Vitamin C Activates the Folate-Mediated One-Carbon Cycle in C2C12 Myoblasts

 ,
, 

Abstract
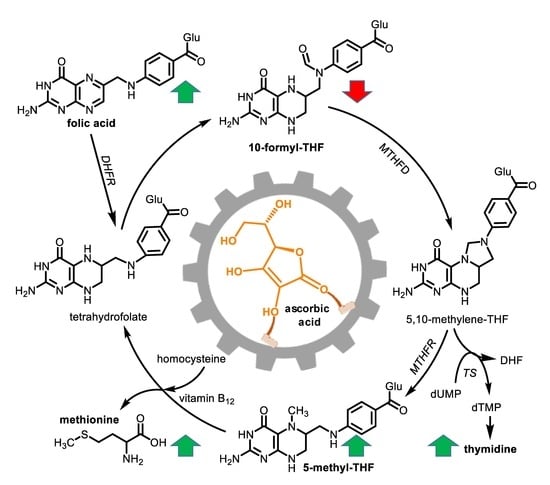
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Culture and Treatment
2.3. LC–MS/MS Analysis
2.4. Metabolomics Data Processing
2.5. Statistical Analysis
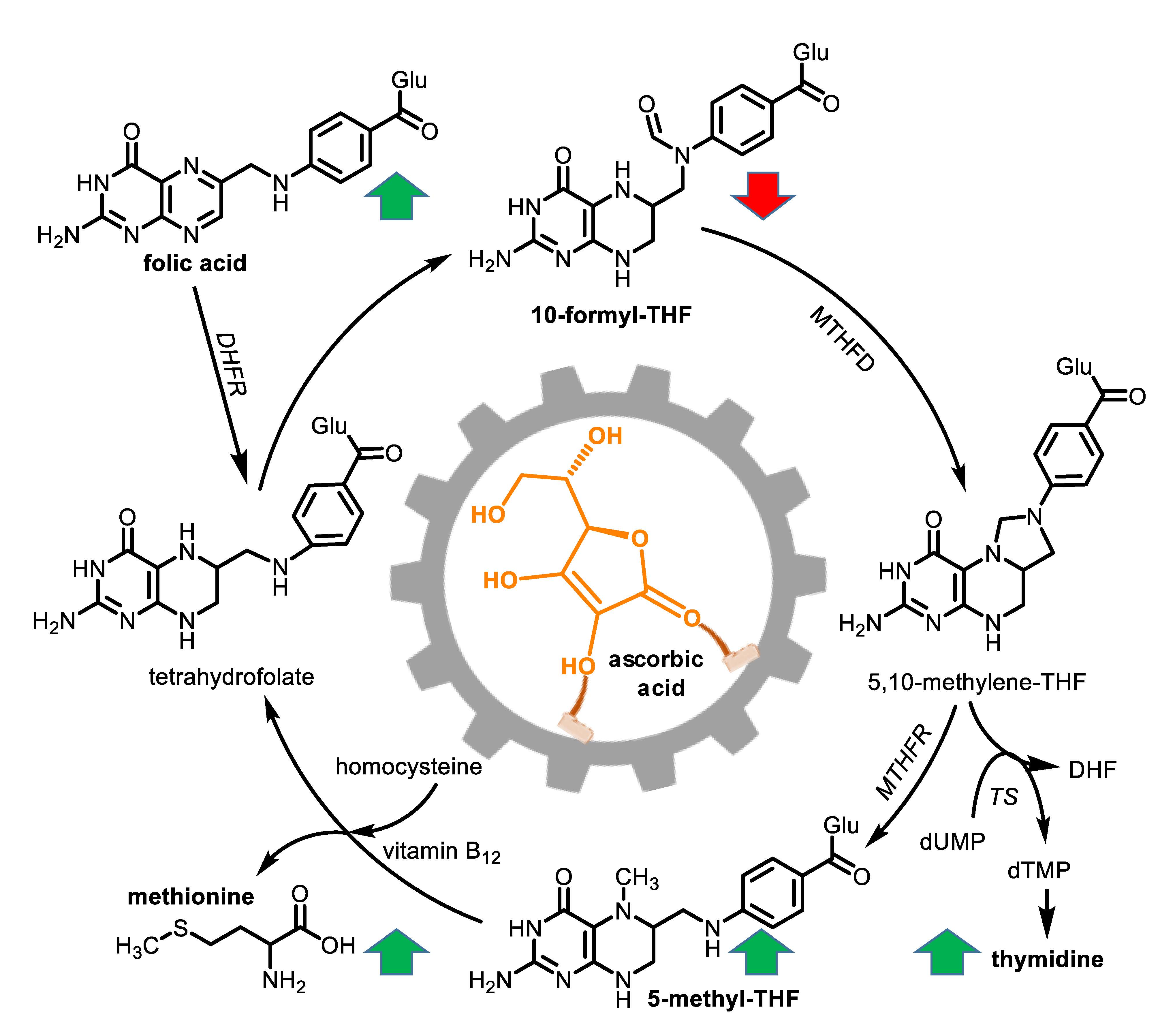
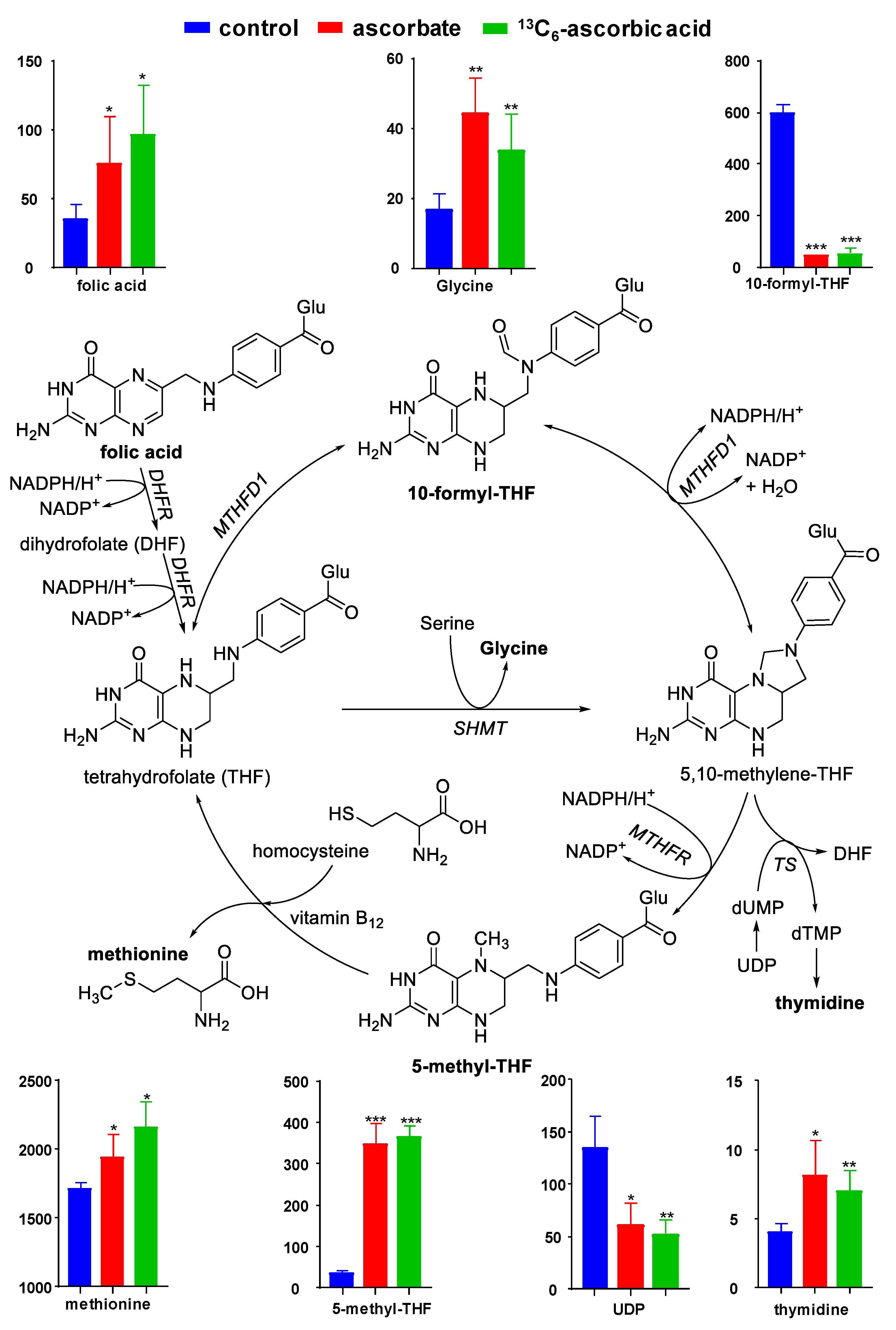
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Higdon, J.V.; Frei, B. Vitamin C: An Introduction, in The Antioxidant Vitamins C and E; Packer, L., Traber, M.G., Kraemer, K., Frei, B., Eds.; AOAC Press: Champaign, IL, USA, 2002; pp. 11–16. [Google Scholar]
- Traber, M.G.; Stevens, J.F. Vitamins C and E: Beneficial effects from a mechanistic perspective. Free Radic Biol. Med. 2011, 51, 1000–1013. [Google Scholar] [CrossRef]
- Kesinger, N.G.; Langsdorf, B.L.; Yokochi, A.F.; Miranda, C.L.; Stevens, J.F. Formation of a vitamin C conjugate of acrolein and its paraoxonase-mediated conversion into 5,6,7,8-tetrahydroxy-4-oxooctanal. Chem. Res. Toxicol. 2010, 23, 836–844. [Google Scholar] [CrossRef][Green Version]
- Kesinger, N.G.; Stevens, J.F. Covalent interaction of ascorbic acid with natural products. Phytochemistry 2009, 70, 1930–1939. [Google Scholar] [CrossRef] [PubMed]
- Kuiper, H.C.; Bruno, R.S.; Traber, M.G.; Stevens, J.F. Vitamin C supplementation lowers urinary levels of 4-hydroperoxy-2-nonenal metabolites in humans. Free Radic Biol. Med. 2011, 50, 848–853. [Google Scholar] [CrossRef] [PubMed]
- Kirkwood, J.S.; Lebold, K.M.; Miranda, C.L.; Wright, C.L.; Miller, G.W.; Tanguay, R.L.; Barton, C.L.; Traber, M.G.; Stevens, J.F. Vitamin C deficiency activates the purine nucleotide cycle in zebrafish. J. Biol. Chem. 2012, 287, 3833–3841. [Google Scholar] [CrossRef] [PubMed]
- Axton, E.R.; Cristobal, E.; Choi, J.; Miranda, C.L.; Stevens, J.F. Metabolomics-Driven Elucidation of Cellular Nitrate Tolerance Reveals Ascorbic Acid Prevents Nitroglycerin-Induced Inactivation of Xanthine Oxidase. Front Pharmacol. 2018, 9, 1085. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.; Gutteridge, C.J.M. Free Radicals in Biology and Medicine, 3rd ed.; Oxford University Press: Oxford, UK, 1999; p. 936. [Google Scholar]
- Lucock, M.; Yates, Z.; Boyd, L.; Naylor, C.; Choi, J.H.; Ng, X.; Skinner, V.; Wai, R.; Kho, J.; Tang, S.; et al. Vitamin C-related nutrient-nutrient and nutrient-gene interactions that modify folate status. Eur. J. Nutr. 2013, 52, 569–582. [Google Scholar] [CrossRef]
- Cafolla, A.; Dragoni, F.; Girelli, G.; Tosti, M.E.; Costante, A.; De Luca, A.M.; Funaro, D.; Scott, C.S. Effect of folic acid and vitamin C supplementation on folate status and homocysteine level: A randomised controlled trial in Italian smoker-blood donors. Atherosclerosis 2002, 163, 105–111. [Google Scholar] [CrossRef]
- Berg, J.M.; Tymoczko, J.L.; Stryer, L. Biochemistry, 5th ed.; W.H. Freeman and Company: New York, NY, USA, 2001. [Google Scholar]
- Verhoef, P.; Stampfer, M.J. Prospective studies of homocysteine and cardiovascular disease. Nutr. Rev. 1995, 53, 283–288. [Google Scholar] [CrossRef]
- Mix, J.A. Do megadoses of vitamin C compromise folic acid’s role in the metabolism of plasma homocysteine? Nutr. Res. 1999, 19, 161–165. [Google Scholar] [CrossRef]
- Drouin, G.; Godin, J.R.; Page, B. The genetics of vitamin C loss in vertebrates. Curr. Genom. 2011, 12, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Kirkwood, J.S.; Legette, L.L.; Miranda, C.L.; Jiang, Y.; Stevens, J.F. A metabolomics-driven elucidation of the anti-obesity mechanisms of xanthohumol. J. Biol. Chem. 2013, 288, 19000–19013. [Google Scholar] [CrossRef] [PubMed]
- Sumner, L.W.; Amberg, A.; Barrett, D.; Beale, M.H.; Beger, R.; Daykin, C.A.; Fan, T.W.; Fiehn, O.; Goodacre, R.; Griffin, J.L.; et al. Proposed minimum reporting standards for chemical analysis Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics 2007, 3, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Viant, M.R.; Kurland, I.J.; Jones, M.R.; Dunn, W.B. How close are we to complete annotation of metabolomes? Curr. Opin. Chem. Biol. 2017, 36, 64–69. [Google Scholar] [CrossRef]
- Chong, J.; Wishart, D.S.; Xia, J. Using MetaboAnalyst 4.0 for Comprehensive and Integrative Metabolomics Data Analysis. Curr. Protoc. Bioinform. 2019, 68, e86. [Google Scholar] [CrossRef]
- Stokes, P.; Melikian, V.; Leeming, R.L.; Portman-Graham, H.; Blair, J.A.; Cooke, W.T. Folate metabolism in scurvy. Am. J. Clin. Nutr. 1975, 28, 126–129. [Google Scholar] [CrossRef]
- Gropper, S.; Smith, J.; Groff, J. Folic acid. In Advanced Nutrition and Human Metabolism; Thomson Wadsworth: Belmont, CA, USA, 2005; pp. 301–309. [Google Scholar]
- Mårtensson, J.; Han, J.; Griffith, O.W.; Meister, A. Glutathione ester delays the onset of scurvy in ascorbate-deficient guinea pigs. Proc. Natl. Acad. Sci. USA 1993, 90, 317–321. [Google Scholar] [CrossRef]
- Chavez, J.; Chung, W.G.; Miranda, C.L.; Singhal, M.; Stevens, J.F.; Maier, C.S. Site-specific protein adducts of 4-hydroxy-2(E)-nonenal in human THP-1 monocytic cells: Protein carbonylation is diminished by ascorbic acid. Chem. Res. Toxicol. 2010, 23, 37–47. [Google Scholar] [CrossRef]
- Miranda, C.L.; Reed, R.L.; Kuiper, H.C.; Alber, S.; Stevens, J.F. Ascorbic acid promotes detoxification and elimination of 4-hydroxy-2(E)-nonenal in human monocytic THP-1 cells. Chem. Res. Toxicol. 2009, 22, 863–874. [Google Scholar] [CrossRef]
- Fan, J.; Ye, J.; Kamphorst, J.J.; Shlomi, T.; Thompson, C.B.; Rabinowitz, J.D. Quantitative flux analysis reveals folate-dependent NADPH production. Nature 2014, 510, 298–302. [Google Scholar] [CrossRef]
- Hilton, J.G.; Cooper, B.A.; Rosenblatt, D.S. Folate polyglutamate synthesis and turnover in cultured human fibroblasts. J. Biol. Chem. 1979, 254, 8398–8403. [Google Scholar] [PubMed]
- Cichowicz, D.J.; Foo, S.K.; Shane, B. Folylpoly-gamma-glutamate synthesis by bacteria and mammalian cells. Mol. Cell Biochem. 1981, 39, 209–228. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.C.; Gladyshev, V.N. The biological significance of methionine sulfoxide stereochemistry. Free Radic Biol. Med. 2011, 50, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Moskovitz, J. Methionine sulfoxide reductases: Ubiquitous enzymes involved in antioxidant defense, protein regulation, and prevention of aging-associated diseases. Biochim. Biophys. Acta 2005, 1703, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Holmgren, A. The thioredoxin antioxidant system. Free Radic Biol. Med. 2014, 66, 75–87. [Google Scholar] [CrossRef]
- Santilli, F.; Davi, G.; Patrono, C. Homocysteine, methylenetetrahydrofolate reductase, folate status and atherothrombosis: A mechanistic and clinical perspective. Vascul. Pharmacol. 2016, 78, 1–9. [Google Scholar] [CrossRef]
- Huo, Y.; Li, J.; Qin, X.; Huang, Y.; Wang, X.; Gottesman, R.F.; Tang, G.; Wang, B.; Chen, D.; He, M.; et al. Efficacy of folic acid therapy in primary prevention of stroke among adults with hypertension in China: The CSPPT randomized clinical trial. JAMA 2015, 313, 1325–1335. [Google Scholar] [CrossRef]
- Krishna, S.M.; Dear, A.; Craig, J.M.; Norman, P.E.; Golledge, J. The potential role of homocysteine mediated DNA methylation and associated epigenetic changes in abdominal aortic aneurysm formation. Atherosclerosis 2013, 228, 295–305. [Google Scholar] [CrossRef]
- Cascalheira, J.F.; Parreira, M.C.; Viegas, A.N.; Faria, M.C.; Domingues, F.C. Serum homocysteine: Relationship with circulating levels of cortisol and ascorbate. Ann. Nutr. Metab. 2008, 53, 67–74. [Google Scholar] [CrossRef]
- Bostom, A.G.; Yanek, L.; Hume, A.L.; Eaton, C.B.; McQuade, W.; Nadeau, M.; Perrone, G.; Jacques, P.F.; Selhub, J. High dose ascorbate supplementation fails to affect plasma homocyst(e)ine levels in patients with coronary heart disease. Atherosclerosis 1994, 111, 267–270. [Google Scholar] [CrossRef]
- Savini, I.; Catani, M.V.; Duranti, G.; Ceci, R.; Sabatini, S.; Avigliano, L. Vitamin C homeostasis in skeletal muscle cells. Free Radic Biol. Med. 2005, 38, 898–907. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | m/z | Retention Time (min) | Formula | Error 1 | FC 2 | p-Value |
|---|---|---|---|---|---|---|
| 5-methyl-THF | 460.1938 | 14.28 | C20H25N7O6 | 0.26 | 9.97 | 8.07 × 10−9 |
| 10-formyl-THF | 474.1731 | 16.96 | C20H23N7O7 | 0.05 | 0.08 | 6.31 × 10−7 |
| Pantothenic acid | 218.1035 | 10.51 | C9H17NO5 | 1.08 | 1.60 | 8.35 × 10−7 |
| Guanidinoacetate | 118.0603 | 4.74 | C3H7N3O2 | 4.32 | 2.67 | 4.17 × 10−6 |
| Methionine sulfoxide | 166.0526 | 4.99 | C5H11NO3S | 4.08 | 0.71 | 4.90 × 10−5 |
| Phosphodimethylethanolamine | 192.0320 | 5.10 | C4H12NO4P | 0.92 | 1.95 | 7.95 × 10−5 |
| PE(22:1/14:0) | 746.5671 | 26.99 | C41H80NO8P | 1.19 | 1.57 | 1.91 × 10−4 |
| Urate | 169.0351 | 5.97 | C5H4N4O3 | 0.63 | 2.69 | 2.63 × 10−4 |
| LysoPC(18:1) | 522.3545 | 25.88 | C26H52NO7P | 0.78 | 1.47 | 4.31 × 10−4 |
| Phenylacetylglycine | 192.0665 | 17.88 | C10H11NO3 | 0.59 | 1.66 | 7.95 × 10−4 |
| PE(18:1/18:0) | 744.5530 | 26.96 | C41H80NO8P | 0.11 | 1.77 | 8.59 × 10−4 |
| Adenosine-5’-diphosphoglucose | 428.0365 | 4.87 | C10H15N5O10P2 | 0.44 | 0.60 | 9.18 × 10−4 |
| 6-keto-PGF1 | 393.2249 | 22.40 | C20H34O6 | 0.34 | 3.37 | 9.93 × 10−4 |
| PE(22:1/14:1) | 744.5529 | 26.99 | C41H78NO8P | 0.33 | 1.43 | 1.22 × 10−3 |
| D-glucose 6-phosphate | 259.0225 | 4.25 | C6H13O9P | 0.16 | 0.38 | 1.31 × 10−3 |
| Adenosine 5’-monophosphate | 346.0558 | 5.41 | C10H14N5O7P | 0.15 | 0.57 | 1.61 × 10−3 |
| Glycerate | 105.0188 | 5.03 | C3H6O4 | 2.27 | 4.38 | 1.65 × 10−3 |
| Uridine diphosphate (UDP) | 405.0094 | 4.07 | C9H14N2O12P2 | 1.36 | 0.42 | 1.73 × 10−3 |
| Thymidine | 241.0831 | 11.03 | C10H14N2O5 | 3.33 | 1.90 | 1.81 × 10−3 |
| Glycocholate | 464.3009 | 23.90 | C26H43NO6 | 6.89 | 1.81 | 1.85 × 10−3 |
| LysoPG(16:0) | 507.2694 | 24.33 | C22H45O9P | 0.42 | 1.38 | 1.96 × 10−3 |
| LysoPE(18:1) | 478.2934 | 24.90 | C23H46NO7P | 2.54 | 1.39 | 1.97 × 10−3 |
| Glycerol 2-phosphate | 171.0060 | 4.44 | C3H9O6P | 0.25 | 1.28 | 2.08 × 10−3 |
| Hippurate | 180.0650 | 17.08 | C9H9NO3 | 1.02 | 1.46 | 2.13 × 10−3 |
| Adenosine 5’-diphosphate | 426.0221 | 4.85 | C10H15N5O10P2 | 0.21 | 0.60 | 2.38 × 10−3 |
| LysoPC(16:1) | 538.3144 | 25.28 | C24H48NO7P | 1.77 | 1.35 | 2.46 × 10−3 |
| Glycine | 76.0385 | 4.79 | C2H5NO2 | 1.84 | 2.31 | 2.86 × 10−3 |
| 5’-CMP | 324.0591 | 4.77 | C9H14N3O8P | 0.03 | 0.65 | 2.97 × 10−3 |
| Adenosine 3’,5’-diphosphate | 450.0186 | 4.97 | C10H15N5O10P2 | 0.20 | 0.58 | 4.39 × 10−3 |
| NAD | 662.1024 | 6.27 | C21H27N7O14P2 | 3.00 | 0.50 | 4.51 × 10−3 |
| Guanosine 5’-monophosphate | 362.0507 | 5.26 | C10H14N5O8P | 2.98 | 0.60 | 4.63 × 10−3 |
| Aminoadipic acid | 162.0755 | 4.92 | C6H11NO4 | 4.20 | 0.78 | 4.70 × 10−3 |
| N-acetyl-D-galactosamine | 220.0826 | 5.16 | C8H15NO6 | 1.29 | 0.46 | 4.80 × 10−3 |
| Deoxyguanosine 5’-monophosphate | 346.0556 | 6.99 | C10H14N5O7P | 0.55 | 0.36 | 5.54 × 10−3 |
| PE(16:0/18:1) | 716.5231 | 26.78 | C39H76NO8P | 0.62 | 1.54 | 6.36 × 10−3 |
| PS(16:0/16:1) | 732.4822 | 26.53 | C38H72NO10P | 0.07 | 0.67 | 7.14 × 10−3 |
| PE(18:0/18:1) | 766.5385 | 27.08 | C41H80NO8P | 1.01 | 1.38 | 8.03 × 10−3 |
| Succinate | 117.0188 | 6.72 | C4H6O4 | 2.85 | 0.64 | 8.12 × 10−3 |
| CMP | 322.0447 | 4.74 | C9H14N3O8P | 0.49 | 0.66 | 8.79 × 10−3 |
| Glycerylphosphorylethanolamine | 214.0486 | 4.49 | C5H14NO6P | 11.19 | 0.75 | 1.19 × 10−2 |
| Phosphocreatine | 212.0427 | 4.97 | C4H10N3O5P | 3.82 | 0.69 | 1.19 × 10−2 |
| PE-NMe(22:5/18:1) | 850.5601 | 29.08 | C46H80NO8P | 0.10 | 1.50 | 1.29 × 10−2 |
| D-ribose 5-phosphate | 229.0119 | 4.46 | C5H11O8P | 0.08 | 0.44 | 1.29 × 10−2 |
| Indolelactic acid | 204.0666 | 18.62 | C11H11NO3 | 0.13 | 1.33 | 1.31 × 10−2 |
| Pyridoxine | 170.0806 | 6.74 | C8H11NO3 | 0.66 | 1.29 | 1.38 × 10−2 |
| Cytidine diphosphate choline | 489.1149 | 5.33 | C14H26N4O11P2 | 0.64 | 0.68 | 1.40 × 10−2 |
| N-acetyl-D-glucosamine | 222.0968 | 5.17 | C8H15NO6 | 0.50 | 0.80 | 1.42 × 10−2 |
| Cytidine 2’,3’-cyclic monophosphate | 306.0486 | 5.17 | C9H12N3O7P | 0.14 | 1.37 | 1.44 × 10−2 |
| Folic acid | 442.1468 | 17.85 | C19H19N7O6 | 0.34 | 2.45 | 1.45 × 10−2 |
| PE(15:0/24:1) | 832.6058 | 28.87 | C44H86NO8P | 0.73 | 1.34 | 1.52 × 10−2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alcazar Magana, A.; Reed, R.L.; Koluda, R.; Miranda, C.L.; Maier, C.S.; Stevens, J.F. Vitamin C Activates the Folate-Mediated One-Carbon Cycle in C2C12 Myoblasts. Antioxidants 2020, 9, 217. https://doi.org/10.3390/antiox9030217
Alcazar Magana A, Reed RL, Koluda R, Miranda CL, Maier CS, Stevens JF. Vitamin C Activates the Folate-Mediated One-Carbon Cycle in C2C12 Myoblasts. Antioxidants. 2020; 9(3):217. https://doi.org/10.3390/antiox9030217
Chicago/Turabian StyleAlcazar Magana, Armando, Ralph L. Reed, Rony Koluda, Cristobal L. Miranda, Claudia S. Maier, and Jan F. Stevens. 2020. "Vitamin C Activates the Folate-Mediated One-Carbon Cycle in C2C12 Myoblasts" Antioxidants 9, no. 3: 217. https://doi.org/10.3390/antiox9030217
APA StyleAlcazar Magana, A., Reed, R. L., Koluda, R., Miranda, C. L., Maier, C. S., & Stevens, J. F. (2020). Vitamin C Activates the Folate-Mediated One-Carbon Cycle in C2C12 Myoblasts. Antioxidants, 9(3), 217. https://doi.org/10.3390/antiox9030217