L-Carnitine in Drosophila: A Review

 ,
, 
 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
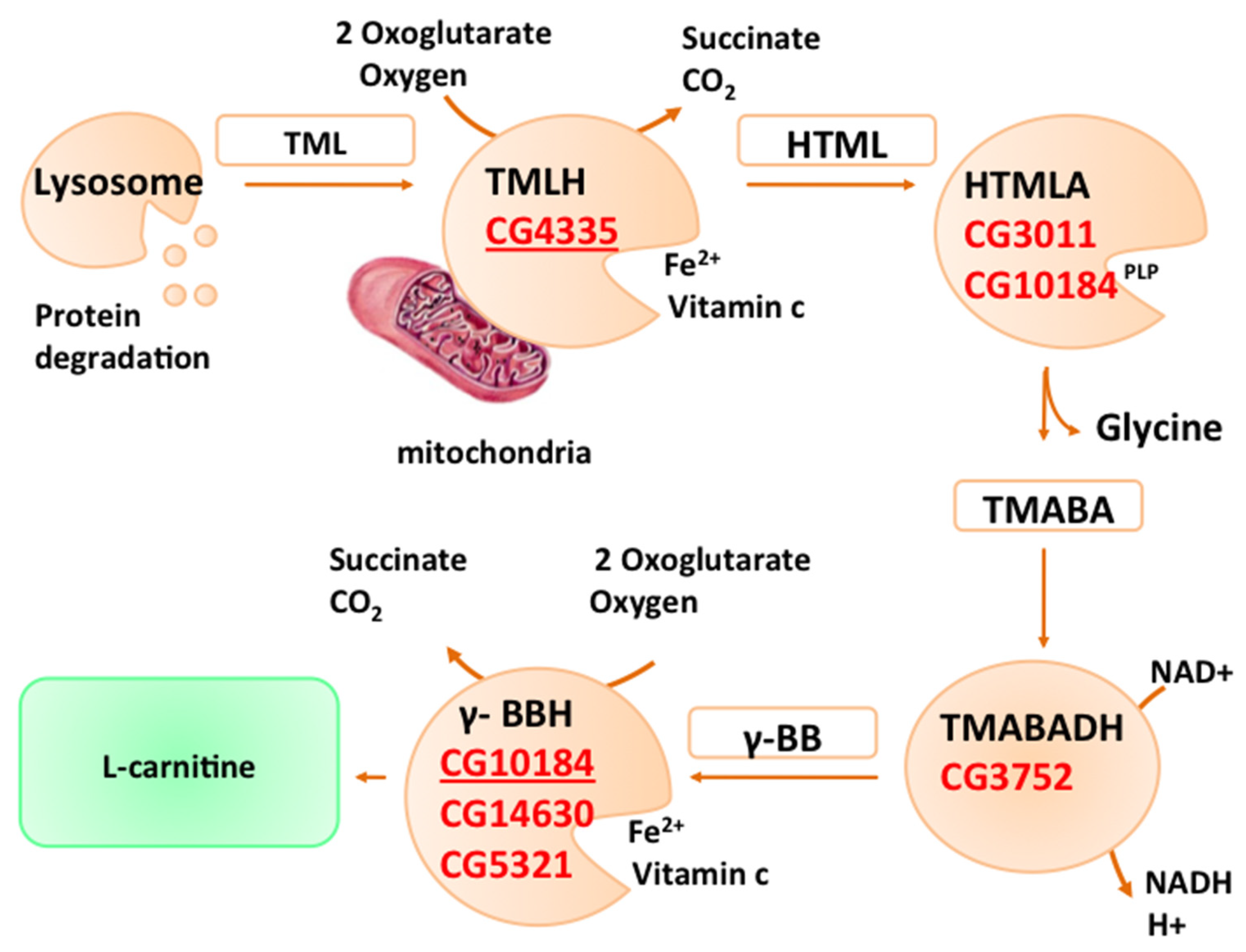
2. L-Carnitine Biosynthesis
2.1. TMLH
2.2. HTMLA
2.3. TMABADH
2.4. γ-BBH H
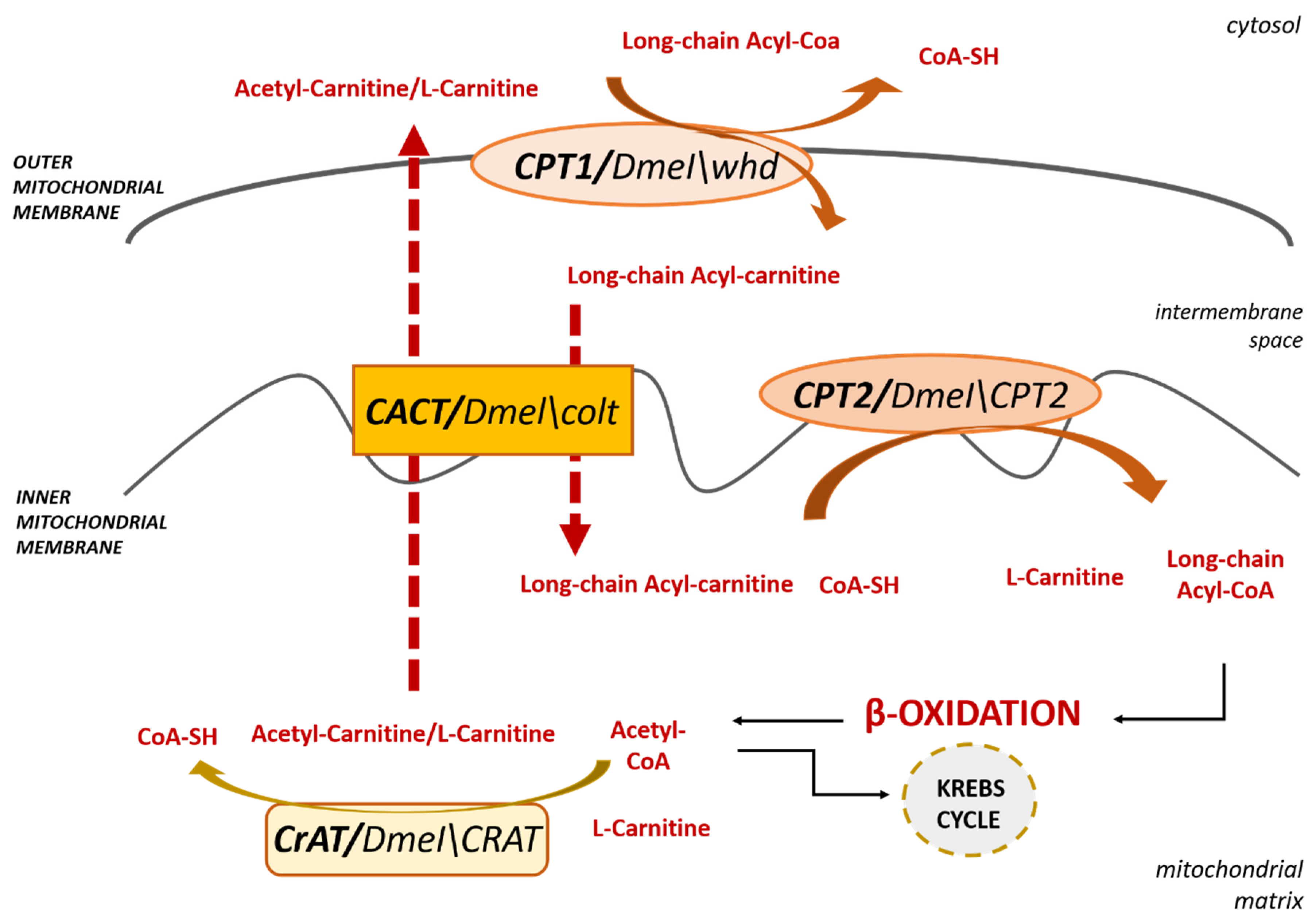
3. L-Carnitine and Fatty Acid Oxidation
3.1. L-Carnitine and Mitochondria
3.1.1. Carnitine PalmitoylTransferase 1
3.1.2. Carnitine Acyl-Carnitine Translocase
3.1.3. Carnitine PalmitoylTransferase 2
3.2. L-Carnitine and Peroxisomes
CrAT and CrOT
4. L-Carnitine Transport through the Plasma Membranes
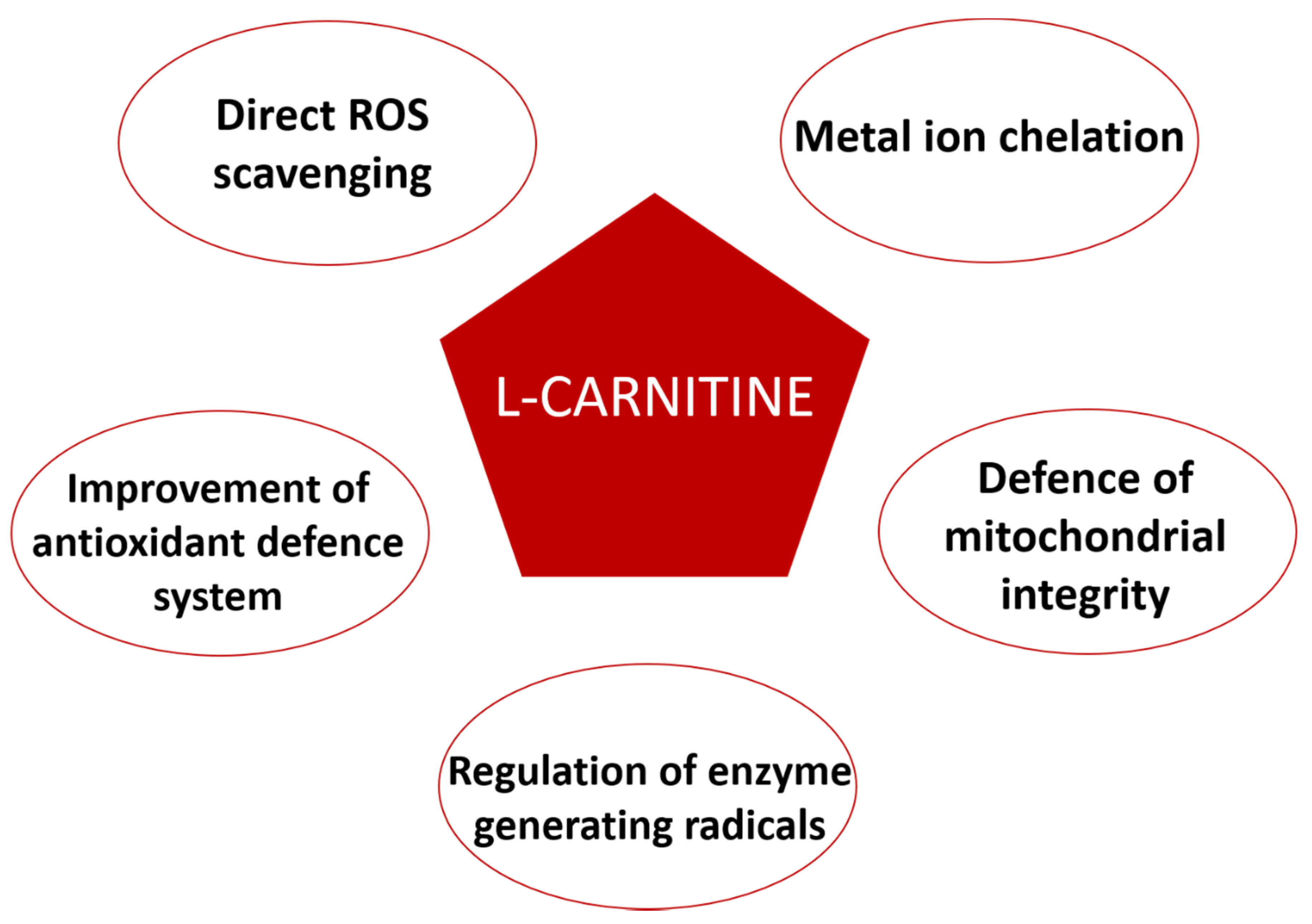
5. L-Carnitine Antioxidant Properties
6. L-Carnitine in Drosophila Brain Physiology
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- McGarry, J.D.; Woeltje, K.F.; Kuwajima, M.; Foster, D.W. Regulation of ketogenesis and the renaissance of carnitine palmitoyltransferase. Diabetes Metab. Rev. 1989, 5, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Bieber, L.L. Carnitine. Annu. Rev. Biochem. 1988, 57, 261–283. [Google Scholar] [CrossRef] [PubMed]
- Ferdinandusse, S.; Mulders, J.; Ijlst, L.; Denis, S.; Dacremont, G.; Waterham, H.R.; Wanders, R.J.A. Molecular cloning and expression of human carnitine octanoyltransferase: Evidence for its role in the peroxisomal beta-oxidation of branched-chain fatty acids. Biochem. Biophys. Res. Commun. 1999, 263, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Clarke, P.R.H.; Bieber, L.L. Isolation and purification of mitochondrial carnitine octanoyltransferase activities from beef heart. J. Biol. Chem. 1981, 256, 9861–9868. [Google Scholar] [PubMed]
- McGarry, J.D.; Brown, N.F. The mitochondrial carnitine palmitoyltransferase system. From concept to molecular analysis. Eur. J. Biochem. 1997, 244, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, R.R.; Gandour, R.D.; Van der Leij, F.R. Molecular enzymology of carnitine transfer and transport. Biochim. Biophys. Acta 2001, 1546, 21–43. [Google Scholar] [CrossRef]
- Console, L.; Giangregorio, N.; Indiveri, C.; Tonazzi, A. Carnitine/acylcarnitine translocase and carnitine palmitoyltransferase 2 form a complex in the inner mi- tochondrial membrane. Mol. Cell. Biochem. 2014, 394, 307–314. [Google Scholar] [CrossRef]
- Longo, N.; Frigeni, M.; Pasquali, M. Carnitine transport and fatty acid oxidation. Biochim. Biophys. Acta 2016, 1863, 2422–2435. [Google Scholar] [CrossRef]
- Carter, A.L.; Abney, T.O.; Lapp, D.F. Biosynthesis and metabolism of carnitine. J. Child Neurol. 1995, 10 (Suppl. S2), S3–S7. [Google Scholar] [CrossRef]
- Ramsay, R.R. The role of the carnitine system in peroxisomal fatty acid oxidation. Am. J. Med. Sci. 1999, 318, 28–35. [Google Scholar] [CrossRef]
- Jakobs, B.S.; Wanders, R.J. Fatty acid β-oxidation in peroxisomes and mitochondria: The first, unequivocal evidence for the involvement of carnitine in shuttling propionyl-CoA from peroxisomes to mitochondria. Biochem. Biophys. Res. Commun. 1995, 213, 1035–1041. [Google Scholar] [CrossRef] [PubMed]
- Verhoeven, N.M.; Roe, D.S.; Kok, R.M.; Wanders, R.J.; Jakobs, C.; Roe, C.R. Phytanic acid and pristanic acid are oxidized by sequential peroxisomal and mitochondrial reactions in cultured fibroblasts. J. Lipid. Res. 1998, 39, 66–74. [Google Scholar] [PubMed]
- Bremer, J. Carnitine-metabolism and functions. Physiol. Rev. 1983, 63, 1420–1480. [Google Scholar] [CrossRef]
- Houten, S.M.; Wanders, R.J. A general introduction to the biochemistry of mitochondrial fatty acid beta-oxidation. J. Inherit. Metab. Dis. 2010, 33, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Duran, M.; Loof, N.E.; Ketting, D.; Dorland, L. Secondary carnitine deficiency. J. Clin. Chem. Clin. Biochem. 1990, 28, 359–363. [Google Scholar] [PubMed]
- Rebouche, C.J. Role of carnitine biosynthesis and renal conservation of carnitine in genetic and acquired disorders of carnitine metabolism. In Carnitine: Pathobiochemical Basics and Clinical Applications; Seim, H., Loster, H., Eds.; Ponte Press: Bochum, Germany, 1996; pp. 111–121. [Google Scholar]
- Bene, J.; Hadzsiev, K.; Melegh, B. Role of carnitine and its derivatives in the development and management of type 2 diabetes. Nutr. Diabetes 2018, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Vaz, F.M.; Wanders, R.J.A. Carnitine biosynthesis in mammals. Biochem. J. 2002, 361, 417–429. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Jiang, W.; Chen, G.; Zhu, W.; Ding, W.; Ge, Z.; Tan, Y.; Ma, T.; Cui, G. L-carnitine treatment of insulin resistance: A systematic review and meta-analysis. Adv. Clin. Exp. Med. Off. Organ Wroc. Med. Univ. 2017, 26, 333–338. [Google Scholar] [CrossRef]
- Pietrocola, F.; Galluzzi, L.; Pedro, J.M.B.-S.; Madeo, F.; Kroemer, G. Acetyl coenzyme A: A central metabolite and second messenger. Cell Metab. 2015, 21, 805–821. [Google Scholar] [CrossRef]
- Madiraju, P.; Pande, S.V.; Prentki, M.; Madiraju, S.R. Mitochondrial acetylcarnitine provides acetyl groups for nuclear histone acetylation. Epigenetics 2009, 4, 399–403. [Google Scholar] [CrossRef]
- Le Borgne, F.; Ravaut, G.; Bernard, A.; Demarquoy, J. L-carnitine protects C2C12 cells against mitochondrial superoxide overproduction and cell death. World J. Biol. Chem. 2017, 8, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Surai, P.F. Antioxidant action of carnitine: Molecular mechanisms and practical applications. EC Vet. Sci. 2015, 2, 66–84. [Google Scholar]
- Ribas, G.S.; Vargas, C.R.; Wajner, M. L-Carnitine supplementation as a potential antioxidant therapy for inherited neurometabolic disorders. Gene 2014, 533, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Komlósi, K.; Havasi, V.; Bene, J.; Süle, N.; Pajor, L.; Nicolai, R.; Benatti, P.; Calvani, M.; Melegh, B. Histopathologic abnormalities of the lymphoreticular tissues in organic cation transporter 2 deficiency: Evidence for impaired B cell maturation. J. Pediatr. 2007, 150, 109–111.e2. [Google Scholar]
- Demarquoy, J. L-Carnitine: Structure and Function. In eLS; John Wiley Sons Ltd.: Chichester, UK, 2011. [Google Scholar]
- Melone, M.A.B.; Valentino, A.; Margarucci, S.; Galderisi, U.; Giordano, A.; Peluso, G. The carnitine system and cancer metabolic plasticity. Cell Death Dis. 2018, 9, 228. [Google Scholar] [CrossRef]
- Qu, Q.; Zeng, F.; Liu, X.; Wang, Q.J.; Deng, F. Fatty acid oxidation and carnitine palmitoyltransferase I: Emerging therapeutic targets in cancer. Cell Death Dis. 2016, 7, e2226. [Google Scholar] [CrossRef]
- Tein, I. Disorders of fatty acid oxidation. Handb. Clin. Neurol. 2013, 113, 1675–1688. [Google Scholar]
- Jernberg, J.N.; Bowman, C.E.; Wolfgang, M.J.; Scafidi, S. Developmental regulation and localization of carnitine palmitoyltransferases (CPTs) in rat brain. J. Neurochem. 2017, 142, 407–419. [Google Scholar] [CrossRef]
- Juraszek, B.; Nałecz, K.A. SLC22A5 (OCTN2) Carnitine Transporter—Indispensable for Cell Metabolism, a Jekyll and Hyde of Human Cancer. Molecules 2020, 25, 14. [Google Scholar] [CrossRef]
- Lamhonwah, A.M.; Baric, I.; Lamhonwah, J.; Grubic, M.; Tein, I. Attention deficit/hyperactivity disorder as an associated feature in OCTN2 deficiency with novel deletion (p.T440-Y449). Clin. Case Rep. 2018, 6, 585–591. [Google Scholar] [CrossRef]
- Celestino-Soper, P.B.; Shaw, C.A.; Sanders, S.J.; Li, J.; Murtha, M.T.; Ercan-Sencicek, A.G.; Davis, L.; Thomson, S.; Gambin, T.; Chinault, A.C. Use of array CGH to detect exonic copy number variants throughout the genome in autism families detects a novel deletion in TMLHE. Hum. Mol. Genet. 2011, 20, 4360–4370. [Google Scholar] [CrossRef] [PubMed]
- Celestino-Soper, P.B.; Violante, S.; Crawford, E.L.; Luo, R.; Lionel, A.C.; Delaby, E.; Cai, G.; Sadikovic, B.; Lee, K.; Lo, C.; et al. A common X-linked inborn error of carnitine biosynthesis may be a risk factor for nondysmorphic autism. Proc. Natl. Acad. Sci. USA 2012, 109, 7974–7981. [Google Scholar] [CrossRef] [PubMed]
- Ziats, M.N.; Comeaux, M.S.; Yang, Y.; Scaglia, F.; Elsea, S.H.; Sun, Q.; Beaudet, A.L.; Schaaf, C.P. Improvement of regressive autism symptoms in a child with TMLHE deficiency following carnitine supplementation. Am. J. Med. Genet. 2015, 167A, 2162–2167. [Google Scholar] [CrossRef] [PubMed]
- Manzo, E.; O’Conner, A.G.; Barrows, J.M.; Shreiner, D.D.; Birchak, G.J.; Zarnescu, D.C. Medium-Chain Fatty Acids, Beta-Hydroxybutyric Acid and Genetic Modulation of the Carnitine Shuttle Are Protective in a Drosophila Model of ALS Based on TDP-43. Front. Mol. Neurosci. 2018, 11, 182. [Google Scholar] [CrossRef]
- Laranjeira, A.; Schulz, J.; Dotti, C.G. Genes Related to Fatty Acid β-Oxidation Play a Role in the Functional Decline of the Drosophila Brain with Age. PLoS ONE 2016, 11, e0161143. [Google Scholar] [CrossRef]
- Di Cristo, F.; Finicelli, M.; Digilio, F.A.; Paladini, S.; Valentino, A.; Scialò, F.; D’Apolito, M.; Saturnino, C.; Galderisi, U.; Giordano, A.; et al. Meldonim improves Huntington’s disease mitochondrial dysfunction by restoring peroxisome proliferator-activated receptor gamma coactivator 1alpha expression. J. Cell. Physiol. 2019, 234, 9233–9246. [Google Scholar] [CrossRef]
- Di Cristo, F.; Calarco, A.; Digilio, F.A.; Sinicropi, M.S.; Rosano, C.; Galderisi, U.; Melone, M.A.B.; Saturnino, C.; Peluso, G. The Discovery of Highly Potent THP Derivatives as OCTN2 Inhibitors: From Structure-Based Virtual Screening to In Vivo Biological Activity. Int. J. Mol. Sci. 2020, 21, 7431. [Google Scholar] [CrossRef]
- Ugur, B.; Chen, K.; Bellen, H.J. Drosophila tools and assays for the study of human diseases. Dis. Models Mech. 2016, 9, 235–244. [Google Scholar] [CrossRef]
- Reiter, L.T.; Potocki, L.; Chien, S.; Gribskov, M.; Bier, E. A systematic analysis of human disease-associated gene sequences in Drosophila melanogaster. Genome Res. 2001, 11, 1114–1125. [Google Scholar] [CrossRef]
- Reiter, L.T.; Bier, E. Using Drosophila melanogaster to uncover human disease gene function and potential drug target proteins. Expert Opin. Ther. Targets 2002, 6, 387–399. [Google Scholar]
- McGurk, L.; Berson, A.; Bonini, N.M. Drosophila as an In Vivo Model for Human Neurodegenerative Disease. Genetics 2015, 1, 377–402. [Google Scholar] [CrossRef] [PubMed]
- Pandey, U.B.; Nichols, C.D. Human Disease Models in Drosophila melanogaster and the Role of the Fly in Therapeutic Drug Discovery. Pharmacol. Rev. 2011, 3, 411–436. [Google Scholar] [CrossRef] [PubMed]
- Tanphaichitr, V.; Horne, D.W.; Broquist, H.P. Lysine, a precursor of carnitine in the rat. J. Biol. Chem. 1971, 246, 6364–6366. [Google Scholar] [PubMed]
- Horne, D.W.; Broquist, H.P. Role of lysine and ε-N-trimethyllysine in carnitine biosynthesis. Studies in Neurospora crassa. J. Biol. Chem. 1973, 248, 2170–2175. [Google Scholar]
- Tanphaichitr, V.; Broquist, H.P. Role of lysine and ε-N-trimethyllysine in carnitine biosynthesis. Studies in the rat. J. Biol. Chem. 1973, 248, 2176–2181. [Google Scholar]
- Paik, W.K.; Kim, S. Protein methylation. Science 1971, 174, 114–119. [Google Scholar] [CrossRef]
- Paik, W.K.; Kim, S. Protein methylation: Chemical, enzymological, biological significance. Adv. Enzymol. Relat. Areas Mol. Biol. 1975, 42, 227–286. [Google Scholar]
- Huszar, G. Tissue-specific biosynthesis of ε-N-monomethyllysine and ε-N-trimethyllysine in skeletal and cardiac muscle myosin: A model for the cell-free study of post-translational amino acid modifications in proteins. J. Mol. Biol. 1975, 94, 311–326. [Google Scholar] [CrossRef]
- Morse, R.K.; Vergnes, J.P.; Malloy, J.; McManus, I.R. Sites of biological methylation of proteins in cultured chick muscle cells. Biochemistry 1975, 14, 4316–4325. [Google Scholar] [CrossRef]
- Labadie, J.; Dunn, W.A.; Aronson, N.N. Hepatic synthesis of carnitine from protein-bound trimethyllysine. Lysoso- mal digestion of methyl-lysine-labeled asialo-fetuin. Biochem. J. 1976, 160, 85–95. [Google Scholar] [CrossRef]
- Vaz, F.M.; van Gool, S.; Ofman, R.; Ijlst, L.; Wanders, R.J. Carnitine biosynthesis: Identification of the cDNA encoding human γ-butyrobetaine hydroxylase. Biochem. Biophys. Res. Commun. 1998, 250, 506–510. [Google Scholar] [CrossRef] [PubMed]
- Strijbis, K.; Vaz, F.M.; Distel, B. Enzymology of the carnitine biosynthesis pathway. IUBMB Life 2010, 62, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Galland, S.; Le Borgne, F.; Bouchard, F.; Georges, B.; Clouet, P.; Grand-Jean, F.; Demarquoy, J. Molecular cloning and characterization of the cDNA encoding the rat liver γ-butyrobetaine hydroxylase. Biochim. Biophys. Acta 1999, 1441, 85–92. [Google Scholar] [CrossRef]
- Vaz, F.M.; Fouchier, S.W.; Ofman, R.; Sommer, M.; Wanders, R.J. Molecular and biochemical characterization of rat γ-trimethylaminobutyraldehyde dehydrogenase and evidence for the involvement of human aldehyde dehydrogenase 9 in carnitine biosynthesis. J. Biol. Chem. 2000, 275, 7390–7394. [Google Scholar] [CrossRef]
- Vaz, F.M.; Ofman, R.; Westinga, K.; Wanders, R.J. Molecular and biochemical characterization of rat ε-N-trimethyllysine hydroxylase, the first enzyme of carnitine biosynthesis. J. Biol. Chem. 2001, 276, 33512–33517. [Google Scholar] [CrossRef]
- Strijbis, K.; Van Roermund, C.W.; Hardy, G.P.; Van Den Burg, J.; Bloem, K.; De Haan, J.; Van Vlies, N.; Wanders, R.J.; Vaz, F.M.; Distel, B. Identification and characterization of a complete carnitine biosynthesis pathway in Candida albicans. FASEB J. 2009, 23, 2349–2359. [Google Scholar] [CrossRef]
- McNeil, J.B.; Flynn, J.; Tsao, N.; Monschau, N.; Stahmann, K.; Haynes, R.H.; Mcintosh, E.M.; Pearlman, R.E. Glycine metabolism in Candida albicans: Characterization of the serine hydroxymethyltrans- ferase (SHM1, SHM2) and threonine aldolase (GLY1) genes. Yeast 2000, 16, 167–175. [Google Scholar] [CrossRef]
- Edgar, A.J. Mice have a transcribed L-threonine aldolase/GLY1 gene, but the human GLY1 gene is a non-processed pseudogene. BMC Genom. 2005, 6, 32. [Google Scholar] [CrossRef]
- Rebouche, C.J.; Engel, A.G. Tissue distribution of carnitine biosynthetic enzymes in man. Biochim. Biophys. Acta. 1980, 630, 22–29. [Google Scholar] [CrossRef]
- Ogawa, H.; Gomi, T.; Fujioka, M. Serine hydroxymethyl- transferase and threonine aldolase: Are they identical? Int. J. Biochem. Cell. Biol. 2000, 32, 289–301. [Google Scholar] [CrossRef]
- Contestabile, R.; Paiardini, A.; Pascarella, S.; Di Salvo, M.L.; D’aguanno, S.; Bossa, F. L-Threonine aldolase, serine hydroxymethyltransferase and fungal alanine racemase. A subgroup of strictly related enzymes specialized for different functions. Eur. J. Bio-Chem. 2001, 268, 6508–6525. [Google Scholar] [CrossRef] [PubMed]
- Hulse, J.D.; Henderson, L.M. Carnitine biosynthesis. Purification of 4-Nh-trimethylaminobutyraldehyde dehydrogenase from beef liver. J. Biol. Chem. 1980, 255, 1146–1151. [Google Scholar] [PubMed]
- Lin, S.W.; Chen, J.C.; Hsu, L.C.; Hsieh, C.L.; Yoshida, A. Human γ-aminobutyraldehyde dehydrogenase (ALDH9): cDNA sequence, genomic organization, polymorphism, chromosomal localization, and tissue expression. Genomics 1996, 34, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Kurys, G.; Shah, P.C.; Kikonygo, A.; Reed, D.; Ambroziak, W.; Pietruszko, R. Human aldehyde dehydrogenase. cDNA cloning and primary structure of the enzyme that catalyzes dehydrogenation of 4-aminobutyraldehyde. Eur. J. Biochem. 1993, 218, 311–320. [Google Scholar] [CrossRef]
- Englard, S.; Blanchard, J.S.; Midelfort, C.F. γ-Butyrobetaine hydroxylase: Stereochemical course of the hydroxylation reaction. Biochemistry 1985, 24, 1110–1116. [Google Scholar] [CrossRef]
- Lindstedt, G.; Lindstedt, S.; Nordin, I. γ-Butyrobetaine hydroxylase in human kidney. Scand. J. Clin. Lab. Investig. 1982, 42, 477–485. [Google Scholar] [CrossRef]
- Roe, C.; Ding, J. Mitochondrial fatty acid oxidation disorders. In The Metabolic and Molecular Bases of Inherited Disease; Scriver, C., Beaudet, A., Sly, W., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 2297–2326. [Google Scholar]
- Drosatos, K.; Schulze, P.C. Cardiac lipotoxicity: Molecular pathways and therapeutic implications. Curr. Heart Fail. Rep. 2013, 10, 109–121. [Google Scholar] [CrossRef]
- Schrader, M.; Costello, J.; Godinho, L.F.; Islinger, M. Peroxisome-mitochondria interplay and disease. J. Inherit. Metab. Dis. 2015, 38, 681–702. [Google Scholar] [CrossRef]
- Wanders, R.J. Peroxisomes in human health and disease: Metabolic pathways, metabolite transport, interplay with other organelles and signal transduction. Subcell. Biochem. 2013, 69, 23–44. [Google Scholar]
- Wanders, R.J. Metabolic functions of peroxisomes in health and disease. Biochimie 2014, 98, 36–44. [Google Scholar] [CrossRef]
- Wanders, R.J.; Ferdinandusse, S.; Brites, P.; Kemp, S. Peroxisomes, lipid metabolism and lipotoxicity. Biochim. Biophys. Acta. 2010, 1801, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Le Borgne, F.; Ben Mohamed, A.; Logerot, M.; Garnier, E.; Demarquoy, J. Changes in carnitine octanoyltransferase activity induce alteration in fatty acid metabolism. Biochem. Biophys. Res. Commun. 2011, 409, 699–704. [Google Scholar] [CrossRef] [PubMed]
- Watkins, P.A.; Maiguel, D.; Jia, Z.; Pevsner, J. Evidence for 26 distinct acyl-coenzyme A synthetase genes in the human genome. J. Lipid Res. 2007, 48, 2736–2750. [Google Scholar] [CrossRef] [PubMed]
- Zammit, V.A. Carnitine palmitoyltransferase 1: Central to cell function. IUBMB Life 2008, 60, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Rufer, A.C.; Thoma, R.; Hennig, M. Structural insight into function and regulation of carnitine palmitoyltransferase. Cell. Mol. Life Sci. CMLS 2009, 66, 2489–2501. [Google Scholar] [CrossRef] [PubMed]
- Cordente, A.G.; Lopez-Vinas, E.; Vazquez, M.I.; Swiegers, J.H.; Pretorius, I.S.; Gomez-Puertas, P.; Hegardt, F.G.; Asins, G.; Serra, D. Redesign of carnitine acetyltransferase specificity by protein engineering. J. Biolchem. 2004, 279, 33899–33908. [Google Scholar] [CrossRef] [PubMed]
- Esser, V.; Brown, N.F.; Cowan, A.T.; Foster, D.W.; McGarry, J.D. Expression of a cDNA isolated from rat brown adipose tissue and heart identifies the product as the muscle isoform of carnitine palmi- toyltransferase I (M-CPT I): M-CPT I is the predominant CPT I isoform expressed in both white (epididymal) and brown adipocytes. J. Biol. Chem. 1996, 271, 6972. [Google Scholar]
- Yamazaki, N.; Shinohara, Y.; Shima, A.; Terada, H. High expression of a novel carnitine palmitoyltransferase-I like protein in rat brown adipose-tissue and heart isolation and characterization of its cDNA clone. FEBS Lett. 1995, 363, 41–45. [Google Scholar] [CrossRef]
- Price, N.; van der Leij, F.; Jackson, V.; Corstorphine, C.; Thomson, R.; Sorensen, A.; Zammit, V. A novel brain-expressed protein related to carnitine palmitoyltransferase I. Genomics 2002, 80, 433–442. [Google Scholar] [CrossRef]
- Lopes-Marques, M.; Delgado, I.L.S.; Ruivo, R.; Torres, Y.; Sainath, S.B.; Rocha, E.; Cunha, I.; Santos, M.M.; Castro, L.F.C. The origin and diversity of Cpt1 genes in vertebrate species. PLoS ONE 2015, 10, 3–5. [Google Scholar] [CrossRef]
- Van der Hoeck, M.D.; Madsen, O.; Keijer, J.; van der Leij, F.R. Evolutionary analysis of the carnitine- and choline acyltransferases suggests distinct evolution of CPT2 versus CPT1 and related variants. BBA Mol. Cell Biol. Lipids 2018, 1863, 909–918. [Google Scholar] [CrossRef] [PubMed]
- Sierra, A.Y.; Gratacos, E.; Carrasco, P.; Clotet, J.; Urena, J.; Serra, D.; Asins, G.; Hegardt, F.G.; Casals, N. CPT1c is localized in endoplasmic reticulum of neurons and has carnitine palmitoyltransferase activity. J. Biol. Chem. 2008, 283, 6878–6885. [Google Scholar] [CrossRef] [PubMed]
- Jackson, V.N.; Cameron, J.M.; Zammit, V.A.; Price, N.T. Sequencing and functional expression of the malonyl-CoA-sensitive carnitine palmitoyltransferase from Drosophila melanogaster. Biochem. J. 1999, 341, 483–489. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Strub, B.R.; Parkes, T.L.; Mukai, S.T.; Bahadorani, S.; Coulthard, A.B.; Hall, N.; Phillips, J.P.; Hilliker, A.J. Mutations of the withered (whd) gene in Drosophila melanogaster confer hypersensitivity to oxidative stress and are lesions of the carnitine palmitoyltransferase I (CPT I) gene. Genome 2008, 51, 409–420. [Google Scholar] [CrossRef]
- Rasmussen, B.B.; Holmbäck, U.C.; Volpi, E.; Morio-Liondore, B.; Paddon-Jones, D.; Wolfe, R.R. Malonyl coenzyme A and the regulation of functional carnitine palmitoyltransferase-1 activity and fat oxidation in human skeletal muscle. J. Clin. Investig. 2002, 110, 1687–1693. [Google Scholar] [CrossRef]
- Olpin, S.E.; Allen, J.; Bonham, J.R.; Clark, S.; Clayton, P.T.; Calvin, J.; Downing, M.; Ives, K.; Jones, S.; Manning, N.J.; et al. Features of carnitine palmitoyltransferase type I deficiency. J. Inherit. Metab. Dis. 2001, 24, 35–42. [Google Scholar] [CrossRef]
- Arockia Rani, J.P.; Panneerselvam, C. Carnitine as a free radical scavenger in aging. Exp. Gerontol. 2001, 36, 1713–1726. [Google Scholar] [CrossRef]
- Indiveri, C.; Tonazzi, A.; Palmieri, F. Identification and purification of the carnitine carrier from rat liver mitochondria. Biochim. Biophys. Acta 1990, 1020, 81–86. [Google Scholar] [CrossRef]
- Indiveri, C.; Iacobazzi, V.; Tonazzi, A.; Giangregorio, N.; Infantino, V.; Convertini, P.; Console, L.; Palmieri, F. The mitochondrial carnitine/acylcarnitine carrier: Function, structure and physiopathology. Mol. Asp. Med. 2011, 32, 223–233. [Google Scholar] [CrossRef]
- Oey, N.A.; Ijlst, L.; van Roermund, C.W.T.; Wijburg, F.A.; Wanders, R.J.A. dif-1 and colt, both implicated in early embryonic development, encode carnitine acylcarnitine translocase. Mol. Genet. Metab. 2005, 85, 121–124. [Google Scholar] [CrossRef]
- Hartenstein, K.; Sinha, P.; Mishra, A.; Schenkel, H.; Torok, I.; Mechler, B.M. The congested-like tracheae gene of Drosophila melanogaster encodes a member of the mitochondrial carrier family required for gas-Wlling of the tracheal system and expansion of the wings after eclosion. Genetics 1997, 147, 1755–1768. [Google Scholar] [PubMed]
- Schulz, J.G.; Laranjeira, A.; Van Huffel, L.; Gärtner, A.; Vilain, S.; Bastianen, J.; Van Veldhoven, P.P.; Dotti, C.G. Glial β-oxidation regulates Drosophila energy metabolism. Sci. Rep. 2015, 5, 7805. [Google Scholar] [CrossRef] [PubMed]
- North, K.N.; Hoppel, C.L.; De Girolami, U.; Kozakewich, H.P.; Korson, M.S. Lethal neonatal deficiency of carnitine palmitoyltransferase II associated with dysgenesis of the brain and kidneys. J. Pediatr. 1995, 127, 414–420. [Google Scholar] [CrossRef]
- Pierce, M.R.; Pridjian, G.; Morrison, S.; Pickoff, A.S. Fatal carnitine palmitoyltransferase II deficiency in a newborn: New phenotypic features. Clin. Pediatr. (Phila.) 1999, 38, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Hug, G.; Bove, K.E.; Soukup, S. Lethal neonatal multiorgan deficiency of carnitine palmitoyltransferase II. N. Engl. J. Med. 1991, 325, 1862–1864. [Google Scholar] [CrossRef] [PubMed]
- Taroni, F.; Verderio, E.; Fiorucci, S.; Cavadini, P.; Finocchiaro, G.; Uziel, G.; Lamantea, E.; Gellera, C.; DiDonato, S. Molecular characterization of inherited carnitine palmitoyltransferase II deficiency. Proc. Natl. Acad. Sci. USA 1992, 89, 8429–8433. [Google Scholar] [CrossRef] [PubMed]
- Clarke, D.; Sokoloff, L. Circulation and Energy Metabolism of the Brain. In Basic Neurochemistry: Molecular, Cellular and Medical Aspects, 6th ed.; Agranoff, B., Albers, R.W., Fisher, S.K., Uhler, M.D., Eds.; Lipincott-Raven: Philadelphia, PA, USA, 1999; Chapter 31. [Google Scholar]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [PubMed]
- Wanders, R.J.A.; Waterham, H.R.; Ferdinandusse, S. Metabolic Interplay between Peroxisomes and Other Subcellular Organelles Including Mitochondria and the Endoplasmic Reticulum. Front. Cell Dev. Biol. 2016, 3, 83. [Google Scholar] [CrossRef]
- Violante, S.; Ijlst, L.; Te Brinke, H.; Koster, J.; de Almeida, I.T.; Wanders, R.J.; Ventura, F.V.; Houten, S.M. Peroxisomes contribute to the acylcarnitine production when the carnitine shuttle is deficient. Biochim. Biophys. Acta 2013, 1831, 1467–1474. [Google Scholar] [CrossRef]
- Theodoulou, F.L.; Holdsworth, M.; Baker, A. Peroxisomal ABC transporters. FEBS Lett. 2006, 580, 1139–1155. [Google Scholar] [CrossRef]
- Chen, H.; Liu, Z.; Huang, X. Drosophila models of peroxisomal biogenesis disorder: Peroxins are required for spermatogenesis and very-long-chain fatty acid metabolism. Hum. Mol. Genet. 2010, 19, 494–505. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, M.; Sato, H.; Okuda, T.; Fujisawa, N.; Kono, N.; Arai, H.; Suzuki, E.; Umeda, M.; Ishikawa, H.O.; Matsuno, K. Drosophila carrying pex3 or pex16 mutations are models of zellweger syndrome that reflect its symptoms associated with the absence of peroxisomes. PLoS ONE 2011, 6, e22984. [Google Scholar] [CrossRef] [PubMed]
- Faust, J.E.; Verma, A.; Peng, C.; McNew, J.A. An inventory of peroxisomal proteins and pathways in Drosophila melanogaster. Traffic 2012, 13, 1378–1392. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.Y.; Jamshidi, N.; Mo, M.L.; Wu, W.; Eraly, S.A.; Dnyanmote, A.; Bush, K.T.; Gallegos, T.F.; Sweet, D.H.; Palsson, B.Ø.; et al. Linkage of organic anion transporter-1 to metabolic pathways through integrated “omics”-driven network and functional analysis. J. Biol. Chem. 2011, 286, 31522–31531. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Nigam, K.B.; Date, R.C.; Bush, K.T.; Springer, S.A.; Saier, M.H.; Wu, W.; Nigam, S.K. Evolutionary Analysis and Classification of OATs, OCTs, OCTNs, and Other SLC22 Transporters: Structure-Function Implications and Analysis of Sequence Motifs. PLoS ONE 2015, 10, e0140569. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Xiong, L.; Xu, Y.; Tian, T.; Wang, T. The -alanine transporter BalaT is required for visual neurotransmission in Drosophila. eLife 2017, 6, e29146. [Google Scholar] [CrossRef] [PubMed]
- Stenesen, E.; Moehlman, A.; Krämer, H. The carcinine transporter CarT is required in Drosophila photoreceptor neurons to sustain histamine recycling. eLife 2015, 4, e10972. [Google Scholar] [CrossRef]
- Gai, Y.; Liu, Z.; Cervantes-Sandoval, I.; Davis, R.L. Drosophila SLC22A Transporter Is a Memory Suppressor Gene that Influences Cholinergic Neurotransmission to the Mushroom Bodies. Neuron 2016, 90, 581–595. [Google Scholar] [CrossRef]
- Eraly, S.A.; Monte, J.C.; Nigam, S. Novel slc22 transporter homologs in fly, worm, and human clarify the phylogeny of organic anion and cation transporters. Physiol. Genom. 2004, 18, 12–24. [Google Scholar] [CrossRef]
- Engelhart, D.C.; Azad, P.; Ali, S.; Granados, J.C.; Haddad, G.G.; Nigam, S.K. Drosophila SLC22 Orthologs Related to OATs, OCTs, and OCTNs Regulate Development and Responsiveness to Oxidative Stress. Int. J. Mol. Sci. 2020, 21, 2002. [Google Scholar] [CrossRef]
- Enomoto, A.; Wempe, M.; Tsuchida, H.; Shin, H.J.; Cha, S.H.; Anzai, N.; Goto, A.; Sakamoto, A.; Niwa, T.; Kanai, Y.; et al. Molecular Identification of a Novel Carnitine Transporter Specific to Human Testis. J. Biol. Chem. 2002, 277, 36262–36271. [Google Scholar] [CrossRef] [PubMed]
- Chintapalli, V.R.; Wang, J.; Dow, J.A.T. Using FlyAtlas to identify better Drosophila melanogaster models of human disease. Nat. Genet. 2007, 39, 715–720. [Google Scholar] [CrossRef] [PubMed]
- Geer, B.W.; Vovis, G.F. The effects of choline and related compounds on the growth and development of Drosophila melanogaster. J. Exp. Zool. 1965, 158, 223–236. [Google Scholar] [CrossRef]
- Gülçin, I. Antioxidant and antiradical activities of L-carnitine. Life Sci. 2006, 78, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Augustyniak, A.; Skrzydlewska, E. The Influence of L-carnitine Supplementation on the Antioxidative Abilities of Serum and the Central Nervous System of Ethanol-Induced Rats. Metab. Brain Dis. 2010, 25, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.L.; Sun, Z.O.; Rehman, R.U.; Wang, H.; Wang, Y.F.; Wang, H. Rosemary Extract-Mediated Lifespan Extension and Attenuated Oxidative Damage in Drosophila melanogaster Fed on High-Fat Diet. J. Food Sci. 2017, 82, 1006–1011. [Google Scholar] [CrossRef] [PubMed]
- Pignatelli, P.; Lenti, L.; Sanguigni, V.; Frati, G.; Simeoni, I.; Gazzaniga, P.P.; Pulcinelli, F.M.; Violi, F. Carnitine inhibits arachidonic acid turnover, platelet function, and oxidative stress. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H41–H48. [Google Scholar] [CrossRef]
- Chaves, M.M.; Rodrigues, A.L.P.; dos Reis, A.P.; Gerzstein, N.C.; Nogueira-Machado, J.A. Correlation between NADPH oxidase and protein kinase C in the ROS production by human granulocytes related to age. Gerontology 2002, 48, 354–359. [Google Scholar] [CrossRef]
- Cao, Y.; Qu, H.J.; Li, P.; Wang, C.B.; Wang, L.X.; Han, Z.W. Single dose administration of L-carnitine improves antioxidant activities in healthy subjects. Tohoku J. Exp. Med. 2011, 224, 209–213. [Google Scholar] [CrossRef]
- Li, J.L.; Wang, Q.Y.; Luan, H.Y.; Kang, Z.C.; Wang, C.B. Effects of L-carnitine against oxidative stress in human hepatocytes: Involvement of peroxisome proliferator-activated receptor alpha. J. Biomed. Sci. 2012, 19, 32. [Google Scholar] [CrossRef]
- Li, J.; Zhang, Y.; Luan, H.; Chen, X.; Han, Y.; Wang, C. L-carnitine protects human hepatocytes from oxidative stress-induced toxicity through Akt-mediated activation of Nrf2 signaling pathway. Can. J. Physiol. Pharmacol. 2016, 94, 517–525. [Google Scholar] [CrossRef]
- Surai, P.F. Carnitine Enigma: From Antioxidant Action to Vitagene Regulation Part 1. Absorption, Metabolism, and Antioxidant Activities. J. Veter Sci. Med. 2015, 3, 14. [Google Scholar] [CrossRef]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 2013, 8, 2003–2014. [Google Scholar] [PubMed]
- Kumaran, S.; Subathra, M.; Balu, M.; Panneerselvam, C. Age-associated decreased activities of mitochondrial electron transport chain complexes in heart and skeletal muscle: Role of L-carnitine. Chem. Biol. Interact. 2004, 148, 11–18. [Google Scholar] [CrossRef]
- Reznick, A.Z.; Kagan, V.E.; Ramsey, R.; Tsuchiya, M.; Khwaja, S.; Serbinova, E.A.; Packer, L. Antiradical effects in L-propionyl carnitine protection of the heart against ischemia-reperfusion injury: The possible role of iron chelation. Arch. Biochem. Biophys. 1992, 296, 394–401. [Google Scholar] [CrossRef]
- Manoharan, S.; Guillemin, G.J.; Abiramasundari, R.S.; Essa, M.M.; Akbar, M.; Akbar, M.D. The Role of Reactive Oxygen Species in the Pathogenesis of Alzheimer’s Disease, Parkinson’s Disease, and Huntington’s Disease: A Mini Review. Oxid. Med. Cell Longev. 2016, 2016, 8590578. [Google Scholar] [CrossRef]
- Sanz, F.J.; Solana-Manrique, C.; Muñoz-Soriano, V.; Calap-Quintana, P.; Moltó, M.D.; Paricio, N. Identification of potential therapeutic compounds for Parkinson’s disease using Drosophila and human cell models. Free Radic. Biol. Med. 2017, 108, 683–691. [Google Scholar] [CrossRef]
- Krishna, G.; Muralidhara, S. Aqueous extract of tomato seeds attenuates rotenone-induced oxidative stress and neurotoxicity in Drosophila melanogaster. J. Sci. Food Agric. 2016, 96, 1745–1755. [Google Scholar] [CrossRef]
- Zhou, Y.; Xue, L.; Gao, L.; Qin, X.; Du, G. Ginger extract extends the lifespan of Drosophila melanogaster through antioxidation and ameliorating metabolic dysfunction. J. Functional Food. 2018, 49, 295–305. [Google Scholar] [CrossRef]
- Knottnerus, S.J.G.; Bleeker, J.C.; Wüst, R.C.I.; Ferdinandusse, S.; IJlst, L.; Wijburg, F.A.; Wanders, R.J.A.; Visser, G.; Houtkooper, R.H. Disorders of mitochondrial long-chain fatty acid oxidation and the carnitine shuttle. Rev. Endocr. Metab. Disord. Mar. 2018, 19, 93–106. [Google Scholar] [CrossRef]
- Houten, S.M.; Violante, S.; Ventura, F.V.; Wanders, R.J.A. The Biochemistry and Physiology of Mitochondrial Fatty Acid β-Oxidation and Its Genetic Disorders. Annu. Rev. Physiol. 2016, 78, 23–44. [Google Scholar] [CrossRef] [PubMed]
- Almannai, M.; Alfadhel, M.; El-Hattab, A.W. Carnitine inborn errors of metabolism. Molecules 2019, 24, 3251. [Google Scholar] [CrossRef] [PubMed]
- Nehlig, A.; de Vasconcelos, A.P. Glucose and ketone body utilization by the brain of neonatal rats. Prog. Neurobiol. 1993, 40, 163–221. [Google Scholar] [CrossRef]
- Patel, A.B.; Lai, J.C.; Chowdhury, G.M.; Hyder, F.; Rothman, D.L.; Shulman, R.G.; Behar, K.L. Direct evidence for activity-dependent glucose phosphorylation in neurons with implications for the astrocyte-to-neuron lactate shuttle. Proc. Natl. Acad. Sci. USA 2014, 111, 5385–5390. [Google Scholar] [CrossRef] [PubMed]
- Ebert, D.; Haller, R.G.; Walton, M.E. Energy contribution of octanoate to intact rat brain metabolism measured by 13C nuclear magnetic resonance spectroscopy. J. Neurosci. 2003, 23, 5928–5935. [Google Scholar] [CrossRef] [PubMed]
- Schonfeld, P.; Reiser, G. Why does brain metabolism not favor burning of fatty acids to provide energy? Reflections on disadvantages of the use of free fatty acids as fuel for brain. J. Cereb. Blood Flow Metab. 2013, 33, 1493–1499. [Google Scholar] [CrossRef]
- Panov, A.; Orynbayeva, Z.; Vavilin, V.; Lyakhovich, V. Fatty acids in energy metabolism of the Central Nervous System. Biomed. Res. Int. 2014, 472459. [Google Scholar] [CrossRef]
- Romano, A.; Koczwara, J.B.; Gallelli, C.A.; Vergara, D.; Di Bonaventura, M.V.M.; Gaetani, S.; Giudetti, A.M. Fats for thoughts: An update on brain fatty acid metabolism. Int. J. Biochem. Cell Biol. 2017, 84, 40–45. [Google Scholar] [CrossRef]
- Hoffman, J.M.; Soltow, Q.A.; Li, S.; Sidik, A.; Jones, D.P.; Promislow, D.E.L. Effects of age, sex, and genotype on high-sensitivity metabolomic profiles in the fruit fly, Drosophila melanogaster. Aging Cell. 2014, 13, 596–604. [Google Scholar] [CrossRef]
- Houtkooper, R.H.; Argmann, C.; Houten, S.M.; Cantó, C.; Jeninga, E.H.; Andreux, P.A.; Thomas, C.; Doenlen, R.; Schoonjans, K.; Auwerx, J. The metabolic footprint of aging in mice. Sci. Rep. 2011, 1, 134. [Google Scholar] [CrossRef]
- Gómez, L.A.; Heath, S.H.; Hagen, T.M. Acetyl-L-carnitine supplementation reverses the age-related decline in carnitine palmitoyltransferase 1 (CPT1) activity in interfibrillar mitochondria without changing the L-carnitine content in the rat heart. Mech. Ageing Dev. 2012, 133, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Lee, S.K.; Paik, D.; Min, K.J. Overexpression of Fatty-Acid-β-Oxidation-Related Genes Extends the Lifespan of Drosophila melanogaster. Oxid. Med. Cell Longev. 2012, 2012, 854502. [Google Scholar] [CrossRef]
- Bruss, M.D.; Khambatta, C.F.; Ruby, M.A.; Aggarwal, I.; Hellerstein, M.K. Calorie restriction increases fatty acid synthesis and whole body fat oxidation rates. Am. J. Physiol. 2010, 298, E108–E116. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Gopalacharyulu, P.; Seppanen-Laakso, T.; Ruskeepaa, A.L.; Aye, C.C.; Carson, B.P.; Mora, S.; Oresic, M.; Teleman, A.A. Insulin signaling regulates fatty acid catabolism at the level of CoA activation. PloS Genet. 2012, 8, e1002478. [Google Scholar] [CrossRef] [PubMed]
- Michalik, L.; Auwerx, J.; Berger, J.P.; Chatterjee, V.K.; Glass, C.K.; Gonzalez, F.J.; Grimaldi, P.A.; Kadowaki, T.; Lazar, M.A.; O’Rahilly, S.; et al. International Union of Pharmacology. LXI. Peroxisome proliferator-activated receptors. Pharmacol. Rev. 2006, 58, 726–741. [Google Scholar] [CrossRef] [PubMed]
- Palanker, L.; Tennessen, J.M.; Lam, G.; Thummel, C.S. Drosophila HNF4 Regulates Lipid Mobilization and b-Oxidation. Cell Metab. 2009, 9, 228–239. [Google Scholar] [CrossRef] [PubMed]
- Guzman, M.; Blazquez, C. Is there an astrocyte-neuron ketone body shuttle? Trends Endocrinol. Metab. 2001, 12, 169–173. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carillo, M.R.; Bertapelle, C.; Scialò, F.; Siervo, M.; Spagnuolo, G.; Simeone, M.; Peluso, G.; Digilio, F.A. L-Carnitine in Drosophila: A Review. Antioxidants 2020, 9, 1310. https://doi.org/10.3390/antiox9121310
Carillo MR, Bertapelle C, Scialò F, Siervo M, Spagnuolo G, Simeone M, Peluso G, Digilio FA. L-Carnitine in Drosophila: A Review. Antioxidants. 2020; 9(12):1310. https://doi.org/10.3390/antiox9121310
Chicago/Turabian StyleCarillo, Maria Rosaria, Carla Bertapelle, Filippo Scialò, Mario Siervo, Gianrico Spagnuolo, Michele Simeone, Gianfranco Peluso, and Filomena Anna Digilio. 2020. "L-Carnitine in Drosophila: A Review" Antioxidants 9, no. 12: 1310. https://doi.org/10.3390/antiox9121310
APA StyleCarillo, M. R., Bertapelle, C., Scialò, F., Siervo, M., Spagnuolo, G., Simeone, M., Peluso, G., & Digilio, F. A. (2020). L-Carnitine in Drosophila: A Review. Antioxidants, 9(12), 1310. https://doi.org/10.3390/antiox9121310