Glutathione Supplementation as an Adjunctive Therapy in COVID-19

 and
and 
Abstract
1. Introduction
2. SARS-CoV-2 Microbiology
3. Transmission
4. COVID-19 Epidemiology
5. Clinical Presentation and Diagnosis
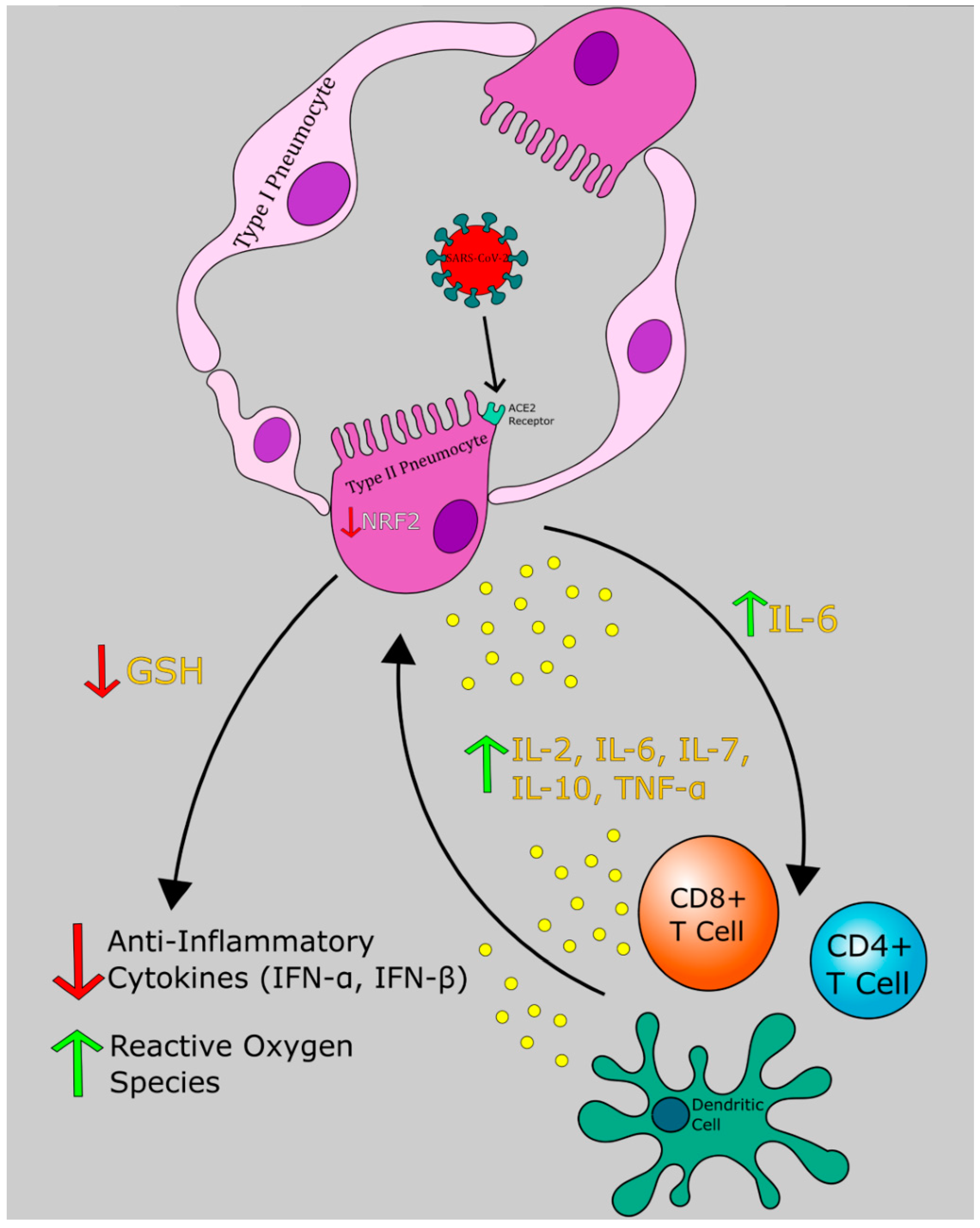
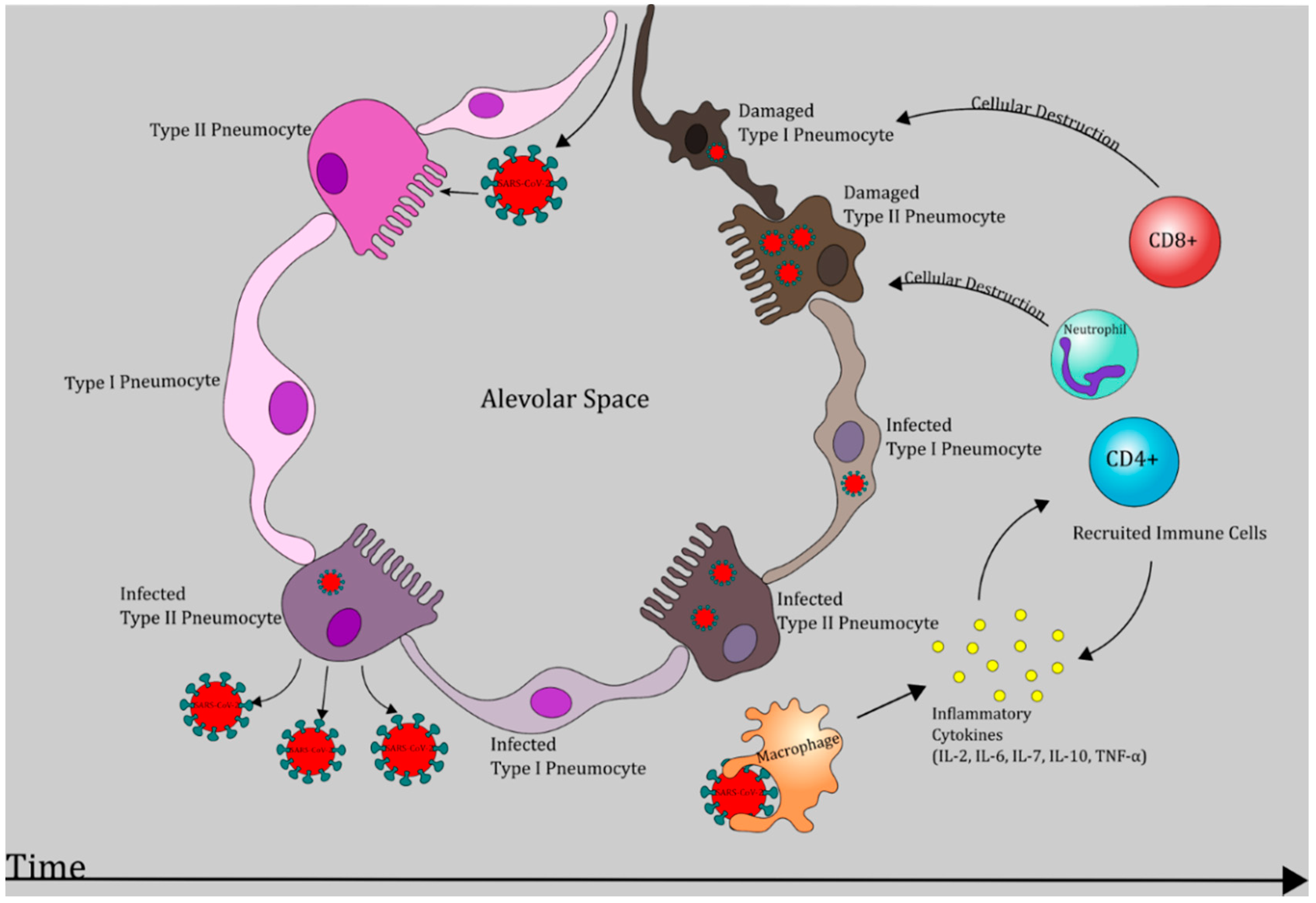
6. Pathogenesis of COVID-19: An Evolving Picture
6.1. Phase I: Respiratory Tract Infection
6.2. Phase II: Acute Inflammation
6.3. Phase II: Resolution or Severe Illness and Hypercoagulable State
6.3.1. Resolution
6.3.2. Severe Illness
7. Interleukin-6 and ARDS
8. Septic Shock and Hypercoagulability in COVID-19
9. Endogenous Glutathione Synthesis and Regulation the Role of Redox Cycle in Reducing Oxidative Stress Redox Cycle, Antioxidant Clearance, and Associated Isoenzymes Involved
10. Glutathione and Tuberculosis
11. Glutathione and Viral Infections
12. GSH and Pulmonary Disease
13. The Role of GSH in Ferroptosis
14. GSH and SARS-CoV-2
15. Conclusions
Funding
Conflicts of Interest
References
- Holshue, M.; DeBolt, C.; Lindquist, S.; Lofy, K.; Wiesman, J.; Bruce, H.; Spitters, C.; Ericson, K.; Wilkerson, S.; Tural, A.; et al. First Case of 2019 Novel Coronavirus in the United States. N. Engl. J. Med. 2020, 382, 929–936. [Google Scholar] [CrossRef]
- Fauci, A.S.; Lane, H.C.; Redfield, R.R. Covid-19-Navigating the Uncharted. N. Engl. J. Med. 2020, 382, 1268–1269. [Google Scholar] [CrossRef] [PubMed]
- De Haan, C.; Kuo, L.; Masters, P.; Vennema, H.; Rottier, P. Coronavirus Particle Assembly: Primary Structure Requirements of the membrane Protein. J. Virol. 1998, 72, 6838–6850. [Google Scholar] [CrossRef]
- Mousavizadeh, L.; Ghasemi, S. Genotype and Phenotype of COVID-19: Their Roles in Pathogenesis. J. Microbiol. Immunol. Infect. 2020. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 497–506. [Google Scholar] [CrossRef]
- Woo, P.; Huang, Y.; Lau, S.; Yuen, K. Coronavirus Genomics and Bioinformatics Analysis. Viruses 2010, 2, 1804–1820. [Google Scholar] [CrossRef]
- Krähling, V.; Stein, D.A.; Spiegel, M.; Weber, F.; Mühlberger, E. Severe acute respiratory syndrome coronavirus triggers apoptosis via protein kinase R but is resistant to its antiviral activity. J. Virol. 2009, 83, 2298–2309. [Google Scholar] [CrossRef]
- Gandhi, R.T.; Lynch, J.B.; del Rio, C. Mild or Moderate Covid-19. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Kolifarhood, G.; Aghaali, M.; Mozafar Saadati, H.; Taherpour, N.; Rahimi, S.; Izadi, N.; Hashemi Nazari, S.S. Epidemiological and Clinical Aspects of COVID-19: A Narrative Review. Arch. Acad. Emerg. Med. 2020, 8, e41. [Google Scholar]
- Jung, S.; Akhmetzhanov, A.; Hayashi, K.; Linton, N.; Yang, Y.; Yuan, B.; Kobayashi, T.; Kinoshita, R.; Nishiura, H. Real-Time Estimation of the Risk of Death from Novel Coronavirus (COVID-19) Infection: Inference Using Exported Cases. J. Clin. Med. 2020, 9, 523. [Google Scholar] [CrossRef]
- Guan, W.; Ni, Z.; Hu, Y.; Liang, W.; Ou, C.; He, J.; Liu, L.; Shan, H.; Lei, C.; Hui, D.S.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Li, R.; Lu, Z.; Huang, Y. Does Comorbidity Increase the Risk of Patients with COVID-19: Evidence from Meta-Analysis. Aging 2020, 12, 6049. [Google Scholar] [CrossRef]
- Lindner, D.; Fitzek, A.; Bräuninger, H.; Aleshcheva, G.; Edler, C.; Meissner, K.; Scherschel, K.; Kirchhof, P.; Escher, F.; Schultheiss, H.; et al. Association of Cardiac Infection with SARS-CoV-2 in Confirmed COVID-19 Autopsy Cases. JAMA Cardiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Puntmann, V.O.; Carerj, M.L.; Wieters, I.; Fahim, M.; Arendt, C.; Hoffmann, J.; Shchendrygina, A.; Escher, F.; Vasa-Nicotera, M.; Zeiher, A.M.; et al. Outcomes of Cardiovascular Magnetic Resonance Imaging in Patients Recently Recovered from Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. 2020. Advance online publication. [Google Scholar] [CrossRef]
- Lauer, S.A.; Grantz, K.H.; Bi, Q.; Jones, F.K.; Zheng, Q.; Meredith, H.R.; Azman, A.S.; Reich, N.G.; Lessler, J. The Incubation Period of Coronavirus Disease 2019 (COVID-19) from Publicly Reported Confirmed Cases: Estimation and Application. Ann. Intern. Med. 2020, 172, 577–582. [Google Scholar] [CrossRef]
- Goyal, P.; Choi, J.; Pinheiro, L.; Schenck, E.; Chen, R.; Jabri, A.; Stalin, M.J.; Campion, T.R.; Nahid, M.; Ringel, J.B.; et al. Clinical Characteristics of Covid-19 in New York City. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Zhong, L.; Deng, J.; Peng, J.; Dan, H.; Zeng, X.; Li, T.; Chen, Q. High expression of ACE2 receptor of 2019-nCoV on the epithelial cells of oral mucosa. Int. J. Oral. Sci. 2020, 12, 8. [Google Scholar] [CrossRef]
- Meng, X.; Deng, Y.; Dai, Z.; Meng, Z. COVID-19 and anosmia: A review based on up-to-date knowledge. Am. J. Otolaryngol. 2020, 41, 102581. [Google Scholar] [CrossRef]
- Luo, Z.; Wang, N.; Liu, P.; Guo, Q.; Ran, L.; Wang, F.; Tang, Y.; Li, Q. Association between chest CT features and clinical course of coronavirus disease 2019. Respir. Med. 2020, 168, 105989. [Google Scholar] [CrossRef]
- Mardani, R.; Ahmadi Vasmehjani, A.; Zali, F.; Gholami, A.; Mousavi Nasab, S.D.; Kaghazian, H.; Kaviani, M.; Ahmadi, N. Laboratory Parameters in Detection of COVID-19 Patients with Positive RT-PCR: A Diagnostic Accuracy Study. Arch. Acad. Emerg. Med. 2020, 8, e43. [Google Scholar]
- Fan, B.; Chong, V.; Chan, S.; Lim, G.; Lim, K.; Tan, G.; Mucheli, S.S.; Kuperan, P.; Ong, K.H. Hematologic parameters in patients with COVID-19 infection. Am. J. Hematol. 2020, 95. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, J.; Yang, Y.; Ma, H.; Li, Z.; Zhang, J.; Cheng, J.; Zhang, X.; Zhao, Y.; Xia, Z.; et al. The role of interleukin-6 in monitoring severe case of coronavirus disease 2019. medRxiv 2020, 12, e12421. [Google Scholar] [CrossRef]
- Wu, C.; Chen, X.; Cai, Y.; Xia, J.; Zhou, X.; Xu, S.; Huang, H.; Zhang, L.; Zhou, X.; Du, C.; et al. Risk Factors Associated With Acute Respiratory Distress Syndrome and Death in Patients With Coronavirus Disease 2019 Pneumonia in Wuhan, China. JAMA Intern. Med. 2020, 180, 934–943. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Kermali, M.; Khalsa, R.K.; Pillai, K.; Ismail, Z.; Harky, A. The role of biomarkers in diagnosis of COVID-19—A systematic review. Life Sci. 2020, 254, 117788. [Google Scholar] [CrossRef]
- Patel, R.; Babady, E.; Theel, E.S.; Storch, G.A.; Pinsky, B.A.; St George, K.; Smith, T.C.; Bertuzzi, S. Report from the American Society for Microbiology COVID-19 International Summit, 23 March 2020: Value of Diagnostic Testing for SARS-CoV-2/COVID-19. Mbio 2020, 11, e00722-20. [Google Scholar] [CrossRef]
- Bao, L.; Deng, W.; Gao, H.; Xiao, C.; Liu, J.; Xue, J.; Lv, Q.; Liu, J.; Yu, P.; Xu, Y.; et al. Lack of Reinfection in Rhesus Macaques Infected with SARS-CoV-2. bioRxiv 2020. [Google Scholar] [CrossRef]
- Tilley, A.E.; Walters, M.S.; Shaykhiev, R.; Crystal, R.G. Cilia dysfunction in lung disease. Annu. Rev. Physiol. 2015, 77, 379–406. [Google Scholar] [CrossRef]
- Cao, W.; Taisheng, L. COVID-19: Towards Understanding of Pathogenesis. Cell Res. 2020, 30, 367–369. [Google Scholar] [CrossRef]
- Mason, R.J. Pathogenesis of COVID-19 from a cell biology perspective. Eur. Respir. J. 2020, 55, 2000607. [Google Scholar] [CrossRef] [PubMed]
- Harada, A.; Sekido, N.; Akahoshi, T.; Wada, T.; Mukaida, N.; Matsushima, K. Essential involvement of interleukin-8 (IL-8) in acute inflammation. J. Leukoc. Biol. 1994, 56, 559–564. [Google Scholar] [CrossRef] [PubMed]
- García, L. Immune Response, Inflammation, and the Clinical Spectrum of COVID-19. Front. Immunol. 2020, 11, 1441. [Google Scholar] [CrossRef]
- Babior, B. The respiratory burst of phagocytes. J. Clin. Investig. 1984, 73, 599–601. [Google Scholar] [CrossRef] [PubMed]
- Polonikov, A. Endogenous deficiency of glutathione as the most likely cause of serious manifestations and death in patients with the novel coronavirus infection (COVID-19): A hypothesis based on literature data and own observations. ACS Infect. Dis. 2020, 6, 1558–1562. [Google Scholar]
- Grant, A.; Ponia, S.S.; Tripathi, S.; Balasubramaniam, V.; Miorin, L.; Sourisseau, M.; Schwarz, M.C.; Sánchez-Seco, M.P.; Evans, M.J.; Best, S.M.; et al. Zika Virus Targets Human STAT2 to Inhibit Type I Interferon Signaling. Cell Host Microbe 2016, 19, 882–890. [Google Scholar] [CrossRef]
- Thurmond, S.; Wang, B.; Song, J.; Hai, R. Suppression of Type I Interferon Signaling by Flavivirus NS5. Viruses 2018, 10, 712. [Google Scholar] [CrossRef]
- Li, J.; Liao, C.; Wang, Q.; Tan, Y.; Luo, R.; Qiu, Y.; Ge, X. The ORF6, ORF8 and nucleocapsid proteins of SARS-CoV-2 inhibit type I interferon signaling pathway. Virus Res. 2020, 286, 198074. [Google Scholar] [CrossRef]
- Mantlo, E.; Bukreyeva, N.; Maruyama, J.; Paessler, S.; Huang, C. Antiviral activities of type I interferons to SARS-CoV-2 infection. Antivir. Res. 2020, 179, 104811. [Google Scholar] [CrossRef]
- Voskoboinik, I.; Whisstock, J.; Trapani, J. Perforin and granzymes: Function, dysfunction and human pathology. Nat. Rev. Immunol. 2015, 15, 388–400. [Google Scholar] [CrossRef]
- Schmitt, N.; Ueno, H. Regulation of human helper T cell subset differentiation by cytokines. Curr. Opin. Immunol. 2015, 34, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.; Lee, K.; Teijaro, J.; Becher, B.; Hamilton, J. GM-CSF-based treatments in COVID-19: Reconciling opposing therapeutic approaches. Nat. Rev. Immunol. 2020, 20, 507–514. [Google Scholar] [CrossRef]
- Croxford, A.L.; Lanzinger, M.; Hartmann, F.J.; Schreiner, B.; Mair, F.; Pelczar, P.; Clausen, B.E.; Jung, S.; Greter, M.; Becher, B. The Cytokine GM-CSF Drives the Inflammatory Signature of CCR2+ Monocytes and Licenses Autoimmunity. Immunity 2015, 43, 502–514. [Google Scholar] [CrossRef] [PubMed]
- Vabret, N.; Britton, G.; Gruber, C.; Hegde, S.; Kim, J.; Kuksin, M.; Levantovsky, R.; Malle, L.; Moreira, A.; Park, M.D.; et al. Immunology of COVID-19: Current State of the Science. Immunity 2020, 52, 910–941. [Google Scholar] [CrossRef]
- Riches, D.; Chan, E.; Winston, B. TNF-α-Induced Regulation and Signalling in Macrophages. Immunobiology 1996, 195, 477–490. [Google Scholar] [CrossRef]
- Gómez-Pastora, J.; Weigand, M.; Kim, J.; Wu, X.; Strayer, J.; Palmer, A.F.; Zborowski, M.; Yazer, M.; Chalmers, J.J. Hyperferritinemia in critically ill COVID-19 patients—Is ferritin the product of inflammation or a pathogenic mediator? Clin. Chim. Acta 2020, 509, 249–251. [Google Scholar] [CrossRef]
- Minagawa, S.; Yoshida, M.; Araya, J.; Hara, H.; Imai, H.; Kuwano, K. Regulated Necrosis in Pulmonary Disease. A Focus on Necroptosis and Ferroptosis. Am. J. Respir. Cell Mol. Biol. 2020, 62, 554–562. [Google Scholar] [CrossRef]
- Cortese-Krott, M.M.; Koning, A.; Kuhnle, G.; Nagy, P.; Bianco, C.L.; Pasch, A.; Wink, D.A.; Fukuto, J.M.; Jackson, A.A.; van Goor, H.; et al. The Reactive Species Interactome: Evolutionary Emergence, Biological Significance, and Opportunities for Redox Metabolomics and Personalized Medicine. Antioxid. Redox Signal. 2017, 27, 684–712. [Google Scholar] [CrossRef]
- Jankun, J.; Landeta, P.; Pretorius, E.; Skrzypczak-Jankun, E.; Lipinski, B. Unusual clotting dynamics of plasma supplemented with iron (III). Int. J. Mol. Med. 2014, 33, 367–372. [Google Scholar]
- Gao, M.; Monian, P.; Pan, Q.; Zhang, W.; Xiang, J.; Jiang, X. Ferroptosis is an autophagic cell death process. Cell Res. 2016, 26, 1021–1032. [Google Scholar] [CrossRef]
- Jiang, Y.; Yang, M.; Sun, X.; Chen, X.; Ma, M.; Yin, X.; Qian, S.; Zhang, Z.; Fu, Y.; Liu, J.; et al. IL-10+ NK and TGF-β+ NK cells play negative regulatory roles in HIV infection. BMC Infect. Dis. 2018, 18, 80. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Liu, J.; Zhang, D.; Xu, Z.; Ji, J.; Wen, C. Cytokine Storm in COVID-19: The Current Evidence and Treatment Strategies. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Venketaraman, V.; Millman, A.; Salman, M.; Swaminathan, S.; Goetz, M.; Lardizabal, A.; Hom, D.; Connell, N.D. Glutathione levels and immune responses in tuberculosis patients. Microb. Pathog. 2008, 44, e255–e261. [Google Scholar] [CrossRef] [PubMed]
- McBride, R.; van Zyl, M.; Fielding, B.C. The coronavirus nucleocapsid is a multifunctional protein. Viruses 2014, 6, 2991–3018. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wu, K.; Wang, D.; Yue, X.; Song, D.; Zhu, Y.; Wu, J. Nucleocapsid protein of SARS-CoV activates interleukin-6 expression through cellular transcription factor NF-kappaB. Virology 2007, 365, 324–335. [Google Scholar] [CrossRef]
- Liao, Q.; Ye, L.; Timani, K.; Zeng, Y.; She, Y.; Ye, L.; WU, Z. Activation of NF-kappaB by the Full-length Nucleocapsid Protein of the SARS Coronavirus. Acta Biochim. Biophys. Sin. 2005, 37, 607–612. [Google Scholar] [CrossRef]
- Wajner, S.M.; Goemann, I.M.; Bueno, A.L.; Larsen, P.R.; Maia, A.L. IL-6 promotes nonthyroidal illness syndrome by blocking thyroxine activation while promoting thyroid hormone inactivation in human cells. J. Clin. Investig. 2011, 121, 1834–1845. [Google Scholar] [CrossRef]
- Ly, J.; Lagman, M.; Saing, T.; Singh, M.K.; Tudela, E.V.; Morris, D.; Anderson, J.; Daliva, J.; Ochoa, C.; Patel, N.; et al. Liposomal Glutathione Supplementation Restores TH1 Cytokine Response to Mycobacterium Tuberculosis Infection in HIV-Infected Individuals. J. Interferon. Cytokine Res. 2015, 35, 875–887. [Google Scholar] [CrossRef]
- Valdivia, A.; Ly, J.; Gonzalez, L.; Hussain, P.; Saing, T.; Islamoglu, H.; Pearce, D.; Ochoa, C.; Venketaraman, V. Restoring Cytokine Balance in HIV-Positive Individuals with Low CD4 T Cell Counts. AIDS Res. Hum. Retrovir. 2017, 33, 905–918. [Google Scholar] [CrossRef]
- Imai, Y.; Kuba, K.; Neely, G.G.; Yaghubian-Malhami, R.; Perkmann, T.; van Loo, G.; Ermolaeva, M.; Veldhuizen, R.; Leung, Y.H.; Wang, H.; et al. Identification of Oxidative Stress and Toll-like Receptor 4 Signaling as a Key Pathway of Acute Lung Injury. Cell 2008, 133, 235–249. [Google Scholar] [CrossRef]
- Guaraldi, G.; Meschiari, M.; Cozzi-Lepri, A.; Milic, J.; Tonelli, R.; Menozzi, M.; Franceschini, E.; Cuomo, G.; Orlando, G.; Borghi, V.; et al. Tocilizumab in patients with severe COVID-19: A retrospective cohort study. Lancet Rheumatol. 2020, 2, e474–e484. [Google Scholar] [CrossRef] [PubMed]
- Middeldorp, S.; Coppens, M.; Haaps, T.; Foppen, M.; Vlaar, A.; Müller, M.; Bouman, C.C.S.; Beenen, L.F.M.; Kootte, R.R.; Heijmans, J.; et al. Incidence of venous thromboembolism in hospitalized patients with COVID-19. J. Thromb. Haemost. 2020, 18, 1995–2002. [Google Scholar] [CrossRef]
- Griffin, D.O.; Jensen, A.; Khan, M.; Chin, J.; Chin, K.; Saad, J.; Parnell, R.; Awwad, C.; Patel, D. Pulmonary Embolism and Increased Levels of d-Dimer in Patients with Coronavirus Disease. Emerg. Infect. Dis. 2020, 26, 1941. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, K.; Wei, H.; Chen, W.; Wang, W.; Jia, L.; Liu, Q.; Zhang, J.; Shan, T.; Peng, Z.; et al. Dynamic relationship between D-dimer and COVID-19 severity. Br. J. Hematol. 2020, 190. [Google Scholar] [CrossRef]
- Sartain, S.; Turner, N.; Moake, J. TNF Regulates Essential Alternative Complement Pathway Components and Impairs Activation of Protein c in Human Glomerular Endothelial Cells. J. Immunol. 2015, 196, 832–845. [Google Scholar] [CrossRef] [PubMed]
- Imai, H.; Matsuoka, M.; Kumagai, T.; Sakamoto, T.; Koumura, T. Lipid Peroxidation-Dependent Cell Death Regulated by GPx4 and Ferroptosis. Curr. Top. Microbiol. Immunol. 2017, 403, 143–170. [Google Scholar] [CrossRef] [PubMed]
- Lu, S. Glutathione synthesis. Biochim. Biophys. Acta Gen. Subj. 2013, 1830, 3143–3153. [Google Scholar] [CrossRef]
- Maiorino, M.; Aumann, K.; Brigelius-Flohé, R.; Doria, D.; Heuvel, J.V.; Mccarthy, J.; Flohé, L. Probing the Presumed Catalytic Triad of Selenium-Containing Peroxidases by Mutational Analysis of Phospholipid Hydroperoxide Glutathione Peroxidase (PHGPx). Biol. Chem. Hoppe-Seyler 1995, 376, 651–660. [Google Scholar] [CrossRef]
- Mills, G.C. Hemoglobincatabolism. I. Glutathione peroxidase, an enzyme which protects hemoglobin from oxidative breakdown. J. Biol. Chem. 1957, 229, 189–197. [Google Scholar]
- Green, R.M.; Seth, A.; Connell, N.D. A peptide permease mutant of Mycobacterium bovis BCG resistant to the toxic peptides glutathione and S-nitrosoglutathione. Infect. Immun. 2000, 68, 429–436. [Google Scholar] [CrossRef]
- Venketaraman, V.; Dayaram, Y.K.; Talaue, M.T.; Connell, N.D. Glutathione and nitrosoglutathione in macrophage defense against Mycobacterium tuberculosis. Infect. Immun. 2005, 73, 1886–1889. [Google Scholar] [CrossRef]
- Morris, D.; Khurasany, M.; Nguyen, T.; Kim, J.; Guilford, F.; Mehta, R.; Gray, D.; Saviola, B.; Venketaraman, V. Glutathione and infection. Biochim. Biophys. Acta 2013, 1830, 3329–3349. [Google Scholar] [CrossRef] [PubMed]
- HIV/AIDS. Available online: https://www.who.int/news-room/fact-sheets/detail/hiv-aids (accessed on 13 September 2020).
- Morris, D.; Guerra, C.; Donohue, C.; Oh, H.; Khurasany, M.; Venketaraman, V. Unveiling the Mechanisms for Decreased Glutathione in Individuals with HIV Infection. Clin. Dev. Immunol. 2012, 1–10. [Google Scholar] [CrossRef]
- Chung, S.S.; Ho, E.C.; Lam, K.S.; Chung, S.K. Contribution of polyol pathway to diabetes-induced oxidative stress. J. Am. Soc. Nephrol. 2003, 14, S233–S236. [Google Scholar] [CrossRef]
- Ozawa, M.; Asano, A. Importance of interpeptide disulfide bond in a viral glycoprotein with hemagglutination and neuraminidase activities. FEBS Lett. 1976, 70, 145–149. [Google Scholar] [CrossRef]
- Sgarbanti, R.; Nencioni, L.; Amatore, D.; Coluccio, P.; Fraternale, A.; Sale, P.; Mammola, C.L.; Carpino, G.; Gaudio, E.; Magnani, M.; et al. Redox regulation of the influenza hemagglutinin maturation process: A new cell-mediated strategy for anti-influenza therapy. Antioxidants Redox Signal. 2011, 15, 593–606. [Google Scholar] [CrossRef] [PubMed]
- Palamara, A.T.; Perno, C.F.; Ciriolo, M.R.; Dini, L.; Balestra, E.; D’Agostini, C.; Di Francesco, P.; Favalli, C.; Rotilio, G.; Garaci, E. Evidence for antiviral activity of glutathione: In vitro inhibition of herpes simplex virus type 1 replication. Antivir. Res. 1995, 27, 237–253. [Google Scholar] [CrossRef]
- Schreck, R.; Albermann, K.; Baeuerle, P.A. Nuclear factor kappa B: An oxidative stress-responsive transcription factor of eukaryotic cells (a review). Free Radic Res. Commun. 1992, 17, 221–237. [Google Scholar] [CrossRef]
- Garaci, E.; Palamara, A.T.; Di Francesco, P.; Favalli, C.; Ciriolo, M.R.; Rotilio, G. Glutathione inhibits replication and expression of viral proteins in cultured cells infected with Sendai virus. Biochem. Biophys. Res. Commun. 1992, 188, 1090–1096. [Google Scholar] [CrossRef]
- Hennet, T.; Peterhans, E.; Stocker, R. Alterations in antioxidant defences in lung and liver of mice infected with influenza A virus. J. Gen. Virol. 1992, 73, 39–46. [Google Scholar] [CrossRef]
- Oda, T.; Akaike, T.; Hamamoto, T.; Suzuki, F.; Hirano, T.; Maeda, H. Oxygen radicals in influenza-induced pathogenesis and treatment with pyran polymer-conjugated SOD. Science 1989, 244, 974–976. [Google Scholar] [CrossRef] [PubMed]
- Ciriolo, M.R.; Palamara, A.T.; Incerpi, S.; Lafavia, E.; Buè, M.C.; De Vito, P.; Garaci, E.; Rotilio, G. Loss of GSH, oxidative stress, and decrease of intracellular pH as sequential steps in viral infection. J. Biol. Chem. 1997, 272, 2700–2708. [Google Scholar] [CrossRef] [PubMed]
- Pérez, L.; Carrasco, L. Involvement of the vacuolar H(+)-ATPase in animal virus entry. J. Gen. Virol. 1994, 75, 2595–2606. [Google Scholar] [CrossRef] [PubMed]
- Cossart, P.; Helenius, A. Endocytosis of viruses and bacteria. Cold Spring Harb. Perspect Biol. 2014, 6, a016972. [Google Scholar] [CrossRef] [PubMed]
- Fung, T.S.; Huang, M.; Liu, D.X. Coronavirus-induced ER stress response and its involvement in regulation of coronavirus-host interactions. Virus Res. 2014, 194, 110–123. [Google Scholar] [CrossRef]
- Fehr, A.R.; Perlman, S. Coronaviruses: An overview of their replication and pathogenesis. Methods Mol. Biol. 2015, 1282, 1–23. [Google Scholar]
- Savarino, A.; Boelaert, J.R.; Cassone, A.; Majori, G.; Cauda, R. Effects of chloroquine on viral infections: An old drug against today’s diseases? Lancet Infect. Dis. 2003, 3, 722–727. [Google Scholar] [CrossRef]
- Al-Bari, M.A.A. Targeting endosomal acidification by chloroquine analogs as a promising strategy for the treatment of emerging viral diseases. Pharmacol. Res. Perspect. 2017, 5, e00293. [Google Scholar] [CrossRef]
- Wessels, U.; Abdelwhab, E.; Veits, J.; Hoffmann, D.; Mamerow, S.; Stech, O.; Hellert, J.; Beer, M.; Mettenleiter, T.; Stech, J. A dual motif in the Hemagglutinin of H5N1 Goose/Guangdong-Like Highly Pathogenic Avian Influenza Virus Strains is Conserved from Their Early Evolution and Increases both Membrane Fusion pH and Virulence. J. Virol. 2018, 92, e00778-18. [Google Scholar] [CrossRef]
- Chu, V.C.; McElroy, L.J.; Chu, V.; Bauman, B.E.; Whittaker, G.R. The avian coronavirus infectious bronchitis virus undergoes direct low-pH-dependent fusion activation during entry into host cells. J. Virol. 2006, 80, 3180–3188. [Google Scholar] [CrossRef]
- Wang, L.F.; Lin, Y.S.; Huang, N.C.; Yu, C.Y.; Tsai, W.L.; Chen, J.J.; Kubota, T.; Matsuoka, M.; Chen, S.R.; Yang, C.S.; et al. Hydroxychloroquine-inhibited dengue virus is associated with host defense machinery. J. Interferon. Cytokine Res. 2015, 35, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Dai, J.; Zhao, X.; Chen, Y.; Wang, G.; Li, K. Chloroquine enhances replication of influenza A virus A/WSN/33 (H1N1) in dose-, time-, and MOI-dependent manners in human lung epithelial cells A549. J. Med. Virol. 2015, 87, 1096–1103. [Google Scholar] [CrossRef] [PubMed]
- Roustit, M.; Guilhaumou, R.; Molimard, M.; Drici, M.D.; Laporte, S.; Montastruc, J.L. French Society of Pharmacology and Therapeutics (SFPT) Chloroquine and hydroxychloroquine in the management of COVID-19: Much kerfuffle but little evidence. Therapie 2020, 75, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Nencioni, L.; Iuvara, A.; Aquilano, K.; Ciriolo, M.R.; Cozzolino, F.; Rotilio, G.; Garaci, E.; Palamara, A.T. Influenza A virus replication is dependent on an antioxidant pathway that involves GSH and Bcl-2. FASEB 2003, 17, 758–760. [Google Scholar] [CrossRef]
- Amatore, D.; Sgarbanti, R.; Aquilano, K.; Baldelli, S.; Limongi, D.; Civitelli, L.; Nencioni, L.; Garaci, E.; Ciriolo, M.R.; Palamara, A.T. Influenza virus replication in lung epithelial cells depends on redox-sensitive pathways activated by NOX4-derived ROS. Cell Microbiol. 2015, 17, 131–145. [Google Scholar] [CrossRef]
- Celestino, I.; Checconi, P.; Amatore, D.; De Angelis, M.; Coluccio, P.; Dattilo, R.; Alunni Fegatelli, D.; Clemente, A.M.; Matarrese, P.; Torcia, M.G.; et al. Differential Redox State Contributes to Sex Disparities in the Response to Influenza Virus Infection in Male and Female Mice. Front. Immunol. 2018, 9, 1747. [Google Scholar] [CrossRef]
- Checconi, P.; De Angelis, M.; Marcocci, M.E.; Fraternale, A.; Magnani, M.; Palamara, A.T.; Nencioni, L. Redox-modulating agents in the treatment of viral infections. Int. J. Mol. Sci. 2020, 21, 4084. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar]
- To, E.E.; Vlahos, R.; Luong, R.; Halls, M.L.; Reading, P.C.; King, P.T.; Chan, C.; Drummond, G.R.; Sobey, C.G.; Broughton, B.; et al. Endosomal NOX2 oxidase exacerbates virus pathogenicity and is a target for antiviral therapy. Nat. Commun. 2017, 8, 69. [Google Scholar] [CrossRef]
- Wiley, D.C.; Skehel, J.J. The structure and function of the hemagglutinin membrane glycoprotein of influenza virus. Annu. Rev. Biochem. 1987, 56, 365–394. [Google Scholar] [CrossRef]
- Lee, C. Therapeutic Modulation of Virus-Induced Oxidative Stress via the Nrf2-Dependent Antioxidative Pathway. Oxid. Med. Cell Longev. 2018, 2018, 6208067. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Ou, J.H. Mechanisms of liver injury. III. Oxidative stress in the pathogenesis of hepatitis C virus. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G847–G851. [Google Scholar] [CrossRef]
- Thoren, F.; Romero, A.; Lindh, M.; Dahlgren, C.; Hellstrand, K. A hepatitis C virus-encoded, nonstructural protein (NS3) triggers dysfunction and apoptosis in lymphocytes: Role of NADPH oxidase-derived oxygen radicals. J. Leukoc Biol. 2004, 76, 1180–1186. [Google Scholar] [CrossRef]
- Anticoli, S.; Amatore, D.; Matarrese, P.; De Angelis, M.; Palamara, A.T.; Nencioni, L. Counteraction of HCV-Induced Oxidative Stress Concurs to Establish Chronic Infection in Liver Cell Cultures. Oxid. Med. Cell. Longev. 2019, 2019, 6452390. [Google Scholar] [CrossRef] [PubMed]
- Fink, K.; Duval, A.; Martel, A.; Soucy-Faulkner, A.; Grandvaux, N. Dual role of NOX2 in respiratory syncytial virus- and sendai virus-induced activation of NF-kappaB in airway epithelial cells. J. Immunol. 2008, 180, 6911–6922. [Google Scholar] [CrossRef] [PubMed]
- Hosakote, Y.M.; Liu, T.; Castro, S.M.; Garofalo, R.P.; Casola, A. Respiratory syncytial virus induces oxidative stress by modulating antioxidant enzymes. Am. J. Respir. Cell Mol. Biol. 2009, 41, 348–357. [Google Scholar] [CrossRef] [PubMed]
- Theodore, M.; Kawai, Y.; Yang, J.; Kleshchenko, Y.; Reddy, S.P.; Villalta, F.; Arinze, I.J. Multiple nuclear localization signals function in the nuclear import of the transcription factor Nrf2. J. Biol. Chem. 2008, 283, 8984–8994. [Google Scholar] [CrossRef] [PubMed]
- Sims, A.C.; Tilton, S.C.; Menachery, V.D.; Gralinski, L.E.; Schäfer, A.; Matzke, M.M.; Webb-Robertson, B.J.M.; Chang, J.; Luna, M.L.; Long, C.E.; et al. Release of severe acute respiratory syndrome coronavirus nuclear import block enhances host transcription in human lung cells. J. Virol. 2013, 87, 3885–3902. [Google Scholar] [CrossRef]
- Chook, Y.M.; Suel, K.E. Nuclear import by karyopherin-betas: Recognition and inhibition. Biochim. Biophys. Acta 2011, 1813, 1593–1606. [Google Scholar] [CrossRef]
- Alvarez, J.A.; Chowdhury, R.; Jones, D.P.; Martin, G.S.; Brigham, K.L.; Binongo, J.N.; Ziegler, T.R.; Tangpricha, V. Vitamin D status is independently associated with plasma glutathione and cysteine thiol/disulphide redox status in adults. Clin. Endocrinol. 2014, 81, 458–466. [Google Scholar] [CrossRef]
- Jain, S.K.; Micinski, D. Vitamin D upregulates glutamate cysteine ligase and glutathione reductase, and GSH formation, and decreases ROS and MCP-1 and IL-8 secretion in high-glucose exposed U937 monocytes. Biochem. Biophys. Res. Commun. 2013, 437, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Han, J.E.; Jones, J.L.; Tangpricha, V.; Brown, M.A.; Brown, L.; Hao, L.; Hebbar, G.; Lee, M.J.; Liu, S.; Ziegler, T.R.; et al. High Dose Vitamin D Administration in Ventilated Intensive Care Unit Patients: A Pilot Double Blind Randomized Controlled Trial. J. Clin. Transl. Endocrinol. 2016, 4, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.; Anderson, G.; Dean, O.; Berk, M.; Galecki, P.; Martin-Subero, M.; Maes, M. The glutathione system: A new drug target in neuroimmune disorders. Mol. Neurobiol. 2014, 50, 1059–1084. [Google Scholar] [CrossRef] [PubMed]
- Nadeem, A.; Siddiqui, N.; Alharbi, N.O.; Alharbi, M.M.; Imam, F. Acute glutathione depletion leads to enhancement of airway reactivity and inflammation via p38MAPK-iNOS pathway in allergic mice. Int. Immunopharmacol. 2014, 22, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Limongi, D.; Baldelli, S.; Checconi, P.; Marcocci, M.E.; De Chiara, G.; Fraternale, A.; Magnani, M.; Ciriolo, M.R.; Palamara, A.T. GSH-C4 Acts as Anti-inflammatory Drug in Different Models of Canonical and Cell Autonomous Inflammation Through NFkappaB Inhibition. Front. Immunol. 2019, 10, 155. [Google Scholar] [CrossRef]
- Schmitt, B.; Vicenzi, M.; Garrel, C.; Denis, F. Effects of N-acetylcysteine, oral glutathione (GSH) and a novel sublingual form of GSH on oxidative stress markers: A comparative crossover study. Redox Biol. 2015, 6, 198–205. [Google Scholar] [CrossRef]
- Pacht, E.R.; Timerman, A.P.; Lykens, M.G.; Merola, A.J. Deficiency of alveolar fluid glutathione in patients with sepsis and the adult respiratory distress syndrome. Chest 1991, 100, 1397–1403. [Google Scholar] [CrossRef]
- Zhang, Q.; Ju, Y.; Ma, Y.; Wang, T. N-acetylcysteine improves oxidative stress and inflammatory response in patients with community acquired pneumonia. Medicine 2018, 97, e13087. [Google Scholar] [CrossRef]
- Ursini, F.; Maiorino, M. Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic. Biol. Med. 2020, 152, 175–185. [Google Scholar] [CrossRef]
- Osaki, T.; Ohshima, M.; Tomita, Y.; Matsugi, N.; Nomura, Y. Clinical and physiological investigations in patients with taste abnormality. J. Oral Pathol. Med. 1996, 25, 38–43. [Google Scholar] [CrossRef]
- Dinc, M.E.; Dalgic, A.; Ulusoy, S.; Dizdar, D.; Develioglu, O.; Topak, M. Does iron deficiency anemia affect olfactory function? Acta Oto-Laryngol. 2016, 136, 754–757. [Google Scholar] [CrossRef] [PubMed]
- Vaira, L.A.; Salzano, G.; Deiana, G.; De Riu, G. Anosmia and Ageusia: Common Findings in COVID-19 Patients. Laryngoscope 2020, 130, 1787. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Dixon, S. Mechanisms of ferroptosis. Cell. Mol. Life Sci. 2016, 73, 2195–2209. [Google Scholar] [CrossRef] [PubMed]
- Mashima, R.; Okuyama, T. The role of lipoxygenases in pathophysiology; new insights and future perspectives. Redox Biol. 2015, 6, 297–310. [Google Scholar] [CrossRef]
- Meng, Y.; Li, T.; Zhou, G.S.; Chen, Y.; Yu, C.H.; Pang, M.X.; Li, W.; Li, Y.; Zhang, W.Y.; Li, X. The angiotensin-converting enzyme 2/angiotensin (1-7)/Mas axis protects against lung fibroblast migration and lung fibrosis by inhibiting the NOX4-derived ROS-mediated RhoA/Rho kinase pathway. Antioxid Redox Signal. 2015, 22, 241–258. [Google Scholar] [CrossRef]
- Khomich, O.A.; Kochetkov, S.N.; Bartosch, B.; Ivanov, A.V. Redox Biology of Respiratory Viral Infections. Viruses 2018, 10, 392. [Google Scholar] [CrossRef]
- Ceriello, A. Hyperglycemia and the worse prognosis of COVID-19. Why a fast blood glucose control should be mandatory. Diabetes Res. Clin. Pract. 2020, 163, 108186. [Google Scholar] [CrossRef]
- Lutchmansingh, F.K.; Hsu, J.W.; Bennett, F.I.; Badaloo, A.V.; McFarlane-Anderson, N.; Gordon-Strachan, G.M.; Wright-Pascoe, R.A.; Jahoor, F.; Boyne, M.S. Glutathione metabolism in type 2 diabetes and its relationship with microvascular complications and glycemia. PLoS ONE 2018, 13, e0198626. [Google Scholar] [CrossRef]
- Pan, F.; Ye, T.; Sun, P.; Gui, S.; Liang, B.; Li, L.; Zheng, D.; Wang, J.; Hesketh, R.L.; Yang, L.; et al. Time course of lung changes at chest CT during recovery from coronavirus disease 2019 (COVID-19). Radiology 2020, 295, 715–721. [Google Scholar] [CrossRef]
- Horowitz, R.I.; Freeman, P.R.; Bruzzese, J. Efficacy of glutathione therapy in relieving dyspnea associated with COVID-19 pneumonia: A report of 2 cases. Respir. Med. Case Rep. 2020, 30, 101063. [Google Scholar] [CrossRef]
- Sinha, R.; Sinha, I.; Calcagnotto, A.; Trushin, N.; Haley, J.; Schell, T.; Richie, J. Oral supplementation with liposomal glutathione elevates body stores of glutathione and markers of immune function. Eur. J. Clin. Nutr. 2017, 72, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Koning, G.; Storm, G. Targeted drug delivery systems for the intracellular delivery of macromolecular drugs. Drug Discov. Today 2003, 8, 482–483. [Google Scholar] [CrossRef]
- Ulrich, A. Biophysical Aspects of Using Liposomes as Delivery Vehicles. Biosci. Rep. 2002, 22, 129–150. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guloyan, V.; Oganesian, B.; Baghdasaryan, N.; Yeh, C.; Singh, M.; Guilford, F.; Ting, Y.-S.; Venketaraman, V. Glutathione Supplementation as an Adjunctive Therapy in COVID-19. Antioxidants 2020, 9, 914. https://doi.org/10.3390/antiox9100914
Guloyan V, Oganesian B, Baghdasaryan N, Yeh C, Singh M, Guilford F, Ting Y-S, Venketaraman V. Glutathione Supplementation as an Adjunctive Therapy in COVID-19. Antioxidants. 2020; 9(10):914. https://doi.org/10.3390/antiox9100914
Chicago/Turabian StyleGuloyan, Vika, Buzand Oganesian, Nicole Baghdasaryan, Christopher Yeh, Manpreet Singh, Frederick Guilford, Yu-Sam Ting, and Vishwanath Venketaraman. 2020. "Glutathione Supplementation as an Adjunctive Therapy in COVID-19" Antioxidants 9, no. 10: 914. https://doi.org/10.3390/antiox9100914
APA StyleGuloyan, V., Oganesian, B., Baghdasaryan, N., Yeh, C., Singh, M., Guilford, F., Ting, Y.-S., & Venketaraman, V. (2020). Glutathione Supplementation as an Adjunctive Therapy in COVID-19. Antioxidants, 9(10), 914. https://doi.org/10.3390/antiox9100914