Understanding the Redox Biology of Selenium in the Search of Targeted Cancer Therapies

 , , and
, , and
Abstract
1. Introduction
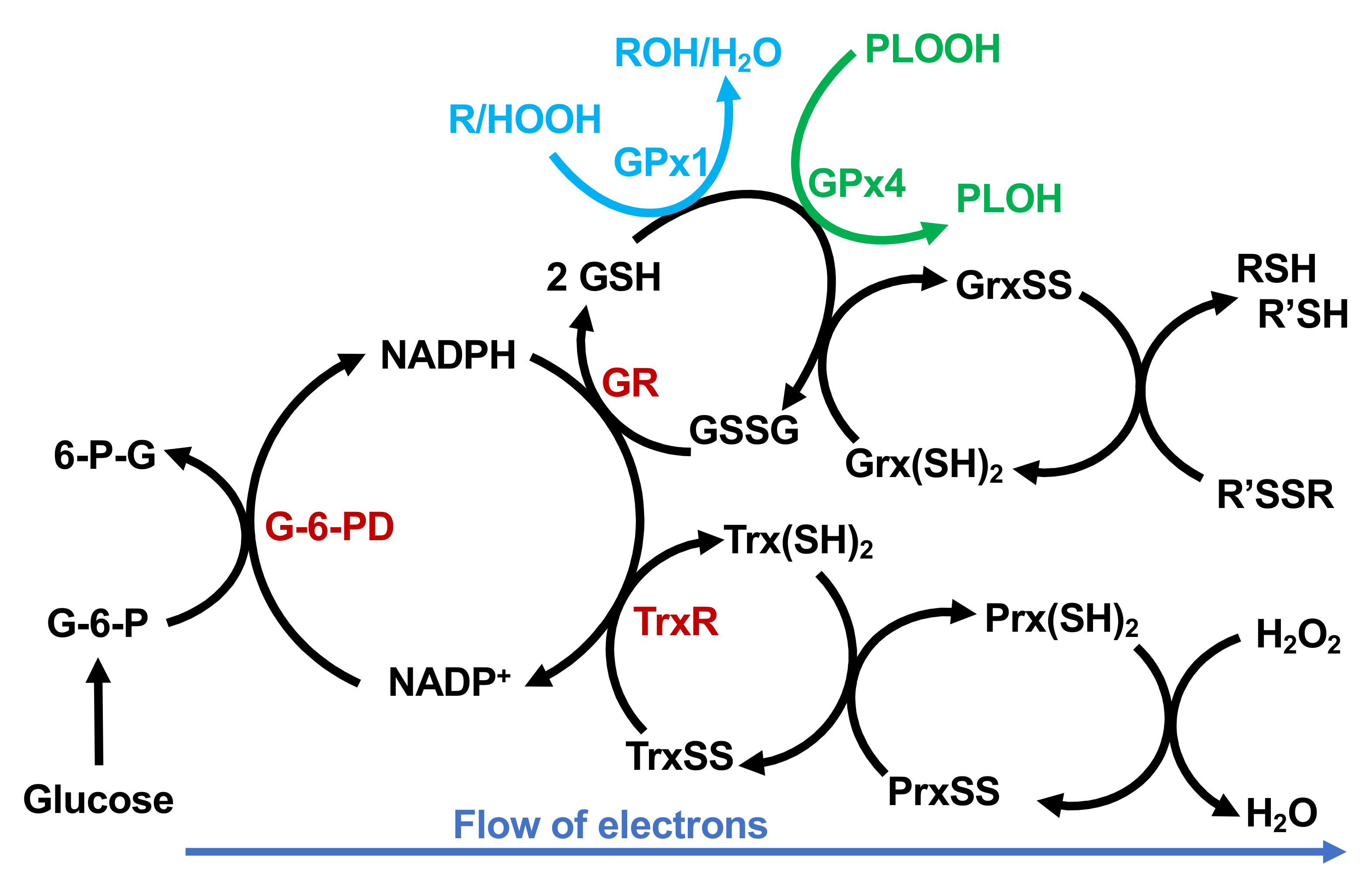
2. Selenoproteins in Redox Biology and Cancer
2.1. Glutathione Peroxidases
2.1.1. GPx1
2.1.2. GPx2
2.1.3. GPx3
2.1.4. GPx4
2.2. Thioredoxin Reductases
2.3. Iodothyronine Deiodinases
2.4. Methionine Sulfoxide Reductases B
2.5. Selenoproteins
2.5.1. SELENOP
2.5.2. SELENOF and SELENOM
2.6. Selenium Deficiency in Cell Culture
3. The Role of Selenium in Cancer Prevention
4. Redox Active Selenium
4.1. Selenium at an Active Site of an Enzyme Is Typically More Reactive Than Its Sulfur Counterpart
4.2. The Pro-oxidant Side of Selenium
4.3. Cancer Therapies Utilizing Selenium
4.4. High Dose Organic Selenium
5. Discussion
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Mills, G.C. Hemoglobin catabolism. I. Glutathione peroxidase, an erythrocyte enzyme which protects hemoglobin from oxidative breakdown. J. Biol. Chem. 1957, 229, 189–197. [Google Scholar] [PubMed]
- Schwarz, K.; Bieri, J.G.; Briggs, G.M.; Scott, M.L. Prevention of exudative diathesis in chicks by factor 3 and selenium. Proc. Soc. Exp. Biol. Med. 1957, 95, 621–625. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, K.; Foltz, C.M. Selenium as an integral part of factor 3 against dietary necrotic liver degeneration. J. Am. Chem. Soc. 1957, 79, 3292–3293. [Google Scholar] [CrossRef]
- Flohe, L.; Gunzler, W.A.; Schock, H.H. Glutathione peroxidase: A selenoenzyme. FEBS Lett. 1973, 32, 132–134. [Google Scholar] [CrossRef]
- Kraus, R.J.; Foster, S.J.; Ganther, H.E. Identification of selenocysteine in glutathione peroxidase by mass spectroscopy. Biochemistry 1983, 22, 5853–5858. [Google Scholar] [CrossRef]
- Medicine, I.O. Dietary Reference Intakes for Vitamin C, Vitamin E, Selenium, and Carotenoids; The National Academies Press: Washington, DC, USA, 2000; p. 528. [Google Scholar]
- Xia, Y.; Hill, K.E.; Li, P.; Xu, J.; Zhou, D.; Motley, A.K.; Wang, L.; Byrne, D.W.; Burk, R.F. Optimization of selenoprotein P and other plasma selenium biomarkers for the assessment of the selenium nutritional requirement: A placebo-controlled, double-blind study of selenomethionine supplementation in selenium-deficient Chinese subjects. Am. J. Clin. Nutr. 2010, 92, 525–531. [Google Scholar] [CrossRef]
- Hurst, R.; Armah, C.N.; Dainty, J.R.; Hart, D.J.; Teucher, B.; Goldson, A.J.; Broadley, M.R.; Motley, A.K.; Fairweather-Tait, S.J. Establishing optimal selenium status: Results of a randomized, double-blind, placebo-controlled trial. Am. J. Clin. Nutr. 2010, 91, 923–931. [Google Scholar] [CrossRef]
- Burk, R.F. Effects of chemical form of selenium on plasma biomarkers in a high-dose human supplementation trial. Cancer Epidemiol. Biomark. Prev. 2006, 15, 804–810. [Google Scholar] [CrossRef]
- Vinceti, M.; Filippini, T.; Del Giovane, C.; Dennert, G.; Zwahlen, M.; Brinkman, M.; Zeegers, M.P.; Horneber, M.; D‘Amico, R.; Crespi, C.M. Selenium for preventing cancer. Cochrane Database Syst. Rev. 2018, 1. [Google Scholar] [CrossRef]
- Fakih, M.G.; Pendyala, L.; Smith, P.F.; Creaven, P.J.; Reid, M.E.; Badmaev, V.; Azrak, R.G.; Prey, J.D.; Lawrence, D.; Rustum, Y.M. A phase I and pharmacokinetic study of fixed-dose selenomethionine and irinotecan in solid tumors. Clin. Cancer Res. 2006, 12, 1237–1244. [Google Scholar] [CrossRef][Green Version]
- Ip, C.; Thompson, H.J.; Zhu, Z.; Ganther, H.E. In vitro and in vivo studies of methylseleninic acid: Evidence that a monomethylated selenium metabolite is critical for cancer chemoprevention. Cancer Res. 2000, 60, 2882–2886. [Google Scholar] [PubMed]
- Letavayova, L.; Vlckova, V.; Brozmanova, J. Selenium: From cancer prevention to DNA damage. Toxicology 2006, 227, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Drake, E.N. Cancer chemoprevention: Selenium as a prooxidant, not an antioxidant. Med. Hypotheses 2006, 67, 318–322. [Google Scholar] [CrossRef] [PubMed]
- Speier, C.; Baker, S.S.; Newburger, P.E. Relationships between in vitro selenium supply, glutathione peroxidase activity, and phagocytic function in the HL-60 human myeloid cell line. J. Biol. Chem. 1985, 260, 8951–8955. [Google Scholar] [PubMed]
- Baker, R.D., Jr.; Baker, S.S.; Rao, R. Selenium deficiency in tissue culture: Implications for oxidative metabolism. J. Pediatr. Gastroenterol. Nutr. 1998, 27, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Leist, M.; Raab, B.; Maurer, S.; Rösick, U.; Brigelius-Flohé, R. Conventional cell culture media do not adequately supply cells with antioxidants and thus facilitate peroxide-induced genotoxicity. Free Radic. Biol. Med. 1996, 21, 297–306. [Google Scholar] [CrossRef][Green Version]
- Stolwijk, J.M.; Falls-Hubert, K.C.; Searby, C.; Wagner, B.A.; Buettner, G.R. Simultaneous detection of the enzyme activities of GPx1 and GPx4 guide optimization of selenium in cell biological experiments. Redox Biol. 2020, 32. [Google Scholar] [CrossRef]
- Avery, J.C.; Hoffmann, P.R. Selenium, selenoproteins, and immunity. Nutrients 2018, 10, 1203. [Google Scholar] [CrossRef]
- Sevanian, A.; Muakkassah-Kelly, S.F.; Montestruque, S. The influence of phospholipase A2 and glutathione peroxidase on the elimination of membrane lipid peroxides. Arch. Biochem. Biophys. 1983, 223, 441–452. [Google Scholar] [CrossRef]
- Weitzel, F.; Ursini, F.; Wendel, A. Phospholipid hydroperoxide glutathione peroxidase in various mouse organs during selenium deficiency and repletion. Biochim. Biophys. Acta (BBA)—Gen. Subj. 1990, 1036, 88–94. [Google Scholar] [CrossRef]
- Maiorino, M.; Gregolin, C.; Ursini, F. Phospholipid hydroperoxide glutathione peroxidase. Methods Enzym. 1990, 186, 448–457. [Google Scholar] [CrossRef]
- Martinez, J.I.; Garcia, R.D.; Galarza, A.M. The kinetic mechanism of glutathione peroxidase from human platelets. Thromb Res. 1982, 27, 197–203. [Google Scholar] [CrossRef]
- Gladyshev, V.N.; Factor, V.M.; Housseau, F.; Hatfield, D.L. Contrasting patterns of regulation of the antioxidant selenoproteins, thioredoxin reductase, and glutathione peroxidase, in cancer cells. Biochem. Biophys. Res. Commun. 1998, 251, 488–493. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Lubos, E.; Loscalzo, J.; Handy, D.E. Glutathione peroxidase-1 in health and disease: From molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 2011, 15, 1957–1997. [Google Scholar] [CrossRef]
- Cheng, W.H.; Ho, Y.S.; Ross, D.A.; Valentine, B.A.; Combs, G.F.; Lei, X.G. Cellular glutathione peroxidase knockout mice express normal levels of selenium-dependent plasma and phospholipid hydroperoxide glutathione peroxidases in various tissues. J. Nutr. 1997, 127, 1445–1450. [Google Scholar] [CrossRef]
- Lei, X.G.; Cheng, W.H.; McClung, J.P. Metabolic regulation and function of glutathione peroxidase-1. Annu. Rev. Nutr. 2007, 27, 41–61. [Google Scholar] [CrossRef]
- Cheng, W.; Fu, Y.X.; Porres, J.M.; Ross, D.A.; Lei, X.G. Selenium-dependent cellular glutathione peroxidase protects mice against a pro-oxidant-induced oxidation of NADPH, NADH, lipids, and protein. FASEB J. 1999, 13, 1467–1475. [Google Scholar] [CrossRef]
- Esworthy, R.S.; Baker, M.A.; Chu, F.F. Expression of selenium-dependent glutathione peroxidase in human breast tumor cell lines. Cancer Res. 1995, 55, 957–962. [Google Scholar]
- Jee, C.D.; Kim, M.A.; Jung, E.J.; Kim, J.; Kim, W.H. Identification of genes epigenetically silenced by CpG methylation in human gastric carcinoma. Eur. J. Cancer 2009, 45, 1282–1293. [Google Scholar] [CrossRef]
- De Haan, J.B.; Bladier, C.; Griffiths, P.; Kelner, M.; O‘Shea, R.D.; Cheung, N.S.; Bronson, R.T.; Silvestro, M.J.; Wild, S.; Zheng, S.S.; et al. Mice with a homozygous null mutation for the most abundant glutathione peroxidase, Gpx1, show increased susceptibility to the oxidative stress-inducing agents paraquat and hydrogen peroxide. J. Biol. Chem. 1998, 273, 22528–22536. [Google Scholar] [CrossRef]
- Baliga, M.S.; Wang, H.; Zhuo, P.; Schwartz, J.L.; Diamond, A.M. Selenium and GPx-1 overexpression protect mammalian cells against UV-induced DNA damage. Biol. Trace Elem. Res. 2007, 115, 227–242. [Google Scholar] [CrossRef]
- Throm, S.L.; Klemsz, M.J. PU. 1 regulates glutathione peroxidase expression in neutrophils. J. Leukoc Biol. 2003, 74, 111–117. [Google Scholar] [CrossRef]
- Borras, C.; Gambini, J.; Gomez-Cabrera, M.C.; Sastre, J.; Pallardo, F.V.; Mann, G.E.; Vina, J. 17beta-oestradiol up-regulates longevity-related, antioxidant enzyme expression via the ERK1 and ERK2[MAPK]/NFkappaB cascade. Aging Cell 2005, 4, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Vina, J.; Borras, C.; Gomez-Cabrera, M.C.; Orr, W.C. Part of the series: From dietary antioxidants to regulators in cellular signalling and gene expression. Role of reactive oxygen species and (phyto)oestrogens in the modulation of adaptive response to stress. Free Radic. Res. 2006, 40, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.; Li, S.; Swaroop, M.; Guan, K.; Oberley, L.W.; Sun, Y. Transcriptional activation of the human glutathione peroxidase promoter by p53. J. Biol. Chem. 1999, 274, 12061–12066. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohé, R.; Kipp, A. Glutathione peroxidases in different stages of carcinogenesis. Biochim. Biophys. Acta (BBA)—Gen. Subj. 2009, 1790, 1555–1568. [Google Scholar] [CrossRef]
- Lee, W.P.; Lee, C.L.; Lin, H.C. Glutathione S-transferase and glutathione peroxidase are essential in the early stage of adriamycin resistance before P-glycoprotein overexpression in HOB1 lymphoma cells. Cancer Chemother. Pharm. 1996, 38, 45–51. [Google Scholar] [CrossRef]
- Lu, Y.P.; Lou, Y.R.; Yen, P.; Newmark, H.L.; Mirochnitchenko, O.I.; Inouye, M.; Huang, M.T. Enhanced skin carcinogenesis in transgenic mice with high expression of glutathione peroxidase or both glutathione peroxidase and superoxide dismutase. Cancer Res. 1997, 57, 1468–1474. [Google Scholar]
- Ho, J.C.; Chan-Yeung, M.; Ho, S.P.; Mak, J.C.; Ip, M.S.; Ooi, G.C.; Wong, M.P.; Tsang, K.W.; Lam, W.K. Disturbance of systemic antioxidant profile in nonsmall cell lung carcinoma. Eur. Respir. J. 2007, 29, 273–278. [Google Scholar] [CrossRef]
- Florian, S.; Krehl, S.; Loewinger, M.; Kipp, A.; Banning, A.; Esworthy, S.; Chu, F.-F.; Brigelius-Flohé, R. Loss of GPx2 increases apoptosis, mitosis, and GPx1 expression in the intestine of mice. Free Radic. Biol. Med. 2010, 49, 1694–1702. [Google Scholar] [CrossRef] [PubMed]
- Emmink, B.L.; Laoukili, J.; Kipp, A.P.; Koster, J.; Govaert, K.M.; Fatrai, S.; Verheem, A.; Steller, E.J.A.; Brigelius-Flohé, R.; Jimenez, C.R.; et al. GPx2 Suppression of H2O2 stress links the formation of differentiated tumor mass to metastatic capacity in colorectal cancer. Cancer Res. 2014, 74, 6717. [Google Scholar] [CrossRef] [PubMed]
- Koyama, H.; Omura, K.; Ejima, A.; Kasanuma, Y.; Watanabe, C.; Satoh, H. Separation of selenium-containing proteins in human and mouse plasma using tandem high-performance liquid chromatography columns coupled with inductively coupled plasma-mass spectrometry. Anal. Biochem. 1999, 267, 84–91. [Google Scholar] [CrossRef]
- Hoffmann, P.R.; Hoge, S.C.; Li, P.A.; Hoffmann, F.W.; Hashimoto, A.C.; Berry, M.J. The selenoproteome exhibits widely varying, tissue-specific dependence on selenoprotein P for selenium supply. Nucleic Acids Res. 2007, 35, 3963–3973. [Google Scholar] [CrossRef]
- Smith, C.V.; Jones, D.P.; Guenthner, T.M.; Lash, L.H.; Lauterburg, B.H. Compartmentation of glutathione: Implications for the study of toxicity and disease. Toxicol. Appl. Pharmacol. 1996, 140, 1–12. [Google Scholar] [CrossRef]
- Jones, D.P.; Carlson, J.L.; Samiec, P.S.; Sternberg, P., Jr.; Mody, V.C., Jr.; Reed, R.L.; Brown, L.A. Glutathione measurement in human plasma. Evaluation of sample collection, storage and derivatization conditions for analysis of dansyl derivatives by HPLC. Clin. Chim. Acta 1998, 275, 175–184. [Google Scholar] [CrossRef]
- Stenzel, J.D.; Welty, S.E.; Benzick, A.E.; Smith, E.O.; Smith, C.V.; Hansen, T.N. Hyperoxic lung injury in Fischer-344 and Sprague-Dawley rats in vivo. Free Radic. Biol. Med. 1993, 14, 531–539. [Google Scholar] [CrossRef]
- Ottaviano, F.G.; Tang, S.-S.; Handy, D.E.; Loscalzo, J. Regulation of the extracellular antioxidant selenoprotein plasma glutathione peroxidase (GPx-3) in mammalian cells. Mol. Cell. Biochem. 2009, 327, 111–126. [Google Scholar] [CrossRef]
- Ren, B.; Huang, W.; Akesson, B.; Ladenstein, R. The crystal structure of seleno-glutathione peroxidase from human plasma at 2.9 A resolution. J. Mol. Biol. 1997, 268, 869–885. [Google Scholar] [CrossRef]
- Freedman, J.E.; Frei, B.; Welch, G.N.; Loscalzo, J. Glutathione peroxidase potentiates the inhibition of platelet function by S-nitrosothiols. J. Clin. Investig. 1995, 96, 394–400. [Google Scholar] [CrossRef]
- Barrett, C.W.; Ning, W.; Chen, X.; Smith, J.J.; Washington, M.K.; Hill, K.E.; Coburn, L.A.; Peek, R.M.; Chaturvedi, R.; Wilson, K.T.; et al. Tumor suppressor function of the plasma glutathione peroxidase gpx3 in colitis-associated carcinoma. Cancer Res. 2013, 73, 1245–1255. [Google Scholar] [CrossRef] [PubMed]
- Conrad, M.; Schneider, M.; Seiler, A.; Bornkamm, G.W. Physiological role of phospholipid hydroperoxide glutathione peroxidase in mammals. Biol. Chem. 2007, 388, 1019–1025. [Google Scholar] [CrossRef] [PubMed]
- Yant, L.J.; Ran, Q.; Rao, L.; Van Remmen, H.; Shibatani, T.; Belter, J.G.; Motta, L.; Richardson, A.; Prolla, T.A. The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radic. Biol. Med. 2003, 34, 496–502. [Google Scholar] [CrossRef]
- Ursini, F.; Heim, S.; Kiess, M.; Maiorino, M.; Roveri, A.; Wissing, J.; Flohe, L. Dual function of the selenoprotein PHGPx during sperm maturation. Science 1999, 285, 1393–1396. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Buettner, G.R. The Pecking Order of Free Radicals and Antioxidants: Lipid Peroxidation, α-Tocopherol, and Ascorbate. Arch. Biochem. Biophys. 1993, 300, 535–543. [Google Scholar] [CrossRef]
- Qian, S.Y.; Wang, H.P.; Schafer, F.Q.; Buettner, G.R. EPR detection of lipid-derived free radicals from PUFA, LDL, and cell oxidations. Free Radic. Biol. Med. 2000, 29, 568–579. [Google Scholar] [CrossRef]
- Girotti, A.W. Mechanisms of lipid peroxidation. J. Free Radic. Biol. Med. 1985, 1, 87–95. [Google Scholar] [CrossRef]
- Wilcox, A.L.; Marnett, L.J. Polyunsaturated fatty acid alkoxyl radicals exist as carbon-centered epoxyallylic radicals: A key step in hydroperoxide-amplified lipid peroxidation. Chem. Res. Toxicol. 1993, 6, 413–416. [Google Scholar] [CrossRef]
- Wagner, B.A.; Buettner, G.R.; Burns, C.P. Vitamin E slows the rate of free radical-mediated lipid peroxidation in cells. Arch. Biochem. Biophys. 1996, 334, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; Yin, H. Role of lipid peroxidation derived 4-hydroxynonenal (4-HNE) in cancer: Focusing on mitochondria. Redox Biol. 2015, 4, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Cheeseman, K.H. Mechanisms and effects of lipid peroxidation. Mol. Asp. Med. 1993, 14, 191–197. [Google Scholar] [CrossRef]
- Wang, H.P.; Schafer, F.Q.; Goswami, P.C.; Oberley, L.W.; Buettner, G.R. Phospholipid hydroperoxide glutathione peroxidase induces a delay in G1 of the cell cycle. Free Radic. Res. 2003, 37, 621–630. [Google Scholar] [CrossRef]
- Wang, H.P.; Qian, S.Y.; Schafer, F.Q.; Domann, F.E.; Oberley, L.W.; Buettner, G.R. Phospholipid hydroperoxide glutathione peroxidase protects against singlet oxygen-induced cell damage of photodynamic therapy. Free Radic. Biol. Med. 2001, 30, 825–835. [Google Scholar] [CrossRef]
- Mustacich, D.; Powis, G. Thioredoxin reductase. Biochem. J. 2000, 346, 1–8. [Google Scholar] [CrossRef]
- Jacob, C.; Giles, G.I.; Giles, N.M.; Sies, H. Sulfur and selenium: The role of oxidation state in protein structure and function. Angew. Chem. Int. Ed. Engl. 2003, 42, 4742–4758. [Google Scholar] [CrossRef]
- Ng, C.F.; Schafer, F.Q.; Buettner, G.R.; Rodgers, V.G. The rate of cellular hydrogen peroxide removal shows dependency on GSH: Mathematical insight into in vivo H2O2 and GPx concentrations. Free Radic. Res. 2007, 41, 1201–1211. [Google Scholar] [CrossRef]
- Rhee, S.G.; Woo, H.A.; Kil, I.S.; Bae, S.H. Peroxiredoxin functions as a peroxidase and a regulator and sensor of local peroxides. J. Biol. Chem. 2012, 287, 4403–4410. [Google Scholar] [CrossRef]
- Kolberg, M.; Strand, K.R.; Graff, P.; Andersson, K.K. Structure, function, and mechanism of ribonucleotide reductases. Biochim. Biophys. Acta 2004, 1699, 1–34. [Google Scholar] [CrossRef]
- Wei, S.J.; Botero, A.; Hirota, K.; Bradbury, C.M.; Markovina, S.; Laszlo, A.; Spitz, D.R.; Goswami, P.C.; Yodoi, J.; Gius, D. Thioredoxin nuclear translocation and interaction with redox factor-1 activates the activator protein-1 transcription factor in response to ionizing radiation. Cancer Res. 2000, 60, 6688–6695. [Google Scholar] [PubMed]
- Tsuji, P.A.; Carlson, B.A.; Anderson, C.B.; Seifried, H.E.; Hatfield, D.L.; Howard, M.T. Dietary Selenium Levels Affect Selenoprotein Expression and Support the Interferon-gamma and IL-6 Immune Response Pathways in Mice. Nutrients 2015, 7, 6529–6549. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Holmgren, A. Selenoproteins. J. Biol. Chem. 2009, 284, 723–727. [Google Scholar] [CrossRef] [PubMed]
- Jakupoglu, C.; Przemeck, G.K.; Schneider, M.; Moreno, S.G.; Mayr, N.; Hatzopoulos, A.K.; De Angelis, M.H.; Wurst, W.; Bornkamm, G.W.; Brielmeier, M.; et al. Cytoplasmic thioredoxin reductase is essential for embryogenesis but dispensable for cardiac development. Mol. Cell. Biol. 2005, 25, 1980–1988. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Chew, E.H.; Holmgren, A. Targeting thioredoxin reductase is a basis for cancer therapy by arsenic trioxide. Proc. Natl. Acad. Sci. USA 2007, 104, 12288–12293. [Google Scholar] [CrossRef]
- Marzano, C.; Gandin, V.; Folda, A.; Scutari, G.; Bindoli, A.; Rigobello, M.P. Inhibition of thioredoxin reductase by auranofin induces apoptosis in cisplatin-resistant human ovarian cancer cells. Free Radic. Biol. Med. 2007, 42, 872–881. [Google Scholar] [CrossRef]
- Nguyen, P.; Awwad, R.T.; Smart, D.D.K.; Spitz, D.R.; Gius, D. Thioredoxin reductase as a novel molecular target for cancer therapy. Cancer Lett. 2006, 236, 164–174. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Holmgren, A.; Bjornstedt, M. Thioredoxin and thioredoxin reductase. Methods Enzym. 1995. [Google Scholar] [CrossRef]
- Nakamura, H.; Nakamura, K.; Yodoi, J. Redox regulation of cellular activation. Annu. Rev. Immunol. 1997, 15, 351–369. [Google Scholar] [CrossRef]
- Lei, H.; Wang, G.; Zhang, J.; Han, Q. Inhibiting TrxR suppresses liver cancer by inducing apoptosis and eliciting potent antitumor immunity. Oncol. Rep. 2018, 40, 3447–3457. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Papp, L.V.; Fang, J.; Rodriguez-Nieto, S.; Zhivotovsky, B.; Holmgren, A. Inhibition of mammalian thioredoxin reductase by some flavonoids: Implications for myricetin and quercetin anticancer activity. Cancer Res. 2006, 66, 4410–4418. [Google Scholar] [CrossRef] [PubMed]
- Bianco, A.C.; Salvatore, D.; Gereben, B.; Berry, M.J.; Larsen, P.R. Biochemistry, cellular and molecular biology, and physiological roles of the iodothyronine selenodeiodinases. Endocr. Rev. 2002, 23, 38–89. [Google Scholar] [CrossRef] [PubMed]
- Gross, M.; Oertel, M.; Köhrle, J. Differential selenium-dependent expression of type I 5′-deiodinase and glutathione peroxidase in the porcine epithelial kidney cell line LLC-PK 1. Biochem. J. 1995, 306, 851–856. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Schomburg, L.; Schweizer, U. Hierarchical regulation of selenoprotein expression and sex-specific effects of selenium. Biochim. Biophys. Acta 2009, 1790, 1453–1462. [Google Scholar] [CrossRef] [PubMed]
- Vanderpas, J. Nutritional epidemiology and thyroid hormone metabolism. Annu. Rev. Nutr. 2006, 26, 293–322. [Google Scholar] [CrossRef]
- Casula, S.; Bianco, A.C. Thyroid hormone deiodinases and cancer. Front. Endocrinol. (Lausanne) 2012, 3, 74. [Google Scholar] [CrossRef]
- Kim, H.Y.; Gladyshev, V.N. Methionine sulfoxide reductases: Selenoprotein forms and roles in antioxidant protein repair in mammals. Biochem. J. 2007, 407, 321–329. [Google Scholar] [CrossRef]
- Fomenko, D.E.; Novoselov, S.V.; Natarajan, S.K.; Lee, B.C.; Koc, A.; Carlson, B.A.; Lee, T.H.; Kim, H.Y.; Hatfield, D.L.; Gladyshev, V.N. MsrB1 (methionine-R-sulfoxide reductase 1) knock-out mice: Roles of MsrB1 in redox regulation and identification of a novel selenoprotein form. J. Biol. Chem. 2009, 284, 5986–5993. [Google Scholar] [CrossRef]
- He, Q.; Li, H.; Meng, F.; Sun, X.; Feng, X.; Chen, J.; Li, L.; Liu, J. Methionine sulfoxide reductase B1 regulates hepatocellular carcinoma cell proliferation and invasion via the mitogen-activated protein kinase pathway and epithelial-mesenchymal transition. Oxid. Med. Cell. Longev. 2018, 2018, 5287971. [Google Scholar] [CrossRef]
- Hill, K.E.; Zhou, J.; McMahan, W.J.; Motley, A.K.; Atkins, J.F.; Gesteland, R.F.; Burk, R.F. Deletion of selenoprotein P alters distribution of selenium in the mouse. J. Biol. Chem. 2003, 278, 13640–13646. [Google Scholar] [CrossRef] [PubMed]
- Schomburg, L.; Schweizer, U.; Holtmann, B.; Flohe, L.; Sendtner, M.; Kohrle, J. Gene disruption discloses role of selenoprotein P in selenium delivery to target tissues. Biochem. J. 2003, 370, 397–402. [Google Scholar] [CrossRef]
- Schweizer, U.; Streckfuss, F.; Pelt, P.; Carlson, B.A.; Hatfield, D.L.; Kohrle, J.; Schomburg, L. Hepatically derived selenoprotein P is a key factor for kidney but not for brain selenium supply. Biochem. J. 2005, 386, 221–226. [Google Scholar] [CrossRef]
- Valentine, W.M.; Abel, T.W.; Hill, K.E.; Austin, L.M.; Burk, R.F. Neurodegeneration in mice resulting from loss of functional selenoprotein P or its receptor apolipoprotein E receptor 2. J. Neuropathol. Exp. Neurol. 2008, 67, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Olson, G.E.; Winfrey, V.P.; Nagdas, S.K.; Hill, K.E.; Burk, R.F. Apolipoprotein E receptor-2 (ApoER2) mediates selenium uptake from selenoprotein P by the mouse testis. J. Biol. Chem. 2007, 282, 12290–12297. [Google Scholar] [CrossRef] [PubMed]
- Olson, G.E.; Winfrey, V.P.; Hill, K.E.; Burk, R.F. Megalin mediates selenoprotein P uptake by kidney proximal tubule epithelial cells. J. Biol. Chem. 2008, 283, 6854–6860. [Google Scholar] [CrossRef]
- Meyer, H.A.; Endermann, T.; Stephan, C.; Stoedter, M.; Behrends, T.; Wolff, I.; Jung, K.; Schomburg, L. Selenoprotein P Status Correlates to Cancer-Specific Mortality in Renal Cancer Patients. PLoS ONE 2012, 7, e46644. [Google Scholar] [CrossRef]
- Renko, K.; Hofmann, P.J.; Stoedter, M.; Hollenbach, B.; Behrends, T.; Kohrle, J.; Schweizer, U.; Schomburg, L. Down-regulation of the hepatic selenoprotein biosynthesis machinery impairs selenium metabolism during the acute phase response in mice. Faseb J. 2009, 23, 1758–1765. [Google Scholar] [CrossRef]
- Nichol, C.; Herdman, J.; Sattar, N.; O’Dwyer, P.J.; St, J.O.R.D.; Littlejohn, D.; Fell, G. Changes in the concentrations of plasma selenium and selenoproteins after minor elective surgery: Further evidence for a negative acute phase response? Clin. Chem. 1998, 44, 1764–1766. [Google Scholar] [CrossRef]
- Meplan, C.; Crosley, L.K.; Nicol, F.; Beckett, G.J.; Howie, A.F.; Hill, K.E.; Horgan, G.; Mathers, J.C.; Arthur, J.R.; Hesketh, J.E. Genetic polymorphisms in the human selenoprotein P gene determine the response of selenoprotein markers to selenium supplementation in a gender-specific manner (the SELGEN study). Faseb J. 2007, 21, 3063–3074. [Google Scholar] [CrossRef]
- Méplan, C.; Nicol, F.; Burtle, B.T.; Crosley, L.K.; Arthur, J.R.; Mathers, J.C.; Hesketh, J.E. Relative Abundance of Selenoprotein P Isoforms in Human Plasma Depends on Genotype, Se Intake, and Cancer Status. Antioxid. Redox Signal. 2009, 11, 2631–2640. [Google Scholar] [CrossRef] [PubMed]
- Kraus, R.J.; Prohaska, J.R.; Ganther, H.E. Oxidized forms of ovine erythrocyte glutathione peroxidase. Cyanide inhibition of a 4-glutathione:4-selenoenzyme. Biochim. Biophys. Acta 1980, 615, 19–26. [Google Scholar] [CrossRef]
- Labunskyy, V.M.; Ferguson, A.D.; Fomenko, D.E.; Chelliah, Y.; Hatfield, D.L.; Gladyshev, V.N. A novel cysteine-rich domain of Sep15 mediates the interaction with UDP-glucose: Glycoprotein glucosyltransferase. J. Biol. Chem. 2005, 280, 37839–37845. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, A.D.; Labunskyy, V.M.; Fomenko, D.E.; Arac, D.; Chelliah, Y.; Amezcua, C.A.; Rizo, J.; Gladyshev, V.N.; Deisenhofer, J. NMR structures of the selenoproteins Sep15 and SelM reveal redox activity of a new thioredoxin-like family. J. Biol. Chem. 2006, 281, 3536–3543. [Google Scholar] [CrossRef]
- Labunskyy, V.M.; Hatfield, D.L.; Gladyshev, V.N. The Sep15 protein family: Roles in disulfide bond formation and quality control in the endoplasmic reticulum. IUBMB Life 2007, 59, 1–5. [Google Scholar] [CrossRef]
- Jiang, H.; Shi, Q.-Q.; Ge, L.-Y.; Zhuang, Q.-F.; Xue, D.; Xu, H.-Y.; He, X.-Z. Selenoprotein M stimulates the proliferative and metastatic capacities of renal cell carcinoma through activating the PI3K/AKT/mTOR pathway. Cancer Med. 2019, 8, 4836–4844. [Google Scholar] [CrossRef]
- Tsuji, P.A.; Naranjo-Suarez, S.; Carlson, B.A.; Tobe, R.; Yoo, M.-H.; Davis, C.D. Deficiency in the 15 kDa selenoprotein inhibits human colon cancer cell growth. Nutrients 2011, 3, 805–817. [Google Scholar] [CrossRef]
- Jaspers, I.; Zhang, W.; Brighton, L.E.; Carson, J.L.; Styblo, M.; Beck, M.A. Selenium deficiency alters epithelial cell morphology and responses to influenza. Free Radic. Biol. Med. 2007, 42, 1826–1837. [Google Scholar] [CrossRef]
- Ebert, R.; Ulmer, M.; Zeck, S.; Meissner-Weigl, J.; Schneider, D.; Stopper, H.; Schupp, N.; Kassem, M.; Jakob, F. Selenium supplementation restores the antioxidative capacity and prevents cell damage in bone marrow stromal cells in vitro. Stem. Cells 2006, 24, 1226–1235. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Yoshida, Y.; Akazawa, T.; Takahashi, K.; Niki, E. Cell death caused by selenium deficiency and protective effect of antioxidants. J. Biol. Chem. 2003. [Google Scholar] [CrossRef]
- Prabhu, K.S.; Zamamiri-Davis, F.; Stewart, J.B.; Thompson, J.T.; Sordillo, L.M.; Reddy, C.C. Selenium deficiency increases the expression of inducible nitric oxide synthase in RAW 264.7 macrophages: Role of nuclear factor-κB in up-regulation. Biochem. J. 2002, 366, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Kristal, A.R.; Darke, A.K.; Morris, J.S.; Tangen, C.M.; Goodman, P.J.; Thompson, I.M.; Meyskens, F.L., Jr.; Goodman, G.E.; Minasian, L.M.; Parnes, H.L.; et al. Baseline selenium status and effects of selenium and vitamin e supplementation on prostate cancer risk. JNCI J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef] [PubMed]
- Klein, E.A.; Thompson, I.M.; Tangen, C.M.; Crowley, J.J.; Lucia, M.S.; Goodman, P.J.; Minasian, L.M.; Ford, L.G.; Parnes, H.L.; Gaziano, J.M.; et al. Vitamin E and the risk of prostate cancer: The Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA 2011, 306, 1549–1556. [Google Scholar] [CrossRef] [PubMed]
- Hatfield, D.L.; Gladyshev, V.N. The outcome of Selenium and Vitamin E Cancer Prevention Trial (SELECT) reveals the need for better understanding of selenium biology. Mol. Interv. 2009, 9, 18–21. [Google Scholar] [CrossRef]
- Muecke, R.; Schomburg, L.; Buentzel, J.; Kisters, K.; Micke, O. Selenium or no selenium—That is the question in tumor patients: A new controversy. Integr. Cancer Ther. 2010, 9, 136–141. [Google Scholar] [CrossRef]
- Rayman, M.P. Food-chain selenium and human health: Emphasis on intake. Br. J. Nutr. 2008, 100, 254–268. [Google Scholar] [CrossRef]
- El-Bayoumy, K. The protective role of selenium on genetic damage and on cancer. Mutat. Res. 2001, 475, 123–139. [Google Scholar] [CrossRef]
- Ip, C.; Lisk, D.J. Enrichment of selenium in allium vegetables for cancer prevention. Carcinogenesis 1994, 15, 1881–1885. [Google Scholar] [CrossRef]
- Ip, C.; Lisk, D.J. Modulation of phase I and phase II xenobiotic-metabolizing enzymes by selenium-enriched garlic in rats. Nutr. Cancer 1997, 28, 184–188. [Google Scholar] [CrossRef]
- Spivack, S.D.; Hurteau, G.J.; Fasco, M.J.; Kaminsky, L.S. Phase I and II Carcinogen Metabolism Gene Expression in Human Lung Tissue and Tumors. Clin. Cancer Res. 2003, 9, 6002. [Google Scholar]
- Smith, L.E.; Denissenko, M.F.; Bennett, W.P.; Li, H.; Amin, S.; Tang, M.S.; Pfeifer, G.P. Targeting of Lung Cancer Mutational Hotspots by Polycyclic Aromatic Hydrocarbons. JNCI J. Natl. Cancer Inst. 2000, 92, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.X.; Zheng, Y.; West, M.; Tang, M.-S. Carcinogens preferentially bind at methylated CpG in the p53 mutational hot spots. Cancer Res. 1998, 58, 2070. [Google Scholar] [PubMed]
- Clark, L.C. Effects of selenium supplementation for cancer prevention in patients with carcinoma of the skin. JAMA 1996, 276, 1957. [Google Scholar] [CrossRef]
- Marshall, J.R.; Tangen, C.M.; Sakr, W.A.; Wood, D.P.; Berry, D.L.; Klein, E.A.; Lippman, S.M.; Parnes, H.L.; Alberts, D.S.; Jarrard, D.F.; et al. Phase III trial of selenium to prevent prostate cancer in men with high-grade prostatic intraepithelial neoplasia: SWOG S9917. Cancer Prev. Res. 2011, 4, 1761–1769. [Google Scholar] [CrossRef]
- Karp, D.D.; Lee, S.J.; Keller, S.M.; Wright, G.S.; Aisner, S.; Belinsky, S.A.; Johnson, D.H.; Johnston, M.R.; Goodman, G.; Clamon, G.; et al. Randomized, double-blind, placebo-controlled, phase III chemoprevention trial of selenium supplementation in patients with resected stage I non-small-cell lung cancer: ECOG 5597. J. Clin. Oncol. 2013, 31, 4179–4187. [Google Scholar] [CrossRef]
- Ip, C.; Ganther, H.E. Activity of methylated forms of selenium in cancer prevention. Cancer Res. 1990, 50, 1206. [Google Scholar] [PubMed]
- Jung, H.J.; Kim, H.L.; Seo, Y.R. Enhanced redox factor 1 (REF1)-modulated p53 stabilization and JNK1 dissociation in response to selenomethionine. Anticancer Res. 2013, 33, 3645–3651. [Google Scholar]
- Fischer, J.L.; Lancia, J.K.; Mathur, A.; Smith, M.L. Selenium protection from DNA damage involves a Ref1/p53/Brca1 protein complex. Anticancer Res. 2006, 26, 899–904. [Google Scholar]
- Seo, Y.R.; Sweeney, C.; Smith, M.L. Selenomethionine induction of DNA repair response in human fibroblasts. Oncogene 2002, 21, 3663–3669. [Google Scholar] [CrossRef]
- Tosatto, S.C.E.; Bosello, V.; Fogolari, F.; Mauri, P.; Roveri, A.; Toppo, S.; Flohé, L.; Ursini, F.; Maiorino, M. The catalytic site of glutathione peroxidases. Antioxid. Redox Signal. 2008, 10, 1515–1526. [Google Scholar] [CrossRef]
- Poole, L.B. The basics of thiols and cysteines in redox biology and chemistry. Free Radic. Biol. Med. 2015, 80, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Misra, S.; Boylan, M.; Selvam, A.; Spallholz, J.; Björnstedt, M. Redox-active selenium compounds—From toxicity and cell death to cancer treatment. Nutrients 2015, 7, 3536–3556. [Google Scholar] [CrossRef]
- Labunskyy, V.M.; Hatfield, D.L.; Gladyshev, V.N. Selenoproteins: Molecular pathways and physiological roles. Physiol. Rev. 2014, 94, 739–777. [Google Scholar] [CrossRef] [PubMed]
- Moghadaszadeh, B.; Beggs, A.H. Selenoproteins and their impact on human health through diverse physiological pathways. Physiology (Bethesda) 2006, 21, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishna, R.; Gundimeda, U. Antioxidant regulation of protein kinase C in cancer prevention. J. Nutr. 2002, 132, 3819s–3823s. [Google Scholar] [CrossRef]
- Ammar, E.M.; Couri, D. Acute toxicity of sodium selenite and selenomethionine in mice after ICV or IV administration. Neurotoxicology 1981, 2, 383–386. [Google Scholar]
- Desta, B.; Maldonado, G.; Reid, H.; Puschner, B.; Maxwell, J.; Agasan, A.; Humphreys, L.; Holt, T. Acute selenium toxicosis in polo ponies. J. Vet. Diagn. Invest. 2011, 23, 623–628. [Google Scholar] [CrossRef]
- Ganther, H.E. Selenium metabolism, selenoproteins and mechanisms of cancer prevention: Complexities with thioredoxin reductase. Carcinogenesis 1999, 20, 1657–1666. [Google Scholar] [CrossRef]
- Morris, J.; Crane, S. Selenium Toxicity from a misformulated dietary supplement, adverse health effects, and the temporal response in the nail biologic monitor. Nutrients 2013, 5, 1024–1057. [Google Scholar] [CrossRef]
- Macfarquhar, J.K. Acute selenium toxicity associated with a dietary supplement. Arch. Intern. Med. 2010, 170, 256. [Google Scholar] [CrossRef]
- Stewart, M.S.; Spallholz, J.E.; Neldner, K.H.; Pence, B.C. Selenium compounds have disparate abilities to impose oxidative stress and induce apoptosis. Free Radic. Biol. Med. 1999, 26, 42–48. [Google Scholar] [CrossRef]
- Spallholz, J.E. On the nature of selenium toxicity and carcinostatic activity. Free Radic. Biol. Med. 1994, 17, 45–64. [Google Scholar] [CrossRef]
- Lafin, J.T.; Sarsour, E.H.; Kalen, A.L.; Wagner, B.A.; Buettner, G.R.; Goswami, P.C. Methylseleninic acid induces lipid peroxidation and radiation sensitivity in head and neck cancer cells. Int. J. Mol. Sci. 2019, 20, 225. [Google Scholar] [CrossRef] [PubMed]
- Seko, Y.; Imura, N. Active oxygen generation as a possible mechanism of selenium toxicity. Biomed. Env. Sci. 1997, 10, 333–339. [Google Scholar]
- Yan, L.; Spallholz, J.E. Generation of reactive oxygen species from the reaction of selenium compounds with thiols and mammary tumor cells. Biochem. Pharmacol. 1993, 45, 429–437. [Google Scholar] [PubMed]
- Kim, H.Y.; Gladyshev, V.N. Methionine sulfoxide reduction in mammals: Characterization of methionine-R-sulfoxide reductases. Mol. Biol. Cell 2004, 15, 1055–1064. [Google Scholar] [CrossRef]
- Kim, I.Y.; Stadtman, T.C. Inhibition of NF-kappaB DNA binding and nitric oxide induction in human T cells and lung adenocarcinoma cells by selenite treatment. Proc. Natl. Acad. Sci. USA 1997, 94, 12904–12907. [Google Scholar] [CrossRef]
- Spyrou, G.; Bjornstedt, M.; Kumar, S.; Holmgren, A. AP-1 DNA-binding activity is inhibited by selenite and selenodiglutathione. FEBS Lett. 1995, 368, 59–63. [Google Scholar] [CrossRef]
- Ghosh, J. Rapid induction of apoptosis in prostate cancer cells by selenium: Reversal by metabolites of arachidonate 5-lipoxygenase. Biochem. Biophys. Res. Commun. 2004, 315, 624–635. [Google Scholar] [CrossRef]
- Bergad, P.L.; Rathbun, W.B. Inhibition of Na, K-ATPase by sodium selenite and reversal by glutathione. Curr. Eye Res. 1986, 5, 919–923. [Google Scholar] [CrossRef]
- Islam, F.; Watanabe, Y.; Morii, H.; Hayaishi, O. Inhibition of rat brain prostaglandin D synthase by inorganic selenocompounds. Arch. Biochem. Biophys. 1991, 289, 161–166. [Google Scholar] [CrossRef]
- Sinha, R.; Medina, D. Inhibition of cdk2 kinase activity by methylselenocysteine in synchronized mouse mammary epithelial tumor cells. Carcinogenesis 1997, 18, 1541–1547. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishna, R.; Chen, Z.H.; Gundimeda, U. Selenocompounds induce a redox modulation of protein kinase C in the cell, compartmentally independent from cytosolic glutathione: Its role in inhibition of tumor promotion. Arch. Biochem. Biophys. 1997, 348, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Park, H.S.; Huh, S.H.; Kim, Y.; Shim, J.; Lee, S.H.; Park, I.S.; Jung, Y.K.; Kim, I.Y.; Choi, E.J. Selenite negatively regulates caspase-3 through a redox mechanism. J. Biol. Chem. 2000, 275, 8487–8491. [Google Scholar] [CrossRef] [PubMed]
- Park, H.S.; Park, E.; Kim, M.S.; Ahn, K.; Kim, I.Y.; Choi, E.J. Selenite inhibits the c-Jun N-terminal kinase/stress-activated protein kinase (JNK/SAPK) through a thiol redox mechanism. J. Biol. Chem. 2000, 275, 2527–2531. [Google Scholar] [CrossRef]
- Liou, G.-Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef]
- Oberley, L.W.; Buettner, G.R. Role of superoxide dismutase in cancer: A review. Cancer Res. 1979, 39, 1141–1149. [Google Scholar]
- Buettner, G.R. Superoxide dismutase in redox biology: The roles of superoxide and hydrogen peroxide. Anticancer. Agents Med. Chem. 2011, 11, 341–346. [Google Scholar] [CrossRef]
- Aykin-Burns, N.; Ahmad, I.M.; Zhu, Y.; Oberley, L.W.; Spitz, D.R. Increased levels of superoxide and H2O2 mediate the differential susceptibility of cancer cells versus normal cells to glucose deprivation. Biochem. J. 2009, 418, 29–37. [Google Scholar] [CrossRef]
- Lu, J.; Kaeck, M.; Jiang, C.; Wilson, A.C.; Thompson, H.J. Selenite induction of DNA strand breaks and apoptosis in mouse leukemic L1210 cells. Biochem. Pharmacol. 1994, 47, 1531–1535. [Google Scholar] [CrossRef]
- Yan, L.; Frenkel, G.D. Inhibition of Cell Attachment by Selenite. Cancer Res. 1992, 52, 5803. [Google Scholar]
- Garberg, P.; Stahl, A.; Warholm, M.; Hogberg, J. Studies of the role of DNA fragmentation in selenium toxicity. Biochem. Pharmacol. 1988, 37, 3401–3406. [Google Scholar] [CrossRef]
- Peyroche, G.; Saveanu, C.; Dauplais, M.; Lazard, M.; Beuneu, F.; Decourty, L.; Malabat, C.; Jacquier, A.; Blanquet, S.; Plateau, P. Sodium Selenide Toxicity Is Mediated by O2-Dependent DNA Breaks. PLoS ONE 2012, 7, e36343. [Google Scholar] [CrossRef] [PubMed]
- Wycherly, B.J.; Moak, M.A.; Christensen, M.J. High dietary intake of sodium selenite induces oxidative DNA damage in rat liver. Nutr. Cancer 2004, 48, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Wallenberg, M.; Misra, S.; Wasik, A.M.; Marzano, C.; Bjornstedt, M.; Gandin, V.; Fernandes, A.P. Selenium induces a multi-targeted cell death process in addition to ROS formation. J. Cell. Mol. Med. 2014, 18, 671–684. [Google Scholar] [CrossRef]
- Brigelius-Flohé, R.; Müller, M.; Lippmann, D.; Kipp, A.P. The Yin and Yang of Nrf2-Regulated Selenoproteins in Carcinogenesis. Int. J. Cell Biol. 2012, 2012, 1–8. [Google Scholar] [CrossRef]
- Motohashi, H.; Yamamoto, M. Nrf2–Keap1 defines a physiologically important stress response mechanism. Trends Mol. Med. 2004, 10, 549–557. [Google Scholar] [CrossRef]
- Johnson, J.A.; Johnson, D.A.; Kraft, A.D.; Calkins, M.J.; Jakel, R.J.; Vargas, M.R.; Chen, P.C. The Nrf2-ARE pathway: An indicator and modulator of oxidative stress in neurodegeneration. Ann. N. Y. Acad. Sci. 2008, 1147, 61–69. [Google Scholar] [CrossRef]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/Small maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef]
- Raghunath, A.; Sundarraj, K.; Nagarajan, R.; Arfuso, F.; Bian, J.; Kumar, A.P.; Sethi, G.; Perumal, E. Antioxidant response elements: Discovery, classes, regulation and potential applications. Redox Biol. 2018, 17, 297–314. [Google Scholar] [CrossRef]
- Brigelius-Flohe, R.; Banning, A. Part of the series: From dietary antioxidants to regulators in cellular signaling and gene regulation. Sulforaphane and selenium, partners in adaptive response and prevention of cancer. Free Radic. Res. 2006, 40, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zeng, L.; Jiang, W.; Fu, Y.; Zheng, W.; Chen, T. Rational design of cancer-targeted selenium nanoparticles to antagonize multidrug resistance in cancer cells. Nanomed. Nanotechnol. Biol. Med. 2015, 11, 947–958. [Google Scholar] [CrossRef] [PubMed]
- Hadrup, N.; Loeschner, K.; Mandrup, K.; Ravn-Haren, G.; Frandsen, H.L.; Larsen, E.H.; Lam, H.R.; Mortensen, A. Subacute oral toxicity investigation of selenium nanoparticles and selenite in rats. Drug Chem. Toxicol. 2019, 42, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Muecke, R.; Micke, O.; Schomburg, L.; Glatzel, M.; Reichl, B.; Kisters, K.; Schaefer, U.; Huebner, J.; Eich, H.T.; Fakhrian, K.; et al. Multicenter, phase III trial comparing selenium supplementation with observation in gynecologic radiation oncology: Follow-up analysis of the survival data 6 years after cessation of randomization. Integr. Cancer Ther. 2014, 13, 463–467. [Google Scholar] [CrossRef]
- Muecke, R.; Schomburg, L.; Glatzel, M.; Berndt-Skorka, R.; Baaske, D.; Reichl, B.; Buentzel, J.; Kundt, G.; Prott, F.J.; Devries, A.; et al. Multicenter, phase 3 trial comparing selenium supplementation with observation in gynecologic radiation oncology. Int. J. Radiat. Oncol. Biol. Phys. 2010, 78, 828–835. [Google Scholar] [CrossRef]
- Garje, R.; An, J.; Sanchez, K.; Greco, A.; Stolwijk, J.; Devor, E.; Rustum, Y.; Zakharia, Y. Current landscape and the potential role of hypoxia-inducible factors and selenium in clear cell renal cell carcinoma treatment. Int. J. Mol. Sci. 2018, 19, 3834. [Google Scholar] [CrossRef]
- Garje, R.; Brown, J.A.; Nepple, K.G.; Dahmoush, L.; Bellizzi, A.; Bonner, J.; Mott, S.L.; Zamba, G.; Laux, D.E.; Milhem, M.M.; et al. Preliminary results of phase I clinical trial of high doses of seleno-L-methionine (SLM) in sequential combination with axitinib in previously treated and relapsed clear cell renal cell carcinoma (ccRCC) patients. J. Clin. Oncol. 2019, 37, 660. [Google Scholar] [CrossRef]
- Brodin, O.; Eksborg, S.; Wallenberg, M.; Asker-Hagelberg, C.; Larsen, E.H.; Mohlkert, D.; Lenneby-Helleday, C.; Jacobsson, H.; Linder, S.; Misra, S.; et al. Pharmacokinetics and toxicity of sodium selenite in the treatment of patients with carcinoma in a phase I clinical trial: The secar study. Nutrients 2015, 7, 4978–4994. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Seshadri, M.; Oven, S.D.; Tóth, K.; Vaughan, M.M.; Rustum, Y.M. Tumor vascular maturation and improved drug delivery induced by methylselenocysteine leads to therapeutic synergy with anticancer drugs. Clin. Cancer Res. 2008, 14, 3926. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Tóth, K.; Sen, A.; Seshadri, M.; Cao, S.; Durrani, F.A.; Faber, E.; Repasky, E.A.; Rustum, Y.M. Inhibition of colon cancer growth by methylselenocysteine-induced angiogenic chemomodulation is influenced by histologic characteristics of the tumor. Clin. Colorectal Cancer 2009, 8, 155–162. [Google Scholar] [CrossRef]
- Cao, S.; Durrani, F.A.; Rustum, Y.M. Selective modulation of the therapeutic efficacy of anticancer drugs by selenium containing compounds against human tumor xenografts. Clin. Cancer Res. 2004, 10, 2561–2569. [Google Scholar] [CrossRef] [PubMed]
- Toth, K.; Chintala, S.; Rustum, Y.M. Constitutive expression of HIF-alpha plays a major role in generation of clear-cell phenotype in human primary and metastatic renal carcinoma. Appl. Immunohistochem. Mol. Morphol. 2014, 22, 642–647. [Google Scholar] [CrossRef]
- Chintala, S.; Najrana, T.; Toth, K.; Cao, S.; Durrani, F.A.; Pili, R.; Rustum, Y.M. Prolyl hydroxylase 2 dependent and Von-Hippel-Lindau independent degradation of Hypoxia-inducible factor 1 and 2 alpha by selenium in clear cell renal cell carcinoma leads to tumor growth inhibition. BMC Cancer 2012, 12, 293. [Google Scholar] [CrossRef] [PubMed]
- Chintala, S.; Toth, K.; Cao, S.; Durrani, F.A.; Vaughan, M.M.; Jensen, R.L.; Rustum, Y.M. Se-methylselenocysteine sensitizes hypoxic tumor cells to irinotecan by targeting hypoxia-inducible factor 1alpha. Cancer Chemother. Pharm. 2010, 66, 899–911. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Du, X.; Rizzi, J.P.; Liberzon, E.; Chakraborty, A.A.; Gao, W.; Carvo, I.; Signoretti, S.; Bruick, R.K.; Josey, J.A.; et al. On-target efficacy of a HIF-2alpha antagonist in preclinical kidney cancer models. Nature 2016, 539, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Hill, H.; Christie, A.; Kim, M.S.; Holloman, E.; Pavia-Jimenez, A.; Homayoun, F.; Ma, Y.; Patel, N.; Yell, P.; et al. Targeting renal cell carcinoma with a HIF-2 antagonist. Nature 2016, 539, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Escudier, B.; Porta, C.; Schmidinger, M.; Rioux-Leclercq, N.; Bex, A.; Khoo, V.; Grunwald, V.; Gillessen, S.; Horwich, A. Renal cell carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-updagger. Ann. Oncol. 2019, 30, 706–720. [Google Scholar] [CrossRef]
- Burk, R.F.; Hill, K.E.; Motley, A.K. Plasma selenium in specific and non-specific forms. BioFactors 2001, 14, 107–114. [Google Scholar] [CrossRef]
- Hondal, R.J.; Motley, A.K.; Hill, K.E.; Burk, R.F. Failure of selenomethionine residues in albumin and immunoglobulin G to protect against peroxynitrite. Arch. Biochem. Biophys. 1999, 371, 29–34. [Google Scholar] [CrossRef]
- Hu, X.; Chandler, J.D.; Orr, M.L.; Hao, L.; Liu, K.; Uppal, K.; Go, Y.-M.; Jones, D.P. Selenium supplementation alters hepatic energy and fatty acid metabolism in mice. J. Nutr. 2018, 148, 675–684. [Google Scholar] [CrossRef]
- Berntsson, R.P.; Alia Oktaviani, N.; Fusetti, F.; Thunnissen, A.M.; Poolman, B.; Slotboom, D.J. Selenomethionine incorporation in proteins expressed in Lactococcus lactis. Protein Sci. 2009, 18, 1121–1127. [Google Scholar] [CrossRef] [PubMed]
- Nair, D.; Radestad, E.; Khalkar, P.; Diaz-Argelich, N.; Schroder, A.; Klynning, C.; Ungerstedt, J.; Uhlin, M.; Fernandes, A.P. Methylseleninic acid sensitizes ovarian cancer cells to T-Cell mediated killing by decreasing PDL1 and VEGF levels. Front. Oncol. 2018, 8, 407. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Jiang, C.; Ip, C.; Rustum, Y.M.; Lu, J. Methylseleninic acid potentiates apoptosis induced by chemotherapeutic drugs in androgen-independent prostate cancer cells. Clin. Cancer Res. 2005, 11, 2379–2388. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mas, A.; Sarkar, B. Role of glutathione in selenite binding by human plasma. Biol. Trace Elem. Res. 1989, 20, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Bothwell, I.R.; Islam, K.; Chen, Y.; Zheng, W.; Blum, G.; Deng, H.; Luo, M. Se-Adenosyl-l-selenomethionine cofactor analogue as a reporter of protein methylation. J. Am. Chem. Soc. 2012, 134, 14905–14912. [Google Scholar] [CrossRef] [PubMed]
- Azad, G.K.; Tomar, R.S. Ebselen, a promising antioxidant drug: Mechanisms of action and targets of biological pathways. Mol. Biol. Rep. 2014, 41, 4865–4879. [Google Scholar] [CrossRef]
- Schewe, T. Molecular actions of ebselen--an antiinflammatory antioxidant. Gen. Pharmacol. 1995, 26, 1153–1169. [Google Scholar] [CrossRef]
- Caeran Bueno, D.; Meinerz, D.F.; Allebrandt, J.; Waczuk, E.P.; Dos Santos, D.B.; Mariano, D.O.; Rocha, J.B. Cytotoxicity and genotoxicity evaluation of organochalcogens in human leucocytes: A comparative study between ebselen, diphenyl diselenide, and diphenyl ditelluride. Biomed. Res. Int. 2013, 2013, 537279. [Google Scholar] [CrossRef] [PubMed]
- Tavadyan, L.A.; Manukyan, Z.H.; Harutyunyan, L.H.; Musayelyan, M.V.; Sahakyan, A.D.; Tonikyan, H.G. antioxidant properties of selenophene, thiophene and their aminocarbonitrile derivatives. Antioxidants (Basel) 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Juang, S.H.; Lung, C.C.; Hsu, P.C.; Hsu, K.S.; Li, Y.C.; Hong, P.C.; Shiah, H.S.; Kuo, C.C.; Huang, C.W.; Wang, Y.C.; et al. D-501036, a novel selenophene-based triheterocycle derivative, exhibits potent in vitro and in vivo antitumoral activity which involves DNA damage and ataxia telangiectasia-mutated nuclear protein kinase activation. Mol. Cancer Ther. 2007, 6, 193–202. [Google Scholar] [CrossRef]
- Fairweather-Tait, S.J.; Bao, Y.; Broadley, M.R.; Collings, R.; Ford, D.; Hesketh, J.E.; Hurst, R. Selenium in human health and disease. Antioxid. Redox Signal. 2011, 14, 1337–1383. [Google Scholar] [CrossRef] [PubMed]
- Schomburg, L.; Orho-Melander, M.; Struck, J.; Bergmann, A.; Melander, O. Selenoprotein-P deficiency predicts cardiovascular disease and death. Nutrients 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Bomer, N.; Grote Beverborg, N.; Hoes, M.F.; Streng, K.W.; Vermeer, M.; Dokter, M.M.; J, I.J.; Anker, S.D.; Cleland, J.G.F.; Hillege, H.L.; et al. Selenium and outcome in heart failure. Eur. J. Heart Fail. 2019. [Google Scholar] [CrossRef] [PubMed]
- Muecke, R.; Micke, O.; Schomburg, L.; Buentzel, J.; Kisters, K.; Adamietz, I.A. Selenium in radiation Oncology-15 years of experiences in Germany. Nutrients 2018, 10. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Selenoprotein | Function | Reference |
|---|---|---|
| GPx1 | Reduction of solubilized organic hydroperoxides and hydrogen peroxide in the water space of cells, cytosol, mitochondria, nucleus | [23,103] |
| GPx2 | Abundantly expressed in tissues of the liver and GI tract. Similar function to GPx1 | [42] |
| GPx3 | Extracellular GPx, regulation of nitric oxide | [45,51] |
| GPx4 | Reduction of lipid hydroperoxides, essential for the termination of lipid peroxidation; prevents ferroptosis | [53,73] |
| GPx6 | Only found in humans, no specific role has yet been identified | [19] |
| TrxR1 | Reduces TrxSS to Trx(SH)2, vitamin C, polyphenols, and other substrates to regulate intracellular redox environment | [67] |
| TrxR2 | Located in mitochondria, control and regulates redox environment | [19] |
| TrxR3 | Abundant in testes, reduces mitochondrial glutathione disulfide | [19] |
| DIO1 | Converts T4 (thyroxine) into T3 (active thyroid hormone) and inactivates T4 to rT3 | [84] |
| DIO2 | Converts T4 into T3 | [84] |
| DIO3 | Inactivates T4 to rT3, as well as T3 to T2 | [84] |
| MSRB | Restores oxidatively damaged methionine (Met-sulfoxide) to native configurations | [89] |
| SELENOF | Oxidoreductase that may assist in disulfide formation and protein folding | [104] |
| SELENOH | Regulates GSH synthesis during embryonic development | [19] |
| SELENOI | Functions as a phospholipid synthase | [19] |
| SELENOK | Abundant in myocytes, function is currently unclear | [19] |
| SELENOM | Abundant in myocytes, function is currently unclear | [105] |
| SELENON | Interacts with ryanodine receptor, mutations result in congenital muscular dystrophy | [73] |
| SELENOO | Suggestive redox function due to Cys-X-X-Sec motif | [19] |
| SELENOP | Plasma selenium transport protein, contains up to 10 Sec, exhibits very low GPx4-like activity when purified | [92,93] |
| SELENOS | Suggested to be involved in ER stress response | [19] |
| SELENOT | Suggested role in Ca2+ mobilization | [19] |
| SELENOV | Suggestive redox function due to Cys-X-X-Sec motif | [19] |
| SELENOW | Expressed in a variety of tissues and may regulate redox state of 14-3-3 proteins | [19] |
| Concentration Supplemented a | Enzyme Studied | Fold Increase b | Cell Line | Reference |
|---|---|---|---|---|
| 4 nM | GPx1 | 3 | Human bronchial epithelial | [109] |
| 50 nM | GPx1, GPx4 | 3–15 c, 3–10 c | L929, HepG2, D10N, THP-1, ECV 304 | [17] |
| 100 nM | GPx1, TrxR | 2, 1.5 | Bone marrow stromal | [110] |
| 100 nM | GPx1, GPx4, TrxR | 2, 2, 1.5 | Jurkat, T-leukemia | [111] |
| 200 nM d | GPx1, GPx4, TrxR | 3, 3, 2 | HepG2, MIA PaCa-2 | [18] |
| 2 µM | GPx1 | 17 | RAW 264.7 macrophage | [112] |
| Compound | IC50a (µM) | Comments | Reference |
|---|---|---|---|
| Sodium selenite | 1–10 | Inorganic source for Se Most widely used Se source Pro-oxidant in high concentrations Administered IV to reach pharmacological doses | [142,166] |
| Sodium selenate | 1–10 | Inorganic source for Se Suggested to play a role in obesity | [191] |
| Seleno-L-methionine | >100 | Organic source for Se Abundant dietary source for Se Can be incorporated in proteins as Se-methionine Relative low toxicity | [137,142,192] |
| Methyl selenocysteine | >100 | Organic source for Se Inhibitor of angiogenesis | [133] |
| Methyl seleninic acid | 1–10 | Synthetic organic Se compound Potential to alter the immune response in tumors Pro-oxidant properties Enhances tumor cell killing | [193,194] |
| Selenium-di-glutathione | 5 | Organic source for Se Product of selenite reduction by GSH | [166,195] |
| Se-DL-cystine | 5–100 | Organic source for Se Both antioxidant and pro-oxidant Exhibit GPx-like activity under reducing conditions | [133,166] |
| Selenocystamine | 25–50 | Organic source for Se Non-catalytic Prevents DNA oxidation | [142] |
| Se-adenosyl-L-selenomethionine | N.D. | Synthetic organic Se compound Proposed substrate for methyltransferases | [196] |
| Ebselen | 5–50 | Synthetic organic Se compound GPx mimic Antioxidant scavenger Anti-inflammatory | [197,198,199] |
| Selenophene | 0.1–10b | Synthetic organic Se compound Antioxidant properties Protect against CCl3 induced liver damage Molecular building block for anti-cancer agents | [200,201] |
| Selenium nanoparticles | <1 | Synthetic inorganic Se particles Much lower apparent toxicity in vivo Induces oxidative stress in vitro | [173,174] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stolwijk, J.M.; Garje, R.; Sieren, J.C.; Buettner, G.R.; Zakharia, Y. Understanding the Redox Biology of Selenium in the Search of Targeted Cancer Therapies. Antioxidants 2020, 9, 420. https://doi.org/10.3390/antiox9050420
Stolwijk JM, Garje R, Sieren JC, Buettner GR, Zakharia Y. Understanding the Redox Biology of Selenium in the Search of Targeted Cancer Therapies. Antioxidants. 2020; 9(5):420. https://doi.org/10.3390/antiox9050420
Chicago/Turabian StyleStolwijk, Jeffrey M., Rohan Garje, Jessica C. Sieren, Garry R. Buettner, and Yousef Zakharia. 2020. "Understanding the Redox Biology of Selenium in the Search of Targeted Cancer Therapies" Antioxidants 9, no. 5: 420. https://doi.org/10.3390/antiox9050420
APA StyleStolwijk, J. M., Garje, R., Sieren, J. C., Buettner, G. R., & Zakharia, Y. (2020). Understanding the Redox Biology of Selenium in the Search of Targeted Cancer Therapies. Antioxidants, 9(5), 420. https://doi.org/10.3390/antiox9050420