Metabolic Dysregulation and Neurovascular Dysfunction in Diabetic Retinopathy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
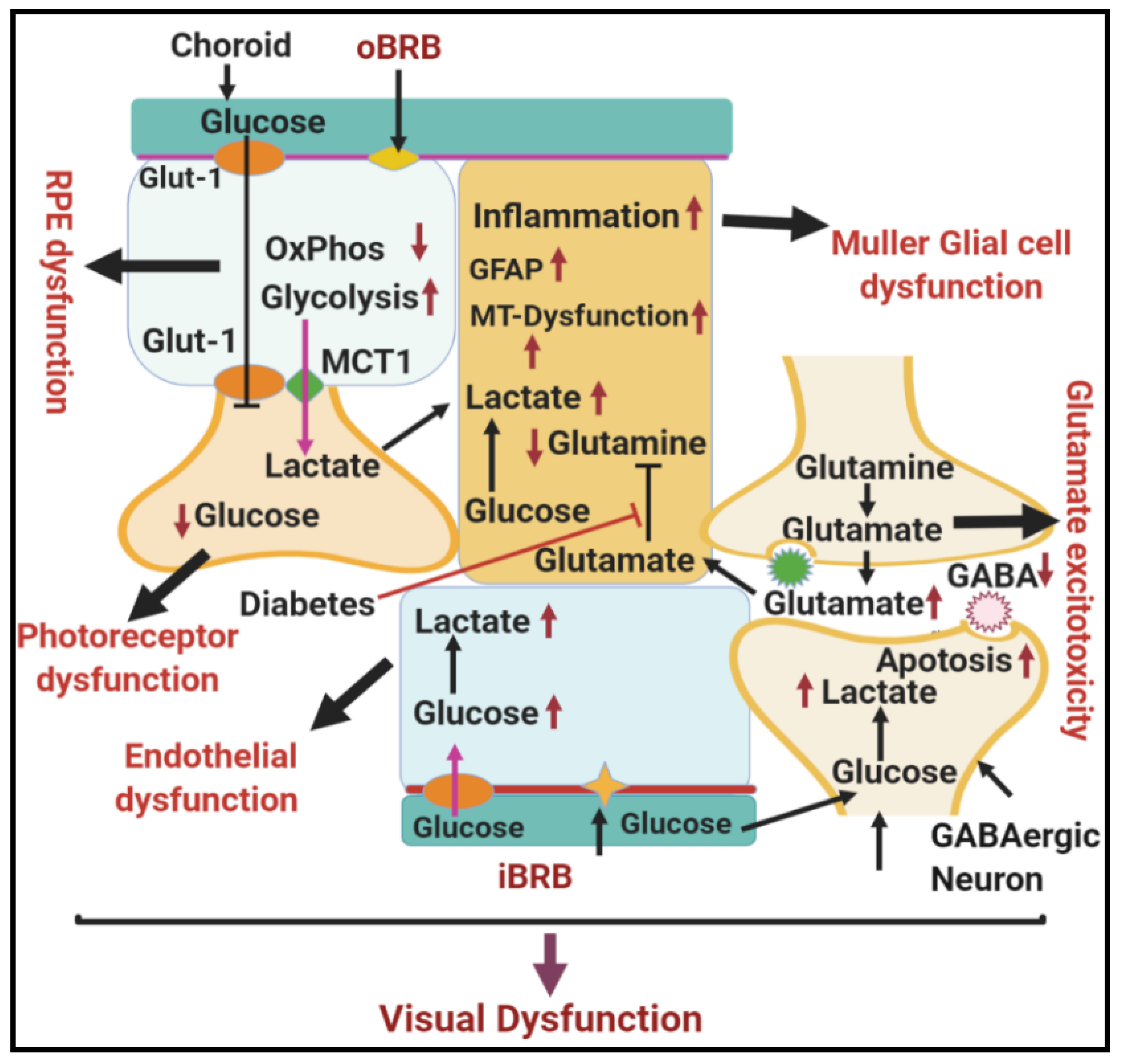
1.1. General Structure and Function of Neurovascular Unit of Retina
1.2. Metabolic Pathways Implicated in DR
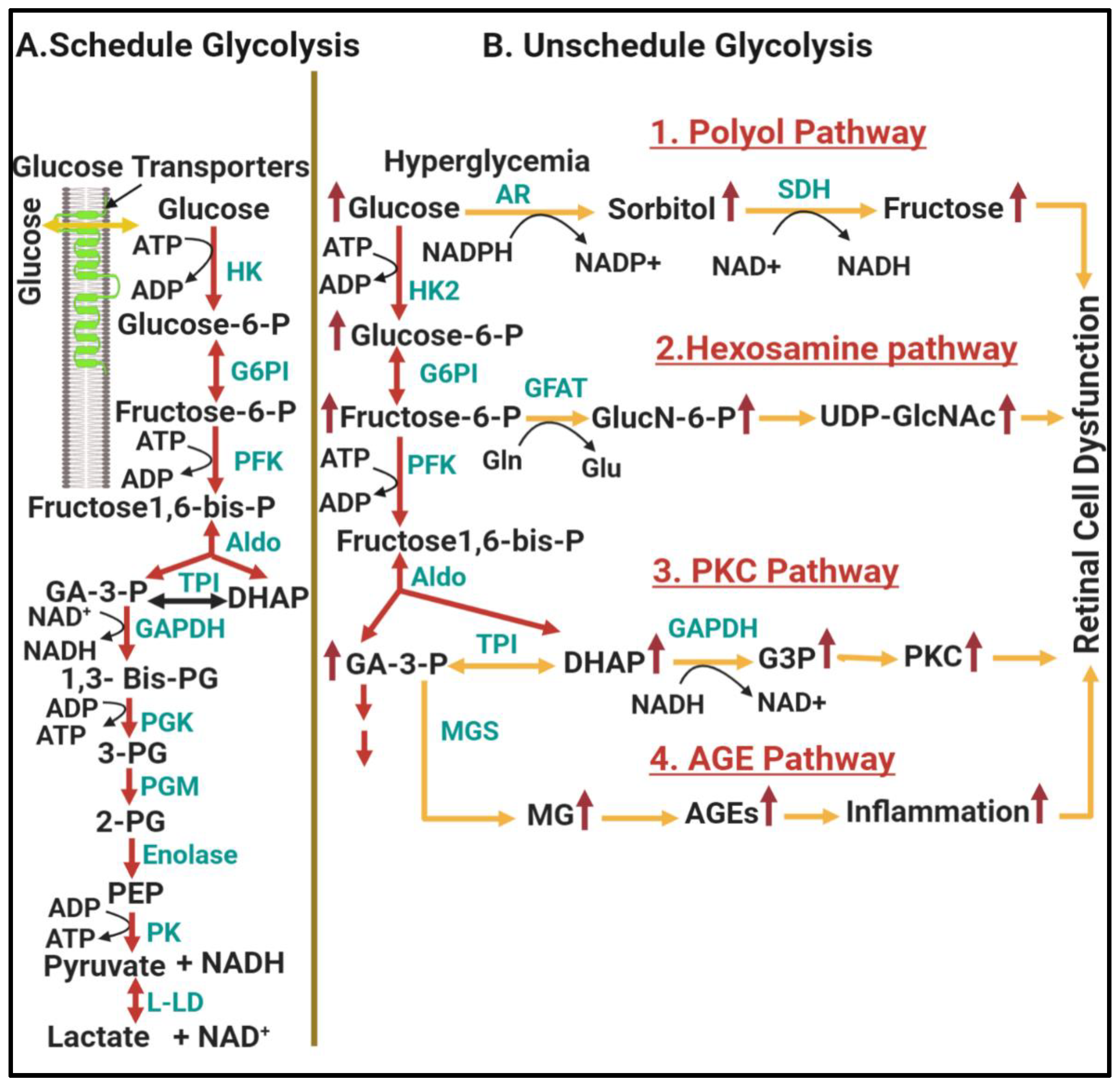
1.2.1. Involvement of Upper Glycolysis in Glycolytic Overload during DR
Shunting into Polyol Pathway
Shunting into Hexosamine Pathway
Shunting into Diacylglycerol (Dag)/Protein Kinase C (PKC) Pathway
Shunting into Glycation End Products (Amadori/AGEs) Pathway
1.2.2. Involvement of Lower Glycolysis Overload in DR
1.3. Other Biochemical and Molecular Changes Related to Dysregulation of Glycolysis in DR
1.3.1. Synergistic Relationship between Hypoxia and Hyperglycemia in DR
1.3.2. Endoplasmic Reticulum-Mitochondria Miscommunication in Diabetic Retinopathy
1.3.3. Mitophagy Dysregulation in Diabetic Retinopathy
2. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Harris Nwanyanwu, K.; Talwar, N.; Gardner, T.W.; Wrobel, J.S.; Herman, W.H.; Stein, J.D. Predicting development of proliferative diabetic retinopathy. Diabetes Care 2013, 36, 1562–1568. [Google Scholar] [CrossRef]
- Klein, R.; Knudtson, M.D.; Lee, K.E.; Gangnon, R.; Klein, B.E. The Wisconsin Epidemiologic Study of Diabetic Retinopathy XXIII: The twenty-five-year incidence of macular edema in persons with type 1 diabetes. Ophthalmology 2009, 116, 497–503. [Google Scholar] [CrossRef]
- Resnikoff, S.; Pascolini, D.; Etya’ale, D.; Kocur, I.; Pararajasegaram, R.; Pokharel, G.P.; Mariotti, S.P. Global data on visual impairment in the year 2002. Bull. World Health Organ. 2004, 82, 844–851. [Google Scholar]
- Sivaprasad, S.; Gupta, B.; Gulliford, M.C.; Dodhia, H.; Mohamed, M.; Nagi, D.; Evans, J.R. Ethnic variations in the prevalence of diabetic retinopathy in people with diabetes attending screening in the United Kingdom (DRIVE UK). PLoS ONE 2012, 7, e32182. [Google Scholar] [CrossRef]
- Simó-Servat, O.; Hernández, C.; Simó, R. Genetics in diabetic retinopathy: Current concepts and new insights. Curr. Genom. 2013, 14, 289–299. [Google Scholar] [CrossRef]
- Yau, J.W.Y.; Rogers, S.L.; Kawasaki, R.; Lamoureux, E.L.; Kowalski, J.W.; Bek, T.; Chen, S.-J.; Dekker, J.M.; Fletcher, A.; Grauslund, J.; et al. Global prevalence and major risk factors of diabetic retinopathy. Diabetes Care 2012, 35, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Early Treatment Diabetic Retinopathy Study Research Group. Grading Diabetic Retinopathy from Stereoscopic Color Fundus Photographs—An Extension of the Modified Airlie House Classification: ETDRS Report Number 10. Ophthalmology 2020, 127, S99–S119. [Google Scholar] [CrossRef] [PubMed]
- The Diabetic Retinopathy Study Research Group. Preliminary report on effects of photocoagulation therapy. Am. J. Ophthalmol. 1976, 81, 383–396. [Google Scholar] [CrossRef]
- Fong, D.S.; Girach, A.; Boney, A. Visual side effects of successful scatter laser photocoagulation surgery for proliferative diabetic retinopathy: A literature review. Retina 2007, 27, 816–824. [Google Scholar] [CrossRef]
- Diabetic Retinopathy Clinical Research Network; Wells, J.A.; Glassman, A.R.; Ayala, A.R.; Jampol, L.M.; Aiello, L.P.; Antoszyk, A.N.; Arnold-Bush, B.; Baker, C.W.; Bressler, N.M.; et al. Aflibercept, bevacizumab, or ranibizumab for diabetic macular edema. N. Engl. J. Med. 2015, 372, 1193–1203. [Google Scholar] [CrossRef] [PubMed]
- Writing Committee for the Diabetic Retinopathy Clinical Research Network; Gross, J.G.; Glassman, A.R.; Jampol, L.M.; Inusah, S.; Aiello, L.P.; Antoszyk, A.N.; Baker, C.W.; Berger, B.B.; Bressler, N.M.; et al. Panretinal Photocoagulation vs Intravitreous Ranibizumab for Proliferative Diabetic Retinopathy: A Randomized Clinical Trial. JAMA 2015, 314, 2137–2146. [Google Scholar] [CrossRef] [PubMed]
- Kuiper, E.J.; Van Nieuwenhoven, F.A.; de Smet, M.D.; van Meurs, J.C.; Tanck, M.W.; Oliver, N.; Klaassen, I.; Van Noorden, C.J.; Goldschmeding, R.; Schlingemann, R.O. The angio-fibrotic switch of VEGF and CTGF in proliferative diabetic retinopathy. PLoS ONE 2008, 3, e2675. [Google Scholar] [CrossRef] [PubMed]
- Gross, J.G.; Glassman, A.R.; Liu, D.; Sun, J.K.; Antoszyk, A.N.; Baker, C.W.; Bressler, N.M.; Elman, M.J.; Ferris, F.L., 3rd; Gardner, T.W.; et al. Five-Year Outcomes of Panretinal Photocoagulation vs Intravitreous Ranibizumab for Proliferative Diabetic Retinopathy: A Randomized Clinical Trial. JAMA Ophthalmol. 2018, 136, 1138–1148. [Google Scholar] [CrossRef]
- Wolfe, J.D.; Shah, A.R.; Yonekawa, Y.; Al Faran, A.; Franklin, M.S.; Abbey, A.M.; Capone, A., Jr. Receiver operating characteristic curve to predict anti-VEGF resistance in retinal vein occlusions and efficacy of Ozurdex. Eur. J. Ophthalmol. 2016, 26, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Krebs, I.; Glittenberg, C.; Ansari-Shahrezaei, S.; Hagen, S.; Steiner, I.; Binder, S. Non-responders to treatment with antagonists of vascular endothelial growth factor in age-related macular degeneration. Br. J. Ophthalmol. 2013, 97, 1443–1446. [Google Scholar] [CrossRef] [PubMed]
- Ehlken, C.; Jungmann, S.; Bohringer, D.; Agostini, H.T.; Junker, B.; Pielen, A. Switch of anti-VEGF agents is an option for nonresponders in the treatment of AMD. Eye 2014, 28, 538–545. [Google Scholar] [CrossRef]
- Fogli, S.; Del Re, M.; Rofi, E.; Posarelli, C.; Figus, M.; Danesi, R. Clinical pharmacology of intravitreal anti-VEGF drugs. Eye 2018, 32, 1010–1020. [Google Scholar] [CrossRef]
- Titchenell, P.M.; Antonetti, D.A. Using the past to inform the future: Anti-VEGF therapy as a road map to develop novel therapies for diabetic retinopathy. Diabetes 2013, 62, 1808–1815. [Google Scholar] [CrossRef]
- Zehetner, C.; Bechrakis, N.E.; Stattin, M.; Kirchmair, R.; Ulmer, H.; Kralinger, M.T.; Kieselbach, G.F. Systemic counterregulatory response of placental growth factor levels to intravitreal aflibercept therapy. Investig. Ophthalmol. Vis. Sci. 2015, 56, 3279–3286. [Google Scholar] [CrossRef]
- Forbes, J.M.; Cooper, M.E. Mechanisms of diabetic complications. Physiol. Rev. 2013, 93, 137–188. [Google Scholar] [CrossRef]
- Chen, Y.; Zhao, X.; Wu, H. Metabolic Stress and Cardiovascular Disease in Diabetes Mellitus: The Role of Protein O-GlcNAc Modification. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1911–1924. [Google Scholar] [CrossRef] [PubMed]
- Gardner, T.W.; Davila, J.R. The neurovascular unit and the pathophysiologic basis of diabetic retinopathy. Graefe’s Arch. Clin. Exp. Ophthalmol. 2017, 255, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Berg, J.M.; Tymoczko, J.L.; Stryer, L. Section 16.1, Glycolysis Is an Energy-Conversion Pathway in Many Organisms. In Biochemistry, 5th ed.; W H Freeman: New York, NY, USA, 2002. Available online: https://www.ncbi.nlm.nih.gov/books/NBK22593/ (accessed on 3 December 2020).
- Lechner, J.; O’Leary, O.E.; Stitt, A.W. The pathology associated with diabetic retinopathy. Vis. Res. 2017, 139, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Masland, R.H. The Fundamental Plan of the Retina Richard; Nature Publishing Group: Berlin, Germany, 2001. [Google Scholar]
- Sun, Y.; Smith, L.E.H. Retinal vasculature in development and diseases. Annu. Rev. Vis. Sci. 2018, 4, 101–122. [Google Scholar] [CrossRef] [PubMed]
- Ban, Y.; Rizzolo, L.J. Regulation of glucose transporters during development of the retinal pigment epithelium. Dev. Brain Res. 2000, 121, 89–95. [Google Scholar] [CrossRef]
- Bergersen, L.; Jóhannsson, E.; Veruki, M.L.; Nagelhus, E.A.; Halestrap, A.; Sejersted, O.M.; Ottersen, O.P. Cellular and subcellular expression of monocarboxylate transporters in the pigment epithelium and retina of the rat. Neuroscience 1999, 90, 319–331. [Google Scholar] [CrossRef]
- Kanow, M.A.; Giarmarco, M.M.; Jankowski, C.S.R.; Tsantilas, K.; Engel, A.L.; Du, J.; Linton, J.D.; Farnsworth, C.C.; Sloat, S.R.; Rountree, A.; et al. Biochemical adaptations of the retina and retinal pigment epithelium support a metabolic ecosystem in the vertebrate eye. eLife 2017. [Google Scholar] [CrossRef]
- Chou, J.; Rollins, S.; Fawzi, A.A. Role of endothelial cell and pericyte dysfunction in diabetic retinopathy: Review of techniques in rodent models. Adv. Exp. Med. Biol. 2014. [Google Scholar] [CrossRef]
- Kur, J.; Newman, E.A.; Chan-Ling, T. Cellular and physiological mechanisms underlying blood flow regulation in the retina and choroid in health and disease. Prog. Retin. Eye Res. 2012, 31, 377–406. [Google Scholar] [CrossRef]
- Klaassen, I.; Van Noorden, C.J.; Schlingemann, R.O. Molecular basis of the inner blood-retinal barrier and its breakdown in diabetic macular edema and other pathological conditions. Prog. Retin. Eye Res. 2013, 34, 19–48. [Google Scholar] [CrossRef]
- Metea, M.R.; Newman, E.A. Signalling within the neurovascular unit in the mammalian retina. Exp. Physiol. 2007, 92, 635–640. [Google Scholar] [CrossRef] [PubMed]
- Antonetti, D.A.; Barber, A.J.; Bronson, S.K.; Freeman, W.M.; Gardner, T.W.; Jefferson, L.S.; Kester, M.; Kimball, S.R.; Krady, J.K.; LaNoue, K.F.; et al. Diabetic retinopathy: Seeing beyond glucose-induced microvascular disease. Diabetes 2006, 55, 2401–2411. [Google Scholar] [CrossRef] [PubMed]
- Lieth, E.; LaNoue, K.F.; Antonetti, D.A.; Ratz, M. Diabetes reduces glutamate oxidation and glutamine synthesis in the retina. Exp. Eye Res. 2000, 70, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Petzold, G.C.; Murthy, V.N. Role of Astrocytes in Neurovascular Coupling. Neuron 2011, 71, 782–797. [Google Scholar] [CrossRef] [PubMed]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef] [PubMed]
- Krady, J.K.; Basu, A.; Allen, C.M.; Xu, Y.; LaNoue, K.F.; Gardner, T.W.; Levison, S.W. Minocycline Reduces Proinflammatory Cytokine Expression, Microglial Activation, and Caspase-3 Activation in a Rodent Model of Diabetic Retinopathy. Diabetes 2005, 54, 1559–1565. [Google Scholar] [CrossRef]
- Elward, K.; Gasque, P. “Eat me” and “don’t eat me” signals govern the innate immune response and tissue repair in the CNS: Emphasis on the critical role of the complement system. Mol. Immunol. 2003, 40, 85–94. [Google Scholar] [CrossRef]
- Ibrahim, A.S.; El-Remessy, A.B.; Matragoon, S.; Zhang, W.; Patel, Y.; Khan, S.; Al-Gayyar, M.M.; El-Shishtawy, M.M.; Liou, G.I. Retinal microglial activation and inflammation induced by amadori-glycated albumin in a rat model of diabetes. Diabetes 2011, 60, 1122–1133. [Google Scholar] [CrossRef]
- Simó, R.; Hernández, C. Neurodegeneration in the diabetic eye: New insights and therapeutic perspectives. Trends Endocrinol. Metab. 2014, 25, 23–33. [Google Scholar] [CrossRef]
- Rui, L. Energy metabolism in the liver. Compr. Physiol. 2014, 4, 177–197. [Google Scholar] [CrossRef]
- Rabbani, N.; Thornalley, P.J. Hexokinase-2 Glycolytic Overload in Diabetes and Ischemia-Reperfusion Injury. Trends Endocrinol. Metab. 2019, 30, 419–431. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, S.; Sima, A.A.F.; Nakajima, T.; Yagihashi, S.; Greene, D.A. Aldose reductase in the BB rat: Isolation, immunological identification and localization in the retina and peripheral nerve. Diabetologia 1987, 30, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Dagher, Z.; Park, Y.S.; Asnaghi, V.; Hoehn, T.; Gerhardinger, C.; Lorenzi, M. Studies of rat and human retinas predict a role for the polyol pathway in human diabetic retinopathy. Diabetes 2004, 53, 2404–2411. [Google Scholar] [CrossRef] [PubMed]
- Drel, V.R.; Pacher, P.; Ali, T.K.; Shin, J.; Julius, U.; El-Remessy, A.B.; Obrosova, I.G. Aldose reductase inhibitor fidarestat counteracts diabetes-associated cataract formation, retinal oxidative-nitrosative stress, glial activation, and apoptosis. Int. J. Mol. Med. 2008, 21, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Hohman, T.C.; Nishimura, C.; Robison, W.G. Aldose reductase and polyol in cultured pericytes of human retinal capillaries. Exp. Eye Res. 1989, 48, 55–60. [Google Scholar] [CrossRef]
- Li, W.; Chan, L.S.; Khatami, M.; Rockey, J.H. Non-competitive inhibition of myo-inositol transport in cultured bovine retinal capillary pericytes by glucose and reversal by Sorbinil. BBA Biomembr. 1986. [Google Scholar] [CrossRef]
- Cheung, N.; Mitchell, P.; Wong, T.Y. Diabetic retinopathy. Lancet 2010, 376, 124–136. [Google Scholar] [CrossRef]
- Barnett, P.A.; Gonzalez, R.G.; Chylack, L.T.; Cheng, H.M. The effect of oxidation on sorbitol pathway kinetics. Diabetes 1986, 35, 426–432. [Google Scholar] [CrossRef]
- Mathebula, S.D. Polyol pathway: A possible mechanism of diabetes complications in the eye. Afr. Vis. Eye Health 2015. [Google Scholar] [CrossRef]
- Szwergold, B.S.; Kappler, F.; Brown, T.R. Identification of fructose 3-phosphate in the lens of diabetic rats. Science 1990, 247, 451–454. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.-J. Redox imbalance stress in diabetes mellitus: Role of the polyol pathway. Anim. Models Exp. Med. 2018, 1, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Buse, M.G. Hexosamines, insulin resistance, and the complications of diabetes: Current status. Am. J. Physiol. Endocrinol. Metab. 2006. [Google Scholar] [CrossRef] [PubMed]
- Mathebula, S.D. Biochemical changes in diabetic retinopathy triggered by hyperglycaemia: A review. Afr. Vis. Eye Health 2018, 77, 1–7. [Google Scholar] [CrossRef]
- Kim, B.J.; Silverman, S.M.; Liu, Y.; Wordinger, R.J.; Pang, I.H.; Clark, A.F. In vitro and in vivo neuroprotective effects of cJun N-terminal kinase inhibitors on retinal ganglion cells. Mol. Neurodegener. 2016, 11, 30. [Google Scholar] [CrossRef]
- Gurel, Z.; Sheibani, N. O-Linked β-N-acetylglucosamine (O-GlcNAc) modification: A new pathway to decode pathogenesis of diabetic retinopathy. Clin. Sci. 2018, 132, 185–198. [Google Scholar] [CrossRef]
- Watanabe, T.; Raff, M.C. Retinal astrocytes are immigrants from the optic nerve. Nature 1988, 332, 834–837. [Google Scholar] [CrossRef]
- Gurel, Z.; Sieg, K.M.; Shallow, K.D.; Sorenson, C.M.; Sheibani, N. Retinal O-linked N-acetylglucosamine protein modifications: Implications for postnatal retinal vascularization and the pathogenesis of diabetic retinopathy. Mol. Vis. 2013, 19, 1047–1059. [Google Scholar]
- Filla, L.A.; Edwards, J.L. Metabolomics in diabetic complications. Mol. Biosyst. 2016. [Google Scholar] [CrossRef]
- Geraldes, P.; King, G.L. Activation of protein kinase C isoforms and its impact on diabetic complications. Circ. Res. 2010, 106, 1319–1331. [Google Scholar] [CrossRef]
- Shin, E.S.; Sorenson, C.M.; Sheibani, N. Diabetes and Retinal Vascular Dysfunction. J. Ophthalmic Vis. Res. 2015, 9, 362–373. [Google Scholar] [CrossRef]
- Cai, J.; Boulton, M. The pathogenesis of diabetic retinopathy: Old concepts and new questions. Eye 2002, 16, 242–260. [Google Scholar] [CrossRef] [PubMed]
- Tarr, J.M.; Kaul, K.; Chopra, M.; Kohner, E.M.; Chibber, R. Pathophysiology of Diabetic Retinopathy. ISRN Ophthalmol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Stitt, A.W. AGEs and diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2010, 51, 4867–4874. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, A.S.; El-Shishtawy, M.M.; Pena, A., Jr.; Liou, G.I. Genistein attenuates retinal inflammation associated with diabetes by targeting of microglial activation. Mol. Vis. 2010, 16, 2033–2042. [Google Scholar] [PubMed]
- Ibrahim, A.S.; El-Shishtawy, M.M.; Zhang, W.; Caldwell, R.B.; Liou, G.I. A((2)A) adenosine receptor (A((2)A)AR) as a therapeutic target in diabetic retinopathy. Am. J. Pathol. 2011, 178, 2136–2145. [Google Scholar] [CrossRef]
- Hammes, H.P.; Brownlee, M.; Edelstein, D.; Saleck, M.; Martin, S.; Federlin, K. Aminoguanidine inhibits the development of accelerated diabetic retinopathy in the spontaneous hypertensive rat. Diabetologia 1994. [Google Scholar] [CrossRef]
- Bierhaus, A.; Humpert, P.M.; Morcos, M.; Wendt, T.; Chavakis, T.; Arnold, B.; Stern, D.M.; Nawroth, P.P. Understanding RAGE, the receptor for advanced glycation end products. J. Mol. Med. 2005, 83, 876–886. [Google Scholar] [CrossRef]
- Milne, R.; Brownstein, S. Advanced glycation end products and diabetic retinopathy. Amino Acids 2013, 20, 3234–3240. [Google Scholar] [CrossRef]
- Zong, H.; Ward, M.; Stitt, A.W. AGEs, RAGE, and diabetic retinopathy. Curr. Diabetes Rep. 2011. [Google Scholar] [CrossRef]
- Xu, J.; Chen, L.J.; Yu, J.; Wang, H.J.; Zhang, F.; Liu, Q.; Wu, J. Involvement of Advanced Glycation End Products in the Pathogenesis of Diabetic Retinopathy. Cell. Physiol. Biochem. 2018, 48, 705–717. [Google Scholar] [CrossRef]
- Fu, D.; Yu, J.Y.; Yang, S.; Wu, M.; Hammad, S.M.; Connell, A.R.; Du, M.; Chen, J.; Lyons, T.J. Survival or death: A dual role for autophagy in stress-induced pericyte loss in diabetic retinopathy. Diabetologia 2016, 59, 2251–2261. [Google Scholar] [CrossRef]
- Du, M.; Wu, M.; Fu, D.; Yang, S.; Chen, J.; Wilson, K.; Lyons, T.J. Effects of modified LDL and HDL on retinal pigment epithelial cells: A role in diabetic retinopathy? Diabetologia 2013, 56, 2318–2328. [Google Scholar] [CrossRef]
- Du, J.; Rountree, A.; Cleghorn, W.M.; Contreras, L.; Lindsay, K.J.; Sadilek, M.; Gu, H.; Djukovic, D.; Raftery, D.; Satrústegui, J.; et al. Phototransduction influences metabolic flux and nucleotide metabolism in mouse retina. J. Biol. Chem. 2016, 291, 4698–4710. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, K.J.; Du, J.; Sloat, S.R.; Contreras, L.; Linton, J.D.; Turner, S.J.; Sadilek, M.; Satrústegui, J.; Hurley, J.B. Pyruvate kinase and aspartate-glutamate carrier distributions reveal key metabolic links between neurons and glia in retina. Proc. Natl. Acad. Sci. USA 2014, 111, 15579–15584. [Google Scholar] [CrossRef] [PubMed]
- Chinchore, Y.; Begaj, T.; Wu, D.; Drokhlyansky, E.; Cepko, C.L. Glycolytic reliance promotes anabolism in photoreceptors. eLife 2017. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.E.; DeWeerd, A.J.; Ildefonso, C.J.; Lewin, A.S.; Ash, J.D. Mitochondrial oxidative stress in the retinal pigment epithelium (RPE) led to metabolic dysfunction in both the RPE and retinal photoreceptors. Redox Biol. 2019, 24, 101201. [Google Scholar] [CrossRef]
- Ola, M.S.; Berkich, D.A.; Xu, Y.; King, M.T.; Gardner, T.W.; Simpson, I.; LaNoue, K.F. Analysis of glucose metabolism in diabetic rat retinas. Am. J. Physiol. Endocrinol. Metab. 2006, 290, E1057–E1067. [Google Scholar] [CrossRef]
- Sas, K.M.; Kayampilly, P.; Byun, J.; Nair, V.; Hinder, L.M.; Hur, J.; Zhang, H.; Lin, C.; Qi, N.R.; Michailidis, G.; et al. Tissue-specific metabolic reprogramming drives nutrient flux in diabetic complications. JCI Insight 2016. [Google Scholar] [CrossRef]
- Kelly, K.; Wang, J.; Zhang, S. The unfolded protein response signaling and retinal Müller cell metabolism. Neural Regen Res. 2018, 13, 1861. [Google Scholar] [CrossRef]
- Bringmann, A.; Pannicke, T.; Grosche, J.; Francke, M.; Wiedemann, P.; Skatchkov, S.N.; Osborne, N.N.; Reichenbach, A. Muller cells in the healthy and diseased retina. Prog. Retin. Eye Res. 2006, 25, 397–424. [Google Scholar] [CrossRef] [PubMed]
- Barber, A.J.; Lieth, E.; Khin, S.A.; Antonetti, D.A.; Buchanan, A.G.; Gardner, T.W. Neural apoptosis in the retina during experimental and human diabetes. Early onset and effect of insulin. J. Clin. Investig. 1998, 102, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Puro, D.G. Diabetes-Induced Dysfunction of the Glutamate Transporter in Retinal Müller Cells. Investig. Ophthalmol. Vis. Sci. 2002, 43, 3109–3116. [Google Scholar]
- Newman, E.A. Functional hyperemia and mechanisms of neurovascular coupling in the retinal vasculature. J. Cereb. Blood Flow Metab. 2013, 33, 1685–1695. [Google Scholar] [CrossRef] [PubMed]
- De Zeeuw, P.; Wong, B.W.; Carmeliet, P. Metabolic adaptations in diabetic endothelial cells. Circ. J. 2015, 79, 934–941. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Kanwar, M.; Kennedy, A. Metabolic memory phenomenon and accumulation of peroxynitrite in retinal capillaries. Exp. Diabetes Res. 2007, 2007, 21976. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Miao, X.; Li, F.; Wang, S.; Liu, Q.; Wang, Y.; Sun, J. Oxidative Stress-Related Mechanisms and Antioxidant Therapy in Diabetic Retinopathy. Oxid. Med. Cell. Longev. 2017. [Google Scholar] [CrossRef]
- Busik, J.V.; Mohr, S.; Grant, M.B. Hyperglycemia-Induced reactive oxygen species toxicity to endothelial cells is dependent on paracrine mediators. Diabetes 2008, 57, 1952–1965. [Google Scholar] [CrossRef]
- Trudeau, K.; Molina, A.J.A.; Guo, W.; Roy, S. High glucose disrupts mitochondrial morphology in retinal endothelial cells: Implications for diabetic retinopathy. Am. J. Pathol. 2010, 177, 447–455. [Google Scholar] [CrossRef]
- Madsen-Bouterse, S.A.; Mohammad, G.; Kanwar, M.; Kowluru, R.A. Role of mitochondrial DNA damage in the development of diabetic retinopathy, and the metabolic memory phenomenon associated with its progression. Antioxid. Redox Signal. 2010, 13, 797–805. [Google Scholar] [CrossRef]
- Cao, R.; Jensen, L.D.E.; Söll, I.; Hauptmann, G.; Cao, Y. Hypoxia-induced retinal angiogenesis in zebrafish as a model to study retinopathy. PLoS ONE 2008, 3, e2748. [Google Scholar] [CrossRef]
- De Gooyer, T.E.; Stevenson, K.A.; Humphries, P.; Simpson, D.A.C.; Gardiner, T.A.; Stitt, A.W. Retinopathy is reduced during experimental diabetes in a mouse model of outer retinal degeneration. Investig. Ophthalmol. Vis. Sci. 2006, 47, 5561–5568. [Google Scholar] [CrossRef] [PubMed]
- Linsenmeier, R.A.; Braun, R.D.; McRipley, M.A.; Padnick, L.B.; Ahmed, J.; Hatchell, D.L.; McLeod, D.S.; Lutty, G.A. Retinal hypoxia in long-term diabetic cats. Investig. Ophthalmol. Vis. Sci. 1998, 39, 1647–1657. [Google Scholar]
- Lai, A.K.W.; Lo, A. Animal Models of Diabetic Retinopathy: Summary and Comparison. J. Diabetes Res. 2013, 2013, 106594. [Google Scholar] [CrossRef] [PubMed]
- Grossniklaus, H.E.; Kang, S.J.; Berglin, L. Animal models of choroidal and retinal neovascularization. Prog. Retin. Eye Res. 2010, 29, 500–519. [Google Scholar] [CrossRef] [PubMed]
- Arden, G. Hypoxia and Oxidative Stress in the Causation of Diabetic Retinopathy. Curr. Diabetes Rev. 2011, 7, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Chronopoulos, A.; Trudeau, K.; Roy, S.; Huang, H.; Vinores, S.A.; Roy, S. High Glucose-induced Altered Basement Membrane Composition and Structure Increases Trans-endothelial Permeability: Implications for Diabetic Retinopathy. Curr. Eye Res. 2011, 36, 747–753. [Google Scholar] [CrossRef]
- Ekberg, N.R.; Eliasson, S.; Li, Y.W.; Zheng, X.; Chatzidionysiou, K.; Falhammar, H.; Gu, H.F.; Catrina, S.-B. Protective Effect of the HIF-1A Pro582Ser Polymorphism on Severe Diabetic Retinopathy. J. Diabetes Res. 2019, 2019, 2936962. [Google Scholar] [CrossRef]
- Sada, K.; Nishikawa, T.; Kukidome, D.; Yoshinaga, T.; Kajihara, N.; Sonoda, K.; Senokuchi, T.; Motoshima, H.; Matsumura, T.; Araki, E. Hyperglycemia Induces Cellular Hypoxia through Production of Mitochondrial ROS Followed by Suppression of Aquaporin-1. PLoS ONE 2016, 11, e0158619. [Google Scholar] [CrossRef]
- Kaur, C. Hypoxia-ischemia and retinal ganglion cell damage. Clin. Ophthalmol. 2008, 2, 879–889. [Google Scholar] [CrossRef]
- Joussen, A.M.; Poulaki, V.; Le, M.L.; Koizumi, K.; Esser, C.; Janicki, H.; Schraermeyer, U.; Kociok, N.; Fauser, S.; Kirchhof, B.; et al. A central role for inflammation in the pathogenesis of diabetic retinopathy. FASEB J. 2004, 18, 1450–1452. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Gong, B.; Hatala, D.A.; Kern, T.S. Retinal Ischemia and Reperfusion Causes Capillary Degeneration: Similarities to Diabetes. Investig. Opthalmol. Vis. Sci. 2007, 48, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.; Itin, A.; Alon, T.; Pe’Er, J.; Gnessin, H.; Chan-Ling, T.; Keshet, E. Development of retinal vasculature is mediated by hypoxia-induced vascular endothelial growth factor (VEGF) expression by neuroglia. J. Neurosci. 1995, 15, 4738–4747. [Google Scholar] [CrossRef] [PubMed]
- Xin, X.; Rodrigues, M.; Umapathi, M.; Kashiwabuchi, F.; Ma, T.; Babapoor-Farrokhran, S.; Wang, S.; Hu, J.; Bhutto, I.; Welsbie, D.S.; et al. Hypoxic retinal Müller cells promote vascular permeability by HIF-1–dependent up-regulation of angiopoietin-like 4. In Proceedings of the Proceedings of the National Academy of Sciences. Proc. Natl. Acad. Sci. USA 2013, 110, E3425–E3434. [Google Scholar] [CrossRef] [PubMed]
- Krock, B.L.; Skuli, N.; Simon, M.C. Hypoxia-Induced Angiogenesis: Good and Evil. Genes Cancer 2011, 2, 1117–1133. [Google Scholar] [CrossRef]
- Das, A.; Stroud, S.; Mehta, A.; Rangasamy, S. New treatments for diabetic retinopathy. Diabetes Obes. Metab. 2015, 17, 219–230. [Google Scholar] [CrossRef]
- Nyengaard, J.R.; Ido, Y.; Kilo, C.; Williamson, J.R. Interactions Between Hyperglycemia and Hypoxia: Implications for Diabetic Retinopathy. Diabetes 2004, 53, 2931–2938. [Google Scholar] [CrossRef]
- Gries, F.A. Alternative therapeutic principles in the prevention of microvascular and neuropathic complications. Diabetes Res. Clin. Pract. 1995, 28, S201–S207. [Google Scholar] [CrossRef]
- Michiels, C.; Arnould, T.; Remacle, J. Endothelial cell responses to hypoxia: Initiation of a cascade of cellular interactions. Biochim. Biophys. Acta (BBA) Bioenerg. 2000, 1497, 1–10. [Google Scholar] [CrossRef]
- Tailor, A.; Granger, D.N. Role of adhesion molecules in vascular regulation and damage. Curr. Hypertens. Rep. 2000, 2, 78–83. [Google Scholar] [CrossRef]
- Levy, A.P.; Levy, N.S.; Loscalzo, J.; Calderone, A.; Takahashi, N.; Yeo, K.-T.; Koren, G.; Colucci, W.S.; Goldberg, M.A. Regulation of Vascular Endothelial Growth Factor in Cardiac Myocytes. Circ. Res. 1995, 76, 758–766. [Google Scholar] [CrossRef] [PubMed]
- Aiello, L.P.; Arrigg, P.G.; Shah, S.T.; Keyt, B.A.; Avery, R.L.; Jampel, H.D.; Pasquale, L.R.; Thieme, H.; King, G.L.; Iwamoto, M.A.; et al. Vascular Endothelial Growth Factor in Ocular Fluid of Patients with Diabetic Retinopathy and Other Retinal Disorders. N. Engl. J. Med. 1994. [Google Scholar] [CrossRef] [PubMed]
- Penn, J.S.; Madan, A.; Caldwell, R.; Bartoli, M.; Hartnett, M. Vascular endothelial growth factor in eye disease. Prog. Retin. Eye Res. 2008, 27, 331–371. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N. Vascular Endothelial Growth Factor. Arter. Thromb. Vasc. Biol. 2009, 29, 789–791. [Google Scholar] [CrossRef]
- Lee, H.K.; Chauhan, S.K.; Kay, E.; Dana, R. Flt-1 regulates vascular endothelial cell migration via a protein tyrosine kinase-7–dependent pathway. Blood 2011, 117, 5762–5771. [Google Scholar] [CrossRef]
- De Vries, C.; Escobedo, J.A.; Ueno, H.; Houck, K.; Ferrara, N.; Williams, L.T. The fms-like tyrosine kinase, a receptor for vascular endothelial growth factor. Science 1992, 255, 989–991. [Google Scholar] [CrossRef]
- Gupta, N.; Mansoor, S.; Sharma, A.; Sapkal, A.; Sheth, J.; Falatoonzadeh, P.; Kuppermann, B.; Kenney, M.C. Diabetic Retinopathy and VEGF. Open Ophthalmol. J. 2013, 7, 4–10. [Google Scholar] [CrossRef]
- Simpson, D.A.; Murphy, G.M.; Bhaduri, T.; Gardiner, T.A.; Archer, D.B.; Stitt, A.W. Expression of the VEGF Gene Family during Retinal Vaso-Obliteration and Hypoxia. Biochem. Biophys. Res. Commun. 1999, 262, 333–340. [Google Scholar] [CrossRef]
- Rodrigues, M.; Xin, X.; Jee, K.; Babapoor-Farrokhran, S.; Kashiwabuchi, F.; Ma, T.; Bhutto, I.; Hassan, S.J.; Daoud, Y.; Baranano, D.; et al. VEGF Secreted by Hypoxic Muller Cells Induces MMP-2 Expression and Activity in Endothelial Cells to Promote Retinal Neovascularization in Proliferative Diabetic Retinopathy. Diabetes 2013, 62, 3863–3873. [Google Scholar] [CrossRef]
- Kaur, C.; Sivakumar, V.; Foulds, W.S. Early Response of Neurons and Glial Cells to Hypoxia in the Retina. Investig. Opthalmol. Vis. Sci. 2006, 47, 1126–1141. [Google Scholar] [CrossRef]
- Lam, T.T.; Abler, A.S.; Tso, M.O.M. Apoptosis and caspases after ischemia-reperfusion injury in rat retina. Investig. Ophthalmol. Vis. Sci. 1999, 40, 967–975. [Google Scholar]
- Kaur, C.; Sivakumar, V.; Foulds, W.S.; Luu, C.D.; Ling, E.-A. Hypoxia-Induced Activation ofN-methyl-D-aspartate Receptors Causes Retinal Ganglion Cell Death in the Neonatal Retina. J. Neuropathol. Exp. Neurol. 2012, 71, 330–347. [Google Scholar] [CrossRef] [PubMed]
- Frank, R.N. Diabetic Retinopathy. N. Engl. J. Med. 2004, 350, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Skondra, D.; Noda, K.; Almulki, L.; Tayyari, F.; Frimmel, S.; Nakazawa, T.; Kim, I.K.; Zandi, S.; Thomas, K.L.; Miller, J.W.; et al. Characterization of Azurocidin as a Permeability Factor in the Retina: Involvement in VEGF-Induced and Early Diabetic Blood-Retinal Barrier Breakdown. Investig. Opthalmol. Vis. Sci. 2008, 49, 726–731. [Google Scholar] [CrossRef][Green Version]
- Simmons, A.B.; Bretz, C.A.; Wang, H.; Kunz, E.; Hajj, K.; Kennedy, C.; Yang, Z.; Suwanmanee, T.; Kafri, T.; Hartnett, M.E. Gene therapy knockdown of VEGFR2 in retinal endothelial cells to treat retinopathy. Angiogenesis 2018. [Google Scholar] [CrossRef]
- Takagi, H.; King, G.L.; Aiello, L.P. Hypoxia upregulates glucose transport activity through an adenosine-mediated increase of GLUT1 expression in retinal capillary endothelial cells. Diabetes 1998, 47, 1480–1488. [Google Scholar] [CrossRef]
- You, Z.-P.; Zhang, Y.-L.; Shi, K.; Shi, L.; Zhang, Y.-Z.; Zhou, Y.; Wang, C.-Y. Suppression of diabetic retinopathy with GLUT1 siRNA. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef]
- Zhang, J.; Li, Y.; Jiang, S.; Yu, H.; An, W. Enhanced endoplasmic reticulum SERCA activity by overexpression of hepatic stimulator substance gene prevents hepatic cells from ER stress-induced apoptosis. Am. J. Physiol. Physiol. 2014, 306, C279–C290. [Google Scholar] [CrossRef]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Rao, R.V.; Ellerby, H.M.; Bredesen, D.E. Coupling endoplasmic reticulum stress to the cell death program. Cell Death Differ. 2004, 11, 372–380. [Google Scholar] [CrossRef]
- Lindholm, D.; Wootz, H.; Korhonen, L. ER stress and neurodegenerative diseases. Cell Death Differ. 2006, 13, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, J.J.; Yu, Q.; Wang, M.; Zhang, S.X. Endoplasmic reticulum stress is implicated in retinal inflammation and diabetic retinopathy. FEBS Lett. 2009, 583, 1521–1527. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Zhang, Y.; Jiang, Y.; Willard, L.; Ortiz, E.; Wark, L.; Medeiros, D.M.; Lin, D. Dietary wolfberry ameliorates retinal structure abnormalities in db/db mice at the early stage of diabetes. Exp. Biol. Med. 2011, 236, 1051–1063. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Li, J.; Chen, Y.; Wang, J.J.; Ratan, R.; Zhang, S.X. Activation of Endoplasmic Reticulum Stress by Hyperglycemia Is Essential for Müller Cell-Derived Inflammatory Cytokine Production in Diabetes. Diabetes 2012, 61, 492–504. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, J.J.; Li, J.; Hosoya, K.I.; Ratan, R.; Townes, T.; Zhang, S.X. Activating transcription factor 4 mediates hyperglycaemia-induced endothelial inflammation and retinal vascular leakage through activation of STAT3 in a mouse model of type 1 diabetes. Diabetologia 2012, 55, 2533–2545. [Google Scholar] [CrossRef]
- Elmasry, K.; Ibrahim, A.S.; Saleh, H.; Elsherbiny, N.; Elshafey, S.; Hussein, K.A.; Al-Shabrawey, M. Role of endoplasmic reticulum stress in 12/15-lipoxygenase-induced retinal microvascular dysfunction in a mouse model of diabetic retinopathy. Diabetologia 2018, 61, 1220–1232. [Google Scholar] [CrossRef]
- Jing, G.; Wang, J.J.; Zhang, S.X. ER Stress and Apoptosis: A New Mechanism for Retinal Cell Death. Exp. Diabetes Res. 2011, 2012, 589589. [Google Scholar] [CrossRef]
- Kim, J.-A.; Wei, Y.; Sowers, J.R. Role of Mitochondrial Dysfunction in Insulin Resistance. Circ. Res. 2008, 102, 401–414. [Google Scholar] [CrossRef]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef]
- Devi, T.S.; Somayajulu, M.; Kowluru, R.A.; Singh, L.P. TXNIP regulates mitophagy in retinal Müller cells under high-glucose conditions: Implications for diabetic retinopathy. Cell Death Dis. 2017, 8, e2777. [Google Scholar] [CrossRef]
- Perrone, L.; Devi, T.S.; Hosoya, K.-I.; Terasaki, T.; Singh, L.P. Thioredoxin interacting protein (TXNIP) induces inflammation through chromatin modification in retinal capillary endothelial cells under diabetic conditions. J. Cell. Physiol. 2009, 221, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Singh, L.P. The Role of Txnip in Mitophagy Dysregulation and Inflammasome Activation in Diabetic Retinopathy: A New Perspective. JOJ Ophthalmol. 2017, 4. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nat. Cell Biol. 2000, 408, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Mishra, M. Oxidative stress, mitochondrial damage and diabetic retinopathy. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2015, 1852, 2474–2483. [Google Scholar] [CrossRef]
- Madsen-Bouterse, S.A.; Kowluru, R.A. Oxidative stress and diabetic retinopathy: Pathophysiological mechanisms and treatment perspectives. Rev. Endocr. Metab. Disord. 2008, 9, 315–327. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Kowluru, A.; Mishra, M.; Kumar, B. Oxidative stress and epigenetic modifications in the pathogenesis of diabetic retinopathy. Prog. Retin. Eye Res. 2015, 48, 40–61. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.X.; Ma, J.H.; Bhatta, M.; Fliesler, S.J.; Wang, J.J. The unfolded protein response in retinal vascular diseases: Implications and therapeutic potential beyond protein folding. Prog. Retin. Eye Res. 2015, 45, 111–131. [Google Scholar] [CrossRef] [PubMed]
- López-Crisosto, C.; Bravo-Sagua, R.; Rodriguez-Peña, M.; Mera, C.; Castro, P.F.; Quest, A.F.G.; Rothermel, B.A.; Cifuentes, M.; Lavandero, S. ER-to-mitochondria miscommunication and metabolic diseases. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2015, 1852, 2096–2105. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R.; Pinton, P.; Carrington, W.; Fay, F.S.; Fogarty, K.E.; Lifshitz, L.M.; Tuft, R.A.; Pozzan, T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 1998, 280, 1763–1766. [Google Scholar] [CrossRef]
- Bravo-Sagua, R.; Torrealba, N.; Paredes, F.; Morales, P.E.; Pennanen, C.; Lopez-Crisosto, C.; Troncoso, R.; Criollo, A.; Chiong, M.; Hill, J.A.; et al. Organelle communication: Signaling crossroads between homeostasis and disease. Int. J. Biochem. Cell Biol. 2014, 50, 55–59. [Google Scholar] [CrossRef]
- Marchi, S.; Patergnani, S.; Pinton, P. The endoplasmic reticulum–mitochondria connection: One touch, multiple functions. Biochim. Biophys. Acta (BBA) Bioenerg. 2014, 1837, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Rizzuto, R.; Hajnoczky, G.; Su, T.-P. MAM: More than just a housekeeper. Trends Cell Biol. 2009, 19, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Kornmann, B. The molecular hug between the ER and the mitochondria. Curr. Opin. Cell Biol. 2013, 25, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Tubbs, E.; Theurey, P.; Vial, G.; Bendridi, N.; Bravard, A.; Chauvin, M.-A.; Ji-Cao, J.; Zoulim, F.; Bartosch, B.; Ovize, M.; et al. Mitochondria-Associated Endoplasmic Reticulum Membrane (MAM) Integrity Is Required for Insulin Signaling and Is Implicated in Hepatic Insulin Resistance. Diabetes 2014, 63, 3279–3294. [Google Scholar] [CrossRef] [PubMed]
- Devi, T.S.; Lee, I.; Huttemann, M.; Kumar, A.; Nantwi, K.D.; Singh, L.P. TXNIP links innate host defense mechanisms to oxidative stress and inflammation in retinal Muller glia under chronic hyperglycemia: Implications for diabetic retinopathy. Exp. Diabetes Res. 2012, 2012, 438238. [Google Scholar] [CrossRef]
- Alfarhan, M.; Jafari, E.; Narayanan, S.P. Acrolein: A Potential Mediator of Oxidative Damage in Diabetic Retinopathy. Biomolecules 2020, 10, 1579. [Google Scholar] [CrossRef]
- Zhang, L.W.; Zhao, H.; Chen, B.H. Reactive oxygen species mediates a metabolic memory of high glucose stress signaling in bovine retinal pericytes. Int. J. Ophthalmol. 2019, 12, 1067–1074. [Google Scholar] [CrossRef]
- Devi, T.S.; Yumnamcha, T.; Yao, F.; Somayajulu, M.; Kowluru, R.A.; Singh, L.P. TXNIP mediates high glucose-induced mitophagic flux and lysosome enlargement in human retinal pigment epithelial cells. Biol. Open 2019. [Google Scholar] [CrossRef]
- Yumnamcha, T.; Devi, T.S.; Singh, L.P. Auranofin Mediates Mitochondrial Dysregulation and Inflammatory Cell Death in Human Retinal Pigment Epithelial Cells: Implications of Retinal Neurodegenerative Diseases. Front. Neurosci. 2019, 13, 1065. [Google Scholar] [CrossRef]
- Jin, S.M.; Youle, R.J. PINK1- and Parkin-mediated mitophagy at a glance. J. Cell Sci. 2012, 125, 795–799. [Google Scholar] [CrossRef]
- Killackey, S.A.; Philpott, D.J.; Girardin, S.E. Mitophagy pathways in health and disease. J. Cell Biol. 2020, 219. [Google Scholar] [CrossRef] [PubMed]
- Bader, V.; Winklhofer, K.F. Mitochondria at the interface between neurodegeneration and neuroinflammation. Semin. Cell Dev. Biol. 2020, 99, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature 2015, 521, 525–528. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, J.C.; Bwiza, C.P.; Lee, C. Mitonuclear genomics and aging. Hum. Genet. 2020, 139, 381–399. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.M.; Tewari, S.; Goldberg, A.F.; Kowluru, R.A. Mitochondrial biogenesis and the development of diabetic retinopathy. Free Radic. Biol. Med. 2011, 51, 1849–1860. [Google Scholar] [CrossRef] [PubMed]
- Hombrebueno, J.R.; Cairns, L.; Dutton, L.R.; Lyons, T.J.; Brazil, D.P.; Moynagh, P.; Curtis, T.M.; Xu, H. Uncoupled turnover disrupts mitochondrial quality control in diabetic retinopathy. JCI Insight. 2019, 4, e129760. [Google Scholar] [CrossRef]
- Perrone, L.; Devi, T.S.; Hosoya, K.I.; Terasaki, T.; Singh, L.P. Inhibition of TXNIP expression in vivo blocks early pathologies of diabetic retinopathy. Cell Death Dis 2010, 1, e65. [Google Scholar] [CrossRef]
- Devi, T.S.; Hosoya, K.I.; Terasaki, T.; Singh, L.P. Critical role of TXNIP in oxidative stress, DNA damage and retinal pericyte apoptosis under high glucose: Implications for diabetic retinopathy. Exp. Cell Res. 2013. [Google Scholar] [CrossRef]
- Singh, L.P. Thioredoxin Interacting Protein (TXNIP) and Pathogenesis of Diabetic Retinopathy. J. Clin. Exp. Ophthalmol. 2013, 4. [Google Scholar] [CrossRef]
- Wang, K.; Zhan, Y.; Chen, B.; Lu, Y.; Yin, T.; Zhou, S.; Zhang, W.; Liu, X.; Du, B.; Wei, X.; et al. Tubeimoside I-induced lung cancer cell death and the underlying crosstalk between lysosomes and mitochondria. Cell Death Dis 2020, 11, 708. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yumnamcha, T.; Guerra, M.; Singh, L.P.; Ibrahim, A.S. Metabolic Dysregulation and Neurovascular Dysfunction in Diabetic Retinopathy. Antioxidants 2020, 9, 1244. https://doi.org/10.3390/antiox9121244
Yumnamcha T, Guerra M, Singh LP, Ibrahim AS. Metabolic Dysregulation and Neurovascular Dysfunction in Diabetic Retinopathy. Antioxidants. 2020; 9(12):1244. https://doi.org/10.3390/antiox9121244
Chicago/Turabian StyleYumnamcha, Thangal, Michael Guerra, Lalit Pukhrambam Singh, and Ahmed S. Ibrahim. 2020. "Metabolic Dysregulation and Neurovascular Dysfunction in Diabetic Retinopathy" Antioxidants 9, no. 12: 1244. https://doi.org/10.3390/antiox9121244
APA StyleYumnamcha, T., Guerra, M., Singh, L. P., & Ibrahim, A. S. (2020). Metabolic Dysregulation and Neurovascular Dysfunction in Diabetic Retinopathy. Antioxidants, 9(12), 1244. https://doi.org/10.3390/antiox9121244