Hydrogen Sulfide and Carnosine: Modulation of Oxidative Stress and Inflammation in Kidney and Brain Axis

 ,
,  ,
,  ,
,  ,
,  , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
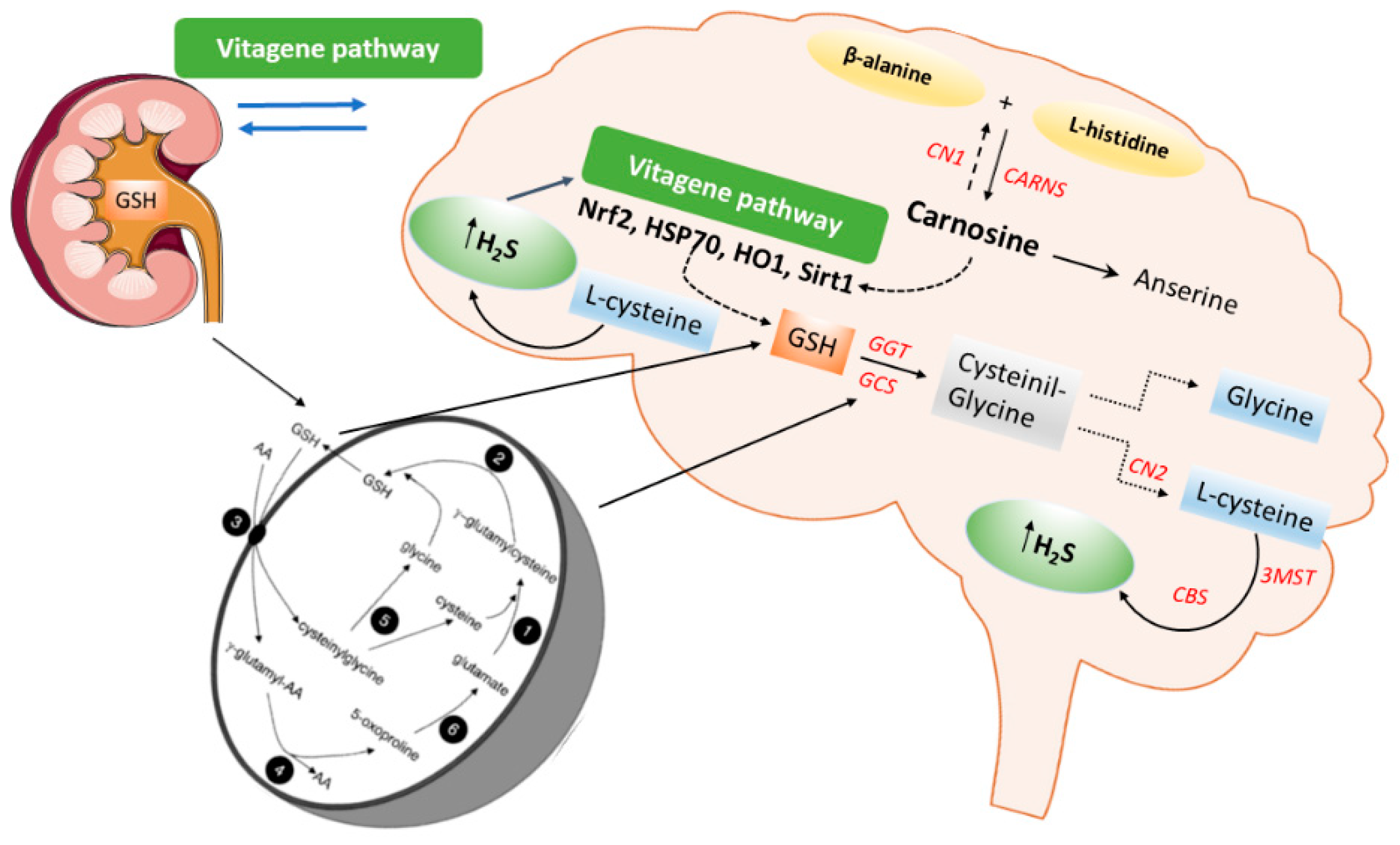
2. Carnosine Signaling in Kidney and Brain
2.1. Carnosine in Cell Lines
2.2. Carnosine in Animal Models
2.3. Carnosine in Clinical Settings
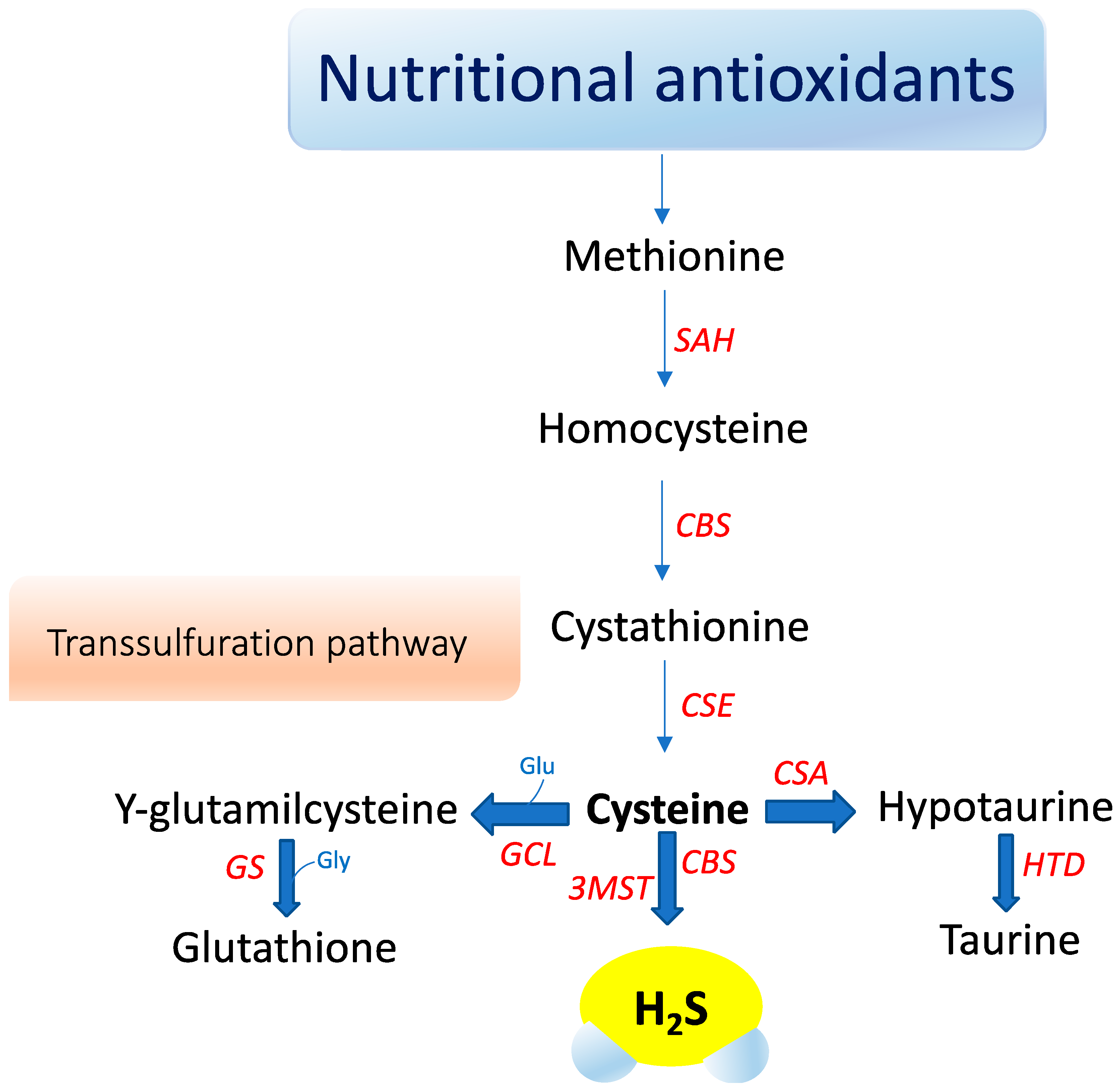
3. Hydrogen Sulfide Signaling
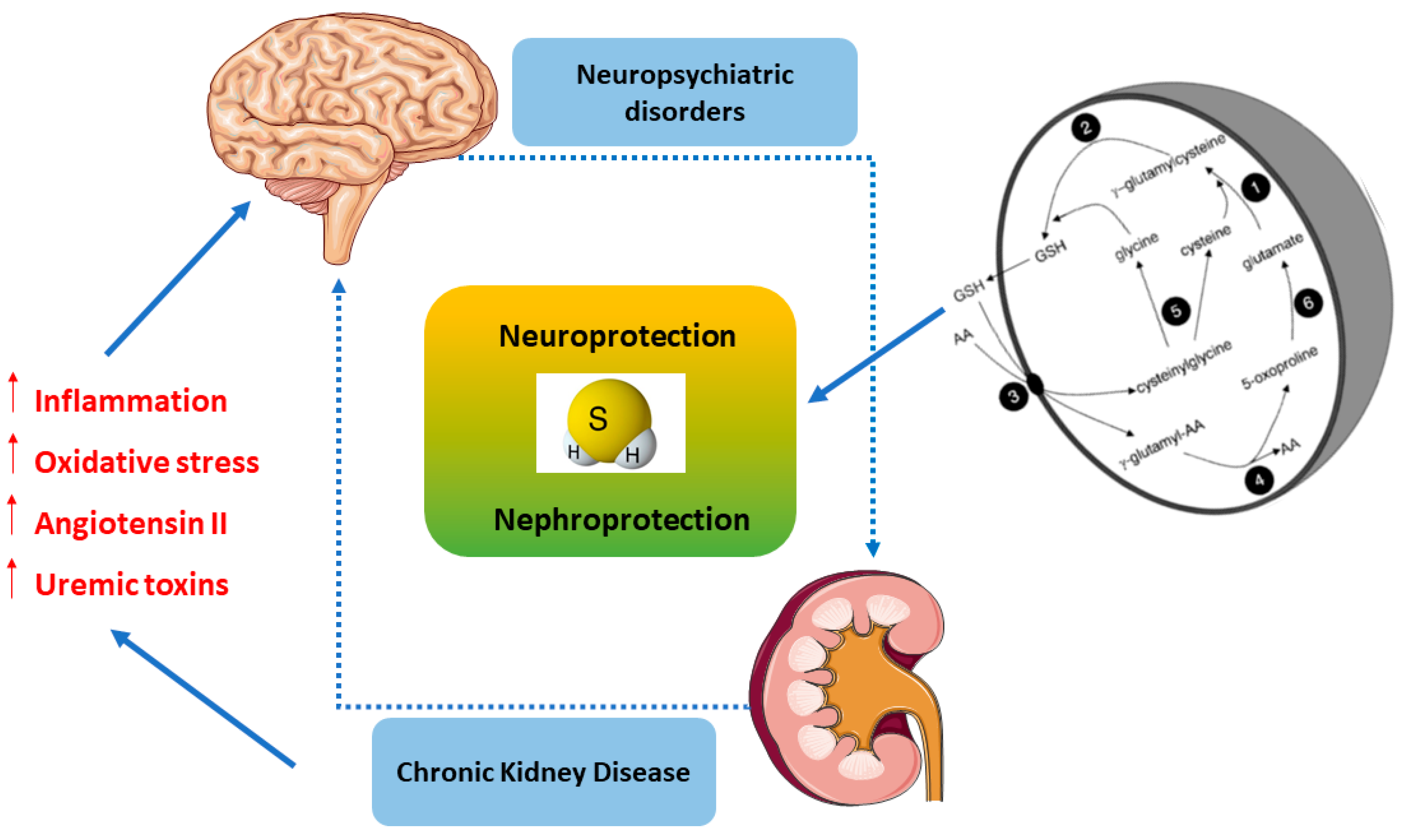
4. H2S as a Signaling Mediator in the Kidney–Brain Axis
4.1. Anti-Inflammatory Role of H2S
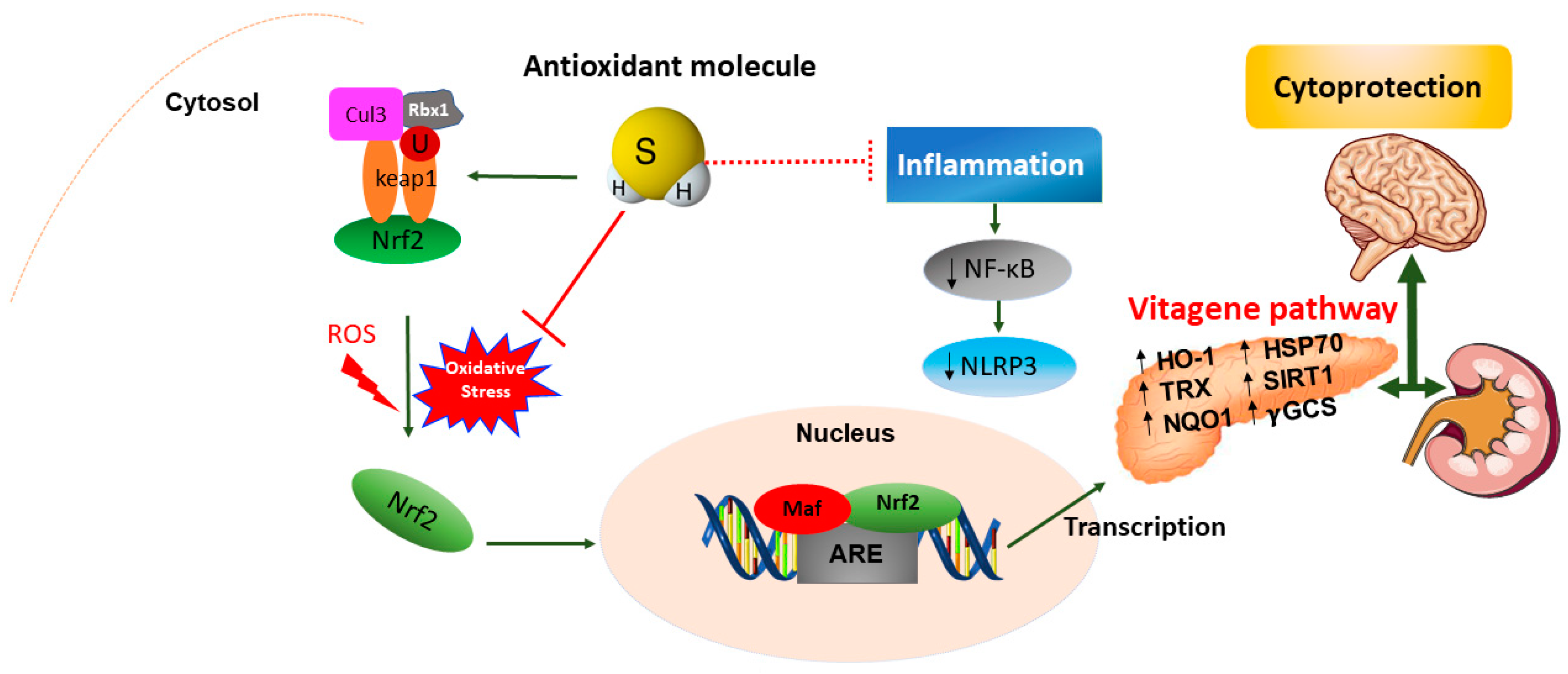
4.2. Antioxidant Role of H2S
5. H2S and Diabetes
6. H2S and Brain
7. H2S Redox Signaling and Resilience
8. Analytical Approaches to Quantify H2S in Biological Matrices
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Calabrese, V.; Cornelius, C.; Dinkova-Kostova, A.T.; Calabrese, E.J.; Mattson, M.P. Cellular Stress Responses, The Hormesis Paradigm, and Vitagenes: Novel Targets for Therapeutic Intervention in Neurodegenerative Disorders. Antioxid. Redox Signal. 2010, 13, 1763–1811. [Google Scholar] [CrossRef]
- Biswas, S.K. Does the Interdependence between Oxidative Stress and Inflammation Explain the Antioxidant Paradox? Oxid. Med. Cell. Longov. 2016, 2016, 5698931. [Google Scholar] [CrossRef] [PubMed]
- Alhamdani, M.; Al-Azzawie, H.F.; Abbas, F.K. Decreased formation of advanced glycation end-products in peritoneal fluid by carnosine and related peptides. Perit. Dial. Int. 2007, 27, 86–89. [Google Scholar] [CrossRef] [PubMed]
- Brings, S.; Fleming, T.; De Buhr, S.; Beijer, B.; Lindner, T.; Wischnjow, A.; Kender, Z.; Peters, V.; Kopf, S.; Haberkorn, U.; et al. A scavenger peptide prevents methylglyoxal induced pain in mice. Biochim. Biophys. Acta 2017, 1863, 654–662. [Google Scholar] [CrossRef]
- Colzani, M.; De Maddis, D.; Casali, G.; Carini, M.; Vistoli, G.; Aldini, G. Reactivity, Selectivity, and Reaction Mechanisms of Aminoguanidine, Hydralazine, Pyridoxamine, and Carnosine as Sequestering Agents of Reactive Carbonyl Species: A Comparative Study. ChemMedChem 2016, 11, 1778–1789. [Google Scholar] [CrossRef] [PubMed]
- Weigand, T.; Singler, B.; Fleming, T.; Nawroth, P.; Klika, K.D.; Thiel, C.; Baelde, H.; Garbade, S.F.; Wagner, A.H.; Hecker, M.; et al. Carnosine Catalyzes the Formation of the Oligo/Polymeric Products of Methylglyoxal. Cell. Physiol. Biochem. 2018, 46, 713–726. [Google Scholar] [CrossRef]
- Hipkiss, A.R. Chapter carnosine and its possible roles in nutrition and health. Adv. Food Nutr. Res. 2009, 57, 87–154. [Google Scholar]
- Hou, W.; Chen, H.J.; Lin, Y.H. Antioxidant peptides with Angiotensin converting enzyme inhibitory activities and applications for Angiotensin converting enzyme purification. J. Agric. Food Chem. 2003, 51, 1706–17093. [Google Scholar] [CrossRef]
- Nakagawa, K.; Ueno, A.; Nishikawa, Y. Interactions between carnosine and captopril on free radical scavenging activity and angiotensin-converting enzyme activity in vitro. Yakugaku Zasshi 2006, 126, 37–42. [Google Scholar] [CrossRef]
- Decker, E.A.; Livisay, S.A.; Zhou, S. A re-evaluation of the antioxidant activity of purified carnosine. Biochemistry 2000, 65, 766–770. [Google Scholar]
- Mozdzan, M.; Szemraj, J.; Rysz, J.; Nowak, D. Antioxidant properties of carnosine re-evaluated with oxidizing systems involving iron and copper ions. Basic Clin. Pharmacol. Toxicol. 2005, 96, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Velez, S.; Nair, N.G.; Reddy, V.P. Transition metal ion binding studies of carnosine and histidine: Biologically relevant antioxidants. Colloids Surf. B Biointerfaces 2008, 66, 291–294. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, V.; Cornelius, C.; Mancuso, C.; Pennisi, G.; Calafato, S.; Bellia, F.; Bates, T.E.; Giuffrida Stella, A.M.; Schapira, T.; Dinkova Kostova, A.T.; et al. Cellular stress response: A novel target for chemoprevention and nutritional neuroprotection in aging, neurodegenerative disorders and longevity. Neuroch Res. 2008, 33, 2444–2471. [Google Scholar] [CrossRef] [PubMed]
- Aldini, G.; Facino, R.M.; Beretta, G.; Carini, M. Carnosine and related dipeptides as quenchers of reactive carbonyl species: From structural studies to therapeutic perspectives. Biofactors 2005, 24, 77–87. [Google Scholar] [CrossRef]
- Grasso, G.I.; Arena, G.; Bellia, F.; Rizzarelli, E.; Vecchio, G. Copper(II)-chelating homocarnosine glycoconjugate as a new multifunctional compound. J. Inorg. Biochem. 2014, 131, 56–63. [Google Scholar] [CrossRef]
- Liu, W.; Wang, D.; Liu, K.; Sun, X. Nrf as a converging node for cellular signaling pathways of gasotransmitters. Med. Hypotheses 2012, 79, 308–310. [Google Scholar] [CrossRef]
- Bogen, K.T. Low dose rese response for in vitro Nrf2 ARE activation in human HepG2 cells. Dose Response 2017, 15, 1559325817699696. [Google Scholar] [CrossRef]
- Jing, X.; Wei, X.; Ren, R.; Wang, L.; Ahang, X.; Lou, H. Neuroprotective effects of tans chinone I against 6OHDA-induced oxidative stress in cellular and mouse model of Parkinson’s Disease through upregulating Nrf2. Neurochem. Res. 2016, 41, 779–786. [Google Scholar] [CrossRef]
- Xiao, J.; Zhu, X.; Kang, B.; Xu, J.; Wu, L.; Hong, J.; Zhang, Y.; Ni, X.; Wang, Z. Hydrogen sulfide attenuates myocardial hypoxia-reoxygenation injury by inhibiting autophage via mTOR activation. Cell Physiol. Biochem. 2015, 37, 2444–2453. [Google Scholar] [CrossRef]
- Zhang, Q.; Liu, S.; Li, T.; Yuan, L.; Liu, H.; Wang, X.; Wang, F.; Wang, S.; Hao, A.; Liu, D.; et al. Preconditioning of bone marrow mesenchymal stem cells with hydrogen sulfide improves their therapeutic potential. Oncotarget 2016, 7, 58089–58104. [Google Scholar] [CrossRef]
- Zhang, J.; Yr, J.; Yuan, C.; Fu, Q.; Zhang, F.; Zhu, X.; Wang, L.; Go, P.; Shu, G.; Jiang, Q.; et al. Exogenous H2S exerts biphasic effects on porcine mammary epithelial cells proliferation through P13K/Akt-mTOR signaling pathway. J. Cell Physiol. 2018, 233, 7071–7081. [Google Scholar] [CrossRef]
- Cai, W.J.; Wang, M.J.; Moore, P.K.; Jin, H.M.; Yao, T.; Zu, Y.C. The novel proangiogenic effect of hydrogen sulfide is dependent on Akt phosphorylation. Cardiovasc. Res. 2017, 76, 29–40. [Google Scholar] [CrossRef]
- Han, Y.; Zeng, F.; Tan, G.; Yang, C.; Tang, H.; Luo, Y.; Feng, J.; Xiong, H.; Guo, Q. Hydrogen sulfide inhibits abnormal proliferation of lymphocytes via AKT/GSK3B signal pathway in systemic lupus erythematosus patients. Cell. Physiol. Biochem. 2013, 31, 795–804. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Chen, Z.; Wu, L. Oxidative stress effects of soluble sulfide on human hepatocytes cell line L02. International. J. Environ. Res. Public Health 2019, 16, 1662. [Google Scholar] [CrossRef]
- Yang, B.; Bai, Y.; Yin, C.; Xing, G.; Wang, S.; Li, F.; Bian, J.; Aschner, M.; Lu, R. Activation of autophagic flux and the Nrf2/ARE signaling pathway by hydrogen sulfide protects against acrylonitrile-induced neurotoxicity in primary rat astrocytes. Arch. Toxicol. 2018, 92, 2093–2108. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liu, D.X.; Wang, F.W.; Zhang, Q.; Du, Z.X.; Zhan, J.M.; Yuan, Q.H.; Ling, E.A.; Hao, A.J. L-cysteine promotes the proliferation and differentiation of neural stem cells via the CBS/H2S pathway. Neuroscience 2013, 237, 106–117. [Google Scholar] [CrossRef]
- Zhao, Y.; Kong, C.; Chen, X.; Wang, Z.; Wan, Z.; Jia, L.; Liu, Q.; Wang, Y.; Li, W.; Cui, J.; et al. Repetitive exposure to low-dose X-irradiation attenuates testicular apoptosis in type diabetic rats, likely via Akt-mediated Nrf2 activation. Mol. Cell. Endocrinol. 2016, 422, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, E.J.; Calabrese, V. Low dose radiation therapy (LDRT) is effective in the treatment of arthritis: Animal model findings. Int. J. Radiat. Biol. 2013, 89, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, E.J.; Calabrese, V. Reduction of arthritic symptoms by low dose radiation therapy (LDRT) is associated with an anti-inflammatory phenotype. Int. J. Radiat. Biol. 2013, 89, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, E.J.; Dhawan, G.; Kapoor, R.; Kozumbo, W.J. Radiotherapy treatment of human inflammatory disease and conditions: Optimal dose. Human Exp. Toxicol. 2019, 38, 888–898. [Google Scholar] [CrossRef] [PubMed]
- Miranda, A.S.; Cordeiro, T.M.; Dos Santos Lacerda Soares, T.M.; Ferreira, R.N.; Simões ESilva, A.C. Kidney-brain axis inflammatory cross-talk: From bench to bedside. Clin. Sci. 2017, 131, 1093–1105. [Google Scholar] [CrossRef]
- Boldyrev, A.A.; Aldini, G.; Derave, W. Physiology and pathophysiology of carnosine. Physiol. Rev. 2013, 93, 1803–1845. [Google Scholar] [CrossRef] [PubMed]
- Abe, H. Role of histidine-related compounds as intracellular proton buffering constituents in vertebrate muscle. Biochemistry 2000, 65, 757–765. [Google Scholar] [PubMed]
- Peters, V.; Schmitt, C.P.; Zschocke, J.; Gross, M.L.; Brismar, K.; Forsberg, E. Carnosine treatment largely prevents alterations of renal carnosine metabolism in diabetic mice. Amino Acids 2012, 42, 2411–2416. [Google Scholar] [CrossRef]
- Peters, V.; Yard, B.; Schmitt, C.P. Carnosine and Diabetic Nephropathy. Curr. Med. Chem. 2020, 27, 1801–1812. [Google Scholar] [CrossRef] [PubMed]
- Pfister, F.; Riedl, E.; Wang, Q.; Vom Hagen, F.; Deinzer, M.; Harmsen, M.C.; Molema, G.; Yard, B.; Feng, Y.; Hammes, H.P. Oral carnosine supplementation prevents vascular damage in experimental diabetic retinopathy. Cell. Physiol. Biochem. 2011, 28, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Mong, M.C.; Chao, C.Y.; Yin, M.C. Histidine and carnosine alleviated hepatic steatosis in mice consumed high saturated fat diet. Eur. J. Pharmacol. 2011, 653, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Kamal, M.A.; Jiang, H.; Hu, Y.; Keep, R.F.; Smith, D.E. Influence of genetic knockout of Pept2 on the in vivo disposition of endogenous and exogenous carnosine in wild-type and Pept2 null mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 296, R986–R991. [Google Scholar] [CrossRef] [PubMed]
- Bauer, K. Carnosine and homocarnosine, the forgotten, enigmatic peptides of the brain. Neuroch Res. 2005, 30, 1339–1345. [Google Scholar] [CrossRef]
- Bellia, F.; Vecchio, G.; Cuzzocrea, S.; Calabrese, V.; Rizzarelli, E. Neuroprotective features of carnosine in oxidative driven diseases. Mol. Aspects Med. 2011, 32, 258–266. [Google Scholar] [CrossRef]
- Teufel, M.; Saudek, V.; Ledig, J.P.; Bernhardt, A.; Boularand, S.; Carreau, A.; Cairns, N.J.; Carter, C.; Cowley, D.J.; Duverger, D.; et al. Sequence identification and characterization of human carnosinase and a closely related non-specific dipeptidase. J. Biol. Chem. 2003, 278, 6251–6531. [Google Scholar] [CrossRef] [PubMed]
- Peters, V.; Klessens, C.Q.; Baelde, H.J.; Singler, B.; Veraar, K.A.M.; Zutinic, A.; Drozak, J.; Zschocke, J.; Schmitt, C.P.; De Heer, E. Intrinsic carnosine metabolism in the human kidney. Amino Acids 2015, 47, 2541–2550. [Google Scholar] [CrossRef] [PubMed]
- Spina-Purrello, V.; Giliberto, S.; Barresi, V.; Nicoletti, V.G.; Giuffrida Stella, A.M.; Rizzarelli, E. Modulation of PARP-and PARP-expression by L-carnosine and trehalose after LPS and INFgamma-induced oxidative stress. Neurochem. Res. 2010, 35, 2144–2153. [Google Scholar] [CrossRef] [PubMed]
- Lopachev, A.V.; Lopacheva, O.M.; Abaimov, D.A.; Koroleva, O.V.; Vladychenskaya, E.A.; Erukhimovich, A.A.; Fedorova, T.N. Neuroprotective Effect of Carnosine on Primary Culture of Rat Cerebellar Cells under Oxidative Stress. Biochemistry 2016, 81, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, M.; Tanaka, K.I.; Kato-Negishi, M. Zinc, Carnosine, and Neurodegenerative Diseases. Nutrients 2018, 10, 147. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, S.; Sato, M.; Matsumoto, T.; Kadooka, K.; Hasegawa, T.; Fujimura, T.; Katakura, Y. Mechanisms of carnosine-induced activation of neuronal cells. Biosci. Biotechnol. Biochem. 2018, 82, 683–688. [Google Scholar] [CrossRef]
- Calabrese, V.; Colombrita, C.; Guagliano, E.; Sapienza, M.; Ravagna, A.; Cardile, V.; Scapagnini, G.; Santoro, A.M.; Mangiameli, A.; Butterfield, D.A.; et al. Protective effect of carnosine during nitrosative stress in astroglial cell cultures. Neurochem. Res. 2005, 30, 797–807. [Google Scholar] [CrossRef]
- Preston, J.E.; Hipkiss, A.R.; Himsworth, D.T.; Romero, I.A.; Abbott, J.N. Toxic effects of beta-amyloid (25–35) on immortalised rat brain endothelial cell: Protection by carnosine, homocarnosine and beta-alanine. Neurosci. Lett. 1998, 242, 105–108. [Google Scholar] [CrossRef]
- Peters, V.; Zschocke, J.; Schmitt, C.P. Carnosinase, diabetes mellitus and the potential relevance of carnosinase deficiency. J. Inherit. Metab. Dis. 2018, 41, 39–47. [Google Scholar] [CrossRef]
- Ansari, F.A.; Mahmood, R. Carnosine and N-acetyl cysteine protect against sodium nitrite-induced oxidative stress in rat blood. Cell Boil. Int. 2018, 42, 281–293. [Google Scholar] [CrossRef]
- Fadda, L.M.; Attia, H.A.; Al-Rasheed, N.M.; Ali, H.M.; Aldossari, M. Attenuation of DNA damage and mRNA gene expression in hypoxic rats using natural antioxidants. J. Biochem. Mol. Toxicol. 2017, 31. [Google Scholar] [CrossRef] [PubMed]
- Iacobini, C.; Menini, S.; Blasetti Fantauzzi, C.; Pesce, C.M.; Giaccari, A.; Salomone, E.; Lapolla, A.; Orioli, M.; Aldini, G.; Pugliese, G. FL-926-16, a novel bioavailable carnosinase-resistant carnosine derivative, prevents onset and stops progression of diabetic nephropathy in db/db mice. Br. J. Pharmacol. 2018, 175, 53–66. [Google Scholar] [CrossRef] [PubMed]
- Caruso, G.; Fresta, C.G.; Martinez-Becerra, F.; Antonio, L.; Johnson, R.T.; De Campos, R.P.S.; Siegel, J.M.; Wijesinghe, M.B.; Lazzarino, G.; Lunte, S.M. Carnosine modulates nitric oxide in stimulated murine RAW 264.7 macrophages. Mol. Cell. Biochem. 2017, 431, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, Z.; Kalaz, E.B.; Aydin, A.F.; Soluk-Tekkesin, M.; Dogru-Abbasoglu, S.; Uysal, M.; Kocak-Toker, N. The effect of carnosine on methylglyoxal-induced oxidative stress in rats. Arch. Physiol. Biochem. 2017, 123, 192–198. [Google Scholar] [CrossRef]
- Aldini, G.; Vistoli, G.; Stefek, M.; Chondrogianni, N.; Grune, T.; Sereikaite, J.; Sadowska-Bartosz, I.; Bartosz, G. Molecular strategies to prevent, inhibit, and degrade advanced glycoxidation and advanced lipoxidation end products. Free Radic. Res. 2013, 47, 93–137. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Bo, S.H.; Lu, X.T.; Xu, A.J.; Zhang, J. Protective effects of carnosine on white matter damage induced by chronic cerebral hypoperfusion. Neural Regen Res. 2016, 11, 1438–1444. [Google Scholar] [CrossRef]
- Zhang, Z.Y.; Sun, B.L.; Yang, M.F.; Li, D.W.; Fang, J.; Zhang, S. Carnosine attenuates early brain injury through its antioxidative and anti-apoptotic effects in a rat experimental subarachnoid hemorrhage model. Cell. Mol. Neurobiol. 2015, 35, 147–157. [Google Scholar] [CrossRef]
- Zhao, J.; Shi, L.; Zhang, L.R. Neuroprotective effect of carnosine against salsolinol-induced Parkinson’s disease. Exp. Ther. Med. 2017, 14, 664–670. [Google Scholar] [CrossRef]
- Ahshin-Majd, S.; Zamani, S.; Kiamari, T.; Kiasalari, Z.; Baluchnejadmojarad, T.; Roghani, M. Carnosine ameliorates cognitive deficits in streptozotocin-induced diabetic rats: Possible involved mechanisms. Peptides 2016, 86, 102–111. [Google Scholar] [CrossRef]
- Baguet, A.; Everaert, I.; Yard, B.; Peters, V.; Zschocke, J.; Zutinic, A.; De Heer, E.; Podgorski, T.; Domaszewska, K.; Derave, W. Does low serum carnosinase activity favor high-intensity exercise capacity? J. Appl. Physiol. 2014, 116, 553–559. [Google Scholar] [CrossRef]
- De Courten, B.; Jakubova, M.; De Courten, M.P.; Kukurova, I.J.; Vallova, S.; Krumpolec, P.; Valkovic, L.; Kurdiova, T.; Garzon, D.; Barbaresi, S.; et al. Ukropcova B: Effects of carnosine supplementation on glucose metabolism: Pilot clinical trial. Obesity 2016, 24, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Houjeghani, S.; Kheirouri, S.; Faraji, E.; Jafarabadi, M.A. L-Carnosine supplementation attenuated fasting glucose, triglycerides, advanced glycation end products, and tumor necrosis factor-alpha levels in patients with type 2 diabetes: A double-blind placebo-controlled randomized clinical trial. Nutr. Res. 2018, 49, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Elbarbary, N.S.; Ismail, E.A.R.; El-Naggar, A.R.; Hamouda, M.H.; El-Hamamsy, M. The effect of weeks carnosine supplementation on renal functional integrity and oxidative stress in pediatric patients with diabetic nephropathy: A randomized placebo-controlled trial. Pediatric Diabetes 2018, 19, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Hauske, S.J.; Zhang, S.; Rodriguez-Niño, A.; Albrecht, T.; Pastene, D.O.; Van den Born, J.; Van Goor, H.; Ruf, S.; Kohlmann, M.; et al. Identification and characterisation of carnostatine (SAN9812), a potent and selective carnosinase (CN1) inhibitor with in vivo activity. Amino Acids 2019, 51, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Peters, V.; Jansen, E.E.; Jakobs, C.; Riedl, E.; Janssen, B.; Yard, B.A.; Wedel, J.; Hoffmann, G.F.; Zschocke, J.; Gotthardt, D.; et al. Anserine inhibits carnosine degradation but in human serum carnosinase (CN1) is not correlated with histidine dipeptide concentration. Clin. Chim. Acta 2011, 412, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Chengappa, K.N.; Turkin, S.R.; DeSanti, S.; Bowie, C.R.; Brar, J.S.; Schlicht, P.J.; Murphy, S.L.; Hetrick, M.L.; Bilder, R.; Fleet, D. A preliminary, randomized, double-blind, placebo-controlled trial of L-carnosine to improve cognition in schizophrenia. Schizoph Res. 2012, 142, 145–152. [Google Scholar] [CrossRef]
- Attanasio, F.; Convertino, M.; Magno, A.; Caflisch, A.; Corazza, A.; Haridas, H.; Esposito, G.; Cataldo, S.; Pignataro, B.; Milardi, D.; et al. Carnosine Inhibits Aβ (42) Aggregation by Perturbing the H-bond Network in and Around the Central Hydrophobic Cluster. Chembiochem 2013, 14, 583–592. [Google Scholar] [CrossRef]
- Cornelli, U. Treatment of Alzheimer’s disease with a cholinesterase inhibitor combined with antioxidants. Neuro-Degener Dis. 2010, 7, 193–202. [Google Scholar] [CrossRef]
- Mehrazad-Saber, Z.; Kheirouri, S.; Noorazar, S.G. Effects of l-Carnosine Supplementation on Sleep Disorders and Disease Severity in Autistic Children: A Randomized, Controlled Clinical Trial. Basic Clin. Pharmacol. Toxicol. 2018, 123, 72–77. [Google Scholar] [CrossRef]
- Masuoka, N.; Yoshimine, C.; Hori, M.; Tanaka, M.; Asada, T.; Abe, K.; Hisatsune, T. Effects of Anserine/Carnosine Supplementation on Mild Cognitive Impairment with APOE4. Nutrients 2019, 11, 1626. [Google Scholar] [CrossRef]
- Katakura, Y.; Totsuka, M.; Imabayashi, E.; Matsuda, H.; Hisatsune, T. Anserine/Carnosine Supplementation Suppresses the Expression of the Inflammatory Chemokine CCL24 in Peripheral Blood Mononuclear Cells from Elderly People. Nutrients 2017, 9, 1199. [Google Scholar] [CrossRef] [PubMed]
- Boldyrev, A.; Fedorova, T.; Stepanova, M.; Dobrotvorskaya, I.; Kozlova, E.; Boldanova, N.; Bagyeva, G.; Ivanova-Smolenskaya, I.; Illarioshkin, S. Carnosine increases efficiency of DOPA therapy of Parkinson’s disease: A pilot study. Rejuvenation Res. 2008, 11, 821–827. [Google Scholar] [CrossRef] [PubMed]
- Fonteh, A.N.; Harrington, R.J.; Tsai, A.; Liao, P.; Harrington, M.G. Free amino acid and dipeptide changes in the body fluids from Alzheimer’s disease subjects. Amino Acids 2007, 32, 213–224. [Google Scholar] [CrossRef]
- Kimura, H.; Shibuya, N.; Kimura, Y. Hydrogen sulfide is a signaling molecule and a cytoprotectant. Antioxid Redox Signal. 2012, 17, 45–57. [Google Scholar] [CrossRef]
- Yang, G.; Zhao, K.; Ju, Y.; Mani, S.; Cao, Q.; Puukila, S.; Khaper, N.; Wu, L.; Wang, R. Hydrogen sulfide protects against cellular senescence via S-sulfhydration of Keap1 and activation of Nrf2. Antioxid. Redox Signal. 2013, 18, 1906–1919. [Google Scholar] [CrossRef]
- Ramazzini, B. Diseases of Workers–De Morbis Artificum Diatriba–1713. Am. J. Public Health 2001, 91, 1380–1382. [Google Scholar] [CrossRef]
- Szabo, C. A timeline of hydrogen sulfide (H2S) research: From environmental toxin to biological mediator. Biochem. Pharmacol. 2018, 149, 5–19. [Google Scholar] [CrossRef]
- Yamamoto, J.; Sato, W.; Kosugi, T.; Yamamoto, T.; Kimura, T.; Taniguchi, S.; Kojima, H.; Maruyama, S.; Imai, E.; Matsuo, S.; et al. Distribution of hydrogen sulfide (H2S)-producing enzymes and the roles of the H2S donor sodium hydrosulfide in diabetic nephropathy. Clin. Exp. Nephrol. 2013, 17, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Gadalla, M.M.; Snyder, S.H. Hydrogen sulfide as a gasotransmitter. J. Neurochem. 2010, 113, 14–26. [Google Scholar] [CrossRef]
- Kabil, O.; Banerjee, R. Enzymology of H2S biogenesis, decay and signaling. Antioxid. Redox Signal. 2014, 20, 770–782. [Google Scholar] [CrossRef]
- Shibuya, N.; Koike, S.; Tanaka, M.; Ishigami-Yuasa, M.; Kimura, Y.; Ogasawara, Y.; Fukui, K.; Nagahara, N.; Kimura, H. A novel pathway for the production of hydrogen sulfide from D-cysteine in mammalian cells. Nat. Commun. 2013, 4, 1366. [Google Scholar] [CrossRef] [PubMed]
- Paul, B.D.; Snyder, S.H. Gasotransmitter hydrogen sulfide signaling in neuronal health and disease. Biochem. Pharmacol. 2018, 149, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Longen, S.; Beck, K.F.; Pfeilschifter, J. H2S-induced thiol-based redox switches: Biochemistry and functional relevance for inflammatory diseases. Pharmacol. Res. 2016, 111, 642–651. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Li, H.; Li, S.; Yang, G. Regulation of cystathionine gamma-lyase/H2S system and its pathological implication. Front. Biosci. 2014, 19, 1355–1369. [Google Scholar] [CrossRef]
- Wang, R. Physiological implications of hydrogen sulfide: A whiff exploration that blossomed. Physiol. Rev. 2012, 92, 791–896. [Google Scholar] [CrossRef]
- Van den Born, J.C.; Frenay, A.R.; Bakker, S.J.; Pasch, A.; Hillebrands, J.L.; Lambers Heerspink, H.J.; Van Goor, H. High urinary sulfate concentration is associated with reduced risk of renal disease progression in type diabetes. Nitric Oxide 2016, 55–56, 18–24. [Google Scholar] [CrossRef]
- Mustafa, A.K.; Gadalla, M.M.; Sen, N.; Kim, S.; Mu, W.; Gazi, S.K.; Barrow, R.K.; Yang, G.; Wang, R.; Snyder, S.H. H2S signals through protein S-sulfhydration. Sci. Signal. 2009, 2, ra72. [Google Scholar] [CrossRef]
- Koike, S.; Ogasawara, Y.; Shibuya, N.; Kimura, H.; Ishii, K. Polysulfide exerts a protective effect against cytotoxicity caused by t-buthylhydroperoxide through Nrf2 signaling in neuroblastoma cells. FEBS Lett. 2013, 587, 3548–3555. [Google Scholar] [CrossRef]
- Meng, G.; Zhao, S.; Xie, L.; Han, Y.; Ji, Y. Protein S-sulfhydration by hydrogen sulfide in cardiovascular system. Br. J. Pharmacol. 2018, 175, 1146–1156. [Google Scholar] [CrossRef]
- Sen, N.; Paul, B.D.; Gadalla, M.M.; Mustafa, A.K.; Sen, T.; Xu, R.; Kim, S.; Snyder, S.H. Hydrogen sulfide-linked sulfhydration of NF-κB mediates its antiapoptotic actions. Mol. Cell. 2012, 45, 13–24. [Google Scholar] [CrossRef]
- Xie, Z.Z.; Liu, Y.; Bian, J.S. Hydrogen Sulfide and Cellular Redox Homeostasis. Oxid. Med. Cell. Longev. 2016, 2016, 6043038. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.L.; Roth, M.B. Hydrogen sulfide increases thermotolerance and lifespan in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2007, 104, 20618–20622. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.M.; Neish, A.S. Redox Signaling Mediated by the Gut Microbiota. Free Radic. Res. 2013, 47, 950–957. [Google Scholar] [CrossRef] [PubMed]
- Youm, Y.H.; Adijiang, A.; Vandanmagsar, B.; Burk, D.; Ravussin, A.; Dixit, V.D. Elimination of the NLRP3-ASC inflammasome protects against chronic obesity-induced pancreatic damage. Endocrinology 2011, 152, 4039–4045. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.S.; Yu, J.T.; Jiang, T.; Zhu, X.C.; Tan, L. The NLRP inflammasome in Alzheimer’s disease. Mol. Neurobiol. 2013, 48, 875–882. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.L.; Pan, L.L.; Long, F.; Wu, W.J.; Yan, D.; Xu, P.; Liu, S.Y.; Qin, M.; Jia, W.W.; Liu, X.H.; et al. Endogenous Hydrogen Sulfide Ameliorates NOX4 Induced Oxidative Stress in LPS-Stimulated Macrophages and Mice. Cell. Physiol. Biochem. 2018, 47, 458–474. [Google Scholar] [CrossRef]
- Wang, P.; Chen, F.; Wang, W.; Zhang, X.D. Hydrogen Sulfide Attenuates High Glucose-Induced Human Retinal Pigment Epithelial Cell Inflammation by Inhibiting ROS Formation and NLRP3 Inflammasome Activation. Mediat. Inflamm. 2019, 2019, 8908960. [Google Scholar] [CrossRef]
- Li, J.; Teng, X.; Jin, S.; Dong, J.; Guo, Q.; Tian, D.; Wu, Y. Hydrogen sulfide improves endothelial dysfunction by inhibiting the vicious cycle of NLRP3 inflammasome and oxidative stress in spontaneously hypertensive rats. J. Hypertens. 2019, 37, 1633–1643. [Google Scholar] [CrossRef]
- Cao, L.; Cao, X.; Zhou, Y.; Nagpure, B.V.; Wu, Z.Y.; Hu, L.F.; Yang, Y.; Sethi, G.; Moore, P.K.; Bian, J.S. Hydrogen sulfide inhibits ATP-induced neuroinflammation and Aβ 1-42 synthesis by suppressing the activation of STAT3 and cathepsin S. Brain Behav. Immun. 2018, 73, 603–614. [Google Scholar] [CrossRef]
- Lin, Z.; Altaf, N.; Li, C.; Chen, M.; Pan, L.; Wang, D.; Xie, L.; Zheng, Y.; Fu, H.; Han, Y.; et al. Hydrogen sulfide attenuates oxidative stress-induced NLRP3 inflammasome activation via S-sulfhydrating c-Jun at Cys269 in macrophages. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 2890–2900. [Google Scholar] [CrossRef]
- Kimura, H. Signaling molecules: Hydrogen sulfide and polysulfide. Antioxid. Redox Signal. 2015, 22, 362–376. [Google Scholar] [CrossRef] [PubMed]
- Mogi, M.; Horiuchi, M. Clinical interaction between brain and kidney in small vessel disease. Cardiol. Res. Pract. 2011, 2011, 306189. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Kiernan, M.C.; Murray, A.; Rosner, M.H.; Ronco, C. Kidney–brain crosstalk in the acute and chronic setting. Nat. Rev. Nephrol. 2015, 11, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Simões, E.; Silva, A.C.; Miranda, A.S.; Rocha, N.P.; Teixeira, A.L. Neuropsychiatric Disorders in Chronic Kidney Disease. Front. Pharmacol. 2019, 16, 10–932. [Google Scholar]
- Etgen, T.; Chonchol, M.; Forstl, H.; Sander, D. Chronic kidney disease and cognitive impairment: A systematic review and meta-analysis. Am. J. Nephrol. 2012, 35, 474–482. [Google Scholar] [CrossRef]
- Lee, H.J.; Lee, D.Y.; Mariappan, M.M.; Feliers, D.; Ghosh-Choudhury, G.; Abboud, H.E.; Gorin, Y.; Kasinath, B.S. Hydrogen sulfide inhibits high glucose-induced NADPH oxidase expression and matrix increase by recruiting inducible nitric oxide synthase in kidney proximal tubular epithelial cells. J. Biol. Chem. 2017, 292, 5665–5675. [Google Scholar] [CrossRef]
- Xia, M.; Chen, L.; Muh, R.W.; Li, P.-L.; Li, N. Production and actions of hydrogen sulfide, a novel gaseous bioactive substance, in the kidneys. J. Pharmacol. Exp. Ther. 2009, 329, 1056–1062. [Google Scholar] [CrossRef]
- Bełtowski, J. Hypoxia in the renal medulla: Implications for hydrogen sulfide signaling. J. Pharmacol. Exp. Ther. 2010, 334, 358–363. [Google Scholar] [CrossRef]
- Hutton, H.L.; Ooi, J.D.; Holdsworth, S.R.; Kitchingm, A.R. The NLRP3 inflammasome in kidney disease and autoimmunity. Nephrology 2016, 21, 736–744. [Google Scholar] [CrossRef]
- Yang, F.; Wang, Z.; Wei, X.; Han, H.; Meng, X.; Zhang, Y.; Shi, W.; Li, F.; Xin, T.; Pang, Q.; et al. NLRP3 deficiency ameliorates neurovascular damage in experimental ischemic stroke. J. Cereb. Blood Flow Metab. 2014, 34, 660–667. [Google Scholar] [CrossRef]
- Granata, S.; Masola, V.; Zoratti, E.; Scupoli, M.T.; Baruzzi, A.; Messa, M.; Sallustio, F.; Gesualdo, L.; Lupo, A.; Zaza, G. NLRP3 inflammasome activation in dialyzed chronic kidney disease patients. PLoS ONE 2015, 10, e0122272. [Google Scholar] [CrossRef] [PubMed]
- Palomo, J.; Dietrich, D.; Martin, P.; Palmer, G.; Gabay, C. The interleukin (IL)-cytokine family–balance between agonists and antagonists in inflammatory diseases. Cytokine 2015, 76, 25–37. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Klionsky, D.J. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef] [PubMed]
- Ghiringhelli, F.; Apetoh, L.; Tesniere, A.; Aymeric, L.; Ma, Y.; Ortiz, C.; Vermaelen, K.; Panaretakis, T.; Mignot, G.; Ullrich, E.; et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1betadependent adaptive immunity against tumors. Nat. Med. 2009, 15, 1170–1178. [Google Scholar] [CrossRef]
- Kim, S.-M.; Lee, S.-H.; Kim, Y.-G.; Kim, S.-Y.; Seo, J.-W.; Choi, Y.-W.; Kim, D.-J.; Jeong, K.-H.; Lee, T.; Ihm, C.-G.; et al. Hyperuricemia-induced NLRP3 activation of macrophages contributes to the progression of diabetic nephropathy. Am. J. Physiol. Renal. Physiol. 2015, 308, F993–F1003. [Google Scholar] [CrossRef]
- Kasimsetty, S.G.; DeWolf, S.E.; Shigeoka, A.A.; McKay, D.B. Regulation of TLR2 and NLRP3 in primary murine renal tubular epithelial cells. Nephron Clin. Pract. 2014, 127, 119–123. [Google Scholar] [CrossRef]
- Abais, J.M.; Zhang, C.; Xia, M.; Liu, Q.; Gehr, T.W.; Boini, K.M.; Li, P. NADPH oxidase-mediated triggering of inflammasome activation in mouse podocytes and glomeruli during hyperhomocysteinemia. Antioxid. Redox Signal. 2013, 18, 1537–1548. [Google Scholar] [CrossRef]
- Ghosh, S.; Wu, M.D.; Shaftel, S.S.; Kyrkanides, S.; LaFerla, F.M.; Olschowka, J.A.; O’Banion, M.K. Sustained interleukin-1beta overexpression exacerbates tau pathology despite reduced amyloid burden in an Alzheimer’s mouse model. J. Neurosci. 2013, 33, 5053–5064. [Google Scholar] [CrossRef]
- Haapakoski, R.; Mathieu, J.; Ebmeier, K.P.; Alenius, H.; Kivimäki, M. Cumulative meta-analysis of interleukins 6 and 1β, tumour necrosis factor α and C-reactive protein in patients with major depressive disorder. Brain Behav. Immun. 2015, 49, 206–215. [Google Scholar] [CrossRef]
- Salama, M.; Farrag, S.M.; Abulasrar, S.A.; Amin, M.M.; Ali, A.A.; Sheashaa, H.; Sobh, M.; Arias-Carrión, O. Up-regulation of TLR-4 in the brain after ischemic kidney-induced encephalopathy in the rat. CNS Neurol. Disord Drug Targets 2013, 12, 583–586. [Google Scholar] [CrossRef]
- Dantzer, R.; Kelley, K.W. Twenty years of research on cytokine-induced sickness behavior. Brain Behav. Immun. 2007, 21, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Tu, C.; Zhao, J.; Ou, D.; Chen, G.; Liu, Y.; Xiao, X. Exogenous hydrogen sulfide protects against global cerebral ischemia/reperfusion injury via its anti-oxidative, anti-inflammatory and anti-apoptotic effects in rats. Brain Res. 2013, 1491, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Aminzadeh, M.A.; Vaziri, N.D. Downregulation of the renal and hepatic hydrogen sulfide (H2S)-producing enzymes and capacity in chronic kidney disease. Nephrol. Dial. Transplant. 2012, 27, 498–504. [Google Scholar] [CrossRef] [PubMed]
- Vandini, E.; Ottani, A.; Zaffe, D.; Calevro, A.; Canalini, F.; Cavallini, G.M.; Rossi, R.; Guarini, S.; Giuliani, D. Mechanisms of Hydrogen Sulfide against the Progression of Severe Alzheimer’s Disease in Transgenic Mice at Different Ages. Pharmacology 2019, 103, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.W.; Feng, W.; Peh, M.T.; Peh, K.; Dymock, B.W.; Moore, P.K. A novel slow-releasing hydrogen sulfide donor, FW1256, exerts anti-inflammatory effects in mouse macrophages and in vivo. Pharmacol. Res. 2016, 113, 533–546. [Google Scholar] [CrossRef]
- Lu, Y.; Gao, L.; Li, L.; Zhu, Y.; Wang, Z.; Shen, H.; Song, K. Hydrogen Sulfide Alleviates Peritoneal Fibrosis via Attenuating Inflammation and TGF-β1 Synthesis. Nephron 2015, 131, 210–219. [Google Scholar] [CrossRef]
- Olas, B. Hydrogen sulfide in signaling pathways. Clin. Chim. Acta 2015, 439, 212–218. [Google Scholar] [CrossRef]
- Liu, Y.; Deng, Y.; Liu, H.; Yin, C.; Li, X.; Gong, Q. Hydrogen sulfide ameliorates learning memory impairment in APP/PS1 transgenic mice: A novel mechanism mediated by the activation of Nrf2 pathway. Pharmacol. Biochem. Behav. 2016, 150–151, 207–216. [Google Scholar] [CrossRef]
- Shi, L.; Liu, X.Y.; Huang, Z.G.; Ma, Z.Y.; Xi, Y.; Wang, L.Y.; Sun, N.L. Endogenous hydrogen sulfide and ERK1/2-STAT3 signaling pathway may participate in the association between homocysteine and hypertension. J. Geriatr. Cardiol. 2019, 16, 822–834. [Google Scholar]
- Hou, X.Y.; Hu, Z.L.; Zhang, D.Z.; Lu, W.; Zhou, J.; Wu, P.F.; Guan, X.L.; Han, Q.Q.; Deng, S.L.; Zhang, H.; et al. Rapid Antidepressant Effect of Hydrogen Sulfide: Evidence for Activation of mTORC1-TrkB-AMPA Receptor Pathways. Antioxid. Redox Signal. 2017, 27, 472–488. [Google Scholar] [CrossRef]
- Shefa, U.; Kim, D.; Kim, M.S.; Jeong, N.Y.; Jung, J. Roles of Gasotransmitters in Synaptic Plasticity and Neuropsychiatric Conditions. Neural. Plast. 2018, 1824713. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Xiong, S.; Zhou, Y.; Wu, Z.; Ding, L.; Zhu, Y.; Wood, M.E.; Whiteman, M.; Moore, P.K.; Bian, J.S. Renal Protective Effect of Hydrogen Sulfide in Cisplatin-Induced Nephrotoxicity. Antioxid. Redox Signal. 2018, 29, 455–470. [Google Scholar] [CrossRef] [PubMed]
- Askari, H.; Abazari, M.F.; Ghoraeian, P.; Torabinejad, S.; Nouri Aleagha, M.; Mirfallah Nassiri, R.; Tahmasebi, F.; Abedi, N.; Rajani, S.F.; Salarian, A.; et al. Ameliorative effects of hydrogen sulfide (NaHS) on chronic kidney disease-induced brain dysfunction in rats: Implication on role of nitric oxide (NO) signaling. Metab. Brain Dis. 2018, 33, 1945–1954. [Google Scholar] [CrossRef] [PubMed]
- Shirazi, M.K.; Azarnezhad, A.; Abazari, M.F.; Poorebrahim, M.; Ghoraeian, P.; Sanadgol, N.; Bokharaie, H.; Heydari, S.; Abbasi, A.; Kabiri, S.; et al. The role of nitric oxide signaling in renoprotective effects of hydrogen sulfide against chronic kidney disease in rats: Involvement of oxidative stress, autophagy and apoptosis. J. Cell. Physiol. 2019, 234, 11411–11423. [Google Scholar] [CrossRef] [PubMed]
- Ozatik, F.Y.; Teksen, Y.; Kadioglu, E.; Ozatik, O.; Bayat, Z. Effects of hydrogen sulfide on acetaminophen-induced acute renal toxicity in rats. Int. Urol. Nephrol. 2019, 51, 745–754. [Google Scholar] [CrossRef]
- Jabbari, B.; Vaziri, N.D. The nature, consequences, and management of neurological disorders in chronic kidney disease. Hemodial. Int. 2018, 22, 150–160. [Google Scholar] [CrossRef]
- Ray, P.D.; Huang, B.W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef]
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants Maintain Cellular Redox Homeostasis by Elimination of Reactive Oxygen Species. Cell. Physiol. Biochem. 2017, 44, 532–553. [Google Scholar] [CrossRef]
- Smeyne, M.; Smeyne, R.J. Glutathione metabolism and Parkinson’s disease. Free Radic. Biol. Med. 2013, 62, 13–25. [Google Scholar] [CrossRef]
- Nezu, M.; Suzuki, N.; Yamamoto, M. Targeting the KEAP1-NRF2 System to Prevent Kidney Disease Progression. Am. J. Nephrol. 2017, 45, 473–483. [Google Scholar] [CrossRef]
- Tabassum, R.; Jeong, N.Y. Potential for therapeutic use of hydrogen sulfide in oxidative stress-induced neurodegenerative diseases. Int. J. Med. Sci. 2019, 16, 1386–1396. [Google Scholar] [CrossRef] [PubMed]
- Dugbartey, G.J. The smell of renal protection against chronic kidney disease: Hydrogen sulfide offers a potential stinky remedy. Pharmacol. Rep. 2018, 70, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Moldogazieva, N.T.; Mokhosoev, I.M.; Feldman, N.B.; Lutsenko, S.V. ROS and RNS signalling: Adaptive redox switches through oxidative/nitrosative protein modifications. Free Radic. Res. 2018, 52, 507–543. [Google Scholar] [CrossRef] [PubMed]
- Galvan, D.L.; Green, N.H.; Danesh, F.R. The hallmarks of mitochondrial dysfunction in chronic kidney disease. Kidney Int. 2017, 92, 1051–1057. [Google Scholar] [CrossRef]
- Yu, M.; Kim, Y.J.; Kang, D.H. Indoxyl sulfate-induced endothelial dysfunction in patients with chronic kidney disease via an induction of oxidative stress. Clin. J. Am. Soc. Nephrol. 2011, 6, 30–39. [Google Scholar] [CrossRef]
- Tbahriti, H.F.; Kaddous, A.; Bouchenak, M.; Mekki, K. Effect of different stages of chronic kidney disease and renal replacement therapies on oxidant-antioxidant balance in uremic patients. Biochem. Res. Int. 2013, 358985. [Google Scholar] [CrossRef]
- Kisic, B.; Miric, D.; Dragojevic, I.; Rasic, J.; Popovic, L. Role of Myeloperoxidase in Patients with Chronic Kidney Disease. Oxid. Med. Cell. Longev. 2016, 2016, 1069743. [Google Scholar] [CrossRef]
- Kimura, Y.; Kimura, H. Hydrogen sulfide protects neurons from oxidative stress. FASEB J. 2004, 18, 1165–1167. [Google Scholar] [CrossRef]
- Kim, H.J.; Vaziri, N.D. Contribution of impaired Nrf2-Keap1 pathway to oxidative stress and inflammation in chronic renal failure. Am. J. Physiol. Physiol. 2010, 298, F662–F671. [Google Scholar] [CrossRef]
- Leaf, D.E.; Body, S.C.; Muehlschlegel, J.D.; McMahon, G.M.; Lichtner, P.; Collard, C.D.; Shernan, S.K.; Fox, A.A.; Waikar, S.S. Length polymorphisms in heme oxygenase-1 and AKI after cardiac surgery. J. Am. Soc. Nephrol. 2016, 27, 3291–3297. [Google Scholar] [CrossRef]
- Vera, M.; Torramade-Moix, S.; Martin-Rodriguez, S.; Cases, A.; Cruzado, J.M.; Rivera, J.; Escolar, G.; Palomo, M.; Diaz-Ricart, M. Antioxidant and Anti-Inflammatory Strategies Based on the Potentiation of Glutathione Peroxidase Activity Prevent Endothelial Dysfunction in Chronic Kidney Disease. Cell Physiol. Biochem. 2018, 51, 1287–1300. [Google Scholar] [CrossRef] [PubMed]
- Pang, P.; Abbott, M.; Abdi, M.; Fucci, Q.A.; Chauhan, N.; Mistri, M.; Proctor, B.; Chin, M.; Wang, B.; Yin, W.; et al. Pre-clinical model of severe glutathione peroxidase-deficiency and chronic kidney disease results in coronary artery thrombosis and depressed left ventricular function. Nephrol. Dial. Transplant. 2018, 33, 923–934. [Google Scholar] [CrossRef] [PubMed]
- Alfieri, C.; Ruzhytska, O.; Vettoretti, S.; Caldiroli, L.; Cozzolino, M.; Messa, P. Native Hypovitaminosis D in CKD Patients: From Experimental Evidence to Clinical Practice. Nutrients 2019, 11, 1918. [Google Scholar] [CrossRef]
- Shao, M.; Wang, S.; Parameswaran, P.K. Hypoalbuminemia: A risk factor for acute kidney injury development and progression to chronic kidney disease in critically ill patients. Int. Urol. Nephrol. 2017, 49, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Escobedo-Monge, M.F.; Ayala-Macedo, G.; Sakihara, G.; Peralta, S.; Almaraz-Gómez, A.; Barrado, E.; Marugán-Miguelsanz, J.M. Effects of Zinc Supplementation on Nutritional Status in Children with Chronic Kidney Disease: A Randomized Trial. Nutrients 2019, 11, 2671. [Google Scholar] [CrossRef]
- Kimura, Y.; Goto, Y.; Kimura, H. Hydrogen sulfide increases glutathione production and suppresses oxidative stress in mitochondria. Antioxid. Redox Signal. 2010, 12, 1–13. [Google Scholar] [CrossRef]
- Tyagi, N.; Moshal, K.S.; Sen, U.; Vacek, T.P.; Kumar, M.; Hughes, W.M.; Kundu, S.; Tyagi, S.C. H2S protects against methionine-induced oxidative stress in brain endothelial cells. Antioxid. Redox Signal. 2009, 11, 25–33. [Google Scholar] [CrossRef]
- Schreier, S.M.; Muellner, M.K.; Steinkellner, H.; Hermann, M.; Esterbauer, H.; Exner, M.; Gmeiner, B.M.; Kapiotis, S.; Laggner, H. Hydrogen sulfide scavenges the cytotoxic lipid oxidation product 4-HNE. Neurotox Res. 2010, 17, 249–256. [Google Scholar] [CrossRef]
- Ishigami, M.; Hiraki, K.; Umemura, K.; Ogasawara, Y.; Ishii, K.; Kimura, H. A source of hydrogen sulfide and a mechanism of its release in the brain. Antioxid. Redox Signal. 2009, 11, 205–214. [Google Scholar] [CrossRef]
- Lee, Z.W.; Low, Y.L.; Huang, S.; Wang, T.; Deng, L.W. The cystathionine γ-lyase/hydrogen sulfide system maintains cellular glutathione status. Biochem. J. 2014, 460, 425–435. [Google Scholar] [CrossRef]
- Kimura, H. Physiological role of hydrogen sulfide and polysulfide in the central nervous system. Neurochem. Int. 2013, 63, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Hu, L.F.; Hu, G.; Bian, J.S. Hydrogen sulfide protects astrocytes against H2O2-induced neural injury via enhancing glutamate uptake. Free Radic. Biol. Med. 2008, 45, 1705–1713. [Google Scholar] [CrossRef] [PubMed]
- Orlowski, M.; Meister, A. The gamma-glutamyl cycle: A possible transport system for amino acids. Proc. Natl. Acad. Sci. USA 1970, 67, 1248–1255. [Google Scholar] [CrossRef] [PubMed]
- Gu, F.; Chauhan, V.; Chauhan, A. Glutathione redox imbalance in brain disorders. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 89–95. [Google Scholar] [CrossRef]
- Luo, H.; Wang, X.; Chen, C.; Wang, J.; Zou, X.; Li, C.; Xu, Z.; Yang, X.; Shi, W.; Zeng, C. Oxidative stress causes imbalance of renal renin angiotensin system (RAS) components and hypertension in obese Zucker rats. J. Am. Heart Assoc. 2015, 4, e001559. [Google Scholar] [CrossRef]
- Tsuruya, K.; Yoshida, H. Brain Atrophy and Cognitive Impairment in Chronic Kidney Disease. Contrib. Nephrol. 2018, 196, 27–36. [Google Scholar]
- Lin, Y.C.; Chang, Y.H.; Yang, S.Y.; Wu, K.D.; Chu, T.S. Update of pathophysiology and management of diabetic kidney disease. J. Formos. Med. Assoc. 2018, 117, 662–675. [Google Scholar]
- Santos, R.A.S.; Sampaio, W.O.; Alzamora, A.C.; Motta-Santos, D.; Alenina, N.; Bader, M.; Campagnole-Santos, M.J. The ACE2/Angiotensin-(1-7)/MAS Axis of the Renin-Angiotensin System: Focus on Angiotensin-(1-7). Physiol. Rev. 2018, 98, 505–553. [Google Scholar] [CrossRef]
- Kaur, P.; Muthuraman, A.; Kaur, M. The implications of angiotensin-converting enzymes and their modulators in neurodegenerative disorders: Current and future perspectives. ACS Chem. Neurosci. 2015, 6, 508–521. [Google Scholar] [CrossRef]
- Kim, S.; Iwao, H. Molecular and cellular mechanisms of angiotensin II-mediated cardiovascular and renal diseases. Pharmacol. Rev. 2000, 52, 11–34. [Google Scholar]
- Lopez-Real, A.; Rey, P.; Soto-Otero, R.; Mendez-Alvarez, E.; Labandeira-Garcia, J.L. Angiotensin-converting enzyme inhibition reduces oxidative stress and protects dopaminergic neurons in a 6-hydroxydopamine rat model of Parkinsonism. J. Neurosci. Res. 2005, 81, 865–873. [Google Scholar] [CrossRef]
- Villapol, S.; Saavedra, J.M. Neuroprotective effects of angiotensin receptor blockers. Am. J. Hypertens. 2015, 28, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.J.; Liu, Y.H.; Khin, E.S.; Bian, J.S. Vasoconstrictive effect of hydrogen sulfide involves downregulation of cAMP in vascular smooth muscle cells. Am. J. Physiol. Cell Physiol. 2008, 295, C1261–C1270. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Feng, X.; Xue, H.; Teng, X.; Jin, S.; Duan, X.; Xiao, L.; Wu, Y. Maternal renovascular hypertensive rats treatment with hydrogen sulfide increased the methylation of AT1b gene in offspring. Am. J. Hypertens. 2017, 30, 1220–1227. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Liu, Y.H.; Goh, H.S.; Wang, J.J.; Yong, Q.C.; Wang, R.; Bian, J.S. Hydrogen sulfide inhibits plasma renin activity. J. Am. Soc. Nephrol. 2010, 21, 993–1002. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.H.; Lu, M.; Xie, Z.Z.; Hua, F.; Xie, L.; Gao, J.H.; Koh, Y.H.; Bian, J.S. Hydrogen sulfide prevents heart failure development via inhibition of renin release from mast cells in isoproterenol-treated rats. Antioxid. Redox Signal. 2014, 20, 759–769. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Organ, C.L.; Kang, J.; Polhemus, D.J.; Trivedi, R.K.; Sharp, T.E.; Jenkins, J.S.; Tao, Y.X.; Xian, M.; Lefer, D.J. Hydrogen sulfide attenuates renin angiotensin and aldosterone pathological signaling to preserve kidney function and improve exercise tolerance in heart failure. JACC Basic Transl. Sci. 2018, 3, 796–809. [Google Scholar] [CrossRef]
- Tain, Y.L.; Hsu, C.N.; Lu, P.C. Early short-term treatment with exogenous hydrogen sulfide postpones the transition from prehypertension to hypertension in spontaneously hypertensive rat. Clin. Exp. Hypertens. 2018, 40, 58–64. [Google Scholar] [CrossRef]
- Zhou, X.; Feng, Y.; Zhan, Z.; Chen, J. Hydrogen sulfide alleviates diabetic nephropathy in a streptozotocin-induced diabetic rat model. J. Biol. Chem. 2014, 289, 28827–28834. [Google Scholar] [CrossRef]
- Karasavvidou, D.; Boutouyrie, P.; Kalaitzidis, R.; Kettab, H.; Pappas, K.; Stagikas, D.; Antonakis, N.; Tsalikakis, D.; Elisaf, M.; Laurent, S. Arterial damage and cognitive decline in chronic kidney disease patients. J. Clin. Hypertens. (Greenwich) 2018, 20, 1276–1284. [Google Scholar] [CrossRef]
- Martinez-Vea, A.; Salvadó, E.; Bardaji, A.; Gutierrez, C.; Ramos, A.; García, C.; Compte, T.; Peralta, C.; Broch, M.; Pastor, R.; et al. Silent cerebral white matter lesions and their relationship with vascular risk factors in middle-aged predialysis patients with CKD. Am. J. Kidney Dis. 2006, 47, 241–250. [Google Scholar] [CrossRef]
- Nakatani, T.; Naganuma, T.; Uchida, J.; Masuda, C.; Wada, S.; Sugimura, T.; Sugimura, K. Silent cerebral infarction in hemodialysis patients. Am. J. Nephrol. 2003, 23, 86–90. [Google Scholar] [CrossRef]
- Seliger, S.L.; Siscovick, D.S.; Stehman-Breen, C.O.; Gillen, D.L.; Fitzpatrick, A.; Bleyer, A.; Kuller, L.H. Moderate renal impairment and risk of dementia among older adults: The Cardiovascular Health Cognition Study. J. Am. Soc. Nephrol. 2004, 15, 1904–1911. [Google Scholar] [CrossRef] [PubMed]
- Moreira, J.M.; Bouissou Morais Soares, C.M.; Teixeira, A.L.; Simoes-e-Silva, A.C.; Kummer, A.M. Anxiety, depression, resilience and quality of life in children and adolescents with pre-dialysis chronic kidney disease. Pediatr. Nephrol. 2015, 30, 2153–2162. [Google Scholar] [CrossRef] [PubMed]
- Duron, E.; Hanon, O. Antihypertensive treatments, cognitive decline, and dementia. J. Alzheimers Dis. 2010, 20, 903–914. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, A.V.; Kiernan, M.C. Neurological complications of chronic kidney disease. Nat. Rev. Neurol. 2009, 5, 542–551. [Google Scholar] [CrossRef]
- De Deyn, P.P.; Vanholder, R.; Eloot, S.; Glorieux, G. Guanidino compounds as uremic (neuro) toxins. Semin. Dial. 2009, 22, 340–345. [Google Scholar] [CrossRef]
- Watanabe, K.; Watanabe, T.; Nakayama, M. Cerebro-renal interactions: Impact of uremic toxins on cognitive function. Neurotoxicology 2014, 44, 184–193. [Google Scholar] [CrossRef]
- De Deyn, P.P.; D’Hooge, R.; Van Bogaert, P.P.; Marescau, B. Endogenous guanidino compounds as uremic neurotoxins. Kidney Int. Suppl. 2001, 78, S77–S83. [Google Scholar] [CrossRef]
- Hénaut, L.; Grissi, M.; Brazier, F.; Assem, M.; Poirot-Leclercq, S.; Lenglet, G.; Boudot, C.; Avondo, C.; Boullier, A.; Choukroun, G.; et al. Cellular and molecular mechanisms associated with ischemic stroke severity in female mice with chronic kidney disease. Sci. Rep. 2019, 9, 6432. [Google Scholar] [CrossRef]
- Adachi, N.; Lei, B.; Deshpande, G.; Seyfried, F.J.; Shimizu, I.; Nagaro, T.; Arai, T. Uraemia suppresses central dopaminergic metabolism and impairs motor activity in rats. Intensive Care Med. 2001, 27, 1655–1660. [Google Scholar] [CrossRef] [PubMed]
- Perna, A.F.; Luciano, M.G.; Ingrosso, D.; Pulzella, P.; Sepe, I.; Lanza, D.; Violetti, E.; Capasso, R.; Lombardi, C.; De Santo, N.G. Hydrogen sulphide-generating pathways in haemodialysis patients: A study on relevant metabolites and transcriptional regulation of genes encoding for key enzymes. Nephrol. Dial. Transplant. 2009, 24, 3756–3763. [Google Scholar] [CrossRef] [PubMed]
- Perna, A.F.; Di Nunzio, A.; Amoresano, A.; Pane, F.; Fontanarosa, C.; Pucci, P.; Vigorito, C.; Cirillo, G.; Zacchia, M.; Trepiccione, F.; et al. Divergent behavior of hydrogen sulfide pools and of the sulfur metabolite lanthionine, a novel uremic toxin, in dialysis patients. Biochimie 2016, 126, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Lanza, D.; Perna, A.F.; Oliva, A.; Vanholder, R.; Pletinck, A.; Guastafierro, S.; Di Nunzio, A.; Vigorito, C.; Capasso, G.; Jankowski, V.; et al. Impact of the uremic milieu on the osteogenic potential of mesenchymal stem cells. PLoS ONE 2015, 10, e0116468. [Google Scholar] [CrossRef] [PubMed]
- Karmin, O.; Siow, Y.L. Metabolic Imbalance of Homocysteine and Hydrogen Sulfide in Kidney Disease. Curr. Med. Chem. 2018, 25, 367–377. [Google Scholar] [PubMed]
- Pushpakumar, S.; Kundu, S.; Sen, U. Hydrogen Sulfide Protects Hyperhomocysteinemia-Induced Renal Damage by Modulation of Caveolin and eNOS Interaction. Sci. Rep. 2019, 9, 2223. [Google Scholar] [CrossRef]
- Kielstein, H.; Suntharalingam, M.; Perthel, R.; Song, R.; Schneider, S.M.; Martens-Lobenhoffer, J.; Jäger, K.; Bode-Böger, S.M.; Kielstein, J.T. Role of the endogenous nitric oxide inhibitor asymmetric dimethylarginine (ADMA) and brain-derived neurotrophic factor (BDNF) in depression and behavioural changes: Clinical and preclinical data in chronic kidney disease. Nephrol. Dial. Transplant. 2015, 30, 1699–1705. [Google Scholar] [CrossRef]
- Monteggia, L.M.; Barrot, M.; Powell, C.M.; Berton, O.; Galanis, V.; Gemelli, T.; Meuth, S.; Nagy, A.; Greene, R.W.; Nestler, E.J. Essential role of brain-derived neurotrophic factor in adult hippocampal function. Proc. Natl. Acad. Sci. USA 2004, 101, 10827–10832. [Google Scholar] [CrossRef]
- Molendijk, M.L.; Spinhoven, P.; Polak, M.; Bus, B.A.A.; Penninx, B.W.J.H.; Elzinga, B.M. Serum BDNF concentrations as peripheral manifestations of depression: Evidence from a systematic review and meta-analyses on associations (N = 9484). Mol. Psychiatry 2014, 19, 791–800. [Google Scholar] [CrossRef]
- Gartner, A.; Staiger, V. Neurotrophin secretion from hippocampal neurons evoked by long-term-potentiation-inducing electrical stimulation patterns. Proc. Natl. Acad. Sci. USA 2002, 99, 6386–6391. [Google Scholar] [CrossRef]
- Karege, F.; Bondolfi, G.; Gervasoni, N.; Schwald, M.; Aubry, J.-M.; Bertschy, G. Low brain-derived neurotrophic factor (BDNF) levels in serum of depressed patients probably results from lowered platelet BDNF release unrelated to platelet reactivity. Biol. Psychiatry 2005, 57, 1068–1072. [Google Scholar] [CrossRef] [PubMed]
- Changli, W.; Armelloni, S.; Zennaro, C.; Wei, C.; Corbelli, A.; Ikehata, M.; Berra, S.; Giardino, L.; Mattinzoli, D.; Watanabe, S.; et al. BDNF repairs podocyte damage by microRNA-mediated increase of actin polymerization. J. Pathol. 2015, 235, 731–744. [Google Scholar]
- Endlich, N.; Lange, T.; Kuhn, J.; Klemm, P.; Kotb, A.M.; Siegerist, F.; Kindt, F.; Lindenmeyer, M.T.; Cohen, C.D.; Kuss, A.W.; et al. BDNF: mRNA expression in urine cells of patients with chronic kidney disease and its role in kidney function. J. Cell. Mol. Med. 2018, 22, 5265–5277. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Zou, W.; Wang, C.Y.; Chen, X.; Tan, H.Y.; Zeng, H.Y.; Zhang, P.; Gu, H.F.; Tang, X.Q. Hydrogen Sulfide Protects against Chronic Unpredictable Mild Stress-Induced Oxidative Stress in Hippocampus by Upregulation of BDNF-TrkB Pathway. Oxid. Med. Cell. Longev. 2016, 2153745. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.M.; Zhou, C.F.; Gao, S.L.; Tian, Y.; Wang, C.Y.; Wang, L.; Tang, X.Q. BDNF-TrkB pathway mediates neuroprotection of hydrogen sulfide against formaldehyde-induced toxicity to PC12 cells. PLoS ONE 2015, 10, e0119478. [Google Scholar] [CrossRef]
- Wei, H.J.; Xu, J.H.; Li, M.H.; Tang, J.P.; Zou, W.; Zhang, P.; Wang, L.; Wang, C.; Tang, X.Q. Hydrogen sulfide inhibits homocysteine-induced endoplasmic reticulum stress and neuronal apoptosis in rat hippocampus via upregulation of the BDNF-TrkB pathway. Acta Pharmacol. Sin. 2014, 35, 707–715. [Google Scholar] [CrossRef]
- Dugbartey, G.J. Diabetic nephropathy: A potential savior with ‘rotten-egg’ smell. Pharmacol. Rep. 2017, 69, 331–339. [Google Scholar] [CrossRef]
- Itani, H.; Liu, X.; Sarsour, E.H.; Goswami, P.C.; Born, E.; Keen, H.L.; Sigmund, C.D. Regulation of renin gene expression by oxidative stress. Hypertension 2009, 53, 1070–1076. [Google Scholar] [CrossRef]
- Wu, L.; Yang, W.; Jia, X.; Yang, G.; Duridanova, D.; Cao, K.; Wang, R. Pancreatic islet overproduction of H2S and suppressed insulin release in Zucker diabetic rats. Lab. Investig. 2009, 89, 59–67. [Google Scholar] [CrossRef]
- Yuan, P.; Xue, H.; Zhou, L.; Qu, L.; Li, C.; Wang, Z.; Ni, J.; Yu, C.; Yao, T.; Huang, Y.; et al. Rescue of mesangial cells from high glucose-induced over-proliferation and extracellular matrix secretion by hydrogen sulfide. Nephrol. Dial. Transplant. 2011, 26, 2119–2126. [Google Scholar] [CrossRef]
- Safar, M.M.; Abdelsalam, R.M. H2S donors attenuate diabetic nephropathy in rats: Modulation of oxidant status and polyol pathway. Pharmacol. Rep. 2015, 67, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Gorin, Y.; Cavaglieri, R.C.; Khazim, K.; Lee, D.Y.; Bruno, F.; Thakur, S.; Fanti, P.; Szyndralewiez, C.; Barnes, J.L.; Block, K.; et al. Targeting NADPH oxidase with a novel dual Nox1/Nox4 inhibitor attenuates renal pathology in type diabetes. Am. J. Physiol. Renal. Physiol. 2015, 308, F1276–F1287. [Google Scholar] [CrossRef]
- Kundu, S.; Pushpakumar, S.B.; Tyagi, A.; Coley, D.; Sen, U. Hydrogen sulfide deficiency and diabetic renal remodeling: Role of matrix metalloproteinase-9. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E1365–E1378. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Mariappan, M.M.; Feliers, D.; Cavaglieri, R.C.; Sataranatarajan, K.; Abboud, H.E.; Choudhury, G.G.; Kasinath, B.S. Hydrogen sulfide inhibits high glucose-induced matrix protein synthesis by activating AMP-activated protein kinase in renal epithelial cells. J. Biol. Chem. 2012, 287, 4451–4461. [Google Scholar] [CrossRef]
- Feliers, D.; Lee, H.J.; Kasinath, B.S. Hydrogen Sulfide in Renal Physiology and Disease. Antioxid. Redox Signal. 2016, 25, 720–731. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Peng, W.; Tao, J.; Lan, Z.; Hei, H.; Tian, L.; Pan, W.; Wang, L.; Zhang, X. Hydrogen Sulfide Inhibits Transforming Growth Factor-β1-Induced EMT via Wnt/Catenin Pathway. PLoS ONE 2016, 11, e0147018. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Li, W.; Chen, Q.; Jiang, Y.; Lu, X.; Zhao, X. Hydrogen sulfide accelerates wound healing in diabetic rats. Int. J. Clin. Exp. Pathol. 2015, 8, 5097–5104. [Google Scholar]
- Hou, C.L.; Wang, M.J.; Sun, C.; Huang, Y.; Jin, S.; Mu, X.P.; Chen, Y.; Zhu, Y.C. Protective Effects of Hydrogen Sulfide in the Ageing Kidney. Oxid. Med. Cell. Longev. 2016, 2016, 7570489. [Google Scholar] [CrossRef]
- Vitvitsky, V.; Thomas, M.; Ghorpade, A.; Gendelman, H.E.; Banerjee, R.A. functional transsulfuration pathway in the brain links to glutathione homeostasis. J. Biol. Chem. 2006, 281, 35785–35793. [Google Scholar] [CrossRef]
- Wang, R. Two’s company, three’s a crowd: Can H2S be the third endogenous gaseous transmitter? FASEB J. 2002, 16, 1792–1798. [Google Scholar] [CrossRef]
- Abe, K.; Kimura, H. The possible role of hydrogen sulfide as an endogenous neuromodulator. J. Neurosci. 1996, 16, 1066–1071. [Google Scholar] [CrossRef] [PubMed]
- Tu, F.P.; Li, J.X.; Li, Q.; Wang, J. Effects of hydrogen sulfide on cognitive dysfunction and NR2B in rats. J. Surg. Res. 2016, 205, 426–431. [Google Scholar] [CrossRef]
- Tang, Z.J.; Zou, W.; Yuan, J.; Zhang, P.; Tian, Y.; Xiao, Z.F.; Li, M.H.; Wei, H.J.; Tang, X.Q. Antidepressant-like and anxiolytic-like effects of hydrogen sulfide in streptozotocin-induced diabetic rats through inhibition of hippocampal oxidative stress. Behav. Pharmacol. 2015, 26, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.F.; Lu, M.; Tiong, C.X.; Dawe, G.S.; Hu, G.; Bian, J.S. Neuroprotective effects of hydrogen sulfide on Parkinson’s disease rat models. Aging Cell 2010, 9, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Vandiver, M.S.; Paul, B.D.; Xu, R.; Karuppagounder, S.; Rao, F.; Snowman, A.M.; Ko, H.S.; Lee, Y.I.; Dawson, V.L.; Dawson, T.M.; et al. Sulfhydration mediates neuroprotective actions of parkin. Nat. Commun. 2013, 4, 1626. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.Q.; Liu, X.Q.; Jiang, P.; Huang, H.; Yan, Y. Plasma levels of endogenous hydrogen sulfide and homocysteine in patients with Alzheimer’s disease and vascular dementia and the significance thereof. Zhonghua Yi Xue Za Zhi 2008, 88, 2246–2249. [Google Scholar]
- Kumar, M.; Ray, R.S.; Sandhir, R. Hydrogen sulfide attenuates homocysteine-induced neurotoxicity by preventing mitochondrial dysfunctions and oxidative damage: In vitro and in vivo studies. Neurochem. Int. 2018, 120, 87–98. [Google Scholar] [CrossRef]
- Yin, W.L.; Yin, W.G.; Huang, B.S.; Wu, L.X. Neuroprotective effects of lentivirus-mediated cystathionine-betasynthase overexpression against 6-OHDA-induced Parkinson’s disease rats. Neurosci. Lett. 2017, 657, 45–52. [Google Scholar] [CrossRef]
- Li, L.; Moore, P.K. Putative biological roles of hydrogen sulfide in health and disease: A breath of not so fresh air? Trends Pharmacol. Sci. 2008, 29, 84–90. [Google Scholar] [CrossRef]
- Halliwell, B. Biochemistry of oxidative stress. Biochem. Soc. Trans. 2007, 35, 1147–1150. [Google Scholar] [CrossRef]
- Amara, I.; Salah, A.; Timoumi, R.; Annabi, E.; Scuto, M.; Trovato, A.; Neffati, F.; Calabrese, V.; Abid-Essefi, S. Effect of di(2-ethylhexyl) phthalate on Nrf2-regulated glutathione homeostasis in mouse kidney. Cell Stress Chaperones. 2020. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Song, T.; Gu, Y.; Zhang, Y.; Cao, S.; Miao, Q.; Zhang, X.; Chen, H.; Gao, Y.; Zhang, L.; et al. Hydrogen sulfide alleviates liver injury via S-sulfhydrated-Keap1/Nrf2/LRP1 pathway. Hepatology 2020, 27. [Google Scholar] [CrossRef] [PubMed]
- Scuto, M.; Di Mauro, P.; Ontario, M.L.; Amato, C.; Modafferi, S.; Ciavardelli, D.; Trovato Salinaro, A.; Maiolino, L.; Calabrese, V. Nutritional Mushroom Treatment in Meniere’s Disease with Coriolus versicolor: A Rationale for Therapeutic Intervention in Neuroinflammation and Antineurodegeneration. Int. J. Mol. Sci. 2019, 21, 284. [Google Scholar] [CrossRef] [PubMed]
- Siracusa, R.; Scuto, M.; Fusco, R.; Trovato, A.; Ontario, M.L.; Crea, R.; Di Paola, R.; Cuzzocrea, S.; Calabrese, V. Anti-inflammatory and Anti-oxidant Activity of Hidrox® in Rotenone-Induced Parkinson’s Disease in Mice. Antioxidants 2020, 9, 824. [Google Scholar] [CrossRef]
- Trovato, A.; Siracusa, R.; Di Paola, R.; Scuto, M.; Ontario, M.L.; Bua, O.; Di Mauro, P.; Toscano, M.A.; Petralia, C.C.T.; Maiolino, L.; et al. Redox modulation of cellular stress response and lipoxin A4 expression by Hericium Erinaceus in rat brain: Relevance to Alzheimer’s disease pathogenesis. Immun. Ageing 2016, 9, 13–23. [Google Scholar] [CrossRef]
- Leak, R.K.; Calabrese, E.J.; Kozumbo, W.J.; Gidday, J.M.; Johnson, T.E.; Mitchell, J.R.; Ozaki, C.K.; Wetzker, R.; Bast, A.; Belz, R.G.; et al. Enhancing and Extending Biological Performance and Resilience. Dose Response 2018, 16, 1559325818784501. [Google Scholar] [CrossRef]
- Hine, C.; Harputlugil, E.; Zhang, Y.; Ruckenstuhl, C.; Lee, B.C.; Brace, L.; Longchamp, A.; Treviño-Villarreal, J.H.; Mejia, P.; Ozaki, C.K.; et al. Endogenous hydrogen sulfide production is essential for dietary restriction benefits. Cell 2015, 160, 132–144. [Google Scholar] [CrossRef]
- Andreadou, I.; Iliodromitis, E.K.; Rassaf, T.; Schulz, R.; Papapetropoulos, A.; Ferdinandy, P. The role of gasotransmitters NO, H2S and CO in myocardial ischaemia/reperfusion injury and cardioprotection by preconditioning, postconditioning and remote conditioning. Br. J. Pharmacol. 2015, 172, 1587–1606. [Google Scholar] [CrossRef]
- Ansari, M.; Kurian, G.A. Mechanism of Hydrogen Sulfide Preconditioning-Associated Protection Against Ischemia-Reperfusion Injury Differs in Diabetic Heart That Develops Myopathy. Cardiovasc. Toxicol. 2020, 20, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Hine, C.; Zhu, Y.; Hollenberg, A.N.; Mitchell, J.R. Dietary and Endocrine Regulation of Endogenous Hydrogen Sulfide Production: Implications for Longevity. Antioxid. Redox Signal. 2018, 28, 1483–1502. [Google Scholar] [CrossRef]
- Calvert, J.W.; Jha, S.; Gundewar, S.; Elrod, J.W.; Ramachandran, A.; Pattillo, C.B.; Kevil, C.G.; Lefer, D.J. Hydrogen sulfide mediates cardioprotection through Nrf2 signaling. Circ. Res. 2009, 105, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Peake, B.F.; Nicholson, C.K.; Lambert, J.P.; Hood, R.L.; Amin, H.; Amin, S.; Calvert, J.W. Hydrogen sulfide preconditions the db/db diabetic mouse heart against ischemia-reperfusion injury by activating Nrf2 signaling in an Erk-dependent manner. Am. J. Physiol. Heart. Circ. Physiol. 2013, 304, H1215–H1224. [Google Scholar] [CrossRef] [PubMed]
- Módis, K.; Ju, Y.; Ahmad, A.; Untereiner, A.A.; Altaany, Z.; Wu, L.; Szabo, C.; Wang, R. S-Sulfhydration of ATP synthase by hydrogen sulfide stimulates mitochondrial bioenergetics. Pharmacol. Res. 2016, 113, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Szabo, C.; Ransy, C.; Módis, K.; Andriamihaja, M.; Murghes, B.; Coletta, C.; Olah, G.; Yanagi, K.; Bouillaud, F. Regulation of mitochondrial bioenergetic function by hydrogen sulfide. Part, I. Biochemical and physiological mechanisms. Br. J. Pharmacol. 2014, 171, 2099–2122. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, E.J.; Mattson, M.P. Hormesis provides a generalized quantitative estimate of biological plasticity. J. Cell Commun. Signal. 2011, 5, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Hildebrandt, T.M.; Grieshaber, M.K. Three enzymatic activities catalyze the oxidation of sulfide to thiosulfate in mammalian and invertebrate mitochondria. FEBS J. 2008, 275, 3352–3361. [Google Scholar] [CrossRef]
- Cooper, C.E.; Brown, G.C. The inhibition of mitochondrial cytochrome oxidase by the gases carbon monoxide, nitric oxide, hydrogen cyanide and hydrogen sulfide: Chemical mechanism and physiological significance. J. Bioenerg. Biomembr. 2008, 40, 533–539. [Google Scholar] [CrossRef]
- Elrod, J.W.; Calvert, J.W.; Morrison, J.; Doeller, J.E.; Kraus, D.W.; Tao, L.; Jiao, X.; Scalia, R.; Kiss, L.; Szabo, C.; et al. Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proc. Natl. Acad. Sci. USA 2007, 104, 15560–15565. [Google Scholar] [CrossRef]
- Calvert, J.W.; Coetzee, W.A.; Lefer, D. Novel Insights into Hydrogen Sulfide Mediated Cytoprotection. Antioxid. Redox Signal. 2010, 12, 1203–1217. [Google Scholar] [CrossRef]
- Xue, H.; Yuan, P.; Ni, J.; Li, C.; Shao, D.; Liu, J.; Shen, Y.; Wang, Z.; Zhou, L.; Zhang, W.; et al. H2S inhibits hyperglycemia-induced intrarenal renin-angiotensin system activation via attenuation of reactive oxygen species generation. PLoS ONE 2013, 8, e74366. [Google Scholar] [CrossRef]
- Yang, R.; Liu, X.F.; Ma, S.F.; Gao, Q.; Li, Z.H.; Jia, Q. Protective effect of hydrogen sulfide on kidneys of type 1 diabetic rats. J. Appl. Physiol. 2016, 32, 181–184. [Google Scholar]
- Papu John, A.S.; Kundu, S.; Pushpakumar, S.; Amin, M.; Tyagi, S.C.; Sen, U. Hydrogen sulfide inhibits Ca2+-induced mitochondrial permeability transition pore opening in type 1-diabetes. Am. J. Physiol. Endocrinol. Metab. 2019, 317, E269–E283. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, V.; Santoro, A.; Trovato Salinaro, A.; Modafferi, S.; Scuto, M.; Albouchi, F.; Monti, D.; Giordano, J.; Zappia, M.; Franceschi, C.; et al. Hormetic approaches to the treatment of Parkinson’s disease: Perspectives and possibilities. J. Neurosci. Res. 2018, 96, 1641–1662. [Google Scholar] [CrossRef] [PubMed]
- Truong, D.H.; Eghbal, M.A.; Hindmarsh, W.; Roth O’Brien, P.J. Molecular mechanisms of hydrogen sulfide toxicity. Drug Metab Rev. 2006, 38, 733–744. [Google Scholar] [CrossRef]
- Giles, G.I.; Tasker, K.M.; Jacob, C. Hypothesis: The role of reactive sulfur species in oxidative stress. Free Radic. Biol. Med. 2001, 31, 1279–1283. [Google Scholar] [CrossRef]
- Olas, B.; Brodek, P.; Kontek, B. The Effect of Hydrogen Sulfide on Different Parameters of Human Plasma in the Presence or Absence of Exogenous Reactive Oxygen Species. Antioxidants 2018, 8, 610. [Google Scholar] [CrossRef]
- Bitar, M.S.; Nader, J.; Al-Ali, W.; Al Madhoun, A.; Arefanian, H.; Al-Mulla, F. Hydrogen Sulfide Donor NaHS Improves Metabolism and Reduces Muscle Atrophy in Type 2 Diabetes: Implication for Understanding Sarcopenic Pathophysiology. Oxid. Med. Cell. Longev. 2018, 2018, 6825452. [Google Scholar] [CrossRef]
- Paul, B.D.; Snyderm, S.H. H2S: A Novel gasotransmitter that signals by sulfhydration. Trends Biochem. Sci. 2015, 40, 687–700. [Google Scholar] [CrossRef]
- Yu, M.; Du, H.; Wang, B.; Chen, J.; Lu, F.; Peng, S.; Sun, Y.; Liu, N.; Sun, X.; Shiyun, D.; et al. Exogenous H2S Induces Hrd1 S-sulfhydration and Prevents CD36 Translocation via VAMP3 Ubiquitylation in Diabetic Hearts. Aging Dis. 2020, 11, 286–300. [Google Scholar] [CrossRef]
- Mimoun, S.; Andriamihaja, M.; Chaumontet, C.; Atanasiu, C.; Benamouzig, R.; Blouin, J.M.; Tomé, D.; Bouillaud, F.; Blachier, F. Detoxification of H2S by differentiated colonic epithelial cells: Implication of the sulfide oxidizing unit and of the cell respiratory capacity. Antioxid. Redox Signal. 2012, 17, 1–10. [Google Scholar] [CrossRef]
- Hourihan, J.M.; Kenna, J.G.; Hayes, J.D. The gasotransmitter hydrogen sulfide induces nrf2-target genes by inactivating the keap1 ubiquitin ligase substrate adaptor through formation of a disulfide bond between cys-226 and cys-613. Antioxid. Redox Signal. 2013, 19, 465–481. [Google Scholar] [CrossRef] [PubMed]
- Gambari, L.; Lisignoli, G.; Cattini, L.; Manferdini, C.; Facchini, A.; Grassi, F. Sodium hydrosulfide inhibits the differentiation of osteoclast progenitor cells via NRF2-dependent mechanism. Pharmacol. Res. 2014, 87, 99–112. [Google Scholar] [CrossRef]
- Cheng, Z.; Shen, X.; Jiang, X.; Shan, H.; Cimini, M.; Fang, P.; Ji, Y.; Park, J.Y.; Drosatos, K.; Yang, X.; et al. Hyperhomocysteinemia potentiates diabetes-impaired EDHF-induced vascular relaxation: Role of insufficient hydrogen sulfide. Redox Biol. 2018, 16, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Bełtowski, J.; Wójcicka, G.; Jamroz-Wiśniewska, A. Hydrogen sulfide in the regulation of insulin secretion and insulin sensitivity: Implications for the pathogenesis and treatment of diabetes mellitus. Biochem. Pharmacol. 2018, 149, 60–76. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Tian, Z.; Sun, Y.; Lu, C.; Liu, N.; Gao, Z.; Zhang, L.; Dong, S.; Yang, F.; Zhong, X.; et al. Exogenous H2S facilitating ubiquitin aggregates clearance via autophagy attenuates type 2 diabetes-induced cardiomyopathy. Cell Death Dis. 2017, 8, e2992. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; An, G.; Lu, X. Hydrogen sulfide attenuates the development of diabetic cardiomyopathy. Clin. Sci. 2015, 128, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Wu, Y.; Meng, M.; Luo, M.; Zhao, H.; Sun, H.; Gao, S. GYY4137 protects against myocardial ischemia/reperfusion injury via activation of the PHLPP-1/Akt/Nrf2 signaling pathway in diabetic mice. J. Surg. Res. 2018, 225, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yuan, Y.Q.; Zhang, L.; Zhang, H.; Zhang, S.W.; Zhang, Y.; Xuan, X.X.; Wang, M.J.; Zhang, J.Y. Exogenous hydrogen sulfide protects against high glucose-induced apoptosis and oxidative stress by inhibiting the STAT3/HIF-1α pathway in H9c2 cardiomyocytes. Exp. Ther. Med. 2019, 18, 3948–3958. [Google Scholar] [CrossRef]
- Wei, W.B.; Hu, X.; Zhuang, X.D.; Liao, L.Z.; Li, W.D. GYY4137, a novel hydrogen sulfide-releasing molecule, likely protects against high glucose-induced cytotoxicity by activation of the AMPK/mTOR signal pathway in H9c2 cells. Mol. Cell Biochem. 2014, 389, 249–256. [Google Scholar] [CrossRef]
- Wu, Z.; Peng, H.; Du, Q.; Lin, W.; Liu, Y. GYY4137, a hydrogen sulfide-releasing molecule, inhibits the inflammatory response by suppressing the activation of nuclear factor-kappa B and mitogen-activated protein kinases in Coxsackie virus B3-infected rat cardiomyocytes. Mol. Med. Rep. 2015, 11, 1837–1844. [Google Scholar] [CrossRef]
- Ye, P.; Gu, Y.; Zhu, Y.R.; Chao, Y.L.; Kong, X.Q.; Luo, J.; Ren, X.M.; Zuo, G.F.; Zhang, D.M.; Chen, S.L. Exogenous hydrogen sulfide attenuates the development of diabetic cardiomyopathy via the FoxO1 pathway. J. Cell Physiol. 2018, 233, 9786–9798. [Google Scholar] [CrossRef] [PubMed]
- Eleftheriadis, T.; Pissas, G.; Nikolaou, E.; Liakopoulos, V.; Stefanidis, I. The H2S-Nrf2-Antioxidant Proteins Axis Protects Renal Tubular Epithelial Cells of the Native Hibernator Syrian Hamster from Reoxygenation-Induced Cell Death. Biology 2019, 8, 74. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Koike, S.; Shibuya, N.; Lefer, D.; Ogasawara, Y.; Kimura, H. 3-Mercaptopyruvate sulfurtransferase produces potential redox regulators cysteine- and glutathione-persulfide (Cys-SSH and GSSH) together with signaling molecules H2S2, H2S3 and H2S. Sci. Rep. 2017, 7, 10459. [Google Scholar] [CrossRef] [PubMed]
- Stubbert, D.; Prysyazhna, O.; Rudyk, O.; Scotcher, J.; Burgoyne, J.R.; Eaton, P. Protein kinase G Ialpha oxidation paradoxically underlies blood pressure lowering by the reductant hydrogen sulfide. Hypertension 2014, 64, 1344–1351. [Google Scholar] [CrossRef]
- Shinkai, Y.; Abiko, Y.; Ida, T.; Miura, T.; Kakehashi, H.; Ishii, I.; Nishida, M.; Sawa, T.; Akaike, T.; Kumagai, Y. Reactive Sulfur Species-Mediated Activation of the Keap1-Nrf2 Pathway by 1,2-Naphthoquinone through Sulfenic Acids Formation under Oxidative Stress. Chem. Res. Toxicol. 2015, 28, 838–847. [Google Scholar] [CrossRef]
- Hatakeyama, Y.; Takahashi, K.; Tominaga, M.; Kimura, H.; Ohta, T. Polysulfide evokes acute pain through the activation of nociceptive TRPA1 in mouse sensory neurons. Mol. Pain. 2015, 11, 24. [Google Scholar] [CrossRef]
- Xie, L.; Gu, Y.; Wen, M.; Zhao, S.; Wang, W.; Ma, Y.; Meng, G.; Han, Y.; Wang, Y.; Liu, G.; et al. Hydrogen Sulfide Induces Keap1 S-sulfhydration and Suppresses Diabetes-Accelerated Atherosclerosis via Nrf2 Activation. Diabetes 2016, 65, 3171–3184. [Google Scholar] [CrossRef]
- Jiang, L.; Jiang, Q.; Yang, S.; Huang, S.; Han, X.; Duan, J.; Pan, S.; Zhao, M.; Guo, S. GYY4137 attenuates LPS-induced acute lung injury via heme oxygenase-1modulation. Pulm. Pharmacol. Ther. 2019, 54, 77–86. [Google Scholar] [CrossRef]
- Zheng, J.; Zhao, T.; Yuan, Y.; Hu, N.; Tang, X. Hydrogen sulfide (H2S) attenuates uranium-induced acute nephrotoxicity through oxidative stress and inflammatory response via Nrf2-NF-kappaB pathways. Chem. Biol. Interact. 2015, 242, 353–362. [Google Scholar] [CrossRef]
- Jha, S.; Calvert, J.W.; Duranski, M.R.; Ramachandran, A.; Lefer, D.J. Hydrogen sulfide attenuates hepatic ischemia-reperfusion injury: Role of antioxidant and antiapoptotic signaling. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H801–H806. [Google Scholar] [CrossRef]
- Trovato, A.; Siracusa, R.; Di Paola, R.; Scuto, M.; Fronte, V.; Koverech, G.; Luca, M.; Serra, A.; Toscano, M.A.; Petralia, A.; et al. Redox modulation of cellular stress response and lipoxin A4 expression by Coriolus versicolor in rat brain: Relevance to Alzheimer’s disease pathogenesis. Neurotoxicology 2016, 53, 350–358. [Google Scholar] [CrossRef]
- Trovato Salinaro, A.; Cornelius, C.; Koverech, G.; Koverech, A.; Scuto, M.; Lodato, F.; Fronte, V.; Muccilli, V.; Reibaldi, M.; Longo, A.; et al. Cellular stress response, redox status, and vitagenes in glaucoma: A systemic oxidant disorder linked to Alzheimer’s disease. Front. Pharmacol. 2014, 6, 5–129. [Google Scholar] [CrossRef]
- Calabrese, V.; Scapagnini, G.; Davinelli, S.; Koverech, G.; Koverech, A.; De Pasquale, C.; Salinaro, A.T.; Scuto, M.; Calabrese, E.J.; Genazzani, A.R. Sex hormonal regulation and hormesis in aging and longevity: Role of vitagenes. J. Cell Commun. Signal. 2014, 8, 369–384. [Google Scholar] [CrossRef] [PubMed]
- Houtkooper, R.H.; Pirinen, E.; Auwerx, J. Sirtuins as regulators of metabolism and healthspan. Nat. Rev. Mol. Cell Biol. 2012, 13, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Tissenbaum, H.A.; Guarente, L. Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature 2001, 410, 227–230. [Google Scholar] [CrossRef] [PubMed]
- Fujitsuka, N.; Asakawa, A.; Morinaga, A.; Amitani, M.S.; Amitani, H.; Katsuura, G.; Sawada, Y.; Sudo, Y.; Uezono, Y.; Mochiki, E.; et al. Increased ghrelin signaling prolongs survival in mouse models of human aging through activation of sirtuin1. Mol. Psychiatry 2016, 21, 1613–1623. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, H.H.; Taha, F.M.; Omar, H.S.; Elwi, H.M.; Abdelnasser, M. Hydrogen sulfide modulates SIRT1 and suppresses oxidative stress in diabetic nephropathy. Mol. Cell. Biochem. 2019, 457, 1–9. [Google Scholar] [CrossRef]
- Jiang, T.; Liu, Y.; Meng, Q.; Lv, X.; Yue, Z.; Ding, W.; Liu, T.; Cui, X. Hydrogen sulfide attenuates lung ischemia-reperfusion injury through SIRT3-dependent regulation of mitochondrial function in type 2 diabetic rats. Surgery 2019, 165, 1014–1026. [Google Scholar] [CrossRef]
- Du, C.; Lin, X.; Xu, W.; Zheng, F.; Cai, J.; Yang, J.; Cui, Q.; Tang, C.; Cai, J.; Xu, G.; et al. Sulfhydrated Sirtuin-1 Increasing Its Deacetylation Activity Is an Essential Epigenetics Mechanism of Anti-Atherogenesis by Hydrogen Sulfide. Antioxid. Redox Signal. 2019, 30, 84–197. [Google Scholar] [CrossRef]
- Guan, R.; Cai, Z.; Wang, J.; Ding, M.; Li, Z.; Xu, J.; Li, Y.; Li, J.; Yao, H.; Liu, W.; et al. Hydrogen sulfide attenuates mitochondrial dysfunction-induced cellular senescence and apoptosis in alveolar epithelial cells by upregulating sirtuin 1. Aging 2019, 11, 11844–11864. [Google Scholar] [CrossRef]
- Wu, L.; Chen, Y.; Wang, C.Y.; Tang, Y.Y.; Huang, H.L.; Kang, X.; Li, X.; Xie, Y.R.; Tang, X.Q. Hydrogen Sulfide Inhibits High Glucose-Induced Neuronal Senescence by Improving Autophagic Flux via Up-regulation of SIRT1. Front. Mol. Neurosci. 2019, 12, 194. [Google Scholar] [CrossRef] [PubMed]
- Purushotham, A.; Schug, T.T.; Xu, Q.; Surapureddi, S.; Guo, X.; Li, X. Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab. 2009, 9, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yu, J.; Chen, Y.; Liu, L.; Xu, M.; Sun, L.; Luo, H.; Wang, Y.; Meng, G. Exogenous Hydrogen Sulfide Supplement Attenuates Isoproterenol-Induced Myocardial Hypertrophy in a Sirtuin 3-Dependent Manner. Oxid. Med. Cell. Longev. 2018, 2018, 9396089. [Google Scholar] [CrossRef] [PubMed]
- Matsuzawa, A. Thioredoxin and redox signaling: Roles of the thioredoxin system in control of cell fate. Arch. Biochem. Biophys. 2017, 617, 101–105. [Google Scholar] [CrossRef]
- Amara, I.; Timoumi, R.; Annabi, E.; Di Rosa, G.; Scuto, M.; Najjar, M.F.; Calabrese, V.; Abid-Essefi, S. Di (2-ethylhexyl) phthalate targets the thioredoxin system and the oxidative branch of the pentose phosphate pathway in liver of Balb/c mice. Environ. Toxicol. 2020, 35, 78–86. [Google Scholar] [CrossRef]
- Mao, Z.; Huang, Y.; Zhang, Z.; Yang, X.; Zhang, X.; Huang, Y.; Sawada, N.; Mitsui, T.; Takeda, M.; Yao, J. Pharmacological levels of hydrogen sulfide inhibit oxidative cell injury through regulating the redox state of thioredoxin. Free Radic. Biol. Med. 2019, 134, 190–199. [Google Scholar] [CrossRef]
- Wedmann, R.; Onderka, C.; Wei, S.; Szijártó, I.A.; Miljkovic, J.L.; Mitrovic, A.; Lange, M.; Savitsky, S.; Yadav, P.K.; Torregrossa, R.; et al. Improved tag-switch method reveals that thioredoxin acts as depersulfidase and controls the intracellular levels of protein persulfidation. Chem. Sci. 2016, 7, 3414–3426. [Google Scholar] [CrossRef]
- Ju, Y.; Wu, L.; Yang, G. Thioredoxin regulation of protein S-desulfhydration. Biochem. Biophys. Rep. 2015, 5, 27–34. [Google Scholar] [CrossRef]
- Krishnan, N.; Fu, C.; Pappin, D.J.; Tonks, N.K. H2S-Induced sulfhydration of the phosphatase PTP1B and its role in the endoplasmic reticulum stress response. Sci. Signal. 2011, 4, ra86. [Google Scholar] [CrossRef]
- Ji, K.; Xue, L.; Cheng, J.; Bai, Y. Preconditioning of H2S inhalation protects against cerebral ischemia/reperfusion injury by induction of HSP70 through PI3K/Akt/Nrf2 pathway. Brain Res. Bull. 2016, 121, 68–74. [Google Scholar] [CrossRef]
- D’Araio, E.; Shaw, N.; Millward, A.; Demaine, A.; Whiteman, M.; Hodgkinson, A. Hydrogen sulfide induces heme oxygenase-in human kidney cells. Acta Diabetol. 2014, 51, 155–157. [Google Scholar] [CrossRef] [PubMed]
- Aziz, N.M.; Elbassuoni, E.A.; Kamel, M.Y.; Ahmed, S.M. Hydrogen sulfide renal protective effects: Possible link between hydrogen sulfide and endogenous carbon monoxide in a rat model of renal injury. Cell Stress Chaperones. 2020, 25, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Xie, K.; Chen, Y.; Wang, Y.; Wang, Y.; Lian, N.; Zhang, K.; Yu, Y. Nrf2/HO-1 signaling pathway participated in the protection of hydrogen sulfide on neuropathic pain in rats. Int. Immunopharmacol. 2019, 75, 105746. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Ye, M. Hydrogen Sulfide Attenuates High Glucose-induced Myocardial Injury in Rat Cardiomyocytes by Suppressing Wnt/beta-catenin Pathway. Curr. Med. Sci. 2019, 39, 938–946. [Google Scholar] [CrossRef]
- Du, Y.; Liu, X.H.; Zhu, H.C.; Wang, L.; Wang, Z.S.; Ning, J.Z.; Xiao, C.C. Hydrogen sulfide treatment protects against renal ischemia-reperfusion injury via induction of heat shock proteins in rats. Iran. J. Basic Med. Sci. 2019, 22, 99–105. [Google Scholar]
- Li, F.; Luo, J.; Wu, Z.; Xiao, T.; Zhang, J.; Yang, J. Effect of hydrogen sulfide on myocardial fibrosis and expression of PKCα and HSP70 in diabetic rats. ZhongNan Da Xue Xue Bao Yi Xue Ban 2015, 40, 1–5. [Google Scholar]
- Liu, M.; Li, Y.; Liang, B.; Li, Z.; Jiang, Z.; Chu, C.; Yang, J. Hydrogen sulphide attenuates myocardial fibrosis in diabetic rats through the JAK/STAT signalling pathway. Int. J. Mol. Med. 2018, 41, 1867–1876. [Google Scholar]
- Corsello, T.; Komaravelli, N.; Casola, A. Role of Hydrogen Sulfide in NRF2-and Sirtuin-Dependent Maintenance of Cellular Redox Balance. Antioxidants 2018, 7, 129. [Google Scholar] [CrossRef]
- Xie, Y.; Zhang, C.; Lai, D.; Sun, Y.; Samma, M.K.; Zhang, J.; Shen, W. Hydrogen sulfide delays GA-triggered programmed cell death in wheat aleurone layers by the modulation of glutathione homeostasis and heme oxygenase-expression. J. Plant Physiol. 2014, 171, 53–62. [Google Scholar] [CrossRef]
- Nagy, P.; Pálinkás, Z.; Nagy, A.; Budai, B.; Tóth, I.; Vasas, A. Chemical aspects of hydrogen sulfide measurements in physiological samples. Biochim. Biophys. Acta 2014, 1840, 876–891. [Google Scholar] [CrossRef]
- Sugahara, S.; Suzuki, M.; Kamiya, H.; Yamamuro, M.; Semura, H.; Senga, Y.; Egawa, M.; Seike, Y. Colorimetric Determination of Sulfide in Microsamples. Anal. Sci. 2016, 32, 1129–1131. [Google Scholar] [CrossRef]
- Furne, J.; Saeed, A.; Levitt, M.D. Whole tissue hydrogen sulfide concentrations are orders of magnitude lower than presently accepted values. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 295, R1479–1485. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.U.; Morris, G.F.; Hidiroglou, M. Rapid estimation of sulfide in rumen and blood with a sulfide-specific ion electrode. Microchem. J. 1980, 25, 388–395. [Google Scholar]
- Doeller, J.E.; Isbell, T.S.; Benavides, G.; Koenitzer, J.; Patel, H.; Patel, R.P.; Lancaster, J.R.; Darley-Usmar, V.; Kraus, D.W. Polarographic measurement of hydrogen sulfide production and consumption by mammalian tissues. Anal. Biochem. 2005, 341, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Karpus, J.; Kabil, O.; Zhang, S.-Y.; Zhu, H.-L.; Banerjee, R.; Zhao, J.; He, C. Selective fluorescent probes for live-cell monitoring of sulphide. Nat. Commun. 2011, 2, 495. [Google Scholar] [CrossRef]
- Wintner, E.; Deckwerth, T.L.; Langston, W.; Bengtsson, A.; Leviten, D.; Hill, P.; Insko, A.M.; Dumpit, R.; VandenEkart, E.; Toombs, C.F.; et al. A monobromobimane-based assay to measure the pharmacokinetic profile of reactive sulphide species in blood. Br. J. Pharmacol. 2010, 160, 941–957. [Google Scholar] [CrossRef]
- Shen, X.; Pattillo, C.B.; Pardue, S.; Bir, S.C.; Wang, R.; Kevil, C.G. Measurement of plasma hydrogen sulfide in vivo and in vitro. Free Radic. Biol. Med. 2011, 50, 1021–1031. [Google Scholar] [CrossRef]
- Shen, X.; Peter, E.A.; Bir, S.; Wang, R.; Kevil, C.G. Analytical measurement of discrete hydrogen sulfide pools in biological specimens. Free Radic. Biol. Med. 2012, 52, 2276–2283. [Google Scholar] [CrossRef]
- Hu, Q.; Wu, D.; Ma, F.; Yang, S.; Tan, B.; Xin, H.; Gu, X.; Chen, X.; Chen, S.; Mao, Y.; et al. Novel Angiogenic Activity and Molecular Mechanisms of ZYZ-803, a Slow-Releasing Hydrogen Sulfide-Nitric Oxide Hybrid Molecule. Antioxid. Redox Signal. 2016, 25, 498–514. [Google Scholar] [CrossRef]
- Togawa, T.; Ogawa, M.; Nawata, M.; Ogasawara, Y.; Kawanabe, K.; Tanabe, S. High performance liquid chromatographic determination of bound sulfide and sulfite and thiosulfate at their low levels in human serum by pre-column fluorescence derivatization with monobromobimane. Chem. Pharm. Bull. 1992, 40, 3000–3004. [Google Scholar] [CrossRef]
- Fahey, R.C.; Newton, G.L.; Dorian, R.; Kosower, E.M. Analysis of biological thiols: Quantitative determination of thiols at the picomole level based upon derivatization with monobromobimanes and separation by cation-exchange chromatography. Anal. Biochem. 1981, 111, 357–365. [Google Scholar] [CrossRef]
- Tan, B.; Jin, S.; Sun, J.; Gu, Z.; Sun, X.; Zhu, Y.; Huo, K.; Cao, Z.; Yang, P.; Xin, X.; et al. New method for quantification of gasotransmitter hydrogen sulfide in biological matrices by LC-MS/MS. Sci. Rep. 2017, 7, 46278. [Google Scholar] [CrossRef] [PubMed]
- Lan, L.; Wu, S.; Meng, X.-G.; Jiang, J.; Zheng, M.-Y.; Fan, G. A simple liquid chromatography tandem mass spectrometric method for fast detection of hydrogen sulfide based on thiolysis of 7-nitro-2,1,3-benzoxadiazole ether. J. Chromatogr. A 2020, 1625, 461243. [Google Scholar] [CrossRef] [PubMed]
- Arndt, S.; Baeza-Garza, C.D.; Logan, A.; Rosa, T.; Wedmann, R.; Prime, T.A.; Martin, J.L.; Saeb-Parsy, K.; Krieg, T.; Filipovic, M.R.; et al. Assessment of H2S in vivo using the newly developed mitochondria-targeted mass spectrometry probe MitoA. J. Biol. Chem. 2017, 292, 7761–7773. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Calabrese, V.; Scuto, M.; Salinaro, A.T.; Dionisio, G.; Modafferi, S.; Ontario, M.L.; Greco, V.; Sciuto, S.; Schmitt, C.P.; Calabrese, E.J.; et al. Hydrogen Sulfide and Carnosine: Modulation of Oxidative Stress and Inflammation in Kidney and Brain Axis. Antioxidants 2020, 9, 1303. https://doi.org/10.3390/antiox9121303
Calabrese V, Scuto M, Salinaro AT, Dionisio G, Modafferi S, Ontario ML, Greco V, Sciuto S, Schmitt CP, Calabrese EJ, et al. Hydrogen Sulfide and Carnosine: Modulation of Oxidative Stress and Inflammation in Kidney and Brain Axis. Antioxidants. 2020; 9(12):1303. https://doi.org/10.3390/antiox9121303
Chicago/Turabian StyleCalabrese, Vittorio, Maria Scuto, Angela Trovato Salinaro, Giuseppe Dionisio, Sergio Modafferi, Maria Laura Ontario, Valentina Greco, Sebastiano Sciuto, Claus Peter Schmitt, Edward J. Calabrese, and et al. 2020. "Hydrogen Sulfide and Carnosine: Modulation of Oxidative Stress and Inflammation in Kidney and Brain Axis" Antioxidants 9, no. 12: 1303. https://doi.org/10.3390/antiox9121303
APA StyleCalabrese, V., Scuto, M., Salinaro, A. T., Dionisio, G., Modafferi, S., Ontario, M. L., Greco, V., Sciuto, S., Schmitt, C. P., Calabrese, E. J., & Peters, V. (2020). Hydrogen Sulfide and Carnosine: Modulation of Oxidative Stress and Inflammation in Kidney and Brain Axis. Antioxidants, 9(12), 1303. https://doi.org/10.3390/antiox9121303