95.32 | 4.68 | 0.95 |
| DOPC + MCI (x = 0.1) | 60.55 | 14.22 | 94.16 | 5.84 | 0.94 |
| DOPC + MCI (x = 0.1) + H2O2 + Fe2+ | 62.31 | 14.09 | 94.40 | 5.60 | 0.95 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sadžak, A.; Mravljak, J.; Maltar-Strmečki, N.; Arsov, Z.; Baranović, G.; Erceg, I.; Kriechbaum, M.; Strasser, V.; Přibyl, J.; Šegota, S. The Structural Integrity of the Model Lipid Membrane during Induced Lipid Peroxidation: The Role of Flavonols in the Inhibition of Lipid Peroxidation. Antioxidants 2020, 9, 430. https://doi.org/10.3390/antiox9050430
Sadžak A, Mravljak J, Maltar-Strmečki N, Arsov Z, Baranović G, Erceg I, Kriechbaum M, Strasser V, Přibyl J, Šegota S. The Structural Integrity of the Model Lipid Membrane during Induced Lipid Peroxidation: The Role of Flavonols in the Inhibition of Lipid Peroxidation. Antioxidants. 2020; 9(5):430. https://doi.org/10.3390/antiox9050430
Chicago/Turabian StyleSadžak, Anja, Janez Mravljak, Nadica Maltar-Strmečki, Zoran Arsov, Goran Baranović, Ina Erceg, Manfred Kriechbaum, Vida Strasser, Jan Přibyl, and Suzana Šegota. 2020. "The Structural Integrity of the Model Lipid Membrane during Induced Lipid Peroxidation: The Role of Flavonols in the Inhibition of Lipid Peroxidation" Antioxidants 9, no. 5: 430. https://doi.org/10.3390/antiox9050430
APA StyleSadžak, A., Mravljak, J., Maltar-Strmečki, N., Arsov, Z., Baranović, G., Erceg, I., Kriechbaum, M., Strasser, V., Přibyl, J., & Šegota, S. (2020). The Structural Integrity of the Model Lipid Membrane during Induced Lipid Peroxidation: The Role of Flavonols in the Inhibition of Lipid Peroxidation. Antioxidants, 9(5), 430. https://doi.org/10.3390/antiox9050430