Endoplasmic Reticulum Stress-Induced Resistance to Doxorubicin Is Reversed by Mulberry Leaf Polyphenol Extract in Hepatocellular Carcinoma through Inhibition of COX-2

Abstract
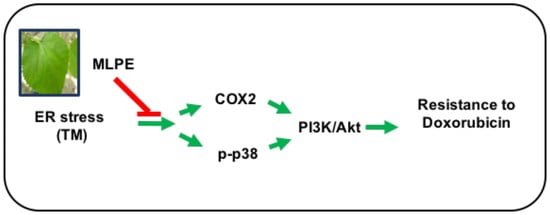
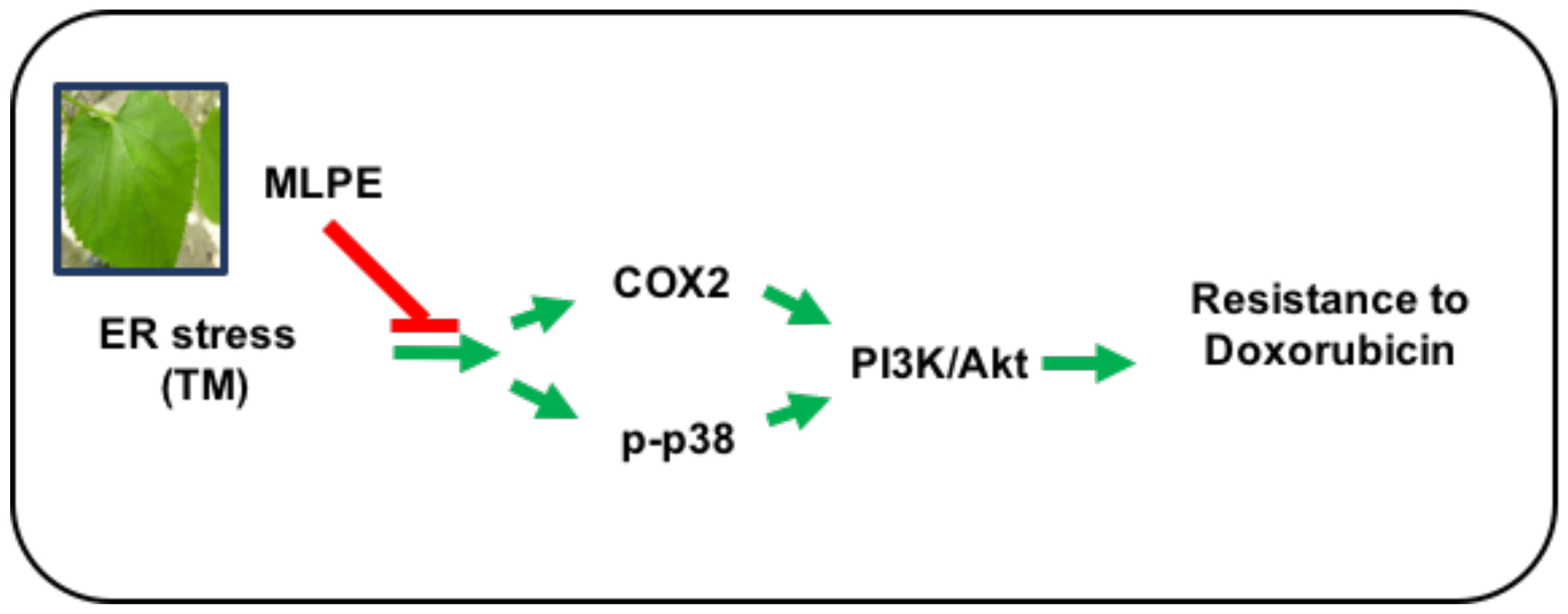{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
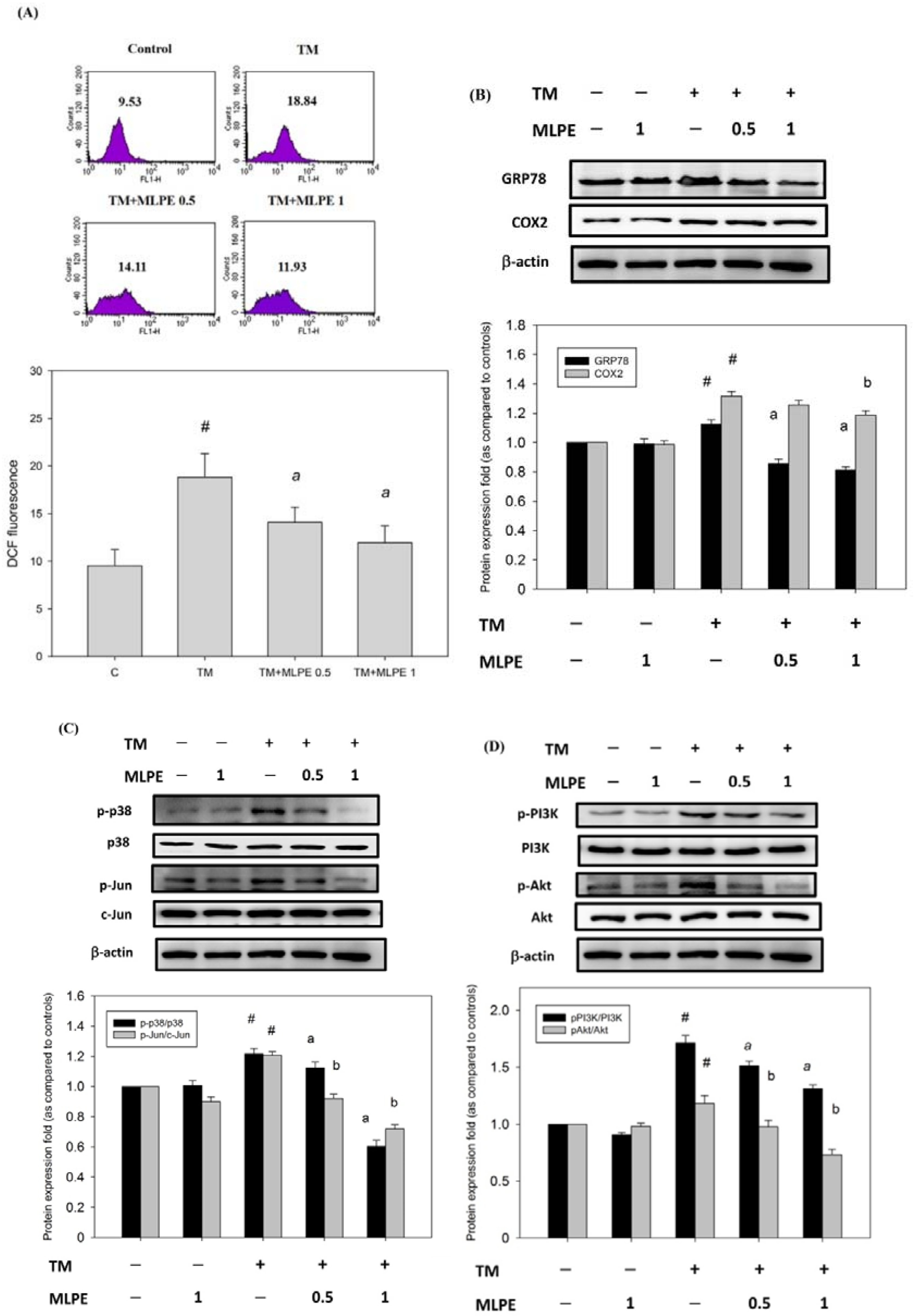{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Preparation of MLPE
2.3. Cell Culture
2.4. MTT Assay
2.5. Fluorescence-Activated Cell Sorting
2.6. Western Blotting
2.7. Statistical Analysis
3. Results
3.1. Analysis of MLPE Using High-Performance Liquid Chromatography
3.2. Effect of DOX and MLPE on Cell Viability in HepG2 Cells
3.3. Induction of ER Stress Protects HCC Cells against Apoptosis Induced by DOX
3.4. Effect of Copretreatment with MLPE and TM on Apoptosis Induced by DOX in HepG2 Cells
3.5. Protection of HCC Cells against DOX-Induced Apoptosis with Pretreatment of TM is Associated with COX-2
3.6. p38/PI3K/Akt Pathway is Involved in the COX-2-Mediated Cytoprotective Effect of ER Stress against DOX-Induced HCC Cell Apoptosis
3.7. Effect of Copretreatment with TM and Celecoxib on Cell Survival in HepG2 Cells
3.8. Effect of Copretreatment with TM and SB203580 on Cell Survival in HepG2 Cells
3.9. Reversing Effect of MLPE on ER Stress-Induced Resistance to DOX in HepG2 Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed]
- Lencioni, R.; Chen, X.P.; Dagher, L.; Venook, A.P. Treatment of intermediate/advanced hepatocellular carcinoma in the clinic: How can outcomes be improved? Oncologist 2010, 15, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Avril, T.; Vauléon, E.; Chevet, E. Endoplasmic reticulum stress signaling and chemotherapy resistance in solid cancers. Oncogenesis 2017, 6, e373. [Google Scholar] [CrossRef] [PubMed]
- Mann, M.J.; Hendershot, L.M. UPR activation alters chemosensitivity of tumor cells. Cancer Biol. Ther. 2006, 5, 736–740. [Google Scholar] [CrossRef]
- Kim, K.M.; Yu, T.K.; Chu, H.H.; Park, H.S.; Jang, K.Y.; Moon, W.S.; Kang, M.J.; Lee, D.G.; Kim, M.H.; Lee, J.H.; et al. Expression of ER stress and autophagy-related molecules in human non-small cell lung cancer and premalignant lesions. Int. J. Cancer 2012, 131, E362–E370. [Google Scholar] [CrossRef] [PubMed]
- Pyrko, P.; Schönthal, A.H.; Hofman, F.M.; Chen, T.C.; Lee, A.S. The unfolded protein response regulator GRP78/BiP as a novel target for increasing chemosensitivity in malignant gliomas. Cancer Res. 2007, 67, 9809–9816. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Nichols, P.; Spicer, D.; Groshen, S.; Mimi, C.Y.; Lee, A.S. GRP78 as a novel predictor of responsiveness to chemotherapy in breast cancer. Cancer Res. 2006, 66, 7849–7853. [Google Scholar] [CrossRef]
- Ranganathan, A.C.; Zhang, L.; Adam, A.P.; Aguirre-Ghiso, J.A. Functional coupling of p38-induced up-regulation of BiP and activation of RNA-dependent protein kinase–like endoplasmic reticulum kinase to drug resistance of dormant carcinoma cells. Cancer Res. 2006, 66, 1702–1711. [Google Scholar] [CrossRef]
- Tsai, H.; Yang, Y.; Wu, A.; Yang, C.; Liu, Y.; Jan, Y.; Lee, C.; Hsiao, Y.; Yeh, C.T.; Shen, C.; et al. Endoplasmic reticulum ribosome-binding protein 1 (RRBP1) overexpression is frequently found in lung cancer patients and alleviates intracellular stress-induced apoptosis through the enhancement of GRP78. Oncogene 2013, 32, 4921. [Google Scholar] [CrossRef]
- Yang, M.Y.; Huang, C.N.; Chan, K.C.; Yang, Y.S.; Peng, C.H.; Wang, C.J. Mulberry leaf polyphenols possess antiatherogenesis effect via inhibiting LDL oxidation and foam cell formation. J. Agric. Food Chem. 2011, 59, 1985–1995. [Google Scholar] [CrossRef]
- Rasheed, Z.; Haqqi, T.M. Endoplasmic reticulum stress induces the expression of COX-2 through activation of eIF2alpha, p38-MAPK and NF-kappaB in advanced glycation end products stimulated human chondrocytes. Biochim. Biophys. Acta 2012, 1823, 2179–2189. [Google Scholar] [CrossRef] [PubMed]
- Cha, W.; Park, S.W.; Kwon, T.K.; Hah, J.H.; Sung, M.W. Endoplasmic reticulum stress response as a possible mechanism of cyclooxygenase-2-independent anticancer effect of celecoxib. Anticancer Res. 2014, 34, 1731–1735. [Google Scholar] [PubMed]
- Hung, J.H.; Su, I.J.; Lei, H.Y.; Wang, H.C.; Lin, W.C.; Chang, W.T.; Huang, W.; Chang, W.C.; Chang, Y.S.; Chen, C.C.; et al. Endoplasmic reticulum stress stimulates the expression of cyclooxygenase-2 through activation of NF-kappaB and pp38 mitogen-activated protein kinase. J. Biol. Chem. 2004, 279, 46384–46392. [Google Scholar] [CrossRef] [PubMed]
- Stagos, D.; Amoutzias, G.D.; Matakos, A.; Spyrou, A.; Tsatsakis, A.M.; Kouretas, D. Chemoprevention of liver cancer by plant polyphenols. Food Chem. Toxicol. 2012, 50, 2155–2170. [Google Scholar] [CrossRef]
- Russo, M.; Russo, G.L.; Daglia, M.; Kasi, P.D.; Ravi, S.; Nabavi, S.F.; Nabavi, S.M. Understanding genistein in cancer: The “good” and the “bad” effects: A review. Food Chem. 2016, 196, 589–600. [Google Scholar] [CrossRef]
- Lee, W.J.; Choi, S.W. Quantitative changes of polyphenolic compounds in mulberry (Morus alba L.) leaves in relation to varieties, harvest period, and heat processing. Prev. Nutr. Food Sci. 2012, 17, 280. [Google Scholar] [CrossRef]
- Azman, K.F.; Amom, Z.; Azlan, A.; Esa, N.M.; Ali, R.M.; Shah, Z.M.; Kadir, K.K.A. Antiobesity effect of Tamarindus indica L. pulp aqueous extract in high-fat diet-induced obese rats. J. Nat. Med. 2012, 66, 333–342. [Google Scholar] [CrossRef]
- Flores, M.B.; Rocha, G.Z.; Damas–Souza, D.M.; Osório–Costa, F.; Dias, M.M.; Ropelle, E.R.; Camargo, J.A.; de Carvalho, R.B.; Carvalho, H.F.; Saad, M.J.; et al. RETRACTED: Obesity-Induced Increase in Tumor Necrosis Factor-α Leads to Development of Colon Cancer in Mice. Gastroenterology 2012, 143, 741–753. [Google Scholar] [CrossRef]
- Ou, T.T.; Hsu, M.J.; Chan, K.C.; Huang, C.N.; Ho, H.H.; Wang, C.J. Mulberry extract inhibits oleic acid-induced lipid accumulation via reduction of lipogenesis and promotion of hepatic lipid clearance. J. Sci. Food Agric. 2011, 91, 2740–2748. [Google Scholar] [CrossRef]
- Wang, X.; Ouyang, Y.Y.; Liu, J.; Zhao, G. Flavonoid intake and risk of CVD: A systematic review and meta-analysis of prospective cohort studies. Br. J. Nutr. 2014, 111, 1–11. [Google Scholar] [CrossRef]
- Godos, J.; Vitale, M.; Micek, A.; Ray, S.; Martini, D.; Del Rio, D.; Riccardi, G.; Galvano, F.; Grosso, G. Dietary Polyphenol Intake, Blood Pressure, and Hypertension: A Systematic Review and Meta-Analysis of Observational Studies. Antioxidants (Basel) 2019, 8, 152. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Luo, J.; Huang, J.; Wen, Q. Flavonoids intake and risk of type 2 diabetes mellitus: A meta-analysis of prospective cohort studies. Medicine (Baltimore) 2018, 97, e0686. [Google Scholar] [CrossRef] [PubMed]
- Grosso, G.; Micek, A.; Godos, J.; Pajak, A.; Sciacca, S.; Galvano, F.; Giovannucci, E.L. Dietary Flavonoid and Lignan Intake and Mortality in Prospective Cohort Studies: Systematic Review and Dose-Response Meta-Analysis. Am. J. Epidemiol. 2017, 185, 1304–1316. [Google Scholar] [CrossRef] [PubMed]
- Grosso, G.; Godos, J.; Lamuela-Raventos, R.; Ray, S.; Micek, A.; Pajak, A.; Sciacca, S.; D’Orazio, N.; Del Rio, D.; Galvano, F. A comprehensive meta-analysis on dietary flavonoid and lignan intake and cancer risk: Level of evidence and limitations. Mol. Nutr. Food Res. 2017, 61. [Google Scholar] [CrossRef] [PubMed]
- Rauf, A.; Imran, M.; Khan, I.A.; Ur-Rehman, M.; Gilani, S.A.; Mehmood, Z.; Mubarak, M.S. Anticancer potential of quercetin: A comprehensive review. Phytother. Res. 2018, 32, 2109–2130. [Google Scholar] [CrossRef] [PubMed]
- Imran, M.; Salehi, B.; Sharifi-Rad, J.; Aslam Gondal, T.; Saeed, F.; Imran, A.; Shahbaz, M.; Tsouh Fokou, P.V.; Umair Arshad, M.; Khan, H.; et al. Kaempferol: A Key Emphasis to Its Anticancer Potential. Molecules 2019, 24, 2277. [Google Scholar] [CrossRef]
- Yan, Y.; Li, J.; Han, J.; Hou, N.; Song, Y.; Dong, L. Chlorogenic acid enhances the effects of 5-fluorouracil in human hepatocellular carcinoma cells through the inhibition of extracellular signal-regulated kinases. Anticancer Drugs 2015, 26, 540–546. [Google Scholar] [CrossRef]
- Deepa, M.; Sureshkumar, T.; Satheeshkumar, P.K.; Priya, S. Antioxidant rich Morus alba leaf extract induces apoptosis in human colon and breast cancer cells by the downregulation of nitric oxide produced by inducible nitric oxide synthase. Nutr. Cancer 2013, 65, 305–310. [Google Scholar] [CrossRef]
- Naowaratwattana, W.; De-Eknamkul, W.; De Mejia, E.G. Phenolic-containing organic extracts of mulberry (Morus alba L.) leaves inhibit HepG2 hepatoma cells through G2/M phase arrest, induction of apoptosis, and inhibition of topoisomerase IIα activity. J. Med. Food 2010, 13, 1045–1056. [Google Scholar] [CrossRef]
- Chen, P.N.; Chu, S.C.; Chiou, H.L.; Kuo, W.H.; Chiang, C.L.; Hsieh, Y.S. Mulberry anthocyanins, cyanidin 3-rutinoside and cyanidin 3-glucoside, exhibited an inhibitory effect on the migration and invasion of a human lung cancer cell line. Cancer Lett. 2006, 235, 248–259. [Google Scholar] [CrossRef]
- Horng, C.T.; Liu, Z.H.; Huang, Y.T.; Lee, H.J.; Wang, C.J. Extract from Mulberry (Morus australis) leaf decelerate acetaminophen induced hepatic inflammation involving downregulation of myeloid differentiation factor 88 (MyD88) signals. J. Food Drug Anal. 2017, 25, 862–871. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.X.; He, W.T.; Pan, L.F.; Feng, H.; Sun, J.L.; Zhang, B.; Yu, L.; Li, L.J. Downregulation of Akt2 attenuates ER stress-induced cytotoxicity through JNK-Wnt pathway in cardiomyocytes. Bioorg. Med. Chem. Lett. 2018, 28, 394–399. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Vu, N.B.; Nguyen, T.T.; Tran, L.C.D.; Do, C.D.; Nguyen, B.H.; Phan, N.K.; Van Pham, P. Doxorubicin and 5-fluorouracil resistant hepatic cancer cells demonstrate stem-like properties. Cytotechnology 2013, 65, 491–503. [Google Scholar] [CrossRef]
- Rasheva, V.I.; Domingos, P.M. Cellular responses to endoplasmic reticulum stress and apoptosis. Apoptosis 2009, 14, 996–1007. [Google Scholar] [CrossRef]
- Haze, K.; Yoshida, H.; Yanagi, H.; Yura, T.; Mori, K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 1999, 10, 3787–3799. [Google Scholar] [CrossRef]
- Tirasophon, W.; Welihinda, A.A.; Kaufman, R.J. A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev. 1998, 12, 1812–1824. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271. [Google Scholar] [CrossRef]
- Kim, B.; Kim, J.; Kim, Y.S. Celecoxib induces cell death on non-small cell lung cancer cells through endoplasmic reticulum stress. Anat. Cell Biol. 2017, 50, 293–300. [Google Scholar] [CrossRef]
- Xu, B.; Wang, Y.; Yang, J.; Zhang, Z.; Zhang, Y.; Du, H. Celecoxib induces apoptosis but up-regulates VEGF via endoplasmic reticulum stress in human colorectal cancer in vitro and in vivo. Cancer Chemother. Pharmacol. 2016, 77, 797–806. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Prescott, S.M. Many actions of cyclooxygenase-2 in cellular dynamics and in cancer. J. Cell Physiol. 2002, 190, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Adhami, V.M.; Subbarayan, M.; MacLennan, G.T.; Lewin, J.S.; Hafeli, U.O.; Fu, P.; Mukhtar, H. Suppression of prostate carcinogenesis by dietary supplementation of celecoxib in transgenic adenocarcinoma of the mouse prostate model. Cancer Res. 2004, 64, 3334–3343. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.S.; Watson, A.J.; Sheng, H.; Helou, R.; Shao, J.; DuBois, R.N. Celecoxib prevents tumor growth in vivo without toxicity to normal gut: Lack of correlation between in vitro and in vivo models. Cancer Res. 2000, 60, 6045–6051. [Google Scholar]
- Kim, D.S.; Kim, J.H.; Lee, G.H.; Kim, H.T.; Lim, J.M.; Chae, S.W.; Chae, H.J.; Kim, H.R. p38 Mitogen-activated protein kinase is involved in endoplasmic reticulum stress-induced cell death and autophagy in human gingival fibroblasts. Biol. Pharm. Bull. 2010, 33, 545–549. [Google Scholar] [CrossRef]
- Hamamura, K.; Goldring, M.B.; Yokota, H. Involvement of p38 MAPK in regulation of MMP13 mRNA in chondrocytes in response to surviving stress to endoplasmic reticulum. Arch. Oral Biol. 2009, 54, 279–286. [Google Scholar] [CrossRef]
- Feng, R.; Zhai, W.L.; Yang, H.Y.; Jin, H.; Zhang, Q.X. Induction of ER stress protects gastric cancer cells against apoptosis induced by cisplatin and doxorubicin through activation of p38 MAPK. Biochem. Biophys. Res. Commun. 2011, 406, 299–304. [Google Scholar] [CrossRef]
- Fan, L.; Song, B.; Sun, G.; Ma, T.; Zhong, F.; Wei, W. Endoplasmic reticulum stress–induced resistance to doxorubicin is reversed by paeonol treatment in human hepatocellular carcinoma cells. PLoS ONE 2013, 8, e62627. [Google Scholar] [CrossRef]
- Chan, K.C.; Ho, H.H.; Huang, C.N.; Lin, M.C.; Chen, H.M.; Wang, C.J. Mulberry leaf extract inhibits vascular smooth muscle cell migration involving a block of small GTPase and Akt/NF-κB signals. J. Agric. Food Chem. 2009, 57, 9147–9153. [Google Scholar] [CrossRef]
- Li, X.M.; Liu, J.; Pan, F.F.; Shi, D.D.; Wen, Z.G.; Yang, P.L. Quercetin and aconitine synergistically induces the human cervical carcinoma HeLa cell apoptosis via endoplasmic reticulum (ER) stress pathway. PLoS ONE 2018, 13, e0191062. [Google Scholar] [CrossRef]
- Gong, C.; Yang, Z.; Zhang, L.; Wang, Y.; Gong, W.; Liu, Y. Quercetin suppresses DNA double-strand break repair and enhances the radiosensitivity of human ovarian cancer cells via p53-dependent endoplasmic reticulum stress pathway. Onco Targets Ther. 2018, 11, 17. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.; Paul, S.; Jakhar, R.; Bhardwaj, M.; Han, J.; Kang, S.C. Novel quercetin derivative TEF induces ER stress and mitochondria-mediated apoptosis in human colon cancer HCT-116 cells. Biomed. Pharmacother. 2016, 84, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Iriti, M.; Kubina, R.; Cochis, A.; Sorrentino, R.; Varoni, E.M.; Kabała-Dzik, A.; Azzimonti, B.; Dziedzic, A.; Rimondini, L.; Wojtyczka, R.D. Rutin, a quercetin glycoside, restores chemosensitivity in human breast cancer cells. Phytother. Res. 2017, 31, 1529–1538. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.K.; Cho, S.G.; Choi, H.S.; Woo, S.M.; Yun, Y.J.; Shin, Y.C.; Ko, S.G. JNK1/2 activation by an extract from the roots of Morus alba L. reduces the viability of multidrug-resistant MCF-7/Dox cells by inhibiting YB-1-dependent MDR1 expression. Evid. Based Complement. Alternat. Med. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, M.-Y.; Wu, C.-H.; Hung, T.-W.; Wang, C.-J. Endoplasmic Reticulum Stress-Induced Resistance to Doxorubicin Is Reversed by Mulberry Leaf Polyphenol Extract in Hepatocellular Carcinoma through Inhibition of COX-2. Antioxidants 2020, 9, 26. https://doi.org/10.3390/antiox9010026
Yang M-Y, Wu C-H, Hung T-W, Wang C-J. Endoplasmic Reticulum Stress-Induced Resistance to Doxorubicin Is Reversed by Mulberry Leaf Polyphenol Extract in Hepatocellular Carcinoma through Inhibition of COX-2. Antioxidants. 2020; 9(1):26. https://doi.org/10.3390/antiox9010026
Chicago/Turabian StyleYang, Mon-Yuan, Cheng-Hsun Wu, Tung-Wei Hung, and Chau-Jong Wang. 2020. "Endoplasmic Reticulum Stress-Induced Resistance to Doxorubicin Is Reversed by Mulberry Leaf Polyphenol Extract in Hepatocellular Carcinoma through Inhibition of COX-2" Antioxidants 9, no. 1: 26. https://doi.org/10.3390/antiox9010026
APA StyleYang, M.-Y., Wu, C.-H., Hung, T.-W., & Wang, C.-J. (2020). Endoplasmic Reticulum Stress-Induced Resistance to Doxorubicin Is Reversed by Mulberry Leaf Polyphenol Extract in Hepatocellular Carcinoma through Inhibition of COX-2. Antioxidants, 9(1), 26. https://doi.org/10.3390/antiox9010026