Melatonin/Nrf2/NLRP3 Connection in Mouse Heart Mitochondria during Aging

 ,
,  , ,
, ,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
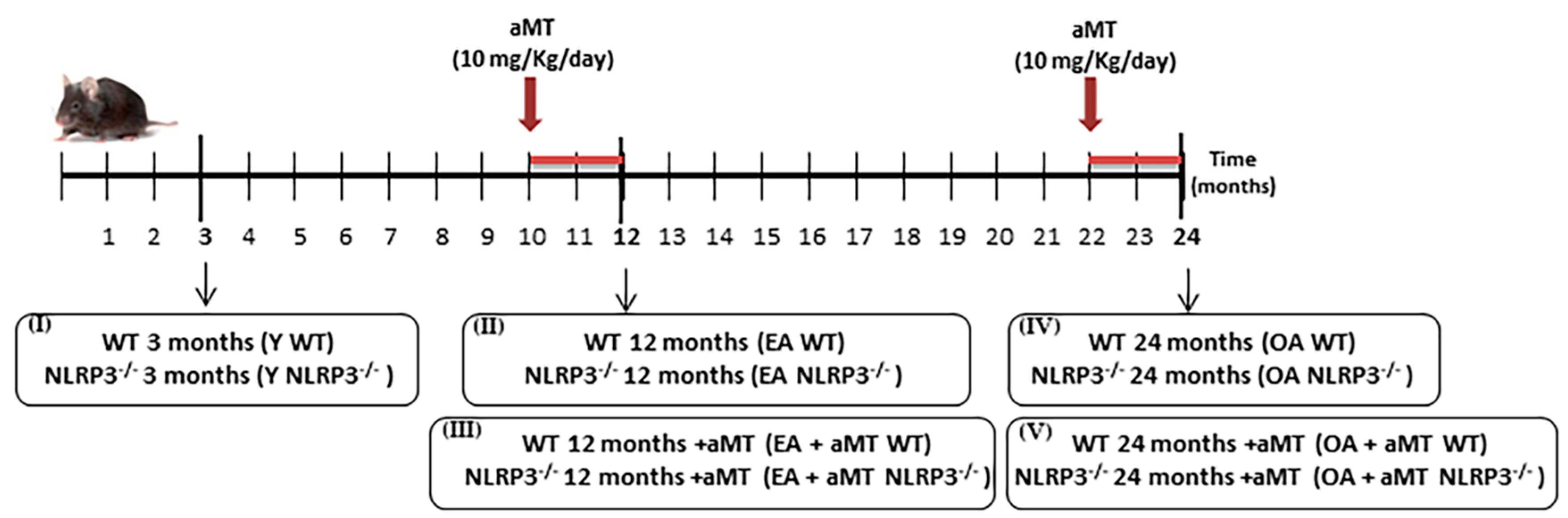
2.1. Animals and Treatment
2.2. Western Blot Analysis
2.3. Transmission Electron Microscopy (TEM)
2.4. Morphometric Analyses
2.5. Statistical Analyses
3. Results
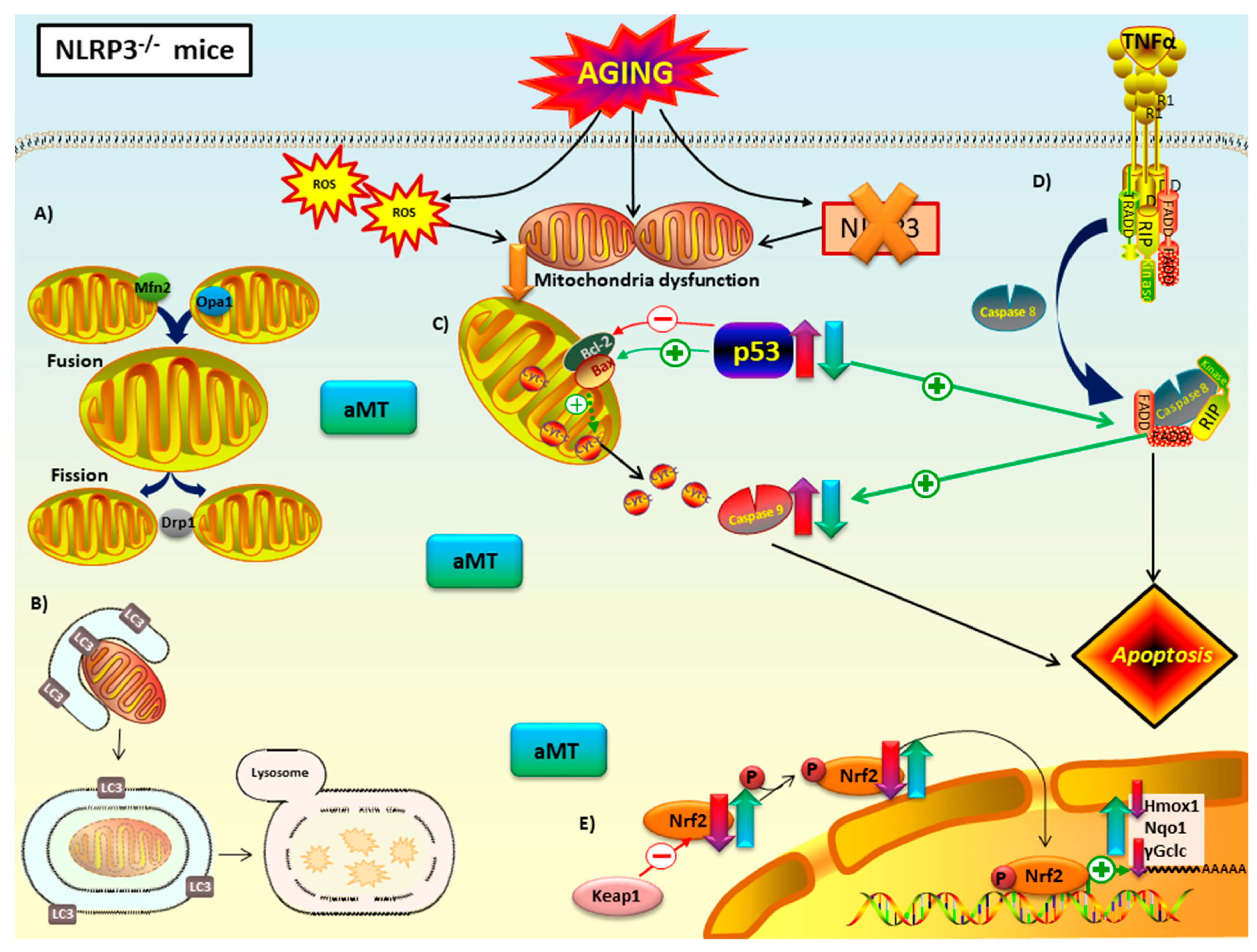
3.1. NLRP3 Deficiency Prevents, and Melatonin Treatment Restores Cardiac Muscle Mitochondrial Dynamics Altered by Aging
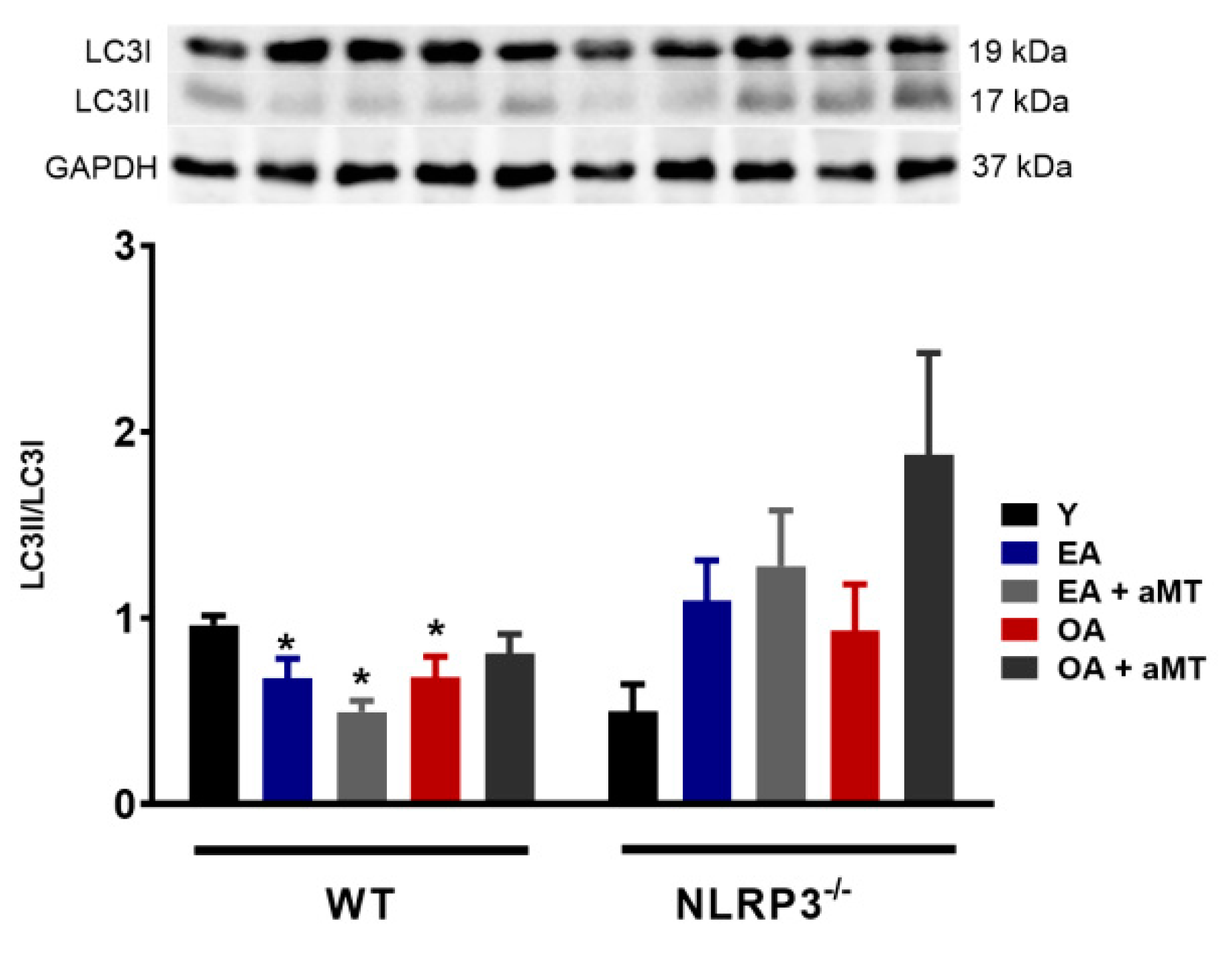
3.2. NLRP3 Deficiency and Melatonin Therapy Had Minimal Effects in Autophagy in Cardiac Muscle during Aging
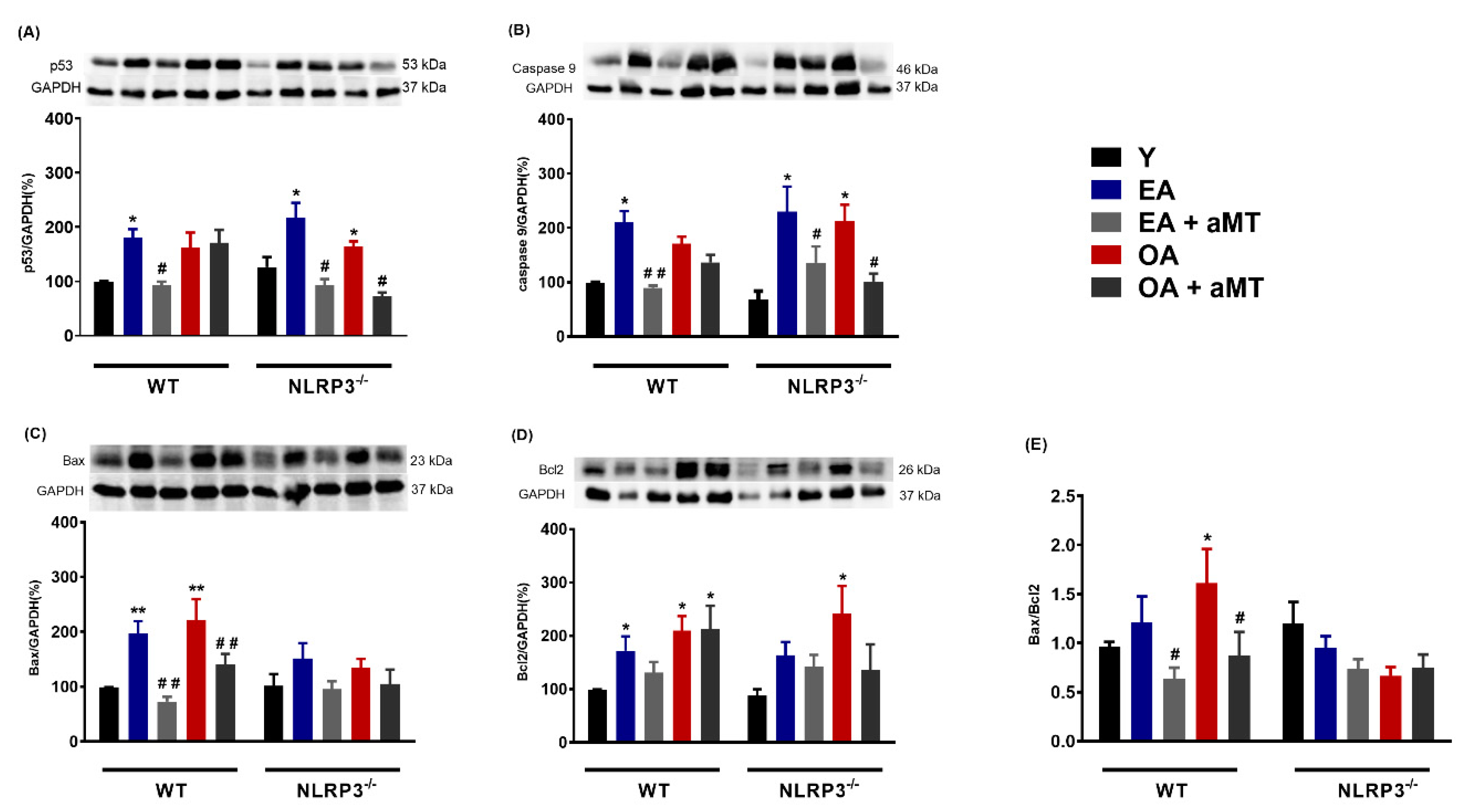
3.3. Melatonin Treatment and, to a Lesser Extent NLRP3 Deficiency, Reduced Apoptosis in Cardiac Muscle during Aging
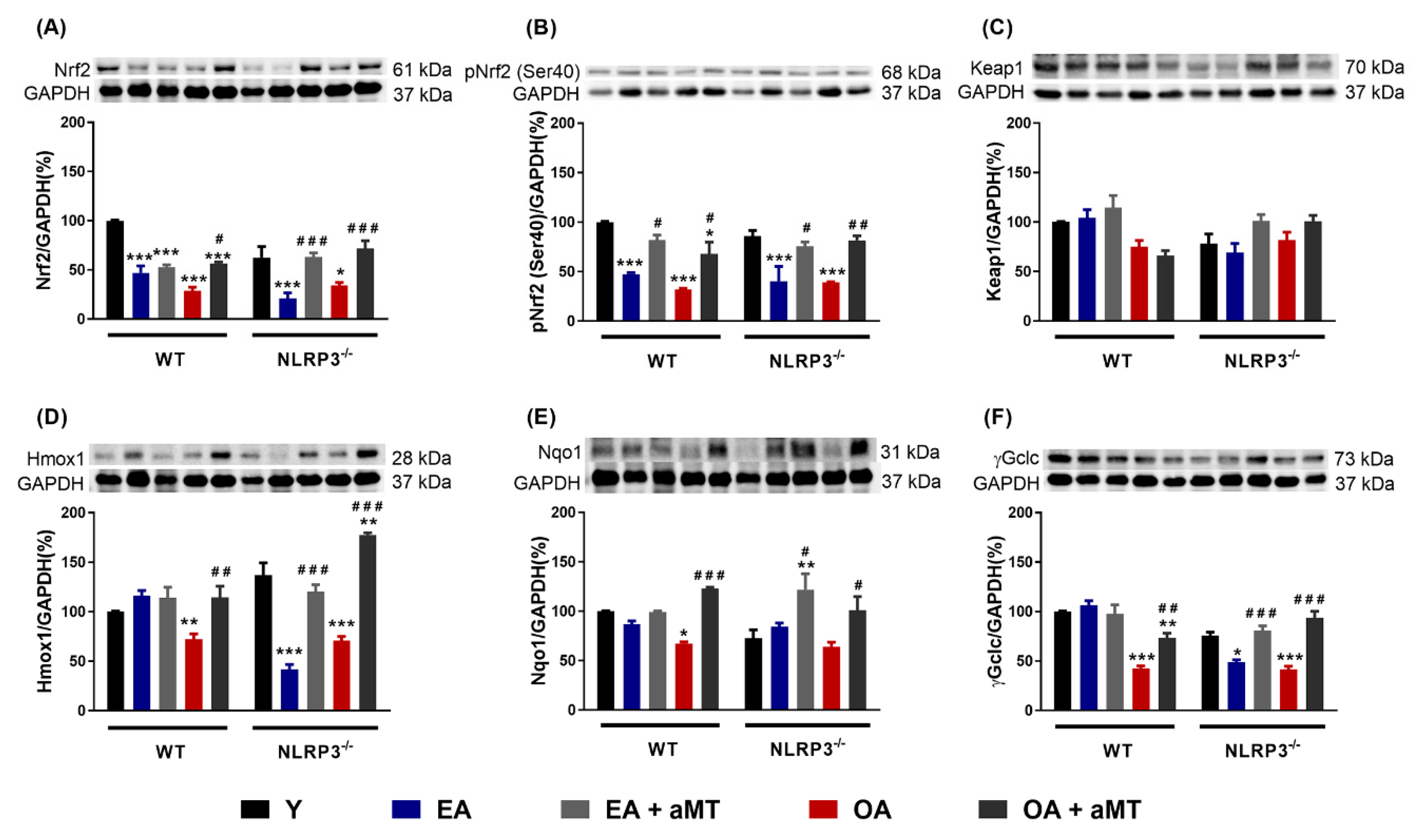
3.4. Melatonin Treatment, but not NLRP3 Deficiency, Recovered the Nrf2-Dependent Antioxidant Capacity in Cardiac Muscle Declined by Aging
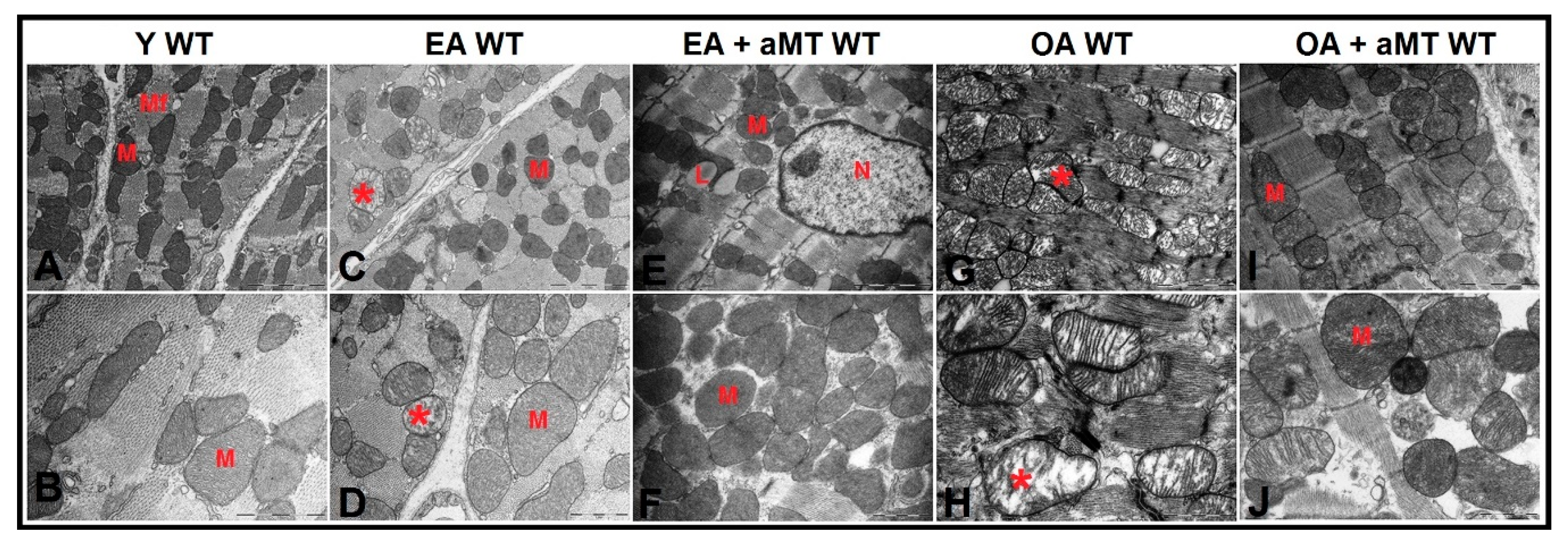
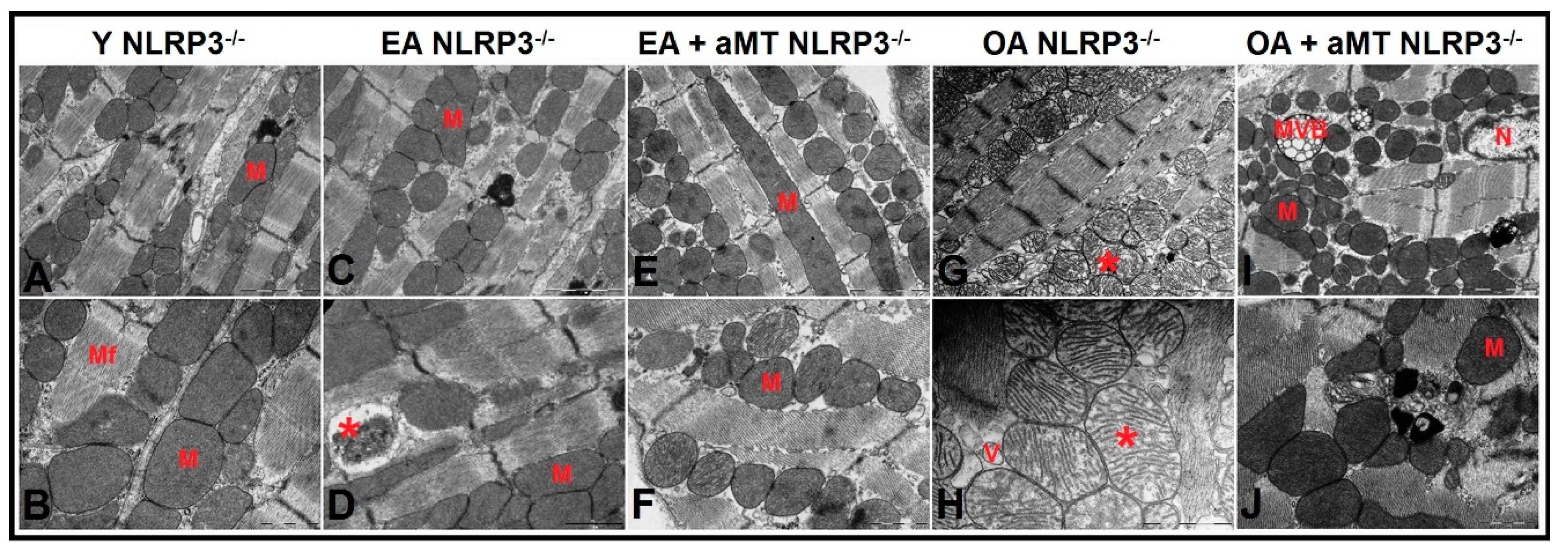
3.5. NLRP3 Deficiency and Melatonin Therapy Improved Mitochondria Ultrastructure Altered by Age in Cardiac Muscle
3.6. Lack of NLRP3 Reduced Mitochondria Number Loss and Mitochondrial Damage, an Effect Shared by Melatonin
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chiao, Y.A.; Rabinovitch, P.S. The Aging Heart. Cold Spring Harb. Perspect. Med. 2015, 5, a025148. [Google Scholar] [CrossRef]
- Gómez-Pardo, E.; Fernández-Alvira, J.M.; Vilanova, M.; Haro, D.; Martínez, R.; Carvajal, I.; Carral, V.; Rodríguez, C.; de Miguel, M.; Bodega, P.; et al. A Comprehensive Lifestyle Peer Group-Based Intervention on Cardiovascular Risk Factors: The Randomized Controlled Fifty-Fifty Program. J. Am. Coll. Cardiol. 2016, 67, 476–485. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-Y.; Du, M.; Dolence, E.K.; Fang, C.X.; Mayer, G.E.; Ceylan-Isik, A.F.; LaCour, K.H.; Yang, X.; Wilbert, C.J.; Sreejayan, N.; et al. Aging induces cardiac diastolic dysfunction, oxidative stress, accumulation of advanced glycation endproducts and protein modification. Aging Cell 2005, 4, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Go, A.S.; Mozaffarian, D.; Roger, V.L.; Benjamin, E.J.; Berry, J.D.; Blaha, M.J.; Dai, S.; Ford, E.S.; Fox, C.S.; Franco, S.; et al. Heart disease and stroke statistics—2014 update: A report from the American Heart Association. Circulation 2014, 129, e28–e292. [Google Scholar] [CrossRef] [PubMed]
- Lakatta, E.G. Cardiovascular ageing in health sets the stage for cardiovascular disease. Heart Lung Circ. 2002, 11, 76–91. [Google Scholar] [CrossRef]
- Forman, K.; Vara, E.; García, C.; Kireev, R.; Cuesta, S.; Acuña-Castroviejo, D.; Tresguerres, J.A.F. Influence of aging and growth hormone on different members of the NFkB family and IkB expression in the heart from a murine model of senescence-accelerated aging. Exp. Gerontol. 2016, 73, 114–120. [Google Scholar] [CrossRef]
- Csiszar, A.; Wang, M.; Lakatta, E.G.; Ungvari, Z. Inflammation and endothelial dysfunction during aging: Role of NF-kappaB. J. Appl. Physiol. 2008, 105, 1333–1341. [Google Scholar] [CrossRef]
- Shi, Y.; Camici, G.G.; Lüscher, T.F. Cardiovascular determinants of life span. Pflugers Arch. 2010, 459, 315–324. [Google Scholar] [CrossRef]
- Ferrucci, L.; Fabbri, E. Inflammageing: Chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 2018, 15, 505–522. [Google Scholar] [CrossRef]
- Franceschi, C.; Campisi, J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2014, 69 (Suppl. 1), S4–S9. [Google Scholar] [CrossRef]
- Rodríguez, M.I.; Carretero, M.; Escames, G.; López, L.C.; Maldonado, M.D.; Tan, D.-X.; Reiter, R.J.; Acuña-Castroviejo, D. Chronic melatonin treatment prevents age-dependent cardiac mitochondrial dysfunction in senescence-accelerated mice. Free Radic. Res. 2007, 41, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, M.I.; Escames, G.; López, L.C.; García, J.A.; Ortiz, F.; López, A.; Acuña-Castroviejo, D. Melatonin administration prevents cardiac and diaphragmatic mitochondrial oxidative damage in senescence-accelerated mice. J. Endocrinol. 2007, 194, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef]
- Escames, G.; López, L.C.; García, J.A.; García-Corzo, L.; Ortiz, F.; Acuña-Castroviejo, D. Mitochondrial DNA and inflammatory diseases. Hum. Genet. 2012, 131, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.N.; Zhang, Y.; Ren, J. Mitophagy, Mitochondrial Dynamics, and Homeostasis in Cardiovascular Aging. Oxid. Med. Cell. Longev. 2019, 2019, 9825061. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Schmidlin, C.J.; Dodson, M.B.; Madhavan, L.; Zhang, D.D. Redox regulation by NRF2 in aging and disease. Free Radic. Biol. Med. 2019, 134, 702–707. [Google Scholar] [CrossRef]
- Zhang, H.; Davies, K.J.A.; Forman, H.J. Oxidative stress response and Nrf2 signaling in aging. Free Radic. Biol. Med. 2015, 88, 314–336. [Google Scholar] [CrossRef]
- Reiter, R.J. Melatonin: The chemical expression of darkness. Mol. Cell. Endocrinol. 1991, 79, C153–C158. [Google Scholar] [CrossRef]
- Acuña-Castroviejo, D.; Escames, G.; Venegas, C.; Díaz-Casado, M.E.; Lima-Cabello, E.; López, L.C.; Rosales-Corral, S.; Tan, D.-X.; Reiter, R.J. Extrapineal melatonin: Sources, regulation, and potential functions. Cell. Mol. Life Sci. 2014, 71, 2997–3025. [Google Scholar] [CrossRef]
- Acuña-Castroviejo, D.; Noguiera-Navarro, M.T.; Reiter, R.J.; Escames, G. Melatonin actions in the heart; more than a hormone. Melatonin Res. 2018, 1, 21–26. [Google Scholar] [CrossRef]
- García, J.A.; Volt, H.; Venegas, C.; Doerrier, C.; Escames, G.; López, L.C.; Acuña-Castroviejo, D. Disruption of the NF-κB/NLRP3 connection by melatonin requires retinoid-related orphan receptor-α and blocks the septic response in mice. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2015, 29, 3863–3875. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Mayo, J.C.; Tan, D.-X.; Sainz, R.M.; Alatorre-Jimenez, M.; Qin, L. Melatonin as an antioxidant: Under promises but over delivers. J. Pineal Res. 2016, 61, 253–278. [Google Scholar] [CrossRef] [PubMed]
- Volt, H.; García, J.A.; Doerrier, C.; Díaz-Casado, M.E.; Guerra-Librero, A.; López, L.C.; Escames, G.; Tresguerres, J.A.; Acuña-Castroviejo, D. Same molecule but different expression: Aging and sepsis trigger NLRP3 inflammasome activation, a target of melatonin. J. Pineal Res. 2016, 60, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Escames, G.; León, J.; Macías, M.; Khaldy, H.; Acuña-Castroviejo, D. Melatonin counteracts lipopolysaccharide-induced expression and activity of mitochondrial nitric oxide synthase in rats. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2003, 17, 932–934. [Google Scholar] [CrossRef]
- López, A.; García, J.A.; Escames, G.; Venegas, C.; Ortiz, F.; López, L.C.; Acuña-Castroviejo, D. Melatonin protects the mitochondria from oxidative damage reducing oxygen consumption, membrane potential, and superoxide anion production. J. Pineal Res. 2009, 46, 188–198. [Google Scholar] [CrossRef]
- Martín, M.; Macías, M.; Escames, G.; León, J.; Acuña-Castroviejo, D. Melatonin but not vitamins C and E maintains glutathione homeostasis in t-butyl hydroperoxide-induced mitochondrial oxidative stress. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2000, 14, 1677–1679. [Google Scholar] [CrossRef]
- Martín, M.; Macías, M.; León, J.; Escames, G.; Khaldy, H.; Acuña-Castroviejo, D. Melatonin increases the activity of the oxidative phosphorylation enzymes and the production of ATP in rat brain and liver mitochondria. Int. J. Biochem. Cell Biol. 2002, 34, 348–357. [Google Scholar] [CrossRef]
- Rahim, I.; Djerdjouri, B.; Sayed, R.K.; Fernández-Ortiz, M.; Fernández-Gil, B.; Hidalgo-Gutiérrez, A.; López, L.C.; Escames, G.; Reiter, R.J.; Acuña-Castroviejo, D. Melatonin administration to wild-type mice and nontreated NLRP3 mutant mice share similar inhibition of the inflammatory response during sepsis. J. Pineal Res. 2017, 63. [Google Scholar] [CrossRef]
- Forman, K.; Vara, E.; García, C.; Kireev, R.; Cuesta, S.; Acuña-Castroviejo, D.; Tresguerres, J.A.F. Beneficial effects of melatonin on cardiological alterations in a murine model of accelerated aging. J. Pineal Res. 2010, 49, 312–320. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Hu, W.; Lu, C.; Ma, Z.; Jiang, S.; Gu, C.; Acuña-Castroviejo, D.; Yang, Y. Targeting NLRP3 (Nucleotide-Binding Domain, Leucine-Rich-Containing Family, Pyrin Domain-Containing-3) Inflammasome in Cardiovascular Disorders. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2765–2779. [Google Scholar] [CrossRef] [PubMed]
- Bachmanov, A.A.; Reed, D.R.; Beauchamp, G.K.; Tordoff, M.G. Food intake, water intake, and drinking spout side preference of 28 mouse strains. Behav. Genet. 2002, 32, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, M.I.; Escames, G.; López, L.C.; López, A.; García, J.A.; Ortiz, F.; Sánchez, V.; Romeu, M.; Acuña-Castroviejo, D. Improved mitochondrial function and increased life span after chronic melatonin treatment in senescent prone mice. Exp. Gerontol. 2008, 43, 749–756. [Google Scholar] [CrossRef]
- Carretero, M.; Escames, G.; López, L.C.; Venegas, C.; Dayoub, J.C.; García, L.; Acuña-Castroviejo, D. Long-term melatonin administration protects brain mitochondria from aging. J. Pineal Res. 2009, 47, 192–200. [Google Scholar] [CrossRef]
- Martín, M.; Macías, M.; Escames, G.; Reiter, R.J.; Agapito, M.T.; Ortiz, G.G.; Acuña-Castroviejo, D. Melatonin-induced increased activity of the respiratory chain complexes I and IV can prevent mitochondrial damage induced by ruthenium red in vivo. J. Pineal Res. 2000, 28, 242–248. [Google Scholar] [CrossRef]
- Khaldy, H.; León, J.; Escames, G.; Bikjdaouene, L.; García, J.J.; Acuña-Castroviejo, D. Circadian rhythms of dopamine and dihydroxyphenyl acetic acid in the mouse striatum: Effects of pinealectomy and of melatonin treatment. Neuroendocrinology 2002, 75, 201–208. [Google Scholar] [CrossRef]
- López, A.; Ortiz, F.; Doerrier, C.; Venegas, C.; Fernández-Ortiz, M.; Aranda, P.; Díaz-Casado, M.E.; Fernández-Gil, B.; Barriocanal-Casado, E.; Escames, G.; et al. Mitochondrial impairment and melatonin protection in parkinsonian mice do not depend of inducible or neuronal nitric oxide synthases. PLoS ONE 2017, 12, e0183090. [Google Scholar] [CrossRef]
- Dimauro, I.; Pearson, T.; Caporossi, D.; Jackson, M.J. A simple protocol for the subcellular fractionation of skeletal muscle cells and tissue. BMC Res. Notes 2012, 5, 513. [Google Scholar] [CrossRef]
- Reynolds, E.S. The use of lead citrate at high pH as an electron-opaque stain in electron microscopy. J. Cell Biol. 1963, 17, 208–212. [Google Scholar] [CrossRef]
- Lee, J.; Giordano, S.; Zhang, J. Autophagy, mitochondria and oxidative stress: Cross-talk and redox signalling. Biochem. J. 2012, 441, 523–540. [Google Scholar] [CrossRef] [PubMed]
- Quarles, E.K.; Dai, D.-F.; Tocchi, A.; Basisty, N.; Gitari, L.; Rabinovitch, P.S. Quality control systems in cardiac aging. Ageing Res. Rev. 2015, 23, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Bonafè, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Guerrero, J.M.; Garcia, J.J.; Acuña-Castroviejo, D. Reactive oxygen intermediates, molecular damage, and aging. Relation to melatonin. Ann. N. Y. Acad. Sci. 1998, 854, 410–424. [Google Scholar] [CrossRef]
- Hardeland, R. Aging, Melatonin, and the Pro- and Anti-Inflammatory Networks. Int. J. Mol. Sci. 2019, 20, 1223. [Google Scholar] [CrossRef] [PubMed]
- Ahuja, P.; Wanagat, J.; Wang, Z.; Wang, Y.; Liem, D.A.; Ping, P.; Antoshechkin, I.A.; Margulies, K.B.; Maclellan, W.R. Divergent mitochondrial biogenesis responses in human cardiomyopathy. Circulation 2013, 127, 1957–1967. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zou, X.; Feng, Z.; Luo, C.; Liu, J.; Li, H.; Chang, L.; Wang, H.; Li, Y.; Long, J.; et al. Evidence for association of mitochondrial metabolism alteration with lipid accumulation in aging rats. Exp. Gerontol. 2014, 56, 3–12. [Google Scholar] [CrossRef]
- Rosca, M.G.; Hoppel, C.L. Mitochondria in heart failure. Cardiovasc. Res. 2010, 88, 40–50. [Google Scholar] [CrossRef]
- Stotland, A.; Gottlieb, R.A. α-MHC MitoTimer mouse: In vivo mitochondrial turnover model reveals remarkable mitochondrial heterogeneity in the heart. J. Mol. Cell. Cardiol. 2016, 90, 53–58. [Google Scholar] [CrossRef]
- Papanicolaou, K.N.; Khairallah, R.J.; Ngoh, G.A.; Chikando, A.; Luptak, I.; O’Shea, K.M.; Riley, D.D.; Lugus, J.J.; Colucci, W.S.; Lederer, W.J.; et al. Mitofusin-2 maintains mitochondrial structure and contributes to stress-induced permeability transition in cardiac myocytes. Mol. Cell. Biol. 2011, 31, 1309–1328. [Google Scholar] [CrossRef]
- Chen, L.; Gong, Q.; Stice, J.P.; Knowlton, A.A. Mitochondrial OPA1, apoptosis, and heart failure. Cardiovasc. Res. 2009, 84, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Mihara, K.; Chen, Y.; Scorrano, L.; Dorn, G.W. Mitochondrial Fission and Fusion Factors Reciprocally Orchestrate Mitophagic Culling in Mouse Hearts and Cultured Fibroblasts. Cell Metab. 2015, 21, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Yasukawa, K.; Oshiumi, H.; Takeda, M.; Ishihara, N.; Yanagi, Y.; Seya, T.; Kawabata, S.; Koshiba, T. Mitofusin 2 inhibits mitochondrial antiviral signaling. Sci. Signal. 2009, 2, ra47. [Google Scholar] [CrossRef] [PubMed]
- Ichinohe, T.; Yamazaki, T.; Koshiba, T.; Yanagi, Y. Mitochondrial protein mitofusin 2 is required for NLRP3 inflammasome activation after RNA virus infection. Proc. Natl. Acad. Sci. USA 2013, 110, 17963–17968. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Jiang, W.; Yan, Y.; Gong, T.; Han, J.; Tian, Z.; Zhou, R. RNA viruses promote activation of the NLRP3 inflammasome through a RIP1-RIP3-DRP1 signaling pathway. Nat. Immunol. 2014, 15, 1126–1133. [Google Scholar] [CrossRef]
- Park, S.; Won, J.-H.; Hwang, I.; Hong, S.; Lee, H.K.; Yu, J.-W. Defective mitochondrial fission augments NLRP3 inflammasome activation. Sci. Rep. 2015, 5, 15489. [Google Scholar] [CrossRef]
- Szabadkai, G.; Simoni, A.M.; Chami, M.; Wieckowski, M.R.; Youle, R.J.; Rizzuto, R. Drp-1-dependent division of the mitochondrial network blocks intraorganellar Ca2+ waves and protects against Ca2+-mediated apoptosis. Mol. Cell 2004, 16, 59–68. [Google Scholar] [CrossRef]
- Horng, T. Calcium signaling and mitochondrial destabilization in the triggering of the NLRP3 inflammasome. Trends Immunol. 2014, 35, 253–261. [Google Scholar] [CrossRef]
- Ma, S.; Dong, Z. Melatonin Attenuates Cardiac Reperfusion Stress by Improving OPA1-Related Mitochondrial Fusion in a Yap-Hippo Pathway-Dependent Manner. J. Cardiovasc. Pharmacol. 2019, 73, 27–39. [Google Scholar] [CrossRef]
- Pei, H.; Du, J.; Song, X.; He, L.; Zhang, Y.; Li, X.; Qiu, C.; Zhang, Y.; Hou, J.; Feng, J.; et al. Melatonin prevents adverse myocardial infarction remodeling via Notch1/Mfn2 pathway. Free Radic. Biol. Med. 2016, 97, 408–417. [Google Scholar] [CrossRef]
- Suwanjang, W.; Abramov, A.Y.; Charngkaew, K.; Govitrapong, P.; Chetsawang, B. Melatonin prevents cytosolic calcium overload, mitochondrial damage and cell death due to toxically high doses of dexamethasone-induced oxidative stress in human neuroblastoma SH-SY5Y cells. Neurochem. Int. 2016, 97, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Chuang, J.-I.; Pan, I.-L.; Hsieh, C.-Y.; Huang, C.-Y.; Chen, P.-C.; Shin, J.W. Melatonin prevents the dynamin-related protein 1-dependent mitochondrial fission and oxidative insult in the cortical neurons after 1-methyl-4-phenylpyridinium treatment. J. Pineal Res. 2016, 61, 230–240. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Pi, H.; Zhang, L.; Zhang, N.; Li, Y.; Zhang, H.; Tang, J.; Li, H.; Feng, M.; Deng, P.; et al. Melatonin prevents abnormal mitochondrial dynamics resulting from the neurotoxicity of cadmium by blocking calcium-dependent translocation of Drp1 to the mitochondria. J. Pineal Res. 2016, 60, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Parameyong, A.; Charngkaew, K.; Govitrapong, P.; Chetsawang, B. Melatonin attenuates methamphetamine-induced disturbances in mitochondrial dynamics and degeneration in neuroblastoma SH-SY5Y cells. J. Pineal Res. 2013, 55, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Parameyong, A.; Govitrapong, P.; Chetsawang, B. Melatonin attenuates the mitochondrial translocation of mitochondrial fission proteins and Bax, cytosolic calcium overload and cell death in methamphetamine-induced toxicity in neuroblastoma SH-SY5Y cells. Mitochondrion 2015, 24, 1–8. [Google Scholar] [CrossRef]
- Ding, M.; Feng, N.; Tang, D.; Feng, J.; Li, Z.; Jia, M.; Liu, Z.; Gu, X.; Wang, Y.; Fu, F.; et al. Melatonin prevents Drp1-mediated mitochondrial fission in diabetic hearts through SIRT1-PGC1α pathway. J. Pineal Res. 2018, 65, e12491. [Google Scholar] [CrossRef]
- Zhou, H.; Zhang, Y.; Hu, S.; Shi, C.; Zhu, P.; Ma, Q.; Jin, Q.; Cao, F.; Tian, F.; Chen, Y. Melatonin protects cardiac microvasculature against ischemia/reperfusion injury via suppression of mitochondrial fission-VDAC1-HK2-mPTP-mitophagy axis. J. Pineal Res. 2017, 63, e12413. [Google Scholar] [CrossRef]
- Ding, M.; Ning, J.; Feng, N.; Li, Z.; Liu, Z.; Wang, Y.; Wang, Y.; Li, X.; Huo, C.; Jia, X.; et al. Dynamin-related protein 1-mediated mitochondrial fission contributes to post-traumatic cardiac dysfunction in rats and the protective effect of melatonin. J. Pineal Res. 2018, 64. [Google Scholar] [CrossRef]
- Cho, H.M.; Ryu, J.R.; Jo, Y.; Seo, T.W.; Choi, Y.N.; Kim, J.H.; Chung, J.M.; Cho, B.; Kang, H.C.; Yu, S.-W.; et al. Drp1-Zip1 Interaction Regulates Mitochondrial Quality Surveillance System. Mol. Cell 2019, 73, 364–376.e8. [Google Scholar] [CrossRef] [PubMed]
- Rana, A.; Oliveira, M.P.; Khamoui, A.V.; Aparicio, R.; Rera, M.; Rossiter, H.B.; Walker, D.W. Promoting Drp1-mediated mitochondrial fission in midlife prolongs healthy lifespan of Drosophila melanogaster. Nat. Commun. 2017, 8, 448. [Google Scholar] [CrossRef] [PubMed]
- Acuña Castroviejo, D.; López, L.C.; Escames, G.; López, A.; García, J.A.; Reiter, R.J. Melatonin-mitochondria interplay in health and disease. Curr. Top. Med. Chem. 2011, 11, 221–240. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; Mariño, G.; Kroemer, G. Autophagy and Aging. Cell 2011, 146, 682–695. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M. Autophagy in neurons: It is not all about food. Trends Mol. Med. 2006, 12, 461–464. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Chong, S.Y.; Lim, A.; Singh, B.K.; Sinha, R.A.; Salmon, A.B.; Yen, P.M. Changes in macroautophagy, chaperone-mediated autophagy, and mitochondrial metabolism in murine skeletal and cardiac muscle during aging. Aging 2017, 9, 583–599. [Google Scholar] [CrossRef]
- Shirakabe, A.; Ikeda, Y.; Sciarretta, S.; Zablocki, D.K.; Sadoshima, J. Aging and Autophagy in the Heart. Circ. Res. 2016, 118, 1563–1576. [Google Scholar] [CrossRef]
- Malicdan, M.C.; Noguchi, S.; Nishino, I. Chapter 19 Monitoring Autophagy in Muscle Diseases. Methods Enzymol. 2009, 453, 379–396. [Google Scholar] [CrossRef]
- Taneike, M.; Yamaguchi, O.; Nakai, A.; Hikoso, S.; Takeda, T.; Mizote, I.; Oka, T.; Tamai, T.; Oyabu, J.; Murakawa, T.; et al. Inhibition of autophagy in the heart induces age-related cardiomyopathy. Autophagy 2010, 6, 600–606. [Google Scholar] [CrossRef]
- Wohlgemuth, S.E.; Julian, D.; Akin, D.E.; Fried, J.; Toscano, K.; Leeuwenburgh, C.; Dunn, W.A., Jr. Autophagy in the Heart and Liver During Normal Aging and Calorie Restriction. Rejuvenation Res. 2007, 10, 281–292. [Google Scholar] [CrossRef]
- Hua, Y.; Zhang, Y.; Ceylan-Isik, A.F.; Wold, L.E.; Nunn, J.M.; Ren, J. Chronic akt activation accentuates aging-induced cardiac hypertrophy and myocardial contractile dysfunction: Role of autophagy. Basic Res. Cardiol. 2011, 106, 1173–1191. [Google Scholar] [CrossRef]
- Dai, D.-F.; Chiao, Y.A.; Marcinek, D.J.; Szeto, H.H.; Rabinovitch, P.S. Mitochondrial oxidative stress in aging and healthspan. Longev. Healthspan 2014, 3, 6. [Google Scholar] [CrossRef]
- Boengler, K.; Schulz, R.; Heusch, G. Loss of cardioprotection with ageing. Cardiovasc. Res. 2009, 83, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ma, Y.; Song, L.; Yu, L.; Zhang, L.; Zhang, Y.; Xing, Y.; Yin, Y.; Ma, H. SIRT3 deficiency exacerbates p53/Parkin-mediated mitophagy inhibition and promotes mitochondrial dysfunction: Implication for aged hearts. Int. J. Mol. Med. 2018, 41, 3517–3526. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Ojala, J.; Kaarniranta, K.; Kauppinen, A. Mitochondrial dysfunction and oxidative stress activate inflammasomes: Impact on the aging process and age-related diseases. Cell. Mol. Life Sci. 2012, 69, 2999–3013. [Google Scholar] [CrossRef]
- Marín-Aguilar, F.; Lechuga-Vieco, A.V.; Alcocer-Gómez, E.; Castejón-Vega, B.; Lucas, J.; Garrido, C.; Peralta-Garcia, A.; Pérez-Pulido, A.J.; Varela-López, A.; Quiles, J.L.; et al. NLRP3 inflammasome suppression improves longevity and prevents cardiac aging in male mice. Aging Cell 2020, 19, e13050. [Google Scholar] [CrossRef]
- Kang, J.-W.; Hong, J.-M.; Lee, S.-M. Melatonin enhances mitophagy and mitochondrial biogenesis in rats with carbon tetrachloride-induced liver fibrosis. J. Pineal Res. 2016, 60, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Chao, H.; Li, Z.; Xu, X.; Liu, Y.; Hou, L.; Liu, N.; Ji, J. Melatonin attenuates traumatic brain injury-induced inflammation: A possible role for mitophagy. J. Pineal Res. 2016, 61, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.; Deng, Y.; Pan, Y.-Y.; Wang, Z.-H.; Ren, J.; Guo, X.-L.; Yuan, X.; Shang, J.; Liu, H.-G. Melatonin protects against chronic intermittent hypoxia-induced cardiac hypertrophy by modulating autophagy through the 5′ adenosine monophosphate-activated protein kinase pathway. Biochem. Biophys. Res. Commun. 2015, 464, 975–981. [Google Scholar] [CrossRef] [PubMed]
- Prieto-Domínguez, N.; Ordóñez, R.; Fernández, A.; Méndez-Blanco, C.; Baulies, A.; Garcia-Ruiz, C.; Fernández-Checa, J.C.; Mauriz, J.L.; González-Gallego, J. Melatonin-induced increase in sensitivity of human hepatocellular carcinoma cells to sorafenib is associated with reactive oxygen species production and mitophagy. J. Pineal Res. 2016, 61, 396–407. [Google Scholar] [CrossRef]
- de Luxán-Delgado, B.; Potes, Y.; Rubio-González, A.; Caballero, B.; Solano, J.J.; Fernández-Fernández, M.; Bermúdez, M.; Rodrigues Moreira Guimarães, M.; Vega-Naredo, I.; Boga, J.A.; et al. Melatonin reduces endoplasmic reticulum stress and autophagy in liver of leptin-deficient mice. J. Pineal Res. 2016, 61, 108–123. [Google Scholar] [CrossRef]
- San-Miguel, B.; Crespo, I.; Sánchez, D.I.; González-Fernández, B.; Ortiz de Urbina, J.J.; Tuñón, M.J.; González-Gallego, J. Melatonin inhibits autophagy and endoplasmic reticulum stress in mice with carbon tetrachloride-induced fibrosis. J. Pineal Res. 2015, 59, 151–162. [Google Scholar] [CrossRef]
- Su, L.-Y.; Li, H.; Lv, L.; Feng, Y.-M.; Li, G.-D.; Luo, R.; Zhou, H.-J.; Lei, X.-G.; Ma, L.; Li, J.-L.; et al. Melatonin attenuates MPTP-induced neurotoxicity via preventing CDK5-mediated autophagy and SNCA/α-synuclein aggregation. Autophagy 2015, 11, 1745–1759. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Chen, J.; Feng, J.; Zhang, R.; Fan, M.; Han, D.; Li, X.; Li, C.; Ren, J.; Wang, Y.; et al. Melatonin Ameliorates the Progression of Atherosclerosis via Mitophagy Activation and NLRP3 Inflammasome Inhibition. Oxid. Med. Cell. Longev. 2018, 2018, 9286458. [Google Scholar] [CrossRef] [PubMed]
- Caballero, B.; Vega-Naredo, I.; Sierra, V.; DeGonzalo-Calvo, D.; Medrano-Campillo, P.; Guerrero, J.M.; Tolivia, D.; Rodríguez-Colunga, M.J.; Coto-Montes, A. Autophagy upregulation and loss of NF-kappaB in oxidative stress-related immunodeficient SAMP8 mice. Mech. Ageing Dev. 2009, 130, 722–730. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.N. Autophagy as a second level protective process in conferring resistance to environmentally-induced oxidative stress. Autophagy 2008, 4, 254–256. [Google Scholar] [CrossRef]
- Houtkooper, R.H.; Mouchiroud, L.; Ryu, D.; Moullan, N.; Katsyuba, E.; Knott, G.; Williams, R.W.; Auwerx, J. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 2013, 497, 451–457. [Google Scholar] [CrossRef]
- Leduc-Gaudet, J.-P.; Picard, M.; St-Jean Pelletier, F.; Sgarioto, N.; Auger, M.-J.; Vallée, J.; Robitaille, R.; St-Pierre, D.H.; Gouspillou, G. Mitochondrial morphology is altered in atrophied skeletal muscle of aged mice. Oncotarget 2015, 6, 17923–17937. [Google Scholar] [CrossRef]
- Moehle, E.A.; Shen, K.; Dillin, A. Mitochondrial proteostasis in the context of cellular and organismal health and aging. J. Biol. Chem. 2019, 294, 5396–5407. [Google Scholar] [CrossRef]
- Sebastián, D.; Sorianello, E.; Segalés, J.; Irazoki, A.; Ruiz-Bonilla, V.; Sala, D.; Planet, E.; Berenguer-Llergo, A.; Muñoz, J.P.; Sánchez-Feutrie, M.; et al. Mfn2 deficiency links age-related sarcopenia and impaired autophagy to activation of an adaptive mitophagy pathway. EMBO J. 2016, 35, 1677–1693. [Google Scholar] [CrossRef]
- Gustafsson, A.B.; Gottlieb, R.A. Mechanisms of apoptosis in the heart. J. Clin. Immunol. 2003, 23, 447–459. [Google Scholar] [CrossRef]
- Kannan, K.; Jain, S.K. Oxidative stress and apoptosis. Pathophysiology 2000, 7, 153–163. [Google Scholar] [CrossRef]
- Aggarwal, B.B. Apoptosis and nuclear factor-κb: A tale of association and dissociation. Biochem. Pharmacol. 2000, 60, 1033–1039. [Google Scholar] [CrossRef]
- D’Oria, R.; Schipani, R.; Leonardini, A.; Natalicchio, A.; Perrini, S.; Cignarelli, A.; Laviola, L.; Giorgino, F. The Role of Oxidative Stress in Cardiac Disease: From Physiological Response to Injury Factor. Oxid. Med. Cell. Longev. 2020, 2020, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Haupt, S.; Berger, M.; Goldberg, Z.; Haupt, Y. Apoptosis—The p53 network. J. Cell Sci. 2003, 116, 4077–4085. [Google Scholar] [CrossRef] [PubMed]
- McDonnell, M.A.; Wang, D.; Khan, S.M.; Vander Heiden, M.G.; Kelekar, A. Caspase-9 is activated in a cytochrome c-independent manner early during TNFalpha-induced apoptosis in murine cells. Cell Death Differ. 2003, 10, 1005–1015. [Google Scholar] [CrossRef] [PubMed]
- D’Sa-Eipper, C.; Leonard, J.R.; Putcha, G.; Zheng, T.S.; Flavell, R.A.; Rakic, P.; Kuida, K.; Roth, K.A. DNA damage-induced neural precursor cell apoptosis requires p53 and caspase 9 but neither Bax nor caspase 3. Development 2001, 128, 137–146. [Google Scholar] [PubMed]
- Cañadas-Lozano, D.; Marín-Aguilar, F.; Castejón-Vega, B.; Ryffel, B.; Navarro-Pando, J.M.; Ruiz-Cabello, J.; Alcocer-Gómez, E.; Bullón, P.; Cordero, M.D. Blockade of the NLRP3 inflammasome improves metabolic health and lifespan in obese mice. GeroScience 2020, 42, 715–725. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Galluzzi, L.; Freije, J.M.P.; Madeo, F.; Kroemer, G. Metabolic Control of Longevity. Cell 2016, 166, 802–821. [Google Scholar] [CrossRef]
- Allam, R.; Lawlor, K.E.; Yu, E.C.-W.; Mildenhall, A.L.; Moujalled, D.M.; Lewis, R.S.; Ke, F.; Mason, K.D.; White, M.J.; Stacey, K.J.; et al. Mitochondrial apoptosis is dispensable for NLRP3 inflammasome activation but non-apoptotic caspase-8 is required for inflammasome priming. EMBO Rep. 2014, 15, 982–990. [Google Scholar] [CrossRef]
- McDonnell, M.A.; Abedin, M.J.; Melendez, M.; Platikanova, T.N.; Ecklund, J.R.; Ahmed, K.; Kelekar, A. Phosphorylation of murine caspase-9 by the protein kinase casein kinase 2 regulates its cleavage by caspase-8. J. Biol. Chem. 2008, 283, 20149–20158. [Google Scholar] [CrossRef]
- Vince, J.E.; De Nardo, D.; Gao, W.; Vince, A.J.; Hall, C.; McArthur, K.; Simpson, D.; Vijayaraj, S.; Lindqvist, L.M.; Bouillet, P.; et al. The Mitochondrial Apoptotic Effectors BAX/BAK Activate Caspase-3 and -7 to Trigger NLRP3 Inflammasome and Caspase-8 Driven IL-1β Activation. Cell Rep. 2018, 25, 2339–2353.e4. [Google Scholar] [CrossRef]
- Acuna-Castroviejo, D.; Escames, G.; Rodriguez, M.I.; Lopez, L.C. Melatonin role in the mitochondrial function. Front. Biosci. 2007, 12, 947–963. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. The biologic clock: The mitochondria? J. Am. Geriatr. Soc. 1972, 20, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Miquel, J.; Economos, A.C.; Fleming, J.; Johnson, J.E. Mitochondrial role in cell aging. Exp. Gerontol. 1980, 15, 575–591. [Google Scholar] [CrossRef]
- Lewis, K.N.; Mele, J.; Hayes, J.D.; Buffenstein, R. Nrf2, a guardian of healthspan and gatekeeper of species longevity. Integr. Comp. Biol. 2010, 50, 829–843. [Google Scholar] [CrossRef]
- Bruns, D.R.; Drake, J.C.; Biela, L.M.; Peelor, F.F.; Miller, B.F.; Hamilton, K.L. Nrf2 Signaling and the Slowed Aging Phenotype: Evidence from Long-Lived Models. Oxid. Med. Cell. Longev. 2015, 2015, 732596. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Abramov, A.Y. The emerging role of Nrf2 in mitochondrial function. Free Radic. Biol. Med. 2015, 88, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Tomobe, K.; Shinozuka, T.; Kuroiwa, M.; Nomura, Y. Age-related changes of Nrf2 and phosphorylated GSK-3β in a mouse model of accelerated aging (SAMP8). Arch. Gerontol. Geriatr. 2012, 54, e1–e7. [Google Scholar] [CrossRef]
- Ma, Q.; Battelli, L.; Hubbs, A.F. Multiorgan Autoimmune Inflammation, Enhanced Lymphoproliferation, and Impaired Homeostasis of Reactive Oxygen Species in Mice Lacking the Antioxidant-Activated Transcription Factor Nrf2. Am. J. Pathol. 2006, 168, 1960–1974. [Google Scholar] [CrossRef]
- Reuland, D.J.; McCord, J.M.; Hamilton, K.L. The role of Nrf2 in the attenuation of cardiovascular disease. Exerc. Sport Sci. Rev. 2013, 41, 162–168. [Google Scholar] [CrossRef]
- Howden, R. Nrf2 and cardiovascular defense. Oxid. Med. Cell. Longev. 2013, 2013, 104308. [Google Scholar] [CrossRef]
- Xu, B.; Zhang, J.; Strom, J.; Lee, S.; Chen, Q.M. Myocardial ischemic reperfusion induces de novo Nrf2 protein translation. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2014, 1842, 1638–1647. [Google Scholar] [CrossRef] [PubMed]
- Strom, J.; Chen, Q.M. Loss of Nrf2 promotes rapid progression to heart failure following myocardial infarction. Toxicol. Appl. Pharmacol. 2017, 327, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Suh, J.H.; Shenvi, S.V.; Dixon, B.M.; Liu, H.; Jaiswal, A.K.; Liu, R.-M.; Hagen, T.M. Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid. Proc. Natl. Acad. Sci. USA 2004, 101, 3381–3386. [Google Scholar] [CrossRef]
- Shih, P.-H.; Yen, G.-C. Differential expressions of antioxidant status in aging rats: The role of transcriptional factor Nrf2 and MAPK signaling pathway. Biogerontology 2007, 8, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Ungvari, Z.; Bailey-Downs, L.; Sosnowska, D.; Gautam, T.; Koncz, P.; Losonczy, G.; Ballabh, P.; de Cabo, R.; Sonntag, W.E.; Csiszar, A. Vascular oxidative stress in aging: A homeostatic failure due to dysregulation of NRF2-mediated antioxidant response. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H363–H372. [Google Scholar] [CrossRef] [PubMed]
- Ungvari, Z.; Bailey-Downs, L.; Gautam, T.; Sosnowska, D.; Wang, M.; Monticone, R.E.; Telljohann, R.; Pinto, J.T.; de Cabo, R.; Sonntag, W.E.; et al. Age-Associated Vascular Oxidative Stress, Nrf2 Dysfunction, and NF-κB Activation in the Nonhuman Primate Macaca mulatta. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2011, 66A, 866–875. [Google Scholar] [CrossRef]
- Zucker, S.N.; Fink, E.E.; Bagati, A.; Mannava, S.; Bianchi-Smiraglia, A.; Bogner, P.N.; Wawrzyniak, J.A.; Foley, C.; Leonova, K.I.; Grimm, M.J.; et al. Nrf2 amplifies oxidative stress via induction of Klf9. Mol. Cell 2014, 53, 916–928. [Google Scholar] [CrossRef]
- Chhunchha, B.; Kubo, E.; Singh, D.P. Sulforaphane-Induced Klf9/Prdx6 Axis Acts as a Molecular Switch to Control Redox Signaling and Determines Fate of Cells. Cells 2019, 8, 1159. [Google Scholar] [CrossRef]
- Xu, X.; Zhang, L.; Ye, X.; Hao, Q.; Zhang, T.; Cui, G.; Yu, M. Nrf2/ARE pathway inhibits ROS-induced NLRP3 inflammasome activation in BV2 cells after cerebral ischemia reperfusion. Inflamm. Res. 2018, 67, 57–65. [Google Scholar] [CrossRef]
- Reiter, R.J.; Tan, D.X.; Manchester, L.C.; Qi, W. Biochemical reactivity of melatonin with reactive oxygen and nitrogen species: A review of the evidence. Cell Biochem. Biophys. 2001, 34, 237–256. [Google Scholar] [CrossRef]
- Rodriguez, C.; Mayo, J.C.; Sainz, R.M.; Antolín, I.; Herrera, F.; Martín, V.; Reiter, R.J. Regulation of antioxidant enzymes: A significant role for melatonin. J. Pineal Res. 2004, 36, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Tomás-Zapico, C.; Coto-Montes, A. A proposed mechanism to explain the stimulatory effect of melatonin on antioxidative enzymes. J. Pineal Res. 2005, 39, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Rahim, I.; Sayed, R.K.; Fernández-Ortiz, M.; Aranda-Martínez, P.; Guerra-Librero, A.; Fernández-Martínez, J.; Rusanova, I.; Escames, G.; Djerdjouri, B.; Acuña-Castroviejo, D. Melatonin alleviates sepsis-induced heart injury through activating the Nrf2 pathway and inhibiting the NLRP3 inflammasome. Naunyn. Schmiedebergs Arch. Pharmacol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Corsetti, G.; Pasini, E.; D’Antona, G.; Nisoli, E.; Flati, V.; Assanelli, D.; Dioguardi, F.S.; Bianchi, R. Morphometric Changes Induced by Amino Acid Supplementation in Skeletal and Cardiac Muscles of Old Mice. Am. J. Cardiol. 2008, 101, S26–S34. [Google Scholar] [CrossRef] [PubMed]
- Shigenaga, M.K.; Hagen, T.M.; Ames, B.N. Oxidative damage and mitochondrial decay in aging. Proc. Natl. Acad. Sci. USA 1994, 91, 10771–10778. [Google Scholar] [CrossRef] [PubMed]
- El’darov, C.M.; Vays, V.B.; Vangeli, I.M.; Kolosova, N.G.; Bakeeva, L.E. Morphometric examination of mitochondrial ultrastructure in aging cardiomyocytes. Biochemistry 2015, 80, 604–609. [Google Scholar] [CrossRef] [PubMed]
- Hefti, M.A.; Harder, B.A.; Eppenberger, H.M.; Schaub, M.C. Signaling pathways in cardiac myocyte hypertrophy. J. Mol. Cell. Cardiol. 1997, 29, 2873–2892. [Google Scholar] [CrossRef]
- Dobaczewski, M.; Chen, W.; Frangogiannis, N.G. Transforming growth factor (TGF)-β signaling in cardiac remodeling. J. Mol. Cell. Cardiol. 2011, 51, 600–606. [Google Scholar] [CrossRef]
- No, M.-H.; Choi, Y.; Cho, J.; Heo, J.-W.; Cho, E.-J.; Park, D.-H.; Kang, J.-H.; Kim, C.-J.; Seo, D.Y.; Han, J.; et al. Aging Promotes Mitochondria-Mediated Apoptosis in Rat Hearts. Life 2020, 10, 178. [Google Scholar] [CrossRef]
- Frenzei, H.; Feimann, J. Age-dependent structural changes in the myocardium of rats. A quantitative light- and electron-microscopic study on the right and left chamber wall. Mech. Ageing Dev. 1984, 27, 29–41. [Google Scholar] [CrossRef]
- Neubauer, S. The failing heart—An engine out of fuel. N. Engl. J. Med. 2007, 356, 1140–1151. [Google Scholar] [CrossRef] [PubMed]
- Sayed, R.K.A.; Fernández-Ortiz, M.; Diaz-Casado, M.E.; Aranda-Martínez, P.; Fernández-Martínez, J.; Guerra-Librero, A.; Escames, G.; López, L.C.; Alsaadawy, R.M.; Acuña-Castroviejo, D. Lack of NLRP3 Inflammasome Activation Reduces Age-Dependent Sarcopenia and Mitochondrial Dysfunction, Favoring the Prophylactic Effect of Melatonin. J. Gerontol. A. Biol. Sci. Med. Sci. 2019, 74, 1699–1708. [Google Scholar] [CrossRef] [PubMed]
- Kates, A.M.; Herrero, P.; Dence, C.; Soto, P.; Srinivasan, M.; Delano, D.G.; Ehsani, A.; Gropler, R.J. Impact of aging on substrate metabolism by the human heart. J. Am. Coll. Cardiol. 2003, 41, 293–299. [Google Scholar] [CrossRef]
- Hyyti, O.M.; Ledee, D.; Ning, X.-H.; Ge, M.; Portman, M.A. Aging impairs myocardial fatty acid and ketone oxidation and modifies cardiac functional and metabolic responses to insulin in mice. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H868–H875. [Google Scholar] [CrossRef]
- Ye, J.; Yu, M.; Zhang, K.; Liu, J.; Wang, Q.; Tao, P.; Jia, K.; Liao, M.; Ning, Z. Tissue-specific expression pattern and histological distribution of NLRP3 in Chinese yellow chicken. Vet. Res. Commun. 2015, 39, 171–177. [Google Scholar] [CrossRef]
- Huang, Z.; Yu, M.; Tong, S.; Jia, K.; Liu, R.; Wang, H.; Li, S.; Ning, Z. Tissue-specific expression of the NOD-like receptor protein 3 in BALB/c mice. J. Vet. Sci. 2014, 15, 173–177. [Google Scholar] [CrossRef]
- Pinto, A.R.; Paolicelli, R.; Salimova, E.; Gospocic, J.; Slonimsky, E.; Bilbao-Cortes, D.; Godwin, J.W.; Rosenthal, N.A. An Abundant Tissue Macrophage Population in the Adult Murine Heart with a Distinct Alternatively-Activated Macrophage Profile. PLoS ONE 2012, 7, e36814. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernández-Ortiz, M.; Sayed, R.K.A.; Fernández-Martínez, J.; Cionfrini, A.; Aranda-Martínez, P.; Escames, G.; de Haro, T.; Acuña-Castroviejo, D. Melatonin/Nrf2/NLRP3 Connection in Mouse Heart Mitochondria during Aging. Antioxidants 2020, 9, 1187. https://doi.org/10.3390/antiox9121187
Fernández-Ortiz M, Sayed RKA, Fernández-Martínez J, Cionfrini A, Aranda-Martínez P, Escames G, de Haro T, Acuña-Castroviejo D. Melatonin/Nrf2/NLRP3 Connection in Mouse Heart Mitochondria during Aging. Antioxidants. 2020; 9(12):1187. https://doi.org/10.3390/antiox9121187
Chicago/Turabian StyleFernández-Ortiz, Marisol, Ramy K. A. Sayed, José Fernández-Martínez, Antonia Cionfrini, Paula Aranda-Martínez, Germaine Escames, Tomás de Haro, and Darío Acuña-Castroviejo. 2020. "Melatonin/Nrf2/NLRP3 Connection in Mouse Heart Mitochondria during Aging" Antioxidants 9, no. 12: 1187. https://doi.org/10.3390/antiox9121187
APA StyleFernández-Ortiz, M., Sayed, R. K. A., Fernández-Martínez, J., Cionfrini, A., Aranda-Martínez, P., Escames, G., de Haro, T., & Acuña-Castroviejo, D. (2020). Melatonin/Nrf2/NLRP3 Connection in Mouse Heart Mitochondria during Aging. Antioxidants, 9(12), 1187. https://doi.org/10.3390/antiox9121187