Molecular Mechanisms That Link Oxidative Stress, Inflammation, and Fibrosis in the Liver


Abstract
1. Introduction
2. Sources of ROS
3. Nitrosative Stress
4. Role of Free Radicals in Liver Fibrosis
4.1. Oxidative Stress in Hepatic Fibrosis
Attenuating Oxidative Stress Produces an Antifibrotic Effect
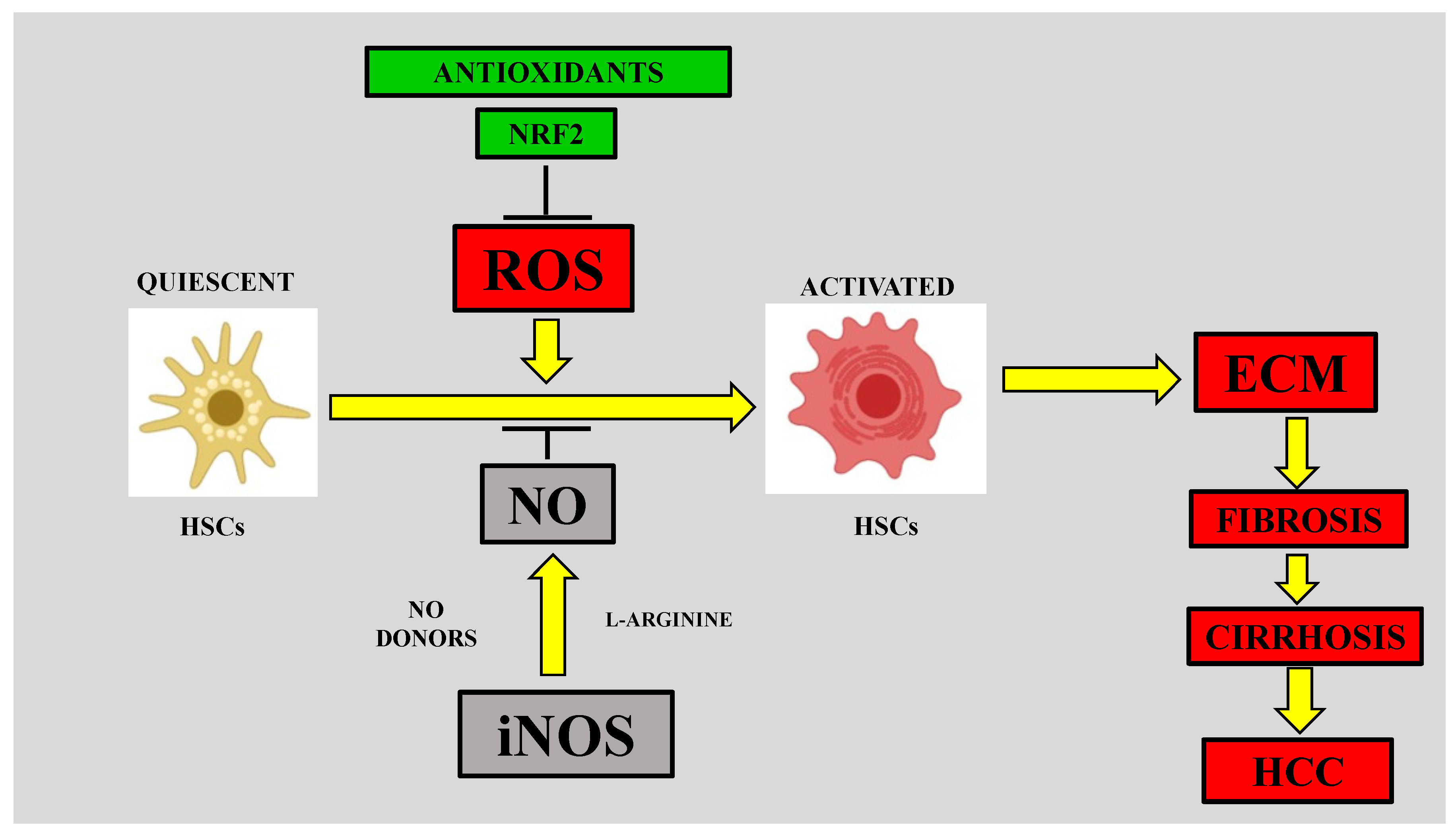
4.2. Role of Nitrosative Stress in Hepatic Fibrosis
4.2.1. Nitrosative Stress and HSC Activation
4.2.2. RNS in the Early Fibrotic Response
4.2.3. RNS in the Late Fibrotic Response
4.2.4. S-nitrosylation and the Liver
4.3. iNOS and Liver Fibrosis
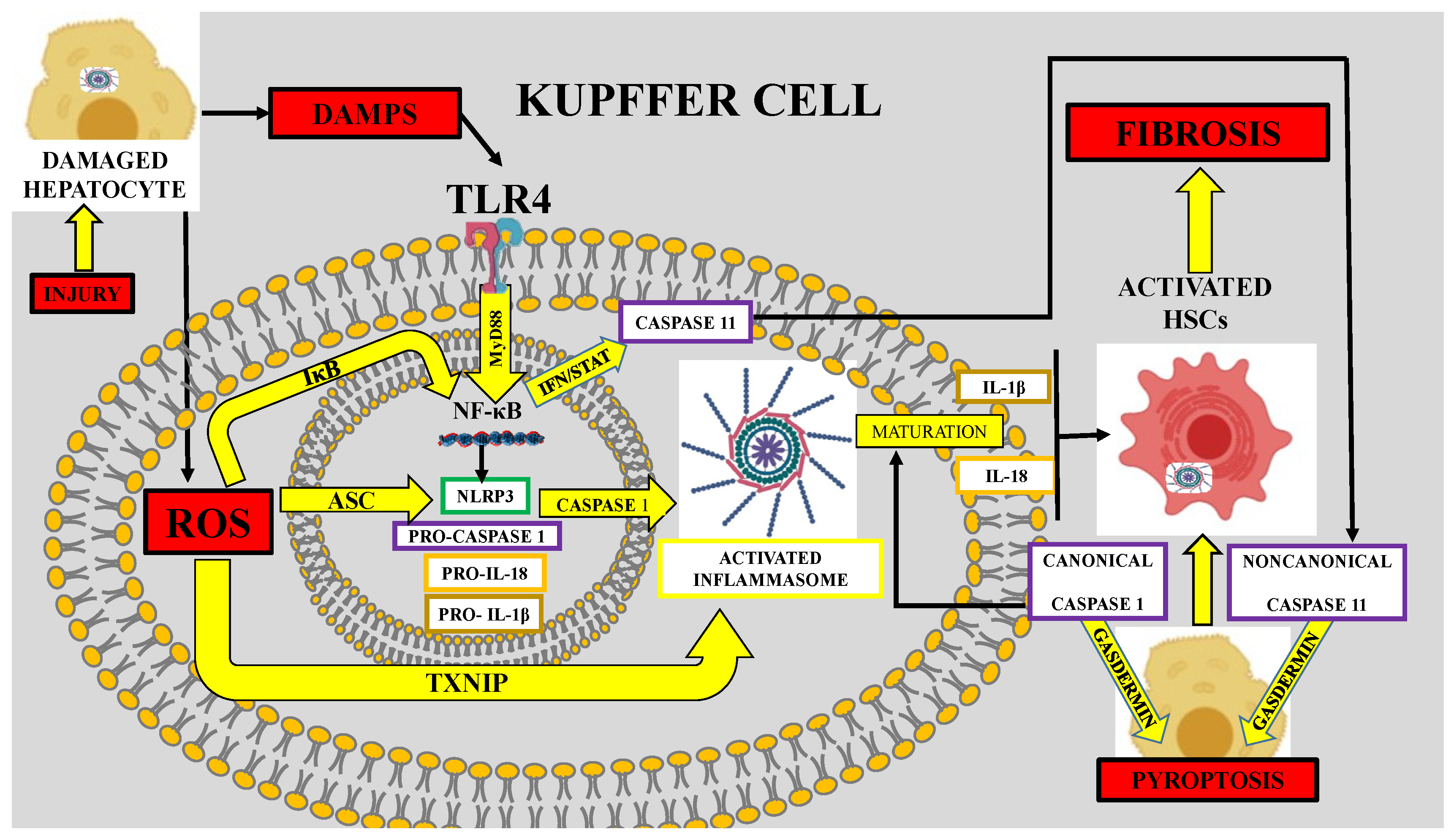
4.3.1. Role of Kupffer Cell iNOS in Liver Fibrosis
4.3.2. Role of Liver Sinusoidal Endothelial Cell (LSEC) iNOS in Liver Fibrosis
4.3.3. Role of HSC iNOS in Liver Fibrosis
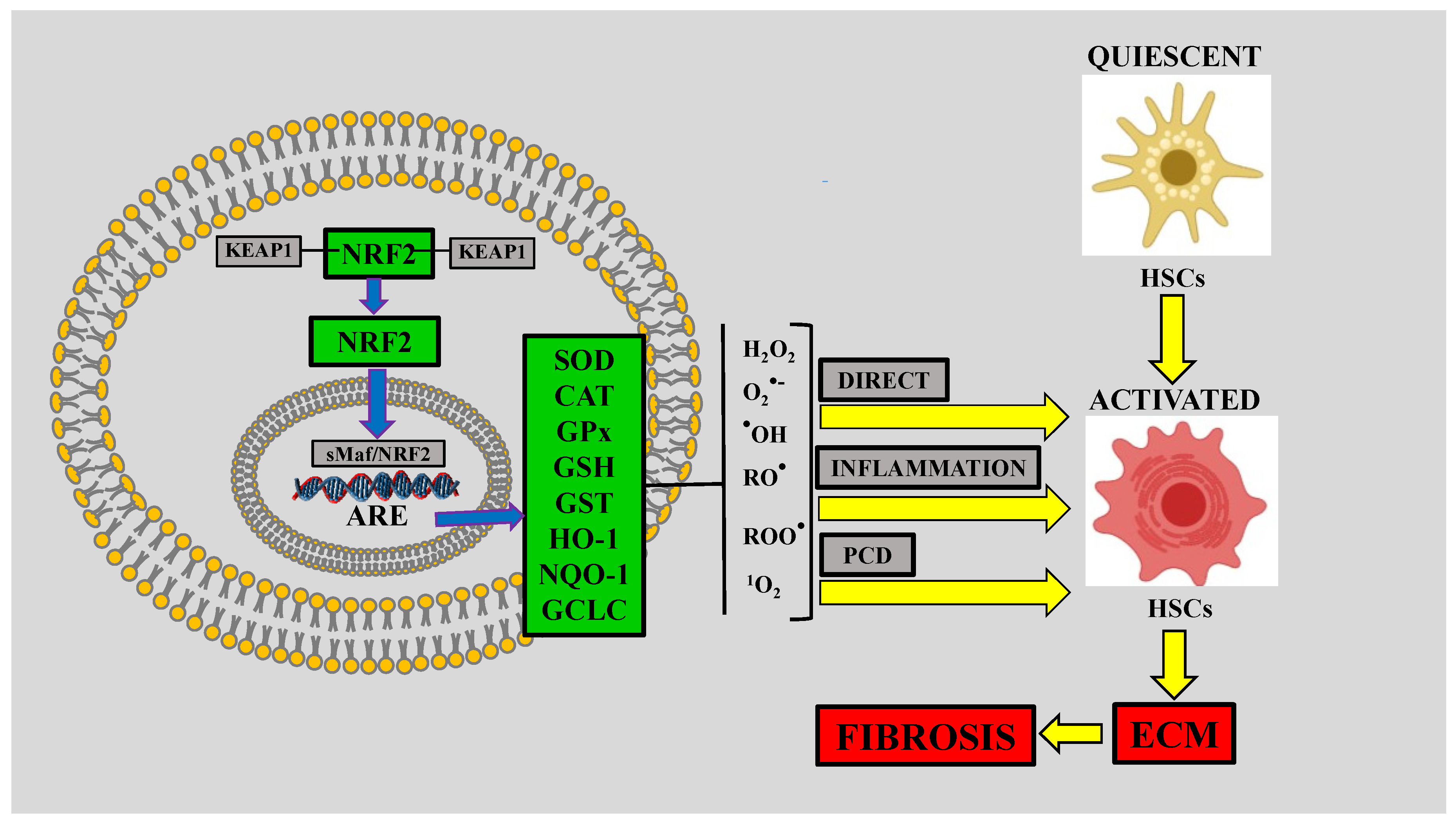
5. Nuclear Factor-E2-Related Factor-2 (NRF2) and Liver Fibrosis
5.1. NRF2
5.2. Role of NRF2 in Alcoholic Liver Disease (ALD)
5.3. NRF2 is Decreased in NASH with Fibrosis
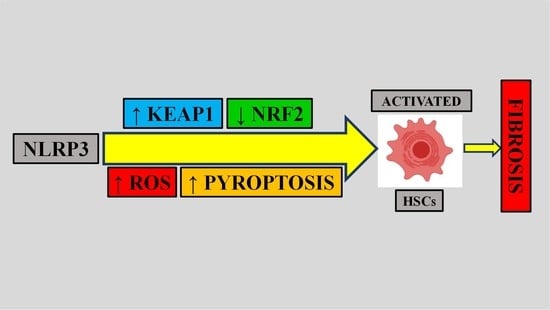
6. Role of Oxidative Stress in Inflammasome-Mediated Fibrosis
7. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Muriel, P. Role of free radicals in liver diseases. Hepatol. Int. 2009, 3, 526–536. [Google Scholar] [CrossRef]
- Casas-Grajales, S.; Muriel, P. The Liver, Oxidative Stress, and Antioxidants. In Liver Pathophysiology; Elsevier BV: Berlin, Germany, 2017; pp. 583–604. [Google Scholar]
- Li, S.; Tan, H.-Y.; Wang, N.; Zhang, Z.-J.; Lao, L.; Wong, C.-W.; Feng, Y. The Role of Oxidative Stress and Antioxidants in Liver Diseases. Int. J. Mol. Sci. 2015, 16, 26087–26124. [Google Scholar] [CrossRef]
- Ramos-Tovar, E.; Muriel, P. Free radicals, antioxidants, nuclear factor-E2-related factor-2 and liver damage. J. Appl. Toxicol. 2020, 40, 151–168. [Google Scholar] [CrossRef]
- Cichoż-Lach, H. Oxidative stress as a crucial factor in liver diseases. World J. Gastroenterol. 2014, 20, 8082–8091. [Google Scholar] [CrossRef]
- Giacco, F.; Brownlee, M. Oxidative Stress and Diabetic Complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef]
- Pham-Huy, L.A.; He, H.; Pham-Huy, C. Free Radicals, Antioxidants in Disease and Health. Int. J. Biomed. Sci. IJBS 2008, 4, 89–96. [Google Scholar]
- Valko, H.M.A.M.T.C.M.; Rhodes, C.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef]
- Brown, G.C.; Borutaite, V. There is no evidence that mitochondria are the main source of reactive oxygen species in mammalian cells. Mitochondrion 2012, 12, 1–4. [Google Scholar] [CrossRef]
- Inoue, M.; Sato, E.F.; Nishikawa, M.; Park, A.-M.; Kira, Y.; Imada, I.; Utsumi, K. Mitochondrial Generation of Reactive Oxygen Species and its Role in Aerobic Life. Curr. Med. Chem. 2003, 10, 2495–2505. [Google Scholar] [CrossRef]
- Lü, J.-M.; Lin, P.H.; Yao, Q.; Chen, C. Chemical and molecular mechanisms of antioxidants: Experimental approaches and model systems. J. Cell. Mol. Med. 2009, 14, 840–860. [Google Scholar] [CrossRef]
- Ushioda, R.; Nagata, K. Redox-Mediated Regulatory Mechanisms of Endoplasmic Reticulum Homeostasis. Cold Spring Harb. Perspect. Biol. 2019, 11, a033910. [Google Scholar] [CrossRef] [PubMed]
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef] [PubMed]
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed]
- Crosas-Molist, E.; Fabregat, I. Role of NADPH oxidases in the redox biology of liver fibrosis. Redox Biol. 2015, 6, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Altenhöfer, S.; Kleikers, P.W.M.; Radermacher, K.A.; Scheurer, P.; Hermans, J.J.R.; Schiffers, P.; Ho, H.; Wingler, K.; Schmidt, H.H.H.W. The NOX toolbox: Validating the role of NADPH oxidases in physiology and disease. Cell. Mol. Life Sci. 2012, 69, 2327–2343. [Google Scholar] [CrossRef]
- Cederbaum, A. Cytochrome P450 and Oxidative Stress in the Liver. In Liver Pathophysiology; Elsevier BV: Berlin, Germany, 2017; pp. 401–419. [Google Scholar]
- Guengerich, F.P. Cytochrome P450 and Chemical Toxicology. Chem. Res. Toxicol. 2008, 21, 70–83. [Google Scholar] [CrossRef]
- Veith, A.; Moorthy, B. Role of cytochrome P450s in the generation and metabolism of reactive oxygen species. Curr. Opin. Toxicol. 2018, 7, 44–51. [Google Scholar] [CrossRef]
- Lu, Y. Cytochrome P450S and Alcoholic Liver Disease. Curr. Pharm. Des. 2018, 24, 1502–1517. [Google Scholar] [CrossRef]
- Jerca, L.; Jerca, O.; Mancaş, G.; Constantinescu, I.; Lupuşoru, R. Mechanism of action and biochemical effects of nitric oxide (NO). J. Prev. Med. 2002, 10, 35–45. [Google Scholar]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric Oxide and Peroxynitrite in Health and Disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef]
- Abrams, G.A.; Trauner, M.; Nathanson, M.H. Nitric oxide and liver disease. Gastroenterologist 1995, 3, 220–233. [Google Scholar]
- Le, T.T.; Thuy, T.; Hai, H.; Kawada, N. Role of Oxidative and Nitrosative Stress in Hepatic Fibrosis. In Liver Pathophysiology; Elsevier BV: Berlin, Germany, 2017; pp. 213–224. [Google Scholar]
- Nordmann, R.; Ribière, C.; Rouach, H. Implication of free radical mechanisms in ethanol-induced cellular injury. Free. Radic. Biol. Med. 1992, 12, 219–240. [Google Scholar] [CrossRef]
- Nanji, A.A.; Zhao, S.; Sadrzadeh, S.M.H.; Dannenberg, A.J.; Tahan, S.R.; Waxman, D.J. Markedly Enhanced Cytochrome P450 2E1 Induction and Lipid Peroxidation Is Associated with Severe Liver Injury in Fish Oil-Ethanol-Fed Rats. Alcohol. Clin. Exp. Res. 1994, 18, 1280–1285. [Google Scholar] [CrossRef]
- Clot, P.; Tabone, M.; Arico, S.; Albano, E. Monitoring oxidative damage in patients with liver cirrhosis and different daily alcohol intake. Gut 1994, 35, 1637–1643. [Google Scholar] [CrossRef][Green Version]
- Aleynik, S.I.; Leo, M.A.; Aleynik, M.K.; Lieber, C.S. Increased circulating products of lipid peroxidation in patients with alcoholic liver disease. Alcohol. Clin. Exp. Res. 1998, 22, 192–196. [Google Scholar] [CrossRef]
- Meagher, E.A.; Barry, O.P.; Burke, A.; Lucey, M.R.; Lawson, J.A.; Rokach, J.; Fitzgerald, G.A. Alcohol-induced generation of lipid peroxidation products in humans. J. Clin. Investig. 1999, 104, 805–813. [Google Scholar] [CrossRef]
- Niemelä, O.; Parkkila, S.; Ylä-Herttuala, S.; Halsted, C.; Witztum, J.L.; Lanca, A.; Israel, Y. Covalent protein adducts in the liver as a result of ethanol metabolism and lipid peroxidation. Lab. Investig. 1994, 70, 537–546. [Google Scholar]
- Casini, A.; Ceni, E.; Salzano, R.; Biondi, P.; Parola, M.; Galli, A.; Foschi, M.; Caligiuri, A.; Pinzani, M.; Surrenti, C. Neutrophil-derived superoxide anion induces lipid peroxidation and stimulates collagen synthesis in human hepatic stellate cells: Role of nitric oxide. Hepatology 1997, 25, 361–367. [Google Scholar] [CrossRef]
- Nieto, N.; Friedman, S.L.; Cederbaum, A.I. Cytochrome P450 2E1-derived Reactive Oxygen Species Mediate Paracrine Stimulation of Collagen I Protein Synthesis by Hepatic Stellate Cells. J. Biol. Chem. 2002, 277, 9853–9864. [Google Scholar] [CrossRef]
- Paik, Y.-H.; Iwaisako, K.; Seki, E.; Inokuchi, S.; Schnabl, B.; Österreicher, C.H.; Kisseleva, T.; Brenner, D.A. The nicotinamide adenine dinucleotide phosphate oxidase (NOX) homologues NOX1 and NOX2/gp91phox mediate hepatic fibrosis in mice. Hepatology 2011, 53, 1730–1741. [Google Scholar] [CrossRef]
- Muriel, P. Nitric oxide protection of rat liver from lipid peroxidation, collagen accumulation, and liver damage induced by carbon tetrachloride. Biochem. Pharmacol. 1998, 56, 773–779. [Google Scholar] [CrossRef]
- Urtasun, R.; Cubero, F.J.; Vera, M.; Nieto, N. Reactive Nitrogen Species Switch on Early Extracellular Matrix Remodeling via Induction of MMP1 and TNFα. Gastroenterology 2009, 136, 1410–1422.e4. [Google Scholar] [CrossRef]
- Aram, G.; Potter, J.J.; Liu, X.; Torbenson, M.S.; Mezey, E. Lack of inducible nitric oxide synthase leads to increased hepatic apoptosis and decreased fibrosis in mice after chronic carbon tetrachloride administration. Hepatology 2008, 47, 2051–2058. [Google Scholar] [CrossRef]
- Ottesen, L.H.; Harry, D.; Frost, M.; Davies, S.; Khan, K.; Halliwell, B.; Moore, K. Increased formation of S-nitrothiols and nitrotyrosine in cirrhotic rats during endotoxemia. Free. Radic. Biol. Med. 2001, 31, 790–798. [Google Scholar] [CrossRef]
- Migita, K.; Maeda, Y.; Abiru, S.; Komori, A.; Yokoyama, T.; Takii, Y.; Nakamura, M.; Yatsuhashi, H.; Eguchi, K.; Ishibashi, H. Peroxynitrite-mediated matrix metalloproteinase-2 activation in human hepatic stellate cells. FEBS Lett. 2005, 579, 3119–3125. [Google Scholar] [CrossRef]
- Anavi, S.; Eisenberg-Bord, M.; Hahn-Obercyger, M.; Genin, O.; Pines, M.; Tirosh, O. The role of iNOS in cholesterol-induced liver fibrosis. Lab. Investig. 2015, 95, 914–924. [Google Scholar] [CrossRef]
- Liu, J.; Wu, K.C.; Lu, Y.-F.; Ekuase, E.; Klaassen, C.D. NRF2 Protection against Liver Injury Produced by Various Hepatotoxicants. Oxidative Med. Cell. Longev. 2013, 2013, 1–8. [Google Scholar] [CrossRef]
- Hernández-Gea, V.; Hilscher, M.; Rozenfeld, R.; Lim, M.P.; Nieto, N.; Werner, S.; Devi, L.A.; Friedman, S.L. Endoplasmic reticulum stress induces fibrogenic activity in hepatic stellate cells through autophagy. J. Hepatol. 2013, 59, 98–104. [Google Scholar] [CrossRef]
- Lamlé, J.; Marhenke, S.; Borlak, J.; Von Wasielewski, R.; Eriksson, C.P.; Geffers, R.; Manns, M.P.; Yamamoto, M.; Vogel, A. Nuclear Factor-Eythroid 2–Related Factor 2 Prevents Alcohol-Induced Fulminant Liver Injury. Gastroenterology 2008, 134, 1159–1168.e2. [Google Scholar] [CrossRef]
- Reichard, J.F.; Petersen, D.R. Involvement of phosphatidylinositol 3-kinase and extracellular-regulated kinase in hepatic stellate cell antioxidant response and myofibroblastic transdifferentiation. Arch. Biochem. Biophys. 2006, 446, 111–118. [Google Scholar] [CrossRef]
- Zarei, M.; Barroso, E.; Palomer, X.; Dai, J.; Rada, P.; Quesada-López, T.; Escolà-Gil, J.C.; Cedó, L.; Zali, M.R.; Molaei, M.; et al. Hepatic regulation of VLDL receptor by PPARβ/δ and FGF21 modulates non-alcoholic fatty liver disease. Mol. Metab. 2018, 8, 117–131. [Google Scholar] [CrossRef]
- Wang, C.; Cui, Y.; Li, C.; Zhang, Y.; Xu, S.; Li, X.; Li, H.; Zhang, X. Nrf2 deletion causes “benign” simple steatosis to develop into nonalcoholic steatohepatitis in mice fed a high-fat diet. Lipids Health Dis. 2013, 12, 165. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Xu, W.; Shao, J.; Zhang, F.; Chen, A.; Zheng, S. Nrf2 induces lipocyte phenotype via a SOCS3-dependent negative feedback loop on JAK2/STAT3 signaling in hepatic stellate cells. Int. Immunopharmacol. 2017, 49, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, A.; Sohail, M.A.; Gomes, D.A.; Hashmi, A.; Nagata, J.; Sutterwala, F.S.; Mahmood, S.; Jhandier, M.N.; Shi, Y.; Flavell, R.A.; et al. Inflammasome-mediated regulation of hepatic stellate cells. Am. J. Physiol. Liver Physiol. 2009, 296, G1248–G1257. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, M.D.; Kono, H.; Yin, M.; Rusyn, I.; Froh, M.; Connor, H.D.; Mason, R.P.; Samulski, R.; Thurman, R.G. Delivery of the Cu/Zn–Superoxide dismutase gene with adenovirus reduces early alcohol-induced liver injury in rats. Gastroenterology 2001, 120, 1241–1250. [Google Scholar] [CrossRef]
- Wheeler, M.D.; Nakagami, M.; Bradford, B.U.; Uesugi, T.; Mason, R.P.; Connor, H.D.; Dikalova, A.; Kadiiska, M.B.; Thurman, R.G. Overexpression of Manganese Superoxide Dismutase Prevents Alcohol-induced Liver Injury in the Rat. J. Biol. Chem. 2001, 276, 36664–36672. [Google Scholar] [CrossRef]
- Muriel, P. The Liver: General Aspects and Epidemiology. In Liver Pathophysiology: Therapies and Antioxidants; Muriel, P., Ed.; Elsevier: Waltham, MA, USA, 2017; pp. 3–22. [Google Scholar]
- Takaki, A.; Uchida, D.; Yamamoto, K. Redox Signaling in NASH. In Liver Pathophysiology; Elsevier BV: Berlin, Germany, 2017; pp. 169–180. [Google Scholar]
- Torok, N.J. Dysregulation of redox pathways in liver fibrosis. Am. J. Physiol. Liver Physiol. 2016, 311, G667–G674. [Google Scholar] [CrossRef]
- Chen, Z.; Tian, R.; She, Z.; Cai, J.; Li, H. Role of oxidative stress in the pathogenesis of nonalcoholic fatty liver disease. Free Radic. Biol. Med. 2020, 152, 116–141. [Google Scholar] [CrossRef]
- Nieto, N. Oxidative-stress and IL-6 mediate the fibrogenic effects of rodent Kupffer cells on stellate cells. Hepatology 2006, 44, 1487–1501. [Google Scholar] [CrossRef]
- Thuy, L.T.T.; Matsumoto, Y.; Van Thuy, T.T.; Hai, H.; Suoh, M.; Urahara, Y.; Motoyama, H.; Fujii, H.; Tamori, A.; Kubo, S.; et al. Cytoglobin Deficiency Promotes Liver Cancer Development from Hepatosteatosis through Activation of the Oxidative Stress Pathway. Am. J. Pathol. 2015, 185, 1045–1060. [Google Scholar] [CrossRef] [PubMed]
- Thuy, L.T.T.; Morita, T.; Yoshida, K.; Wakasa, K.; Iizuka, M.; Ogawa, T.; Mori, M.; Sekiya, Y.; Momen, S.; Motoyama, H.; et al. Promotion of Liver and Lung Tumorigenesis in DEN-Treated Cytoglobin-Deficient Mice. Am. J. Pathol. 2011, 179, 1050–1060. [Google Scholar] [CrossRef] [PubMed]
- Thuy, L.T.T.; Hai, H.; Kawada, N. Role of cytoglobin, a novel radical scavenger, in stellate cell activation and hepatic fibrosis. Clin. Mol. Hepatol. 2020, 26, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Arauz, J.; Rivera-Espinoza, Y.; Shibayama, M.; Favari, L.; Flores-Beltrán, R.E.; Muriel, P. Nicotinic acid prevents experimental liver fibrosis by attenuating the prooxidant process. Int. Immunopharmacol. 2015, 28, 244–251. [Google Scholar] [CrossRef]
- Arauz, J.; Zarco, N.; Segovia, J.; Shibayama, M.; Tsutsumi, V.; Muriel, P. Caffeine prevents experimental liver fibrosis by blocking the expression of TGF-β. Eur. J. Gastroenterol. Hepatol. 2014, 26, 164–173. [Google Scholar] [CrossRef]
- Casas-Grajales, S.; Reyes-Gordillo, K.; Cerda-García-Rojas, C.M.; Tsutsumi, V.; Lakshman, M.R.; Muriel, P. Rebaudioside A administration prevents experimental liver fibrosis: An in vivo and in vitro study of the mechanisms of action involved. J. Appl. Toxicol. 2019, 39, 1118–1131. [Google Scholar] [CrossRef]
- Galicia-Moreno, M.; Favari, L.; Muriel, P. Trolox mitigates fibrosis in a bile duct ligation model. Fundam. Clin. Pharmacol. 2011, 27, 308–318. [Google Scholar] [CrossRef]
- Aldaba-Muruato, L.R.; Moreno Gil, M.; Shibayama, M.; Tsutsumi, V.; Muriel, P. Protective effects of allopurinol against acute liver damage and cirrhosis induced by carbon tetrachloride: Modulation of NF-κB, cytokine production and oxidative stress. Biochim. Biophys. Acta 2012, 1820, 65–75. [Google Scholar] [CrossRef]
- Aldaba-Muruato, L.; Moreno, M.G.; Hernández-Mercado, E.; Shibayama, M.; Muriel, P. Secondary biliary cirrhosis in the rat is prevented by decreasing NF-κ B nuclear translocation and TGF-β expression using allopurinol, an inhibitor of xanthine oxidase. Can. J. Physiol. Pharmacol. 2012, 90, 1469–1478. [Google Scholar] [CrossRef]
- Hernández-Aquino, E.; Zarco, N.; Casas-Grajales, S.; Ramos-Tovar, E.; Flores-Beltrán, R.E.; Arauz, J.; Shibayama, M.; Favari, L.; Tsutsumi, V.; Segovia, J.; et al. Naringenin prevents experimental liver fibrosis by blocking TGFβ-Smad3 and JNK-Smad3 pathways. World J. Gastroenterol. 2017, 23, 4354–4368. [Google Scholar] [CrossRef]
- Casas-Grajales, S.; Vázquez-Flores, L.F.; Ramos-Tovar, E.; Hernández-Aquino, E.; Flores-Beltrán, R.E.; Cerda-García-Rojas, C.M.; Camacho, J.; Shibayama, M.; Tsutsumi, V.; Muriel, P. Quercetin reverses experimental cirrhosis by immunomodulation of the proinflammatory and profibrotic processes. Fundam. Clin. Pharmacol. 2017, 31, 610–624. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Tovar, E.; Hernández-Aquino, E.; Casas-Grajales, S.; Buendia-Montaño, L.D.; Galindo-Gómez, S.; Camacho, J.; Tsutsumi, V.; Muriel, P. Stevia Prevents Acute and Chronic Liver Injury Induced by Carbon Tetrachloride by Blocking Oxidative Stress through Nrf2 Upregulation. Oxidative Med. Cell. Longev. 2018, 2018, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Tovar, E.; Flores-Beltrán, R.E.; Galindo-Gómez, S.; Vera-Aguilar, E.; Diaz-Ruiz, A.; Montes, S.; Camacho, J.; Tsutsumi, V.; Muriel, P. Stevia rebaudiana tea prevents experimental cirrhosis via regulation of NF-κB, Nrf2, transforming growth factor beta, Smad7, and hepatic stellate cell activation. Phyther. Res. 2018, 32, 2568–2576. [Google Scholar] [CrossRef]
- Ramos-Tovar, E.; Buendia-Montaño, L.D.; Galindo-Gómez, S.; Hernández-Aquino, E.; Tsutsumi, V.; Muriel, P. Stevia prevents experimental cirrhosis by reducing hepatic myofibroblasts and modulating molecular profibrotic pathways. Hepatol. Res. 2019, 49, 212–223. [Google Scholar] [CrossRef] [PubMed]
- Casas-Grajales, S.; Alvarez-Suarez, D.; Ramos-Tovar, E.; Dayana Buendía-Montaño, L.; Reyes-Gordillo, K.; Camacho, J.; Tsutsumi, V.; Lakshman, M.R.R.; Muriel, P.; Casas-Grajales, S.; et al. Stevioside inhibits experimental fibrosis by down-regulating profibrotic Smad pathways and blocking hepatic stellate cell activation. Basic Clin. Pharmacol. Toxicol. 2019, 124, 670–680. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Tovar, E.; Casas-Grajales, S.; Hernández-Aquino, E.; Flores-Beltrán, R.E.; Galindo-Gómez, S.; Vera-Aguilar, E.; Diaz-Ruiz, A.; Montes, S.; Camacho, J.; Tsutsumi, V.; et al. Cirrhosis induced by thioacetamide is prevented by stevia. Molecular mechanisms. J. Funct. Foods 2019, 52, 552–564. [Google Scholar] [CrossRef]
- Casas-Grajales, S.; Ramos-Tovar, E.; Chávez-Estrada, E.; Alvarez-Suarez, D.; Hernández-Aquino, E.; Reyes-Gordillo, K.; Cerda-García-Rojas, C.M.; Camacho, J.; Tsutsumi, V.; Lakshman, M.R.; et al. Antioxidant and immunomodulatory activity induced by stevioside in liver damage: In vivo, in vitro and in silico assays. Life Sci. 2019, 224, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Mato, J.; Martínez-Chantar, M.; Noureddin, M.; Lu, S. One-Carbon Metabolism in Liver Health and Disease. In Liver Pathophysiology; Elsevier BV: Berlin, Germany, 2017; pp. 761–765. [Google Scholar]
- Lucena, M.I.; Andrade, R.J.; de la Cruz, J.P.; Rodriguez-Mendizabal, M.; Blanco, E.; de la Cuesta, F.S. Effects of silymarin MZ-80 on oxidative stress in patients with alcoholic cirrhosis. Int. J. Clin. Pharmacol. Ther. 2002, 40, 2–8. [Google Scholar] [CrossRef]
- Ferenci, P.; Dragosics, B.; Dittrich, H.; Frank, H.; Benda, L.; Lochs, H.; Meryn, S.; Base, W.; Schneider, B. Randomized controlled trial of silymarin treatment in patients with cirrhosis of the liver. J. Hepatol. 1989, 9, 105–113. [Google Scholar] [CrossRef]
- Vargas-Pozada, E.E.; Muriel, P. Herbal medicines for the liver. Eur. J. Gastroenterol. Hepatol. 2020, 32, 148–158. [Google Scholar] [CrossRef]
- Liu, J.; Garcia-Cardena, G.; Sessa, W.C. Biosynthesis and Palmitoylation of Endothelial Nitric Oxide Synthase: Mutagenesis of Palmitoylation Sites, Cysteines-15 and/or -26, Argues against Depalmitoylation-Induced Translocation of the Enzyme. Biochemistry 1995, 34, 12333–12340. [Google Scholar] [CrossRef] [PubMed]
- Alderton, W.K.; Cooper, C.E.; Knowles, R.G. Nitric oxide synthases: Structure, function and inhibition. Biochem. J. 2001, 357, 593. [Google Scholar] [CrossRef] [PubMed]
- Kolluru, G.K.; Siamwala, J.H.; Chatterjee, S. eNOS phosphorylation in health and disease. Biochimie 2010, 92, 1186–1198. [Google Scholar] [CrossRef] [PubMed]
- Ignarro, L.J. Haem-Dependent Activation of Guanylate Cyclase and Cyclic GMP Formation by Endogenous Nitric Oxide: A Unique Transduction Mechanism for Transcellular Signaling. Pharmacol. Toxicol. 1990, 67, 1–7. [Google Scholar] [CrossRef]
- Mayer, B.; Hemmens, B. Biosynthesis and action of nitric oxide in mammalian cells. Trends Biochem. Sci. 1997, 22, 477–481. [Google Scholar] [CrossRef]
- Stuehr, D.J. Mammalian nitric oxide synthases. Biochim. Biophys. Acta (BBA) Bioenerg. 1999, 1411, 217–230. [Google Scholar] [CrossRef]
- Clemens, M.G. Does altered regulation of ECNOS in sinusoidal endothelial cells determine increased intrahepatic resistance leading to portal hypertension? Hepatology 1998, 27, 1745–1747. [Google Scholar] [CrossRef]
- Lowenstein, C.J.; Glatt, C.S.; Bredt, D.S.; Snyder, S.H. Cloned and expressed macrophage nitric oxide synthase contrasts with the brain enzyme. Proc. Natl. Acad. Sci. USA 1992, 89, 6711–6715. [Google Scholar] [CrossRef]
- Rockey, D.C.; Chung, J.J. Regulation of inducible nitric oxide synthase in hepatic sinusoidal endothelial cells. Am. J. Physiol. Content 1996, 271, G260–G267. [Google Scholar] [CrossRef]
- Rockey, D.C.; Chung, J.J.; McKee, C.M.; Noble, P.W. Stimulation of inducible nitric oxide synthase in rat liver by hyaluronan fragments. Hepatology 1998, 27, 86–92. [Google Scholar] [CrossRef]
- Curran, R.D.; Billiar, T.R.; Stuehr, D.J.; Ochoa, J.B.; Harbrecht, B.G.; Flint, S.G.; Simmons, R.L. Multiple Cytokines Are Required to Induce Hepatocyte Nitric Oxide Production and Inhibit Total Protein Synthesis. Ann. Surg. 1990, 212, 462–471. [Google Scholar] [CrossRef] [PubMed]
- Geller, D.A.; Lowenstein, C.J.; Shapiro, R.A.; Nussler, A.K.; Di Silvio, M.; Wang, S.C.; Nakayama, D.K.; Simmons, R.L.; Snyder, S.H.; Billiar, T.R. Molecular cloning and expression of inducible nitric oxide synthase from human hepatocytes. Proc. Natl. Acad. Sci. USA 1993, 90, 3491–3495. [Google Scholar] [CrossRef] [PubMed]
- Helyar, L.; Bundschuh, D.S.; Laskin, J.D.; Laskin, D.L. Induction of hepatic ito cell nitric oxide production after acute endotoxemia. Hepatology 1994, 20, 1509–1515. [Google Scholar] [CrossRef] [PubMed]
- Loughran, P.; Xu, L.; Billiar, T. Nitric Oxide and the Liver. In Liver Pathophysiology; Elsevier BV: Berlin, Germany, 2017; pp. 799–816. [Google Scholar]
- Ridnour, L.A.; Thomas, D.D.; Mancardi, D.; Espey, M.G.; Miranda, K.M.; Paolocci, N.; Feelisch, M.; Fukuto, J.; Wink, D.A. The chemistry of nitrosative stress induced by nitric oxide and reactive nitrogen oxide species. Putting perspective on stressful biological situations. Biol. Chem. 2004, 385, 1–10. [Google Scholar] [CrossRef]
- Squadrito, G.L.; Pryor, W.A. Oxidative chemistry of nitric oxide: The roles of superoxide, peroxynitrite, and carbon dioxide. Free. Radic. Biol. Med. 1998, 25, 392–403. [Google Scholar] [CrossRef]
- Dröge, W. Free Radicals in the Physiological Control of Cell Function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef]
- Rockey, D.C.; Chung, J.J. Inducible nitric oxide synthase in rat hepatic lipocytes and the effect of nitric oxide on lipocyte contractility. J. Clin. Investig. 1995, 95, 1199–1206. [Google Scholar] [CrossRef]
- Bauer, M.; Bauer, I.; Sonin, N.V.; Kresge, N.; Baveja, R.; Yokoyama, Y.; Harding, D.; Zhang, J.X.; Clemens, M.G. Functional significance of endothelin B receptors in mediating sinusoidal and extrasinusoidal effects of endothelins in the intact rat liver. Hepatology 2000, 31, 937–947. [Google Scholar] [CrossRef]
- Zhang, J.X.; Pegoli, W.; Clemens, M.G. Endothelin-1 induces direct constriction of hepatic sinusoids. Am. J. Physiol. Content 1994, 266, G624–G632. [Google Scholar] [CrossRef]
- Tan, A.S.; Berridge, M.V. Superoxide produced by activated neutrophils efficiently reduces the tetrazolium salt, WST-1 to produce a soluble formazan: A simple colorimetric assay for measuring respiratory burst activation and for screening anti-inflammatory agents. J. Immunol. Methods 2000, 238, 59–68. [Google Scholar] [CrossRef]
- Harbrecht, B.G.; Wu, B.; Watkins, S.C.; Billiar, T.R.; Peitzman, A.B. Inhibition of nitric oxide synthesis during severe shock but not after resuscitation increases hepatic injury and neutrophil accumulation in hemorrhaged rats. Shock 1997, 8, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Fukatsu, K.; Saito, H.; Han, I.; Furukawa, S.; Lin, M.-T.; Matsuda, T.; Ikeda, S.; Inoue, T.; Yasuhara, H.; Muto, T. Nitric Oxide Donor Decreases Neutrophil Adhesion in both Lung and Peritoneum during Peritonitis. J. Surg. Res. 1998, 74, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Bataller, R.; Ginès, P.; Nicolás, J.M.; Görbig, M.N.; Garcia–Ramallo, E.; Gasull, X.; Bosch, J.; Arroyo, V.; Rodés, J. Angiotensin II induces contraction and proliferation of human hepatic stellate cells. Gastroenterology 2000, 118, 1149–1156. [Google Scholar] [CrossRef]
- Failli, P.; DeFranco, R.M.; Caligiuri, A.; Gentilini, A.; Romanelli, R.G.; Marra, F.; Batignani, G.; Guerra, C.T.; Laffi, G.; Gentilini, P.; et al. Nitrovasodilators inhibit platelet-derived growth factor-induced proliferation and migration of activated human hepatic stellate cells. Gastroenterology 2000, 119, 479–492. [Google Scholar] [CrossRef] [PubMed]
- Svegliati-Baroni, G.; Saccomanno, S.; Van Goor, H.; Jansen, P.; Benedetti, A.; Moshage, H. Involvement of reactive oxygen species and nitric oxide radicals in activation and proliferation of rat hepatic stellate cells. Liver Int. 2001, 21, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ferrini, M.; Vernet, D.; Magee, T.; Shahed, A.; Qian, A.; Rajfer, J.; Gonzalez-Cadavid, N.F. Antifibrotic Role of Inducible Nitric Oxide Synthase. Nitric Oxide 2002, 6, 283–294. [Google Scholar] [CrossRef]
- Witte, M.H.; Borgs, P.; Way, D.L.; Ramirez, G.; Bernas, M.J.; Witte, C.L. Alcohol, hepatic sinusoidal microcirculation, and chronic liver disease. Alcohol 1992, 9, 473–480. [Google Scholar] [CrossRef]
- Schwentker, A.; Vodovotz, Y.; Weller, R.; Billiar, T.R. Nitric oxide and wound repair: Role of cytokines? Nitric Oxide 2002, 7, 1–10. [Google Scholar] [CrossRef]
- Chu, A.; Prasad, J. Up-regulation by human recombinant transforming growth factor β-1 of collagen production in cultured dermal fibroblasts is mediated by the inhibition of nitric oxide signaling. J. Am. Coll. Surg. 1999, 188, 271–280. [Google Scholar] [CrossRef]
- Kolpakov, V.; Gordon, D.; Kulik, T.J. Nitric Oxide–Generating Compounds Inhibit Total Protein and Collagen Synthesis in Cultured Vascular Smooth Muscle Cells. Circ. Res. 1995, 76, 305–309. [Google Scholar] [CrossRef]
- Cao, M.; Westerhausen-Larson, A.; Niyibizi, C.; Kavalkovich, K.; Georgescu, H.I.; Rizzo, C.F.; Hebda, P.A.; Stefanovic-Racic, M.; Evans, C.H. Nitric oxide inhibits the synthesis of type-II collagen without altering Col2A1 mRNA abundance: Prolyl hydroxylase as a possible target. Biochem. J. 1997, 324, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Genovese, T.; Cuzzocrea, S.; Di Paola, R.; Failla, M.; Mazzon, E.; Sortino, M.A.; Frasca, G.; Gili, E.; Crimi, N.; Caputi, A.P.; et al. Inhibition or knock out of Inducible nitric oxide synthase result in resistance to bleomycin-induced lung injury. Respir. Res. 2005, 6, 58. [Google Scholar] [CrossRef] [PubMed]
- Thornton, F.J.; Schäffer, M.R.; Witte, M.B.; Moldawer, L.L.; Mackay, S.L.; Abouhamze, A.; Tannahill, C.L.; Barbul, A. Enhanced Collagen Accumulation Following Direct Transfection of the Inducible Nitric Oxide Synthase Gene in Cutaneous Wounds. Biochem. Biophys. Res. Commun. 1998, 246, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Koruk, M.H.; Ülya, A.H.; Akçay, F.; Onuk, M.D. Antioxidant capacity and nitric oxide in patients with hepatic cirrhosis. Ann. Clin. Lab. Sci. 2002, 32, 252–256. [Google Scholar] [PubMed]
- Koshy, A.; De Gottardi, A.; Ledermann, M.; Saegesser, H.; Shaw, S.G.; Zimmermann, A.; Reichen, N. Endothelial nitric oxide synthase is not essential for the development of fibrosis and portal hypertension in bile duct ligated mice. Liver Int. 2005, 25, 1044–1052. [Google Scholar] [CrossRef]
- Moreno, M.G.; Muriel, P. Inducible nitric oxide synthase is not essential for the development of fibrosis and liver damage induced by CCl4 in mice. J. Appl. Toxicol. 2006, 26, 326–332. [Google Scholar] [CrossRef]
- Fernando, V.; Zheng, X.; Walia, Y.; Sharma, V.; Letson, J.; Furuta, S. S-Nitrosylation: An Emerging Paradigm of Redox Signaling. Antioxidants 2019, 8, 404. [Google Scholar] [CrossRef]
- Hess, D.T.; Matsumoto, A.; Kim, S.-O.; Marshall, H.E.; Stamler, J.S. Protein S-nitrosylation: Purview and parameters. Nat. Rev. Mol. Cell Biol. 2005, 6, 150–166. [Google Scholar] [CrossRef]
- Selvakumar, B.; Jenkins, M.A.; Hussain, N.K.; Huganir, R.L.; Traynelis, S.F.; Snyder, S.H. S-nitrosylation of AMPA receptor GluA1 regulates phosphorylation, single-channel conductance, and endocytosis. Proc. Natl. Acad. Sci. USA 2012, 110, 1077–1082. [Google Scholar] [CrossRef]
- Martínez-Ruiz, A. S-nitrosylation: A potential new paradigm in signal transduction. Cardiovasc. Res. 2004, 62, 43–52. [Google Scholar] [CrossRef]
- Sun, J.; Steenbergen, C.; Murphy, E. S -Nitrosylation: NO-Related Redox Signaling to Protect Against Oxidative Stress. Antioxid. Redox Signal. 2006, 8, 1693–1705. [Google Scholar] [CrossRef]
- Takahara, T.; Furui, K.; Funaki, J.; Nakayama, Y.; Itoh, H.; Miyabayashi, C.; Sato, H.; Seiki, M.; Ooshima, A.; Watanabe, A. Increased expression of matrix metalloproteinase-II in experimental liver fibrosis in rats. Hepatology 1995, 21, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Arthur, M.J.P. Fibrogenesis II. Metalloproteinases and their inhibitors in liver fibrosis. Am. J. Physiol. Liver Physiol. 2000, 279, G245–G249. [Google Scholar] [CrossRef] [PubMed]
- Iwakiri, Y.; Kim, M.Y. Nitric oxide in liver diseases. Trends Pharmacol. Sci. 2015, 36, 524–536. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Checa, J.C. Alcohol-induced liver disease: When fat and oxidative stress meet. Ann. Hepatol. 2004, 2, 69–75. [Google Scholar] [CrossRef]
- Bai, J.; Cederbaum, A.I. Overexpression of CYP2E1 in Mitochondria Sensitizes HepG2 Cells to the Toxicity Caused by Depletion of Glutathione. J. Biol. Chem. 2005, 281, 5128–5136. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Ruiz, C.; Fernandez-Checa, J.C. Mitochondrial glutathione: Hepatocellular survival–death switch. J. Gastroenterol. Hepatol. 2006, 21, S3–S6. [Google Scholar] [CrossRef] [PubMed]
- Braet, F.; Wisse, E. Structural and functional aspects of liver sinusoidal endothelial cell fenestrae: A review. Comp. Hepatol. 2002, 1, 1. [Google Scholar] [CrossRef]
- Xie, G.; Choi, S.S.; Syn, W.-K.; Michelotti, G.A.; Swiderska, M.; Karaca, G.; Chan, I.S.; Chen, Y.; Diehl, A.M. Hedgehog signalling regulates liver sinusoidal endothelial cell capillarisation. Gut 2013, 62, 299–309. [Google Scholar] [CrossRef]
- Lee, Y.A.; Wallace, M.C.; Friedman, S.L. Pathobiology of liver fibrosis: A translational success story. Gut 2015, 64, 830–841. [Google Scholar] [CrossRef]
- Miyao, M.; Kotani, H.; Ishida, T.; Kawai, C.; Manabe, S.; Abiru, H.; Tamaki, K. Pivotal role of liver sinusoidal endothelial cells in NAFLD/NASH progression. Lab. Investig. 2015, 95, 1130–1144. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Wang, X.; Wang, L.; Wang, L.; Atkinson, R.D.; Kanel, G.C.; Gaarde, W.A.; Deleve, L.D. Role of Differentiation of Liver Sinusoidal Endothelial Cells in Progression and Regression of Hepatic Fibrosis in Rats. Gastroenterology 2012, 142, 918–927.e6. [Google Scholar] [CrossRef] [PubMed]
- Ruart, M.; Chavarria, L.; Campreciós, G.; Suárez-Herrera, N.; Montironi, C.; Guixé-Muntet, S.; Bosch, J.; Friedman, S.L.; Garcia-Pagán, J.C.; Hernández-Gea, V. Impaired endothelial autophagy promotes liver fibrosis by aggravating the oxidative stress response during acute liver injury. J. Hepatol. 2019, 70, 458–469. [Google Scholar] [CrossRef] [PubMed]
- Witek, R.P.; Yang, L.; Liu, R.; Jung, Y.; Omenetti, A.; Syn, W.; Choi, S.S.; Cheong, Y.; Fearing, C.M.; Agboola, K.M.; et al. Liver Cell–Derived Microparticles Activate Hedgehog Signaling and Alter Gene Expression in Hepatic Endothelial Cells. Gastroenterology 2009, 136, 320–330.e2. [Google Scholar] [CrossRef]
- Iwakiri, Y. Nitric oxide in liver fibrosis: The role of inducible nitric oxide synthase. Clin. Mol. Hepatol. 2015, 21, 319–325. [Google Scholar] [CrossRef]
- La Mura, V.; Pasarín, M.; Rodriguez-Vilarrupla, A.; García-Pagán, J.C.; Bosch, J.; Abraldes, J.G. Liver sinusoidal endothelial dysfunction after LPS administration: A role for inducible-nitric oxide synthase. J. Hepatol. 2014, 61, 1321–1327. [Google Scholar] [CrossRef]
- Schwabe, R.F.; Wells, R.G. Origin and Function of Myofibroblasts in the Liver. Semin. Liver Dis. 2015, 35, 097–106. [Google Scholar] [CrossRef]
- Wells, R.G. The Portal Fibroblast: Not Just a Poor Man’s Stellate Cell. Gastroenterology 2014, 147, 41–47. [Google Scholar] [CrossRef]
- Li, H.; Mittal, A.; Makonchuk, D.Y.; Bhatnagar, S.; Kumar, A. Matrix metalloproteinase-9 inhibition ameliorates pathogenesis and improves skeletal muscle regeneration in muscular dystrophy. Hum. Mol. Genet. 2009, 18, 2584–2598. [Google Scholar] [CrossRef]
- Maher, J.M.; Cheng, X.; Slitt, A.L.; Dieter, M.Z.; Klaassen, C.D. Induction of the multidrug resistance-associated protein family of transporters by chemical activators of receptor-mediated pathways in mouse liver. Drug Metab. Dispos. 2005, 33, 956–962. [Google Scholar] [CrossRef]
- Kobayashi, M.; Yamamoto, M. Molecular Mechanisms Activating the Nrf2-Keap1 Pathway of Antioxidant Gene Regulation. Antioxid. Redox Signal. 2005, 7, 385–394. [Google Scholar] [CrossRef]
- Kim, K.M.; Ki, S.H. Nrf2: A Key Regulator of Redox Signaling in Liver Diseases. In Liver Pathophysiology; Muriel, P., Ed.; Academic Press: Cambridge, MA, USA, 2017; pp. 355–374. [Google Scholar]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Motohashi, H.; Yamamoto, M. Nrf2–Keap1 defines a physiologically important stress response mechanism. Trends Mol. Med. 2004, 10, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.-L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef] [PubMed]
- Hirotsu, Y.; Katsuoka, F.; Funayama, R.; Nagashima, T.; Nishida, Y.; Nakayama, K.; Engel, J.D.; Yamamoto, M. Nrf2–MafG heterodimers contribute globally to antioxidant and metabolic networks. Nucleic Acids Res. 2012, 40, 10228–10239. [Google Scholar] [CrossRef] [PubMed]
- Itoha, K.; Chibabc, T.; Takahashia, S.; Ishiia, T.; Igarashia, K.; Katoha, Y.; Oyaked, T.; Hayashid, N.; Satohe, K.; Hatayamae, I.; et al. An Nrf2/Small Maf Heterodimer Mediates the Induction of Phase II Detoxifying Enzyme Genes through Antioxidant Response Elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef]
- Nguyen, T.; Sherratt, P.J.; Nioi, P.; Yang, C.S.; Pickett, C.B. Nrf2 Controls Constitutive and Inducible Expression of ARE-driven Genes through a Dynamic Pathway Involving Nucleocytoplasmic Shuttling by Keap1. J. Biol. Chem. 2005, 280, 32485–32492. [Google Scholar] [CrossRef]
- Wu, D.; Cederbaum, A.I. Oxidative Stress and Alcoholic Liver Disease. Semin. Liver Dis. 2009, 29, 141–154. [Google Scholar] [CrossRef]
- Shin, S.M.; Yang, J.H.; Ki, S.H. Role of the Nrf2-ARE Pathway in Liver Diseases. Oxidative Med. Cell. Longev. 2013, 2013, 1–9. [Google Scholar] [CrossRef]
- Sun, J.; Fu, J.; Li, L.; Chen, C.; Wang, H.; Hou, Y.; Xu, Y.; Pi, J. Nrf2 in alcoholic liver disease. Toxicol. Appl. Pharmacol. 2018, 357, 62–69. [Google Scholar] [CrossRef]
- Wu, K.C.; Liu, J.; Klaassen, C.D. Role of Nrf2 in preventing ethanol-induced oxidative stress and lipid accumulation. Toxicol. Appl. Pharmacol. 2012, 262, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zhuge, J.; Wang, X.; Bai, J.; Cederbaum, A.I. Cytochrome P450 2E1 contributes to ethanol-induced fatty liver in mice. Hepatology 2008, 47, 1483–1494. [Google Scholar] [CrossRef] [PubMed]
- Cho, I.J.; Kim, J.W.; Jung, J.J.; Sung, S.H.; Kim, J.; Lee, N.; Ku, S.K. Hepatoprotective Effects of Hoveniae Semen Cum Fructus Extracts in ethanol intoxicated mice. J. Exerc. Nutr. Biochem. 2016, 20, 50–65. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.-K.; Han, J.-M.; Kim, H.-G.; Lee, J.-S.; Lee, J.-S.; Wang, J.-H.; Son, S.-W.; Park, H.-J.; Son, C.-G. Aqueous extract of Artemisia capillaris exerts hepatoprotective action in alcohol–pyrazole-fed rat model. J. Ethnopharmacol. 2013, 147, 662–670. [Google Scholar] [CrossRef]
- Gao, Y.; Chu, S.-F.; Xia, C.-Y.; Zhang, Z.; Zhang, S.; Chen, N.-H. Rg1 Attenuates alcoholic hepatic damage through regulating AMP-activated protein kinase and nuclear factor erythroid 2-related factor 2 signal pathways. J. Asian Nat. Prod. Res. 2016, 18, 765–778. [Google Scholar] [CrossRef]
- Li, Y.-P. Caffeic acid phenethyl ester inhibits liver fibrosis in rats. World J. Gastroenterol. 2015, 21, 3893–3903. [Google Scholar] [CrossRef]
- Mani, V.; Arivalagan, S.; Siddique, A.I.; Nalini, N. Antioxidant and anti-inflammatory role of zingerone in ethanol-induced hepatotoxicity. Mol. Cell. Biochem. 2016, 421, 169–181. [Google Scholar] [CrossRef]
- Rabelo, A.C.S.; Lúcio, K.D.P.; Araujo, C.M.; De Araujo, G.R.; Miranda, P.H.D.A.; Carneiro, A.C.A.; Ribeiro, E.M.D.C.; Silva, B.D.M.; De Lima, W.G.; Costa, D.C. Baccharis trimera protects against ethanol induced hepatotoxicity in vitro and in vivo. J. Ethnopharmacol. 2018, 215, 1–13. [Google Scholar] [CrossRef]
- Xu, D.; Xu, M.; Jeong, S.; Qian, Y.; Wu, H.; Xia, Q.; Kong, X. The Role of Nrf2 in Liver Disease: Novel Molecular Mechanisms and Therapeutic Approaches. Front. Pharmacol. 2019, 9, 1428. [Google Scholar] [CrossRef]
- Zhou, D.; Lin, Z.; Huang, C.; Xie, D.; Zhan, S.; Kong, L.; Wang, Y.; Li, J. Cytochrome P4502E1 inhibitor, a potential oxidative stress regulator in liver diseases. Hepatology Res. 2014, 44, 591–592. [Google Scholar] [CrossRef]
- Chambel, S.S.; Santos-Gonçalves, A.; Duarte, T.L. The Dual Role of Nrf2 in Nonalcoholic Fatty Liver Disease: Regulation of Antioxidant Defenses and Hepatic Lipid Metabolism. BioMed Res. Int. 2015, 2015, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Chowdhry, S.; Nazmy, M.H.; Meakin, P.J.; Dinkova-Kostova, A.T.; Walsh, S.V.; Tsujita, T.; Dillon, J.F.; Ashford, M.L.J.; Hayes, J.D. Loss of Nrf2 markedly exacerbates nonalcoholic steatohepatitis. Free. Radic. Biol. Med. 2010, 48, 357–371. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, H.; Okada, K.; Shoda, J.; Warabi, E.; Ishige, K.; Ueda, T.; Taguchi, K.; Yanagawa, T.; Nakahara, A.; Hyodo, I.; et al. Deletion of nuclear factor-E2-related factor-2 leads to rapid onset and progression of nutritional steatohepatitis in mice. Am. J. Physiol. Liver Physiol. 2010, 298, G283–G294. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-K.J.; Yeager, R.L.; Tanaka, Y.; Klaassen, C.D. Enhanced expression of Nrf2 in mice attenuates the fatty liver produced by a methionine- and choline-deficient diet☆. Toxicol. Appl. Pharmacol. 2010, 245, 326–334. [Google Scholar] [CrossRef]
- Okada, K.; Warabi, E.; Sugimoto, H.; Horie, M.; Gotoh, N.; Tokushige, K.; Hashimoto, E.; Utsunomiya, H.; Takahashi, H.; Ishii, T.; et al. Deletion of Nrf2 leads to rapid progression of steatohepatitis in mice fed atherogenic plus high-fat diet. J. Gastroenterol. 2013, 48, 620–632. [Google Scholar] [CrossRef]
- Prestigiacomo, V.; Suter-Dick, L. Nrf2 protects stellate cells from Smad-dependent cell activation. PLoS ONE 2018, 13, e0201044. [Google Scholar] [CrossRef]
- Yates, M.S.; Tauchi, M.; Katsuoka, F.; Flanders, K.C.; Liby, K.T.; Honda, T.; Gribble, G.W.; Johnson, D.A.; Johnson, J.A.; Burton, N.C.; et al. Pharmacodynamic characterization of chemopreventive triterpenoids as exceptionally potent inducers of Nrf2-regulated genes. Mol. Cancer Ther. 2007, 6, 154–162. [Google Scholar] [CrossRef]
- Yates, M.S.; Tran, Q.T.; Dolan, P.M.; Osburn, W.O.; Shin, S.; McCulloch, C.C.; Silkworth, J.B.; Taguchi, K.; Yamamoto, M.; Williams, C.R.; et al. Genetic versus chemoprotective activation of Nrf2 signaling: Overlapping yet distinct gene expression profiles between Keap1 knockout and triterpenoid-treated mice. Carcinogenesis 2009, 30, 1024–1031. [Google Scholar] [CrossRef]
- Saha, P.K.; Reddy, V.T.; Konopleva, M.; Andreeff, M.; Chan, L. The Triterpenoid 2-Cyano-3,12-dioxooleana-1,9-dien-28-oic-acid Methyl Ester Has Potent Anti-diabetic Effects in Diet-induced Diabetic Mice and Lepr db/db Mice. J. Biol. Chem. 2010, 285, 40581–40592. [Google Scholar] [CrossRef]
- Jeong, W.-S.; Keum, Y.-S.; Chen, C.; Jain, M.R.; Shen, G.; Kim, J.H.; Li, W.; Kong, A.-N.T. Differential Expression and Stability of Endogenous Nuclear Factor E2-related Factor 2 (Nrf2) by Natural Chemopreventive Compounds in HepG2 Human Hepatoma Cells. J. Biochem. Mol. Biol. 2005, 38, 167–176. [Google Scholar] [CrossRef]
- Oh, C.J.; Kim, J.-Y.; Min, A.-K.; Park, K.-G.; Harris, R.A.; Kim, H.-J.; Lee, I.K. Sulforaphane attenuates hepatic fibrosis via NF-E2-related factor 2-mediated inhibition of transforming growth factor-β/Smad signaling. Free. Radic. Biol. Med. 2012, 52, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Knorr, J.; Wree, A.; Tacke, F.; Feldstein, A.E. The NLRP3 Inflammasome in Alcoholic and Nonalcoholic Steatohepatitis. Semin. Liver Dis. 2020, 1, 1–9. [Google Scholar] [CrossRef]
- Biswas, S.K. Does the Interdependence between Oxidative Stress and Inflammation Explain the Antioxidant Paradox? Oxidative Med. Cell. Longev. 2016, 2016, 1–9. [Google Scholar] [CrossRef]
- Wu, X.; Dong, L.; Lin, X.; Li, J. Relevance of the NLRP3 Inflammasome in the Pathogenesis of Chronic Liver Disease. Front. Immunol. 2017, 8, 1728. [Google Scholar] [CrossRef] [PubMed]
- Abais, J.M.; Xia, M.; Zhang, Y.; Boini, K.M.; Li, P.-L. Redox Regulation of NLRP3 Inflammasomes: ROS as Trigger or Effector? Antioxid. Redox Signal. 2015, 22, 1111–1129. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Lin, S.; Wan, B.; Velani, B.; Zhu, Y. Pyroptosis in Liver Disease: New Insights into Disease Mechanisms. Aging Dis. 2019, 10, 1094–1108. [Google Scholar] [CrossRef] [PubMed]
- Alyaseer, A.A.A.; De Lima, M.H.S.; Braga, T.T. The Role of NLRP3 Inflammasome Activation in the Epithelial to Mesenchymal Transition Process During the Fibrosis. Front. Immunol. 2020, 11, 883. [Google Scholar] [CrossRef]
- Pacana, T.; Sanyal, A. Vitamin E and nonalcoholic fatty liver disease. Curr. Opin. Clin. Nutr. Metab. Care 2012, 15, 641–648. [Google Scholar] [CrossRef]
- Morimoto, M.; Hagbjörk, A.-L.; Nanji, A.; Ingelman-Sundberg, M.; Lindros, K.; Fu, P.; Albano, E.; French, S. Role of cytochrome P4502E1 in alcoholic liver disease pathogenesis. Alcohol 1993, 10, 459–464. [Google Scholar] [CrossRef]
- Morimoto, M.; Reitz, R.C.; Morin, R.J.; Nguyen, K.; Ingelman-Sundberg, M.; French, S.W. CYP-2E1 inhibitors partially ameliorate the changes in hepatic fatty acid composition induced in rats by chronic administration of ethanol and a high fat diet. J. Nutr. 1995, 125, 2953–2964. [Google Scholar]
- Gouillon, Z.Q.; Lucas, D.; Li, J.; Hagbjork, A.L.; French, B.A.; Fu, P.; Fang, C.; Ingelman-Sundberg, M.; Donohue, T.M.; French, S.W. Inhibition of ethanol-induced liver disease in the intragastric feeding rat model by chlormethiazole (44545). Exp. Biol. Med. 2000, 224, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Mookerjee, R.; Wiesenthal, A.; Icking, A.; Hodges, S.J.; Davies, N.; Schilling, K.; Sen, S.; Williams, R.; Novelli, M.; Müller-Esterl, W.; et al. Increased Gene and Protein Expression of the Novel eNOS Regulatory Protein NOSTRIN and a Variant in Alcoholic Hepatitis. Gastroenterology 2007, 132, 2533–2541. [Google Scholar] [CrossRef] [PubMed]
- Shen, F.; Wang, Z.; Liu, W.; Liang, Y. Ethyl pyruvate can alleviate alcoholic liver disease through inhibiting Nrf2 signaling pathway. Exp. Ther. Med. 2018, 15, 4223–4228. [Google Scholar] [CrossRef] [PubMed]
- Thomas, H. A critical role for the NLRP3 inflammasome in NASH. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 197. [Google Scholar] [CrossRef]
- Mridha, A.R.; Wree, A.; Robertson, A.A.; Yeh, M.M.; Johnson, C.D.; Van Rooyen, D.M.; Haczeyni, F.; Teoh, N.C.-H.; Savard, C.; Ioannou, G.N.; et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J. Hepatol. 2017, 66, 1037–1046. [Google Scholar] [CrossRef]
- Barreyro, F.J.; Holod, S.; Inocchietto, P.V.F.; Camino, A.M.; Aquino, J.B.; Avagnina, A.; Carreras, M.C.; Poderoso, J.J.; Gores, G.J. The pan-caspase inhibitor Emricasan (IDN-6556) decreases liver injury and fibrosis in a murine model of non-alcoholic steatohepatitis. Liver Int. 2015, 35, 953–966. [Google Scholar] [CrossRef]
- Ratziu, V.; Sheikh, M.Y.; Sanyal, A.J.; Lim, J.K.; Conjeevaram, H.; Chalasani, N.; Abdelmalek, M.; Bakken, A.; Renou, C.; Palmer, M.; et al. A phase 2, randomized, double-blind, placebo-controlled study of GS-9450 in subjects with nonalcoholic steatohepatitis. Hepatology 2012, 55, 419–428. [Google Scholar] [CrossRef]
- Witek, R.P.; Stone, W.C.; Karaca, F.G.; Syn, W.-K.; Pereira, T.A.; Agboola, K.M.; Omenetti, A.; Jung, Y.; Teaberry, V.; Choi, S.S.; et al. Pan-caspase inhibitor VX-166 reduces fibrosis in an animal model of nonalcoholic steatohepatitis. Hepatology 2009, 50, 1421–1430. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Study | Model | Observed Effects | Ref. |
|---|---|---|---|
| In vivo | Acute and chronic ethanol administration | ROS participates in the pathological processes of fatty liver and fibrosis development | [25,26] |
| Clinical | Alcohol abuse | Elevated oxidative markers | [27,28,29] |
| Ex vivo | Elevated alcohol consumption | Protein adducts | [30] |
| In vitro | Stimulated neutrophils | ROS increases production of procollagen mRNA | [31] |
| In vitro | HSC and primary hepatocytes pyrazole-treated rats | Changes in collagen protein by ROS-dependent mechanisms | [32] |
| In vivo | Mice deficient in NOX | Exhibited attenuated ROS production | [33] |
| In vivo | NO inhibition | Increased collagen deposition and liver damage | [34] |
| In vitro | HSC incubated with ONOO− | NO plays no role during the late phase of fibrogenesis | [35] |
| In vivo | iNOS deficiency in CCl4-induced fibrosis | Decreased collagen synthesis; inhibited ONOO− formation and HSC apoptosis | [36] |
| In vivo | Endotoxemia | Nitrosative stress. | [37] |
| In vitro | Peroxynitrite donor | Peroxynitrite may contribute to the activation of pro-MMP-2 | [38] |
| In vivo | Blockade of iNOS | Induced antifibrotic effects | [39] |
| In vivo | Lacking KEAP1 | Minor susceptibility to hepatic injury | [40] |
| In vivo | NRF2-null mice stimulated with ethanol | Developed macrovascular steatosis and fulminant liver injury | [41,42,43,44] |
| In vitro | Tert-butylhydroquinone | Stimulated HSC activation and increased fibrogenic gene expression | [43] |
| In vivo | Deletion of NRF2 | Rapid onset and progression of nutritional steatohepatitis in mice | [45] |
| Ex vivo and in vitro | Human fibrotic liver and transient transfections of NRF2 via plasmids | Correlation between Nrf2 in HSCs and development of hepatic fibrosis NRF2 induced lipocyte phenotype in HSCs | [46] |
| In vitro and in vivo | Monosodium urate crystals, CCl4, and TAA | Inflammasome-mediated regulation of hepatic stellate cells | [47] |
| In vitro and in vivo | Monosodium urate crystals, H202 and NLRP3 activators, and Txnip–/– mice | TXNIP deficiency impaired activation of the NLRP3 inflammasome and subsequent secretion of interleukin 1β | [48] |
| Target | Model | Molecule | Effect | Ref. |
|---|---|---|---|---|
| ROS | NASH | Vitamin E | Protected the structural components of the cell membrane from peroxidation | [176] |
| ROS | ALD and NALD | Silibinin | Increased glutathione concentrations, reversed fibrosis, and stimulated regeneration | [74,75] |
| CYP2E1 | ALD | Diallyl sulfide and phenethyl isothiocyanate | Prevented the production of lipid peroxide and the accumulation of important fatty acids and reduced the pathology score | [177,178] |
| CYP2E1 | ALD | Chlormethiazole | Reduced the proteasome proteolytic enzyme activity induced by ethanol feeding | [179] |
| NO | ALD | Nostrin | Decreased enzymatic activity of endothelial nitric oxide synthase | [180] |
| NRF2 | ALD | Ethyl pyruvate | Increased anti-inflammatory factors | [181,182] |
| NRF2 | NAFLD and NASH | NRF2 activators | Prevented inflammation, triglyceride accumulation, and fibrosis in the liver | [159,163,165,166,167,168,169]. |
| NLPR3 | NASH | MCC950 | Normalized hepatic caspase 1 and IL-1β expression, plasma IL-1β, MCP-1 and IL-6, lowered ALT/AST, and reduced the severity of liver inflammation | [183] |
| Caspases | NASH | Emricasan | Ameliorated liver injury and fibrosis | [184] |
| Caspases | NASH | GS-9450 | Reduced ALT levels in NASH patients | [185] |
| Caspases | NASH | VX-166 | Inhibited collagen gene expression and reduced hepatic accumulation of cells expressing the myofibroblast marker α-SMA | [186] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramos-Tovar, E.; Muriel, P. Molecular Mechanisms That Link Oxidative Stress, Inflammation, and Fibrosis in the Liver. Antioxidants 2020, 9, 1279. https://doi.org/10.3390/antiox9121279
Ramos-Tovar E, Muriel P. Molecular Mechanisms That Link Oxidative Stress, Inflammation, and Fibrosis in the Liver. Antioxidants. 2020; 9(12):1279. https://doi.org/10.3390/antiox9121279
Chicago/Turabian StyleRamos-Tovar, Erika, and Pablo Muriel. 2020. "Molecular Mechanisms That Link Oxidative Stress, Inflammation, and Fibrosis in the Liver" Antioxidants 9, no. 12: 1279. https://doi.org/10.3390/antiox9121279
APA StyleRamos-Tovar, E., & Muriel, P. (2020). Molecular Mechanisms That Link Oxidative Stress, Inflammation, and Fibrosis in the Liver. Antioxidants, 9(12), 1279. https://doi.org/10.3390/antiox9121279